Rare Co-occurrence of Beta-Thalassemia and Pseudoxanthoma elasticum: Novel Biomolecular Findings
 Federica Boraldi
Federica Boraldi Francesco Demetrio Lofaro
Francesco Demetrio Lofaro  Daniela Quaglino
Daniela Quaglino- Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy
A number of beta-thalassemia patients, independently from the type of beta-thalassemia (β0 or β+) and blood transfusion requirements, may develop, after puberty, dermal, cardiovascular, and ocular complications associated with an ectopic mineralization phenotype similar to that observed in another rare genetic disorder, namely, Pseudoxanthoma elasticum (PXE). To date, the causes of these alterations in beta-thalassemia patients are not known, but it has been suggested that they could be the consequence of oxidative stress-driven epigenetic regulatory mechanisms producing an ABCC6 down-regulation. Since, in the last years, several genes have been associated to the ectopic mineralization phenotype, this study, for the first time, applied, on beta-thalassemia patients with ectopic mineralization phenotype, a multigene testing strategy. Selection of genes to be analyzed was done on the basis of (i) their genetic involvement in calcification diseases or (ii) their role in calcium-phosphate equilibrium. Although, due to the rarity of these conditions, a limited number of patients was analyzed, the detection of pathogenic variants represents the proof of concept that PXE and beta-thalassemia traits co-occur on a genetic basis and that, in addition to causative mutations, functional polymorphisms may further influence connective tissue manifestations. The use of a multigene-based next-generation sequencing represents a useful time- and cost-effective approach, allowing to identify sequence variants that might improve prognostic assessment and better management of these patients, especially in the current era of precision medicine aiming to identify individual optimal care based on a unique personal profile.
Introduction
Beta-thalassemia (β-thal) (OMIM 613985) is a genetic disorder caused by mutations in the β-hemoglobin (HBB) gene that determines either a reduced (β+) or absent (β0) synthesis of the β-globin chains. In addition to hematological findings, according to the literature, up to 10% of β-thal patients (1) exhibit clinical manifestation similar to those observed in Pseudoxanthoma elasticum (PXE) (OMIM 264800), a rare genetic disease in which ABCC6 gene mutations cause the mineralization of elastic fibers responsible for skin, ocular, and cardiovascular complications that generally start after puberty and continuously progress with age (2).
A first study failed to detect ABCC6 gene mutations in 10 β-thal patients with soft connective tissue complications (3) and in the light of these data, the involvement, in these patients, of ABCC6 gene mutations in the pathogenesis of PXE-like alterations was ruled out. Therefore, PXE-like manifestations in β-thal patients have been hypothesized to be the consequence of oxidative stress-driven epigenetic regulatory mechanisms causing ABCC6 down-regulation (4). However, in the recent years, the expanding knowledge on the mechanisms controlling the mineralization process has revealed that, in addition to ABCC6, several other genes can be responsible for ectopic mineralization phenotypes (2), but, to our knowledge, a multigene approach has not been applied to patients in which beta-thalassemia is associated with PXE-like connective tissue manifestation.
The aim of this study was to define a multigene next-generation sequencing to identify pathogenic variants in three unrelated β-thal patients exhibiting soft connective tissue manifestations.
Materials and Methods
Patients
In the present study, three unrelated beta-thalassemia female patients with skin papules and/or skin laxity typical of PXE (Figure 1A) and by ocular angioid streaks (Figure 1B) have been investigated.
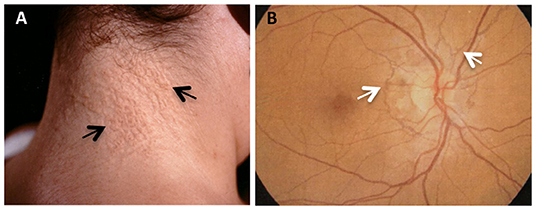
Figure 1. PXE clinical features in beta-thalassemia patients. (A) Skin papules (arrows) typically located on the neck; (B) fluoroangiography showing angioid streaks (arrows) as irregular lines spreading from the optic nerve to the retinal periphery.
