Abstract
Peptide therapeutics have recently gained momentum in antiviral therapy due to their increased potency and cost-effectiveness. Interaction of the HIV-1 envelope gp120 with the host CD4 receptor is a critical step for viral entry, and therefore the CD4-binding site (CD4bs) of gp120 is a potential hotspot for blocking HIV-1 infection. The present study aimed to design short peptides from well-characterized CD4bs targeting broadly neutralizing antibodies (bNAbs), which could be utilized as bNAb mimetics for viral neutralization. Co-crystallized structures of HIV-1 gp120 in complex with CD4bs-directed bNAbs were used to derive hexameric peptides using the Rosetta Peptiderive protocol. Based on empirical insights into co-crystallized structures, peptides derived from the heavy chain alone were considered. The peptides were docked with both HIV-1 subtype B and C gp120, and the stability of the peptide–antigen complexes was validated using extensive Molecular Dynamics (MD) simulations. Two peptides identified in the study demonstrated stable intermolecular interactions with SER365, GLY366, and GLY367 of the PHE43 cavity in the CD4 binding pocket, and with ASP368 of HIV-1 gp120, thereby mimicking the natural interaction between ASP368gp120 and ARG59CD4–RECEPTOR. Furthermore, the peptides featured favorable physico-chemical properties for virus neutralization suggesting that these peptides may be highly promising bNAb mimetic candidates that may be taken up for experimental validation.
Graphical flow chart of methodology. 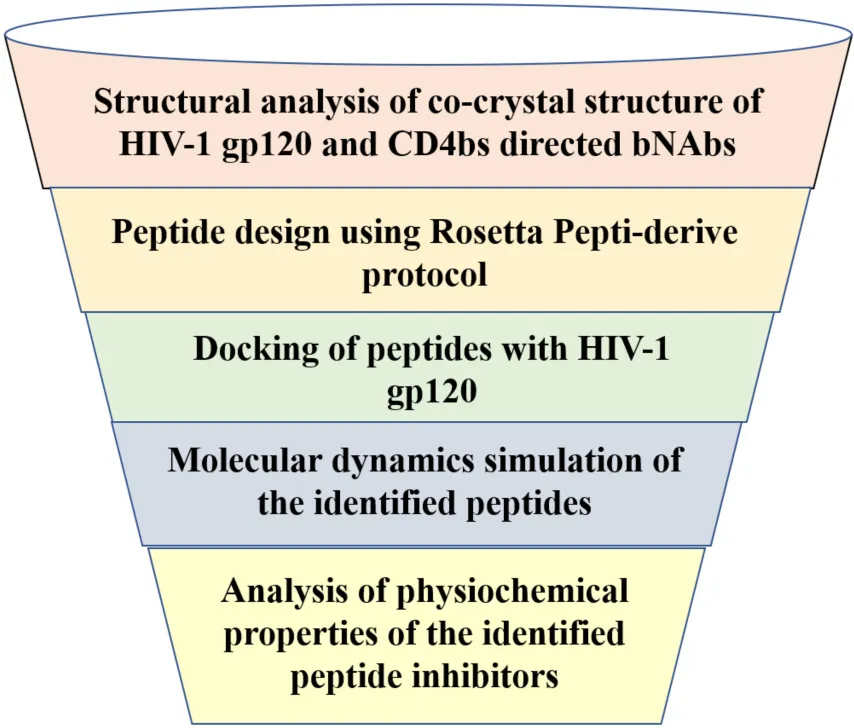
Introduction
Human Immuno-deficiency Virus (HIV), the causative agent of Acquired Immuno-Deficiency Syndrome (AIDS), continues to be a tenacious global public health challenge. According to the UNAIDS 2021 report, there were 37.7 million people living with HIV (PLHIV), of which 27.5 million people were on Anti-Retroviral Treatment (ART) and 1.5 million people were newly infected with HIV in 20201. Though 40 years have passed since the discovery of HIV, a preventive vaccine against HIV continues to be a dream of the future (1, 2). However, the introduction of combinatorial Anti-Retroviral Therapy (cART)/Highly Active ART (HAART) has revolutionized the treatment of HIV infection and contributed significantly to viral suppression in infected individuals and control of transmission (3, 4). However, the emergence of drug resistance and the establishment of long-lived latent reservoirs remain major obstacles to the cure of HIV infection and elimination of the disease (5, 6).
In recent years, broadly neutralizing antibodies (bNAbs) that can neutralize diverse HIV-1 strains by targeting vulnerable epitopes on the HIV-1 envelope and thereby block HIV-1 infection have gained attention as potential adjuncts to antiretroviral therapy (7, 8). Recent studies have demonstrated that the administration of bNAbs is effective in suppressing viremia (9) and protecting against lentiviral infection in animal models (10, 11), thus providing valuable insights for the design of effective HIV-1 vaccines (12, 13). Very recently, researchers have directed their attention towards the development of therapeutic proteins and peptides targeting HIV, due to their advantages such as specificity and selective nature of action as compared to drugs and antibodies (14–16). Enfuvirtide (also known as Fuzeon or T20), an FDA-approved peptide-based drug, prevents the completion of HIV fusion events and has been used in combination with other anti-retroviral drugs for treating HIV infection (17). However, the drug has limited clinical application due to the emergence of resistant HIV-1 strains (12, 18).
Selective interaction of the HIV-1 envelope glycoprotein (gp120) with the CD4 molecule which serves as the primary cellular receptor, and one of the chemokine receptors CCR5/CXCR4 or both, constitutes a crucial step in HIV-1 infection (19–21). Regardless of the genomic and antigenic variation between HIV-1 strains, the CD4 binding site (CD4bs) is known to be well-conserved among the different HIV-1 subtypes and is reported to be one of the potential targets of neutralizing antibodies (22–24). The CD4bs is centered in a cavity formed at the interface of the gp120 outer and inner domains, where the hydrophobic residues present in the deep pocket constitute the point of contact with Phe-43 of the CD4 receptor (also called the Phe43 cavity) (25, 26). In addition, Arg59 of the CD4 receptor forms a salt bridge with D368 of gp120 to stabilize the CD4 binding site interaction (27, 28).