The diagnosis of beta-thalassemia was based on the usual hematological criteria (peripheral blood evaluation and hemoglobin electrophoresis) and on the presence of mutations in HBB gene. Two patients (#1 and #3) were diagnosed with β-thalassemia Major and were carrier of a pathogenic variant in intron 1 (IVS1-110G>A in the homozygous state) and a stop codon mutation in exon 2 (c.C118T, p.Gln40* in the homozygous state), respectively. Both patients required blood transfusion and chelation therapy. The third patient (#2) was diagnosed with β-thalassemia and was carried of a pathogenic variant in intron 1 (IVS1-110G>A in the heterozygous condition). This patient never required blood transfusion.
Clinical laboratory tests (i.e., Ca, P, intact PTH, and ALPL) were within normal ranges.
The study was done, with informed consent, in accordance with the basic principles of the Declaration of Helsinki and approval by the local Ethical Committee (Comitato Etico Provinciale di Modena) (n. 358/17).
Targeted Exome Sequencing
DNA was extracted from whole blood using standard methods and quantified and quality tested using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Samples were subjected to target exome sequencing. A DNA custom panel was designed with AmpliSeq Custom DNA Panel for Illumina and sequenced on NextSeq500 (Illumina).
Raw reads were quality trimmed at both ends with ERNE-filter v1.4.3 (5) using default parameters and minimum read length of 40 bp.
Reads were aligned to the human reference genome (GRCh37/hg19 assembly) and only reads, mapping to unique positions, were retained. Variant discovery and functional annotation were performed by the software tool ANNOVAR (6).
Analyses were performed focusing on a panel of 19 genes, 12 of them are responsible for calcification-related diseases and 7 of them play a role in calcium-phosphate equilibrium (7–13) (Figures 2A,B and Table S1).
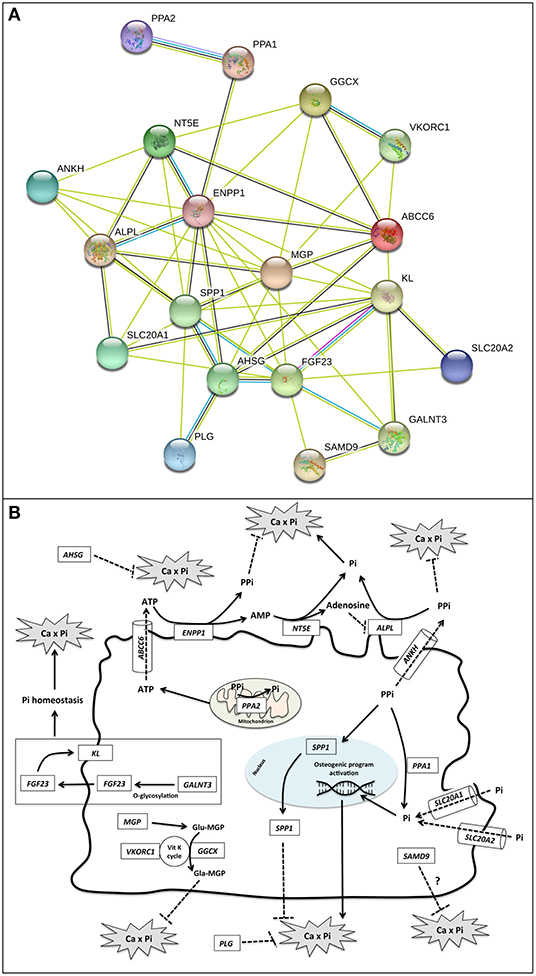
Figure 2. (A) Gene–protein interaction network. Gene relationships were obtained by STRING 11 (Search Tool for the Retrieval of Interacting Genes/Proteins) software. STRING parameters included: Active Prediction Methods: Textmining (yellow), Experiments (violet), Databases (light blue), and Co-expression (black); max number of interactors: none. ABCC6 = Multidrug resistance-associated protein 6; ALPL = Alkaline phosphatase, tissue-non-specific isozyme; AHSG, Alpha-2-HS-glycoprotein; ANKH, Progressive ankylosis protein homolog; ENPP1, Ectonucleotide pyrophosphatase/phosphodiesterase family member 1; FGF23, Fibroblast growth factor 23; GALNT3, Polypeptide N-acetylgalactosaminyltransferase 3; GGCX, Vitamin K-dependent gamma-carboxylase; KL, Klotho; MGP = Matrix Gla protein; NT5E, 5'-nucleotidase; PLG, Plasminogen; PPA1, Inorganic pyrophosphatase; PPA2, Inorganic pyrophosphatase 2; SAMD9, Sterile alpha motif domain-containing protein 9; SLC20A1, Sodium-dependent phosphate transporter 1; SLC20A2, Sodium-dependent phosphate transporter 2; SPP1, Osteopontin; VKORC1, Vitamin K epoxide reductase complex subunit 1. (B) Schematic representation of functional roles of calcification-related genes. Localization, substrate specificity, and/or transported molecules are reported for the investigated genes.
Filtering of Genetic Variants
Analyses were carried out primarily looking at coding and splice site regions excluding reference sequences and variants that do not change the translated amino acid sequence (i.e., synonymous). Thereafter, the frequency of each sequence variant was assessed querying databases as dbSNP138 build, 1,000 Genomes Project (1000g2012apr_all), and ESP6500si_all in order to discriminate common variants, with a minor allele frequency ≥1%, and rare sequence variants.
In Silico Analysis for Functional Prediction of Variants
For rare sequence variants not yet reported in the literature as pathogenic, the pathogenicity score was predicted by Sorting Intolerant From Tolerant (SIFT) (14), Polymorphism Phenotyping (Polyphen2) (15), and Panther (16), whereas the Genomic Evolutionary Rate Profiling (GERP++) (17) method and the Phylogenetic P-values (PhyloP) (18) were used to estimate the level of single-nucleotide variation conservation. To predict protein stability I-Mutant and MUpro were used (19).
Sanger Sequencing
All pathogenic variants were confirmed by conventional Sanger sequencing. Primer sequences were designed by NCBI Primer-BLAST software, and purified PCR products were directly sequenced using an Applied Biosystems 3100 DNA sequencer (20).
Results and Discussion
Unexpectedly, rare sequence variants were detected in ABCC6 and ENPP1 genes (Table 1 and Figure S1).
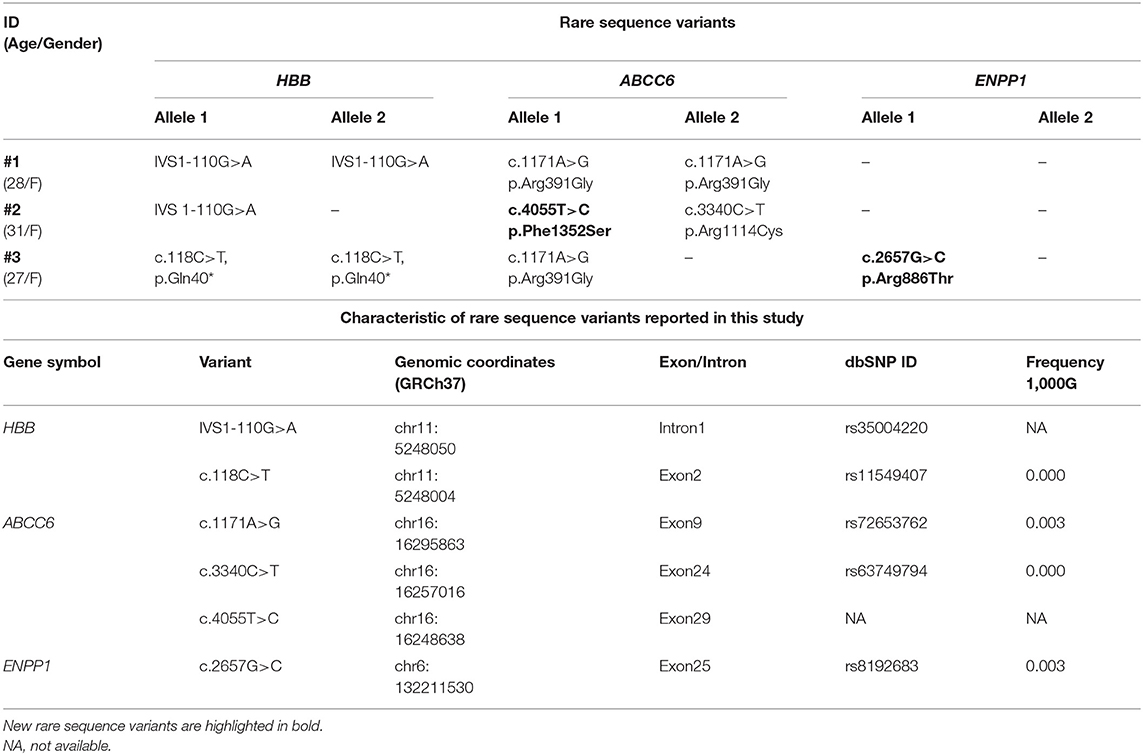
Table 1. Clinical/demographic data of patients (#1, #2, and #3) and rare sequence variants found in HBB, ABCC6, and ENPP1 genes.
ABCC6 is well-known as a PXE causing gene, encoding for a transmembrane protein (MRP6) probably involved in the extrusion of ATP (Figure 2B) (21).