As early as 1999, Vita et al. reported that oligo-peptides targeting the CD4bs could inhibit the binding of gp120 with the CD4 receptor and thereby prevent HIV infection (29). The present study is based on the hypothesis that short peptides derived from the paratope of broadly neutralizing antibodies might function as potent mimics of these antibodies. This is based on earlier reports that ultra-short peptides of size up to seven amino acids have several useful features including biocompatibility, tunability, non-immunogenicity, biodegradability, and most importantly, efficient survival against proteolytic degradation in the gastrointestinal tract, as compared to longer peptides (30). We chose ultra-short peptides of 6-amino acids length (hexamers) for our study. Taking advantage of the available HIV-1 gp120-neutralizing antibody crystal structure complexes, we made an attempt to identify hexameric peptides from the paratope of neutralizing antibodies and characterized them using in silico methods like Molecular modeling, interacting interface analysis, and Molecular Dynamic (MD) simulation to understand their usefulness as therapeutic tools for HIV.
Materials and methods
Selection of co-crystal structures of broadly neutralizing antibody with HIV-1 envelope gp120
A number of CD4bs-directed neutralizing antibodies have been identified and reported. Based on their mode of recognition and B-cell ontogeny, CD4bs antibodies fall into two categories: VH-gene restricted antibodies derived from the heavy chain germline genes VH1-2 or VH1-46, and CDRH3 dominated antibodies in which the antibody binding interfaces are dominated by the complementary-determining region three (CDR3) (13, 31, 32). The CD4bs directed bNAbs used for this study included VRC01 and 8ANC131, considered to be the first identified members of the VH-gene restricted “VRC01-class and 8ANC131-class” bNAbs (33) since the co-crystal structures of these antibodies with HIV-1B and C envelopes were available. VRC01 (VH1-2) and 8ANC131 (VH1-46) are both potent bNAbs found to be capable of neutralizing about 91 and 78% of the HIV-1 strains, respectively (34). The co-crystal structures of 8ANC131 with the HIV-1 subtype B envelope YU-2 gp120 (PDB ID: 4RWY 2.13 Å resolution) and VRC01 with the HIV-1 subtype C envelope ZM176.66 gp120 (PDB ID: 4LST 2.55 Å resolution) were downloaded from PDB (Protein Data Bank) (Figure 1). Both co-crystal structures included the heavy and light chains of the respective antibodies complexed with HIV-1 envelope gp120. The Fab (Fragment antigen-binding) regions of the antibodies were bound to the CD4bs in HIV-1 gp120.
FIGURE 1
Design of short linear peptides targeting the CD4-binding site
The Rosetta Peptiderive is a computational tool designed to predict possible inhibitory peptides from the crystal structures of protein complexes based on their interacting interface, was used to identify short linear peptides that would target the CD4bs and bring about virus neutralization. This tool is hosted online in ROSIE (Rosetta Online Server that Includes Everyone) web interface and can be accessed at https://rosie.rosettacommons.org/peptiderive. The antigen (HIV-1 gp120)–antibody (bNAb) complex was uploaded on the Rosetta peptiderive tool in PDB format, with optimal parameters defining the Receptor and Partner. The tool automatically refines the antigen–antibody complex by removing local clashes and extracts potential peptide fragments of specified window size. The binding energies of the identified peptide–antigen complexes were calculated using the Rosetta energy function (35). Peptides with the most significant binding scores were shortlisted, and their position, sequence, interface score and relative interface score were obtained (36). Intermolecular interactions of the identified peptide-antigen complexes were visualized in the PDBsum webserver (37) and CHIMERA (38).
Docking of peptides with human immunodeficiency virus-1 gp120
To validate the binding of the identified peptides with HIV-1 gp120, peptide–antigen docking was performed using HADDOCK (High Ambiguity Driven protein-protein DOCKing) webserver (Version 2.2) in the EASY interface available at https://wenmr.science.uu.nl/haddock2.4/. The antigen and peptides were docked by generating Ambiguous Interaction Restraints (AIR) with the interface residues identified from the PDBsum analysis of the Rosetta peptiderive complexes (39, 40). The docked structures were summarized in clusters, and each cluster was assigned a HADDOCK score, cluster size, RMSD from the overall lowest energy conformations, Z-score and buried surface area along with bonding energies (Vander Waal’s, electrostatic, desolvation, and restraints violation energies). The best-docked complex (topmost cluster suggested by HADDOCK) replicating the desired residual interactions was identified and selected for further analysis. The binding affinity (ΔG) and dissociation constant (Kd) of the docked complexes were calculated using the PRODIGY webserver, available at https://wenmr.science.uu.nl/prodigy/. This webserver predicts binding affinities based on inter-molecular contacts within a distance cut-off of 5.5 Å (41, 42).
Molecular dynamics simulations of the peptides with human immunodeficiency virus-1 envelopes
The peptide-HIV-1 envelope complexes identified using Rosetta peptiderive were subjected to Molecular dynamics (MD) simulations to deduce their dynamic behavior under physiologically simulated conditions (43). MD simulations were performed using the DESMOND software package (44) with OPLS_2005 as a force field and implemented as in Muthukumaran et al. (45). To begin with, the system was built in an auto-calculated cubic box and solvated with explicit Single Point Charge (SPC) water molecules. The solvated system was energy minimized and the MD run was carried out for 200 ns by implementing an NPT ensemble with a sampling interval of 10 ps. During the MD run, the whole system was maintained at an equilibrium of 300 K temperature and 1 atm pressure. Analytical tools available in DESMOND were used to infer the Root Mean Square Deviation (RMSD) of the protein backbone, the Root Mean Square Fluctuation (RMSF) of the residues, the radius of gyration, and other structural transitions throughout the simulations.
Molecular mechanics-poisson boltzmann surface area calculation for the top-scoring stable neutralizing peptide-antigen complexes
The binding free energy (ΔG) of the final frames of stable neutralizing peptide–antigen complexes obtained from the MD simulation was calculated by implementing MM-PBSA (Molecular Mechanics-Poisson Boltzmann Surface Area) protocol in farPPI (fast amber rescoring for Protein–Protein interaction Inhibitors) webserver, available at http://cadd.zju.edu.cn/farppi/. Precise binding energies of the docked poses were evaluated by the MM-PBSA method which combines energy calculations based on implicit solvent and molecular mechanics model (46). Among the MM-PBSA procedures, PB3 based approach was found to be highly accurate as compared to the other approaches in farPPI, as two force fields, GAFF2 and ff14SB, were applied to the peptide and antigen, respectively (47, 48). Hence, this method was adopted to score the binding free energy of the peptide–antigen complexes.