ENPP1 encodes for an ectonucleotide-pyrophosphatase/phosphodiesterase-1 that utilizes adenosine triphosphate to release pyrophosphate (PPi) (2, 22), a potent inhibitor of ectopic calcification (Figure 2B).
Note that ABCC6 and ENPP1 have been both recently recognized as PXE causing genes (22).
Patient #1 was homozygous for ABCC6Arg391Gly (22), patient #2 was compound heterozygous for ABCC6Phe1352Ser and ABCC6Arg1114Cys (22), and patient #3 was heterozygous for ABCC6Arg391Gly and for ENPP1Arg886Thr (Table 1).
Of these variants, ABCC6Phe1352Ser and ENPP1Arg886Thr were never found in the literature, but, for both missense variants, the prediction scores implied a probably damaging effect (Table S2).
Present data underline the importance of ABCC6 and also provide evidence that, in one patient, an ABCC6 pathogenic allele is associated with a new rare sequence variant in the ENPP1 gene (c.2657G > C, p.Arg886Thr), where a substitution of the highly conserved arginine in position 886 with a threonine can affect the structural stability and the functional properties of the protein due to different polarity, charges, and size of these two amino acids. In the light of these data, a digenic inheritance of PXE manifestation could be suggested, as already shown for ABCC6 and GGCX (23).
Sequence variants were never detected in the following genes: AHSG, ANKH, GALNT3, KL, PLG, PPA1, SLC20A1, SLC20A2, and VKORC1. However, the absence of rare sequence variants and/or single-nucleotide polymorphisms (SNPs) in these genes does not exclude a priori their possible role in the development of ectopic mineralization phenotypes, and therefore, in perspective, studies on a larger cohort of patients would better elucidate this aspect.
SNPs were detected in other genes (Table 2 and Table S3).
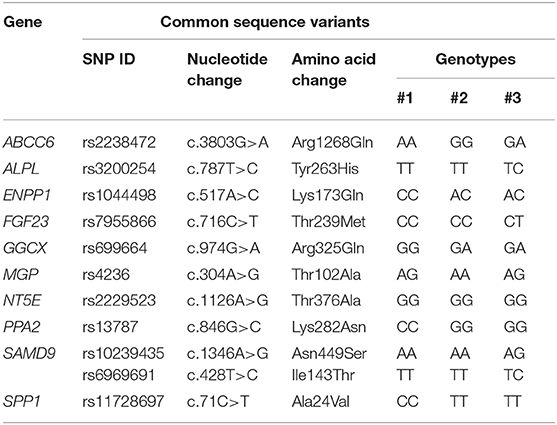
Table 2. Single-nucleotide polymorphisms (SNPs) found in at least one patient.
Interestingly, two of these polymorphisms have been reported in the literature to modulate the ectopic mineralization phenotype: rs1044498 in ENPP1 (24) and rs7955866 in FGF23 (25).
ENPP1Lys173Gln is a functional polymorphism found in all three patients in the homozygous (#1) or heterozygous state (#2 and #3). This polymorphism (c.517A>C; p.Lys173Gln) has been demonstrated to reduce enzyme activity, thus causing diminished calcification inhibitory properties of inorganic pyrophosphate (PPi) (24) and consequently predisposing to cardiovascular complications as coronary calcification and earlier onset of peripheral arterial disease.
Moreover, it could be suggested that rare sequence variants and/or functional polymorphisms in ENPP1 can act in synergy with epigenetic regulatory mechanisms. The oxidative stress condition, in fact, can down-regulate, by epigenetic mechanisms, the activity of ENPP1 (26) and this finding is of special interest in β-thal patients who are known to be characterized by an altered redox balance (27).
As far as FGF23Thr239Met, this SNP, found in patient #3 in the heterozygous condition, has been reported to influence FGF23 biological function, providing an increased stability of the protein, thus influencing phosphate homeostasis and favoring kidney stone formation (25).
Interestingly, the ABCC6Arg1268Gln sequence variant was detected in two patients, in the homozygous (#1) and in the heterozygous state (#3). Although considered in early studies to be pathogenic, given its frequency (19%), it has been later considered as a functional polymorphism contributing to the development and/or severity of angioid streaks (28, 29) and therefore cannot be disregarded in a prognostic assessment.
As far as rs699664 in GGCX, rs4236 in MGP, and rs2229523 in NT5E genes, data in the literature (30–32) indicate that SNPs observed in our patients determine either an enzyme activity similar to wildtype (GGCXArg325Gln and NT5EThr376Ala) or absent association with increased risk of calcification (MGPThr102Ala) and therefore their functional contribution to the ectopic mineralization phenotype can be likely irrelevant.
To our knowledge, other SNPs found in ALPL (rs3200254), PPA2 (rs13787), SAMD9 (rs10239435 and rs6969691), and SSP1 (rs11728697) genes have not been investigated for their possible functional role, and therefore, at present, pathogenic hypotheses cannot be put forward.
Evidence is provided that there are patients in which, independently from the type of beta-thalassemia (β0 or β+) and blood transfusion requirements, PXE clinical manifestations can occur on a genetic basis.
These data are partially in contrast to previous observations suggesting the absence of ABCC6 pathogenic variants in β-thal patients (3). This discrepancy could be due to (i) the continuously increasing number of recognized pathogenic variants (>350) since the discovery of ABCC6 as the causative PXE gene and (ii) the demonstrated pathogenic role of other genes in addition to ABCC6 (2, 22).
Conclusion
Considering the current longer life expectancy of β-thal patients, the development of complications that appear and progress with age should deserve higher consideration in both clinical and genetic evaluations. In the light of present data, searching for sequence variants in calcification-related genes also at the pre-symptomatic stage is recommended for a better management of β-thal patients and of their possible complications. Moreover, although this condition may affect a limited number of patients, genetic tests, given their increasing widespread distribution, can easily provide high-throughput and informative data to make a more accurate diagnostic and prognostic assessment, especially in the current era of precision medicine, distinguishing a given patient from other patients with similar clinical phenotype.
Data Availability Statement
All datasets for this study are included in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by Comitato Etico Provinciale di Modena. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
FB designed the study and wrote the manuscript. FL, SC, and PM collected samples and performed the research. FB and FL analyzed the results. DQ supervised the research and critically revised the manuscript. All authors approved the final submitted version of the manuscript.