KDeep absolute binding affinity calculation for the most stable neutralizing peptide-antigen complexes
In addition to MM-PBSA, absolute binding affinity (ΔG) of the topmost neutralizing peptide–antigen complexes was calculated using KDeep, a protein–ligand affinity predictor tool available at https://playmolecule.com/Kdeep/. This predictor works based on a machine learning approach using a state-of-the-art 3D convolutional neural network (49). The input was voxelized into pharmacophore features like aromaticity, hydrophobicity, total excluded volume, etc., and passed onto the DCNN (Deep Convolutional Neural Network) model, which is pre-trained by the PDBbind benchmark (v.2006). Based on the implemented algorithm, the binding affinity of the identified neutralizing peptide–antigen complexes was calculated as discussed by Karlov et al. (50) and Varela-rial et al. (51).
Additional computational predictions
The identified peptides were subjected to alanine scanning using Bude Alanine Scan2 (52, 53) and Robetta Alanine scan3 (54) webservers to infer the energetically significant amino acids at the peptide–antigen interface. This prediction helps to prioritize key residues in the identified peptides. Toxicity and physico-chemical properties of the peptides were predicted using ToxinPred4 (55) and the peptide analyzing tool provided by Thermo-fisher Scientific5.
Results
Neutralizing peptides derived from the CD4-binding site-directed neutralizing antibodies
Four hexameric peptides were derived through structure-based sequence inference from the 8ANC131 and VRC01 neutralizing antibody-HIV-1 gp120 complexes using the Rosetta peptiderive protocol as shown in Figures 2, 3. From the hot segments in the bNAbs (that contribute to the most significant binding interaction with the HIV-1 envelope gp120 protein), two peptides were identified from each of the two antigen–antibody complexes. These included the peptide Arg-Asp-Arg-Ser-Thr-Gly (RDRSTG) from the H chain of 8ANC131, which had an interface score of –9.447 and contributed to 29% of binding energy, and the peptide Glu-Tyr-Ser-Ser-Thr-Pro (EYSSTP) from the L chain, which had an interface score of –4.554 and contributed to 101% of binding energy. Two other hexamers were derived from the PDB crystal structure of VRC01-HIV-1C envelope, namely, Val-Asn-Tyr-Ala-Arg-Pro (VNYARP) from the H chain, which had an interface score of –9.982 and contributed to 30% of binding energy, and QQYEFF (Gln-Gln-Tyr-Glu-Phe-Phe) from the L chain, which had an interface score of –6.839 and contributed to 68% of binding energy (Table 1). In general, the peptides derived from the heavy chain of the antibodies gave comparatively lower interface scores than peptides derived from the light chain, signifying better binding affinity of the former. Among the four peptides, RDRSTG peptide having an interface score of –9.447 showed the most significant binding to the HIV-1 envelope.
FIGURE 2
FIGURE 3
TABLE 1
| PDB ID (Co-crystal structure) | Peptide sequence | Receptor (Envelope gp120) | Antibody chain (H-Heavy/ L-Light) | Position in neutralizing antibody (Crystal structure) | Interface score | Total interface score (REU) | Relative interface score (%) |
| 4RWY (8ANC131- subtype B gp120) | RDRSTG | A | H | 71–76 | –9.447 | –33.11 | 28.54 |
| EYSSTP | A | L | 90–95 | –4.554 | –4.49 | 101.32 | |
| 4LST (VRC01- subtype C gp120) | VNYARP | G | H | 57–62 | –9.982 | –33.57 | 29.73 |
| QQYEFF | G | L | 89–91, 96–98 | –6.839 | –10.03 | 68.23 |
Peptides derived from neutralizing antibodies and their interface scores.
*Highlighted peptides contribute significantly to binding with the respective antigen.
Molecular docking of peptides with antigens
Structural analysis of the 8ANC131-subtype B gp120 (PDB ID: 4RWY) and VRC01-subtype C gp120 (PDB ID: 4LST) complexes revealed close interaction between the antibody Heavy chains and the HIV-1 gp120 CD4-binding site, while the light chains protruded beyond the CD4bs, particularly the D Loop and V5 regions (Figure 4). Therefore, we excluded the peptides derived from the light chains as they did not engage our target, i.e., the CD4bs. Residues 365–371 of HIV-1 gp120 were found to be the key residues involved in making critical contacts with Phe43 and Arg59 residues of the CD4 receptor (56). The VRC01 antibody showed a non-bonded interaction with Ser365gp120, Gly366gp120, and Gly367gp120 of the Phe43 cavity, while in 8ANC131, Gly366gp120 and Gly367gp120 were found to be involved in the interaction (57). Furthermore, ASP368gp120 was observed to mediate the interaction with ARG718ANC131/VRC01 by forming hydrogen bonds and salt bridges, which mimicked the natural interaction between ARG59CD4 RECEPTOR and ASP368gp120 (25, 31, 34) (Figure 5).
FIGURE 4
FIGURE 5
We also performed intermolecular interaction analysis of the Rosetta-derived peptide-antigen complexes and observed similar interactions as seen in the PDB crystal structures (Supplementary Figure 1). Among the peptides derived from the antibody heavy chains, RDRSTG was found to form two hydrogen bonds with ASP368 (2.75 Å and 2.82 Å) and MET426 (2.72 Å and 3.20 Å), and one hydrogen bond with GLY431 (2.76 Å); the other crystal structure residues were found to have non-bonded contacts in the vicinity of <5 Å (LEU122, VAL430, TRP427 and LYS432). The VNYARP peptide formed two hydrogen bonds with GLY458 (3.08 Å and 3.12 Å) and one hydrogen bond with ARG456 (2.73 Å) and THR467 (3.00 Å). In addition, salt bridges were also observed at ASP461 and GLU466. Other non-bonded contacting residues were ASN280, THR465, GLY366, SER365, ASN460 and ASP457. Based on these observations, we docked the neutralizing antibody 8ANC131-derived peptide RDRSTG with subtype C (ZM176.66) gp120, and the VRC01-derived peptides VNYARP with subtype B (YU-2) gp120, to examine the closeness of the interaction patterns (especially ASP368 and SER365) with that seen in the native crystal structures. To revalidate the observed interactions in the Rosetta derived complexes, we performed re-docking of the peptide RDRSTG with subtype B (YU-2) gp120 and VNYARP with subtype C (ZM176.66) gp120. (Positions of residues are different in PDB crystal structures and 2D LIGPLOT—Supplementary Table 1; Residues stated here are in accordance with the crystal structure but different from that in LIGPLOT).
Docking with subtype B gp120
The RDRSTG peptide derived from 8ANC131 was found to form hydrogen bonds with ASP368 (2.96 Å, 2.62 Å), TRP427 (2.81 Å, 2.97 Å), GLY198, GLU370, ASN425, MET426, GLU429 and LYS432, with a binding affinity (ΔG) of –9.1 kCal/mol and Kd of 2.2E-07. The peptide VNYARP derived from VRC01 showed hydrogen bond interactions with ASP368 (2.68 Å) and GLY431 (2.86 Å) with a binding affinity (ΔG) of –8.6 kCal/mol and Kd of 5.0E-07 (Supplementary Figure 2).
Docking with subtype C gp120
The RDRSTG peptide featured interactions at positions SER365 (2.75 Å and 2.69 Å), GLY366, ASP457 (2.78 Å and 2.67 Å), GLY458 and ASN460, with a binding affinity (ΔG) of –8.8 kCal/mol and Kd of 3.4E-07. In the case of VNYARP-subtype C gp120 re-docking, hydrogen bond interactions were observed at ASN280 (2.87 Å and 3.10 Å), LYS360, HIS364, ASP457, ASP461 (2.65 Å, 3.23 Å), THR465, GLU466, THR467 and ARG469, thus concurring with the Rosetta peptiderive prediction. However, the main residue SER365 was noticed to form a non-bonded contact with a binding affinity (ΔG) of –9.9 kCal/mol and Kd of 5.9E-08 (Supplementary Figure 3). The redocking study demonstrated the predictive accuracy of the methods implemented.
Molecular dynamics simulation analysis of the peptide-antigen complexes
To start with, the HIV-1 subtype B and subtype C gp120 antigens (without peptides) were subjected to a production run of 200 ns, and trajectory analysis was performed. The system of subtype B gp120 antigen comprised of 49,311 atoms with 14,688 water molecules in the neutralized state, while the subtype C gp120 antigen system comprised of 48,478 atoms with 14,405 water molecules in the neutralized state. The RMSD plot of both antigens revealed that the Cα deviations were stable and within the range of 3 Å, and were found to converge toward the final stages of simulation (Supplementary Figure 4). The RMSF plot identified the peaks which represent the regions/residues that fluctuated the most during the simulation: 175–200 (4.7 Å) and 225–250 (5.0 Å) regions in subtype B gp120, and 250–275 (4.0 Å) and 300–337 (4.2 Å) regions in subtype C gp120 (Supplementary Figure 5).
We then performed molecular dynamics simulation of the peptide–gp120 complexes. The simulation system of subtype B gp120-RDRSTG solvated complex comprised of 49,305 atoms with 14,654 water molecules, and was neutralized by adding one cl– ion (1.241 mM). On trajectory analysis, the protein–ligand RMSD plot revealed that the complex converged at 10 ns with a 0.6 Å difference between the peptide and antigen-bound state (Figure 6). The ligand RMSD value was in the range of 3.0 Å with reference to the backbone of the antigen and was found to be well-bound to the binding regions. The RMSF plot revealed that RDRSTG (Supplementary Figure 6) interacted well at regions 50–100, 220–250, 250–300 and 300–337, despite fluctuations. Fluctuations posed by the peptide throughout the simulation were inferred from the ligand RMSF plot (Supplementary Figure 7), where it was found to be stable in the range of 4 Å. The structural compactness of the peptide was measured based on the radius of gyration (rGyr). This analysis revealed that the peptide RDRSTG maintained its compactness up to 150 ns in the range of 1 Å (Supplementary Figure 8). The bonded interactions between the antigenic residues and the RDRSTG peptide were analyzed from the ligand–protein contacts plot (Figure 7), wherein it was found that for about 79 and 52% of the duration of the run, Asp229 (ASP368) and Glu274 (GLU429) interacted by means of hydrogen bonds, ionic bonds and water bridges, respectively (Supplementary Figure 10).
FIGURE 6
FIGURE 7
Subtype C gp120-VNYARP peptide was made up of 48,414 atoms with 14,350 water molecules in the neutralized state. The antigen–peptide RMSD plot inferred that the complex converged at 75 ns with a 0.6 Å difference between the peptide and antigen-bound states (Figure 6). However, the peptide VNYARP evolved to make stable interactions between 85 and 160 ns in the vicinity of <3 Å. The ligand RMSD value was in the range of 2.0 Å with a major fluctuation at 75 ns. Residues in the region 150–180, 200–250, and 300–339 were found to sustain bonded interactions with the peptide as per the RMSF plot (Supplementary Figure 6). The ligand RMSF plot inferred that the peptide is stable as the fluctuations were within the range of 4 Å (Supplementary Figure 7). The rGyr analysis revealed a minimum deviation of 6.0–6.5 Å, indicating that the peptide sustained high compactness during the entire simulation process (Supplementary Figure 9). With regard to peptide–antigen contacts (Figure 8), Gly305 (GLY458) was found to interact 96% of the time during the entire run by means of hydrogen bonds and water bridges. Asp304 (ASP457—70%), Asp227 (ASP368—67%), Gly226 (GLY367—62%) and Ser224 (SER365—62%) formed hydrogen bond interactions and water bridges, with the exception of Asp227 (ASP368), where an additional ionic interaction featured. The least interacting residue was Arg303 (ARG456), which revealed sustained binding (hydrogen bonds and water bridges) around 53% of the 200 ns production run (Supplementary Figure 11).
FIGURE 8
The MD trajectories revealed RDRDTG and VNYARP peptides to be highly stable in terms of bonded interactions during the 200 ns of simulation. The dynamic evolution of the peptides RDRSTG and VNYARP are illustrated in Figures 9, 10. The MD trajectory analyses revealed that the peptides RDRSTG and VNYARP were stable binders, as they feature stable contacts with the key residues namely, SER365, GLY366, GLY367, and ASP368 across the production run (Supplementary Figure 12). The binding free energies (ΔG) of the complexes (RDRSTG-subtype B gp120 and VNYARP-subtype C gp120) were calculated over the MD simulation trajectory for the frames sampled at an interval of 20 ns and subjected to MM-PBSA (PB3) using the far-ppi server and binding affinity calculation using Kdeep, respectively. MM-PBSA calculations of RDRSTG-subtype B gp120 and VNYARP-subtype C gp120 complexes gave an average of –13.58 ± 2.85 (Mean ± SD) kCal/mol and –16.04 ± 8.77 (Mean ± SD) kCal/mol, respectively. Similarly, KDeep calculations gave an average of –9.32 ± 0.80 (Mean ± SD) kCal/mol for RDRSTG-subtype B gp120 and –10.18 ± 0.63 (Mean ± SD) kCal/mol for VNYARP-subtype C gp120, respectively (Supplementary Figure 17).