Funding
This study was supported by grants from FCRMO2015.0306 (FB) and from PXE Italia Onlus E96C18000600007 (DQ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2019.00322/full#supplementary-material
References
1. Aessopos A, Savvides P, Stamatelos G, Rombos I, Tassiopoulos T, Karagiorga M, et al. Pseudoxanthoma elasticum-like skin lesions and angioid streaks in beta-thalassemia. Am J Hematol. (1992) 41:159–64. doi: 10.1002/ajh.2830410304
2. Li Q, Jiang Q, Uitto J. Ectopic mineralization disorders of the extracellular matrix of connective tissue: molecular genetics and pathomechanisms of aberrant calcification. Matrix Biol. (2014) 33:23–8. doi: 10.1016/j.matbio.2013.06.003
3. Hamlin N, Beck K, Bacchelli B, Cianciulli P, Pasquali-Ronchetti I, Le Saux O. Acquired Pseudoxanthoma elasticum-like syndrome in beta-thalassaemia patients. Br J Haematol. (2003) 122:852–4. doi: 10.1046/j.1365-2141.2003.04484.x
4. Le Saux O, Martin L, Aherrahrou Z, Leftheriotis G, Váradi A, Brampton CN. The molecular and physiological roles of ABCC6: more than meets the eye. Front Genet. (2012) 3:289. doi: 10.3389/fgene.2012.00289
5. Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS ONE. (2013) 8:e85024. doi: 10.1371/journal.pone.0085024
6. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
7. Jahnen-Dechent W, Schinke T, Trindl A, Müller-Esterl W, Sablitzky F, Kaiser S, et al. Cloning and targeted deletion of the mouse fetuin gene. J Biol Chem. (1997) 272:31496–503. doi: 10.1074/jbc.272.50.31496
8. Mignemi NA, Yuasa M, Baker CE, Moore SN, Ihejirika RC, Oelsner WK, et al. Plasmin prevents dystrophic calcification after muscle injury. J Bone Miner Res. (2017) 32:294–308. doi: 10.1002/jbmr.2973
9. Polewski MD, Johnson KA, Foster M, Millán JL, Terkeltaub R. Inorganic pyrophosphatase induces type I collagen in osteoblasts. Bone. (2010) 46:81–90. doi: 10.1016/j.bone.2009.08.055
10. Li X, Yang H-Y, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. (2006) 98:905–12. doi: 10.1161/01.RES.0000216409.20863.e7
11. Crouthamel MH, Lau WL, Leaf EM, Chavkin NW, Wallingford MC, Peterson DF, et al. Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: redundant roles for PiT-1 and PiT-2. Arterioscler Thromb Vasc Biol. (2013) 33:2625–32. doi: 10.1161/ATVBAHA.113.302249
12. Hunter GK. Role of osteopontin in modulation of hydroxyapatite formation. Calcif Tissue Int. (2013) 93:348–54. doi: 10.1007/s00223-013-9698-6
13. Boraldi F, Annovi G, Bartolomeo A, Quaglino D. Fibroblasts from patients affected by Pseudoxanthoma elasticum exhibit an altered PPi metabolism and are more responsive to pro-calcifying stimuli. J Dermatol Sci. (2014) 74:72–80. doi: 10.1016/j.jdermsci.2013.12.008
14. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
15. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
16. Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. (2019) 47:D419–26. doi: 10.1093/nar/gky1038
17. Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. (2005) 15:901–13. doi: 10.1101/gr.3577405
18. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. (2010) 20:110–21. doi: 10.1101/gr.097857.109
19. Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. (2006) 62:1125–32. doi: 10.1002/prot.20810
20. Boraldi F, Costa S, Rabacchi C, Ciani M, Vanakker O, Quaglino D. Can APOE and MTHFR polymorphisms have an influence on the severity of cardiovascular manifestations in Italian Pseudoxanthoma elasticum affected patients? Mol Genet Metab Rep. (2014) 1:477–82. doi: 10.1016/j.ymgmr.2014.11.002