FIGURE 9
FIGURE 10
The Alanine scan analysis for RDRSTG-Subtype B gp120 and VNYARP-Subtype C gp120 complexes using Robetta and Bude scan identified the cumulative energetically important amino acids in the peptides across the binding interface as R, D, R, S and T in the RDRSTG peptide and V, N, Y and R in the VNYARP peptide. The binding affinities of the alanine mutated peptides are provided in Supplementary Table 2. The results of the physico-chemical analysis are provided in Table 2. Further, the peptides were found to be non-toxic.
TABLE 2
| Peptide | Alanine scan | ToxinPred | Hydrophobicity | Charge | GRAVY | MW Avg. g/mol | MW Mono-isotopic | Theoretical PI | |
| Bude | Robetta | ||||||||
| RDRSTG |  |  | Non-toxic | 2.66 | +1 | –2.40 | 690.7193 | 690.3409 | 10.9 |
| VNYARP |  |  | Non-toxic | 6.63 | +1 | –0.82 | 718.8183 | 718.3763 | 9.9 |
Predicted physico-chemical properties of the neutralizing antibody heavy chain derived peptides.
*Highlighted letters indicate hot spot residues in the alanine scan. MW, Molecular weight.
Discussion
The CD4-binding site of the HIV-1 envelope has been a key target of therapeutics for many years. However, not a single drug targeting the CD4bs has been approved by the US FDA to date (58). With the discovery of broadly neutralizing antibodies, various new approaches have been explored to improve treatment strategies for HIV infection. Despite advancements witnessed in the treatment of HIV, the development of immune therapeutics still remains a cumbersome, time-consuming, and highly expensive process. In recent decades, peptide therapeutics have gained significance in the field of medicine, for being highly specific and efficacious, with good tolerability and safety profiles (59). The interest in peptide therapeutics has been mitigated by certain limitations; these include the relatively short half-life, physiological instability, and difficulty in oral administration (60). However, there have been ongoing efforts to eliminate the obstacles in utilizing peptides, through half-life extension and stability enhancement under physiological conditions (61). Numerous studies have demonstrated the usefulness of short inhibitory peptides in the treatment of several diseases, particularly cancer (62–65). More recently, peptide therapeutics have also shown promise for the treatment of HIV infection (12, 16).
A number of studies in the past have attempted to identify potent peptide inhibitors targeting the CD4bs (29, 66–70), but without much success. This is because a successful inhibitor should not only block the binding of the HIV envelope to the CD4 receptor but should also efficiently block co-receptor interaction which is important for HIV-1 entry into the target cell (71). This kind of inhibition is actually accomplished very well by neutralizing antibodies, which target specific epitopes on the virus and lead to virus neutralization, thereby preventing HIV infection. Modern methods in computer-aided drug design have catalyzed the ability to reduce cost and time which limits the development of novel therapeutics (72).
Andrianov et al. (73) utilized a computer-aided strategy to screen a public web-oriented virtual screening platform (pepMMsMIMIC) to identify a few promising peptidomimetic candidates from the broadly neutralizing antibody VRC01 (73). In a similar line, we undertook an in-depth analysis of the co-crystal structures of the bNAb 8ANC131-subtype B YU-2 gp120 and VRC01-subtype C ZM176.66 gp120 complexes and inferred that the contacts made by each CD4bs-directed broadly neutralizing antibody with the HIV-1 gp120 were highly variable. However, it was observed that the heavy chain of the CD4bs-directed neutralizing antibodies engaged well with the CD4bs, i.e., the Phe43 cavity, which is highly conserved among the different bNAbs. Based on earlier studies as well as our analysis of the co-crystal structures of the antibody-antigen complexes, we decided to narrow down on hexameric peptides that would be short and at the same time target the critical residues in the CD4bs. Subsequently, potential hexamers were derived from the crystal structures of 8ANC131-subtype B gp120 and VRC01-subtype C gp120. Two peptides were predicted from each crystal structure, one from the heavy chain and another from the light chain. Only the peptides derived from heavy chains were taken up for further computational evaluations as they bound best to the CD4bs. The heavy chain derived peptides were docked with subtype B and subtype C envelopes, to identify interactions with the key residues in the CD4bs. Based on the 2D-interaction plot of the crystallized complexes, peptides RDRSTG and VNYARP, derived from the heavy chain of 8ANC131 and VRC01, respectively, were shortlisted as they interacted with the key residues of the CD4bs mentioned earlier. Molecular dynamics simulation of the RDRSTG-subtype B gp120 and VNYARP-subtype C gp120 complexes across the 200 ns trajectory (frames sampled at an interval of 20 ns) revealed that the peptides RDRSTG and VNYARP precisely target the binding site of the CD4 receptor (Phe43 and Arg59 contacts) and interact with the critical residues through hydrogen bonds and Vander Waal’s interactions with an average binding free energy (ΔG) (MM-PBSA) of –13.58 ± 2.85 (Mean ± SD) kCal/mol and –16.04 ± 8.77 (Mean ± SD) kCal/mol, respectively. The sampled frames were also subjected to KDeep calculation, wherein, the peptides RDRSTG and VNYARP scored a significant average binding affinity (ΔG) of –9.32 ± 0.80 (Mean ± SD) kCal/mol and –10.18 ± 0.63 (Mean ± SD) kCal/mol, respectively. In the case of VNYARP, one of the frames at the 60th ns gave a higher MMPBSA value (ΔG = +3.18 kCal/mol) due to a major conformation change; however, the lower binding free energy state was quickly regained around the 80th ns.
The energetically significant amino acids in the topmost stable peptide-antigen complexes of RDRSTG-subtype B gp120 and VNYARP-subtype C gp120 were found to be R, D, R, S, T, V, N, Y and R, as inferred from the cumulative results of the alanine scan (52, 53) and Robetta analyses (54, 74). The two peptides were also predicted to possess favorable physicochemical properties including non-toxicity, hydrophobicity of 2.66 and 6.63, and GRAVY (Grand Average of Hydropathy) of –2.40 and –0.82 (75, 76), respectively (Table 2) making these highly promising therapeutic candidates. A striking finding to be noted is that the RDRSTG peptide is derived from the site that is involved in the critical interaction between ARG59CD4–RECEPTOR and ASP368gp120. This could be the likely reason for this peptide standing out as the best CD4bs-targeting neutralizing peptide, as compared to all other peptides.
We further analyzed the co-crystal structures of other VH-gene-restricted (VRC01-class and 8ANC131-class) and CDR-H3-dominated antibodies with gp120 envelope for their residual interactions. The VRC01-class antibodies 3BNC117 (PDB ID: 4JPV), N6 (PDB ID: 5TE7) and NIH45-46 Fab (PDB ID: 4JDV) revealed interactions between the conserved ARG71HC/Heavy Chain residue and ASP368gp120. In addition, these antibodies also interacted with SER365gp120, GLY366gp120 and ASP368gp120 through Leu44CD4 and Lys46CD4 (10, 57, 77). In case of 8ANC131-class antibodies (1B2530; PDB ID: 4YFL) and CDR-H3 dominated antibody (CH103; PDB ID: 4JAN), the key contacts were ASP368gp120 through ARG72HC and ARG97HC, respectively. These antibodies also showed interaction with residues of the PHE43 cavity in gp120 (34, 78). Given these observations, we speculate that peptides derived from these neutralizing antibodies could also be explored for the identification of novel neutralizing peptide mimetics against HIV.
The binding of HIV-1 gp120 with the CD4 receptor on the target cell triggers a conformational change that uncovers epitopes called CD4-induced (CD4i) epitopes that bind to the chemokine co-receptors on the host cell, either CCR5 or CXCR4. Since the binding of the candidate bNAb mimetics to the CD4bs prevents conformational changes in the HIV-1 gp120 and obsoletes binding to the co-receptor, the process of viral entry into the target cells is also inhibited. Thus, the peptide mimetics identified in this study hold promise as highly potent candidates for HIV therapeutics.
Conclusion
Using modern computational tools the present study identified two short, hexameric peptides from the heavy chain of two well-characterized CD4bs-targeting bNAbs, 8ANC131 and VRC01, that hold promise as potential therapeutic candidates that can be exploited for the treatment of HIV-infected persons. This study is the first of its kind to identify short peptides that can bind to and possibly neutralize HIV-1. Given the potential of the identified candidate peptides to function as mimetics of HIV-1 broadly neutralizing antibodies, in vitro studies are in progress to validate their efficacy in HIV-1 neutralization in our laboratory (20).
Statements
Data availability statement
The original analyses presented in this study are included in the article/Supplementary material. Further inquiries can be directed to the corresponding authors.
Author contributions
UV and LH: conceptualization, resources, writing—review and editing, and supervision. SV: methodology, formal analysis, investigation, data curration, and writing—original draft preparation. SV, UV, and LH: validation. LH: project administration. All the authors have read and agreed to the published version of the manuscript.
Acknowledgments
The authors thank the funding agency, Regional Centre for Biotechnology (RCB)/Department of Biotechnology (DBT) for awarding her fellowship (DBT/2020/NIRT/1321).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.1036874/full#supplementary-material
Footnotes
2.^https://pragmaticproteindesign.bio.ed.ac.uk/balas/
3.^https://robetta.bakerlab.org/queue.jsp
4.^http://crdd.osdd.net/raghava/toxinpred/
5.^https://www.thermofisher.com/in/en/home/life-science/protein-biology/peptides-proteins/custom-peptide-synthesis-services/peptide-analyzing-tool.html
References
1.
EulerZSchuitemakerH. Cross-reactive broadly neutralizing antibodies: timing is everything.Front Immun. (2012) 3:215. 10.3389/fimmu.2012.00215
2.
BiWXuWChengLXueJWangQYuFet alIgG Fc-binding motif-conjugated HIV-1 fusion inhibitor exhibits improved potency and in vivo half-life: potential application in combination with broad neutralizing antibodies.PLoS Pathog. (2019) 15:e1008082. 10.1371/journal.ppat.1008082
3.
AttaMGDe SeigneuxSLucasGM. Clinical pharmacology in HIV therapy.CJASN. (2019) 14:435–44. 10.2215/CJN.02240218
4.
GhosnJTaiwoBSeedatSAutranBKatlamaC. HIV.Lancet. (2018) 392:685–97. 10.1016/S0140-6736(18)31311-4
5.
SimonVHoDDAbdool KarimQ. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment.Lancet. (2006) 368:489–504. 10.1016/S0140-6736(06)69157-5
6.
AbnerEJordanA. HIV “shock and kill” therapy: in need of revision.Antiviral Res. (2019) 166:19–34. 10.1016/j.antiviral.2019.03.008
7.
HaynesBFBurtonDRMascolaJR. Multiple roles for HIV broadly neutralizing antibodies.Sci Transl Med. (2019) 11:eaaz2686. 10.1126/scitranslmed.aaz2686
8.
SpencerDAShapiroMBHaigwoodNLHessellAJ. Advancing HIV broadly neutralizing antibodies: from discovery to the clinic.Front Public Health. (2021) 9:690017. 10.3389/fpubh.2021.690017
9.
MishraNSharmaSDobhalAKumarSChawlaHSinghRet alBroadly neutralizing plasma antibodies effective against autologous circulating viruses in infants with multivariant HIV-1 infection.Nat Commun. (2020) 11:4409. 10.1038/s41467-020-18225-x
10.
HuangJKangBHIshidaEZhouTGriesmanTShengZet alIdentification of a CD4-binding-site antibody to HIV that evolved near-pan neutralization breadth.Immunity. (2016) 45:1108–21. 10.1016/j.immuni.2016.10.027
11.
CaskeyMKleinFNussenzweigMC. Broadly neutralizing anti-HIV-1 monoclonal antibodies in the clinic.Nat Med. (2019) 25:547–53. 10.1038/s41591-019-0412-8
12.
ChupraditKMoonmuangSNangolaSKitideeKYasamutUMougelMet alCurrent peptide and protein candidates challenging HIV therapy beyond the vaccine era.Viruses. (2017) 9:281. 10.3390/v9100281
13.
DingCPatelDMaYMannJFSWuJGaoY. Employing broadly neutralizing antibodies as a human immunodeficiency virus prophylactic & therapeutic application.Front Immunol. (2021) 12:697683. 10.3389/fimmu.2021.697683
14.
VetrivelUNagarajanHThirumudiI. Design of inhibitory peptide targeting Toxoplasma gondii RON4-human β−tubulin interactions by implementing structural bioinformatics methods.J Cell Biochem. (2018) 119:3236–46. 10.1002/jcb.26480
15.