21. Jansen RS, Küçükosmanoglu A, de Haas M, Sapthu S, Otero JA, Hegman IEM, et al. ABCC6 prevents ectopic mineralization seen in Pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci USA. (2013) 110:20206–11. doi: 10.1073/pnas.1319582110
22. Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, et al. Generalized arterial calcification of infancy and Pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. (2012) 90:25–39. doi: 10.1016/j.ajhg.2011.11.020
23. Li Q, Grange DK, Armstrong NL, Whelan AJ, Hurley MY, Rishavy MA, et al. Mutations in the GGCX and ABCC6 genes in a family with Pseudoxanthoma elasticum-like phenotypes. J Invest Dermatol. (2009) 129:553–63. doi: 10.1038/jid.2008.271
24. Stella J, Buers I, van de Wetering K, Höhne W, Rutsch F, Nitschke Y. Effects of different variants in the ENPP1 gene on the functional properties of ectonucleotide pyrophosphatase/phosphodiesterase family member 1. Hum Mutat. (2016) 37:1190–201. doi: 10.1002/humu.23057
25. Rendina D, Esposito T, Mossetti G, De Filippo G, Gianfrancesco F, Perfetti A, et al. A functional allelic variant of the FGF23 gene is associated with renal phosphate leak in calcium nephrolithiasis. J Clin Endocrinol Metab. (2012) 97:E840–4. doi: 10.1210/jc.2011-1528
26. Mori D, Matsui I, Shimomura A, Hashimoto N, Matsumoto A, Shimada K, et al. Protein carbamylation exacerbates vascular calcification. Kidney Int. (2018) 94:72–90. doi: 10.1016/j.kint.2018.01.033
27. Boraldi F, Garcia-Fernandez M, Paolinelli-Devincenzi C, Annovi G, Schurgers L, Vermeer C, et al. Ectopic calcification in β-thalassemia patients is associated with increased oxidative stress and lower MGP carboxylation. Biochim Biophys Acta. (2013) 1832:2077–84. doi: 10.1016/j.bbadis.2013.07.017
28. Germain DP, Perdu J, Remones V, Jeunemaitre X. Homozygosity for the R1268Q mutation in MRP6, the Pseudoxanthoma elasticum gene, is not disease-causing. Biochem Biophys Res Commun. (2000) 274:297–301. doi: 10.1006/bbrc.2000.3101
29. Li Q, Sadowski S, Uitto J. Angioid streaks in Pseudoxanthoma elasticum: role of the p.R1268Q mutation in the ABCC6 gene. J Invest Dermatol. (2011) 131:782–5. doi: 10.1038/jid.2010.384
30. Tie J-K, Carneiro JDA, Jin D-Y, Martinhago CD, Vermeer C, Stafford DW. Characterization of vitamin K-dependent carboxylase mutations that cause bleeding and nonbleeding disorders. Blood. (2016) 127:1847–55. doi: 10.1182/blood-2015-10-677633
31. Sheng K, Zhang P, Lin W, Cheng J, Li J, Chen J. Association of Matrix Gla protein gene (rs1800801, rs1800802, rs4236) polymorphism with vascular calcification and atherosclerotic disease: a meta-analysis. Sci Rep. (2017) 7:8713. doi: 10.1038/s41598-017-09328-5
Keywords: genetic testing, beta-thalassemia, PXE, ABCC6, ENPP1
Citation: Boraldi F, Lofaro FD, Costa S, Moscarelli P and Quaglino D (2020) Rare Co-occurrence of Beta-Thalassemia and Pseudoxanthoma elasticum: Novel Biomolecular Findings. Front. Med. 6:322. doi: 10.3389/fmed.2019.00322
Received: 23 October 2019; Accepted: 16 December 2019;
Published: 23 January 2020.
Edited by:
John Joseph Strouse, Duke University, United StatesReviewed by:
Ciprian Tomuleasa, Iuliu Haţieganu University of Medicine and Pharmacy, RomaniaLuisa Anelli, University of Bari Aldo Moro, Italy
Copyright © 2020 Boraldi, Lofaro, Costa, Moscarelli and Quaglino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Quaglino, daniela.quaglino@unimore.it