UsmaniSSKumarRBhallaSKumarVRaghavaGPS. In silico tools and databases for designing peptide-based vaccine and drugs. In: DonevReditor.Advances in Protein Chemistry and Structural Biology.Amsterdam: Elsevier (2018). p. 221–63. 10.1016/bs.apcsb.2018.01.006
16.
PuJWangQXuWLuLJiangS. Development of protein- and peptide-based HIV entry inhibitors targeting gp120 or gp41.Viruses. (2019) 11:705. 10.3390/v11080705
17.
JamjianMCMcNichollIR. Enfuvirtide: first fusion inhibitor for treatment of HIV infection.Am J Health Syst Pharm. (2004) 61:1242–7. 10.1093/ajhp/61.12.1242
18.
LalezariJPTrottierBChungJSalgoM. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV Infection in North and South America.N Engl J Med. (2003) 348:2175–85. 10.1056/NEJMoa035026
19.
GeorgievISGordon JoyceMZhouTKwongPD. Elicitation of HIV-1-neutralizing antibodies against the CD4-binding site.Curr Opin HIV AIDS. (2013) 8:382–92. 10.1097/COH.0b013e328363a90e
20.
BashirTPatgaonkarMKumarCSPasiAReddyKVR. HbAHP-25, an in-silico designed peptide, inhibits HIV-1 entry by blocking gp120 binding to CD4 receptor.PLoS One. (2015) 10:e0124839. 10.1371/journal.pone.0124839
21.
LandaisEMoorePL. Development of broadly neutralizing antibodies in HIV-1 infected elite neutralizers.Retrovirology. (2018) 15:61. 10.1186/s12977-018-0443-0
22.
LiHGuanYSzczepanskaAMoreno-VargasAJCarmonaATRobinaIet alSynthesis and anti-HIV activity of trivalent CD4-mimetic miniproteins.Bioorgan Med Chem. (2007) 15:4220–8. 10.1016/j.bmc.2007.03.064
23.
LynchRMTranLLouderMKSchmidtSDCohenMChavi 001 Clinical Team Members et al.The development of CD4 binding site antibodies during HIV-1 infection.J Virol. (2012) 86:7588–95. 10.1128/JVI.00734-12
24.
PanceraMZhouTDruzAGeorgievISSotoCGormanJet alStructure and immune recognition of trimeric pre-fusion HIV-1 Env.Nature. (2014) 514:455–61. 10.1038/nature13808
25.
CurreliFBelovDSRameshRRPatelNAltieriAKurkinAVet alDesign, synthesis and evaluation of small molecule CD4-mimics as entry inhibitors possessing broad spectrum anti-HIV-1 activity.Bioorgan Med Chem. (2016) 24:5988–6003. 10.1016/j.bmc.2016.09.057
26.
Parker MillerEFinkelsteinMTErdmanMCSethPCFeraDA. Structural update of neutralizing epitopes on the HIV envelope, a moving target.Viruses. (2021) 13:1774. 10.3390/v13091774
27.
KasslerKMeierJEichlerJStichtH. Structural basis for species selectivity in the HIV-1 gp120-CD4 interaction: restoring affinity to gp120 in murine CD4 mimetic peptides.Adv Bioinformat. (2011) 2011:1–12. 10.1155/2011/736593
28.
MeierJKasslerKStichtHEichlerJ. Peptides presenting the binding site of human CD4 for the HIV-1 envelope glycoprotein gp120.Beilstein J Org Chem. (2012) 8:1858–66. 10.3762/bjoc.8.214
29.
VitaCDrakopoulouEVizzavonaJRochetteSMartinLMénezAet alRational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein.Proc Natl Acad Sci USA. (1999) 96:13091–6. 10.1073/pnas.96.23.13091
30.
ApostolopoulosVBojarskaJChaiT-TElnagdySKaczmarekKMatsoukasJet alA global review on short peptides: frontiers and perspectives.Molecules. (2021) 26:430. 10.3390/molecules26020430
31.
ScheidJFMouquetHUeberheideBDiskinRKleinFOliveiraTYKet alSequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding.Science. (2011) 333:1633–7. 10.1126/science.1207227
32.
BonsignoriMZhouTShengZChenLGaoFJoyceMGet alMaturation pathway from germline to broad HIV-1 neutralizer of a CD4-mimic antibody.Cell. (2016) 165:449–63. 10.1016/j.cell.2016.02.022
33.
WibmerCKMoorePLMorrisL. HIV broadly neutralizing antibody targets.Curr Opin HIV AIDS. (2015) 10:135–43. 10.1097/COH.0000000000000153
34.
ZhouTLynchRMChenLAcharyaPWuXDoria-RoseNAet alStructural repertoire of HIV-1-neutralizing antibodies targeting the CD4 supersite in 14 donors.Cell. (2015) 161:1280–92. 10.1016/j.cell.2015.05.007
35.
LyskovSChouF-CConchúirSÓDerBSDrewKKurodaDet alServerification of molecular modeling applications: the rosetta online server that includes everyone (ROSIE).PLoS One. (2013) 8:e63906. 10.1371/journal.pone.0063906
36.
SedanYMarcuOLyskovSSchueler-FurmanO. Peptiderive server: derive peptide inhibitors from protein–protein interactions.Nucleic Acids Res. (2016) 44:W536–41. 10.1093/nar/gkw385
37.
LaskowskiRAJabłońskaJPravdaLVaøekováRSThorntonJM. PDBsum: structural summaries of PDB entries.Protein Sci. (2018) 27:129–34. 10.1002/pro.3289
38.
PettersenEFGoddardTDHuangCCCouchGSGreenblattDMMengECet alChimera?A visualization system for exploratory research and analysis.J Comput Chem. (2004) 25:1605–12. 10.1002/jcc.20084
39.
van ZundertGCPRodriguesJPGLMTrelletMSchmitzCKastritisPLKaracaEet alThe HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes.J Mol Biol. (2016) 428:720–5. 10.1016/j.jmb.2015.09.014
40.
HonoratoRVKoukosPIJiménez-GarcíaBTsaregorodtsevAVerlatoMGiachettiAet alStructural biology in the clouds: the WeNMR-EOSC ecosystem.Front Mol Biosci. (2021) 8:729513. 10.3389/fmolb.2021.729513
41.
VangoneABonvinAM. Contacts-based prediction of binding affinity in protein–protein complexes.Elife. (2015) 4:e07454. 10.7554/eLife.07454
42.
XueLCRodriguesJPKastritisPLBonvinAMVangoneA. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes.Bioinformatics. (2016) 32:3676–8. 10.1093/bioinformatics/btw514
43.
ChowEKlepeisJLRendlemanCADrorROShawDE. 9.6 new technologies for molecular dynamics simulations. In: EgelmanEeditor.Comprehensive Biophysics.Amsterdam: Elsevier (2012). p. 86–104. 10.1016/B978-0-12-374920-8.00908-5
44.
BowersKJChowDEXuHDrorROEastwoodMPGregersenBAet alScalable algorithms for molecular dynamics simulations on commodity clusters.Proceedings of the ACM/IEEE SC 2006 Conference (SC’06).Tampa, FL: IEEE (2006). p. 43–43. 10.1109/SC.2006.54
45.
MuthukumaranSSulochanaKNUmashankarV. Structure based design of inhibitory peptides targeting ornithine decarboxylase dimeric interface and in vitro validation in human retinoblastoma Y79 cells.J Biomol Struct Dyn. (2021) 39:5261–75. 10.1080/07391102.2020.1785331
46.
WangZWangXLiYLeiTWangELiDet alfarPPI: a webserver for accurate prediction of protein-ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods.Bioinformatics. (2019) 35:1777–9. 10.1093/bioinformatics/bty879
47.
SinghNChaputLVilloutreixBO. Virtual screening web servers: designing chemical probes and drug candidates in the cyberspace.Brief Bioinformat. (2021) 22:1790–818. 10.1093/bib/bbaa034
48.
UmashankarVDeshpandeSHHegdeHVSinghIChattopadhyayD. Phytochemical moieties from indian traditional medicine for targeting dual hotspots on SARS-CoV-2 spike protein: an integrative in-silico approach.Front Med. (2021) 8:672629. 10.3389/fmed.2021.672629
49.
JiménezJŠkalièMMartínez-RosellGDe FabritiisG. KDEEP?: protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks.J Chem Inf Model. (2018) 58:287–96. 10.1021/acs.jcim.7b00650
50.
KarlovDSSosninSFedorovMVPopovP. graphDelta: MPNN scoring function for the affinity prediction of protein–ligand complexes.ACS Omega. (2020) 5:5150–9. 10.1021/acsomega.9b04162
51.
Varela-RialAMaryanowIMajewskiMDoerrSSchapinNJiménez-LunaJet alPlayMolecule glimpse: understanding protein–ligand property predictions with interpretable neural networks.J Chem Inf Model. (2022) 62:225–31. 10.1021/acs.jcim.1c00691
52.
IbarraAABartlettGJHegedüsZDuttSHoborFHornerKAet alPredicting and experimentally validating hot-spot residues at protein–protein interfaces.ACS Chem Biol. (2019) 14:2252–63. 10.1021/acschembio.9b00560
53.
WoodCWIbarraAABartlettGJWilsonAJWoolfsonDNSessionsRB. BAlaS: fast, interactive and accessible computational alanine-scanning using BudeAlaScan.Bioinformatics. (2020) 36:2917–9. 10.1093/bioinformatics/btaa026
54.
KortemmeTKimDEBakerD. Computational alanine scanning of protein-protein interfaces.Sci STKE. (2004) 2004:l2. 10.1126/stke.2192004pl2
55.
GuptaSKapoorPChaudharyKGautamAKumarR. Open source drug discovery consortium, raghava GPS. In silico approach for predicting toxicity of peptides and proteins.PLoS One. (2013) 8:e73957. 10.1371/journal.pone.0073957
56.
PrévostJTolbertWDMedjahedHSherburnRTMadaniNZoubchenokDet alThe HIV-1 Env gp120 inner domain shapes the Phe43 cavity and the CD4 binding site.mBio. (2020) 11:e00280-20. 10.1128/mBio.00280-20.
57.
ZhouTZhuJWuXMoquinSZhangBAcharyaPet alMultidonor Analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies.Immunity. (2013) 39:245–58. 10.1016/j.immuni.2013.04.012
58.
CurreliFBelovDSKwonYDRameshRFurimskyAMO’LoughlinKet alStructure-based lead optimization to improve antiviral potency and ADMET properties of phenyl-1H-pyrrole-carboxamide entry inhibitors targeted to HIV-1 gp120.Eur J Med Chem. (2018) 154:367–91. 10.1016/j.ejmech.2018.04.062
59.
FosgerauKHoffmannT. Peptide therapeutics: current status and future directions.Drug Discovery Today. (2015) 20:122–8. 10.1016/j.drudis.2014.10.003
60.
LauJLDunnMK. Therapeutic peptides: historical perspectives, current development trends, and future directions.Bioorgan Med Chem. (2018) 26:2700–7. 10.1016/j.bmc.2017.06.052
61.
MuttenthalerMKingGFAdamsDJAlewoodPF. Trends in peptide drug discovery.Nat Rev Drug Discov. (2021) 20:309–25. 10.1038/s41573-020-00135-8
62.
AmitCMuralikumarSJanakiSLakshmipathyMThereseKLUmashankarVet alDesigning and enhancing the antifungal activity of corneal specific cell penetrating peptide using gelatin hydrogel delivery system.IJN. (2019) 14:605–22. 10.2147/IJN.S184911
63.
CabriWCantelmiPCorbisieroDFantoniTFerrazzanoLMartelliGet alTherapeutic peptides targeting PPI in clinical development: overview, mechanism of action and perspectives.Front Mol Biosci. (2021) 8:697586. 10.3389/fmolb.2021.697586
64.
BaigMHAhmadKSaeedMAlharbiAMBarretoGEAshrafGMet alPeptide based therapeutics and their use for the treatment of neurodegenerative and other diseases.Biomed Pharmacother. (2018) 103:574–81. 10.1016/j.biopha.2018.04.025
65.
ChenRP. From nose to brain: the promise of peptide therapy for Alzheimer’s Disease and other neurodegenerative diseases.J Alzheimers Dis Parkinson. (2017) 7:314. 10.4172/2161-0460.1000314
66.
FerrerMHarrisonSC. Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries.J Virol. (1999) 73:5795–802. 10.1128/JVI.73.7.5795-5802.1999
67.
MartinLStricherFMisséDSironiFPugnièreMBarthePet alRational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes.Nat Biotechnol. (2003) 21:71–6. 10.1038/nbt768
68.
StricherFHuangCDescoursADuquesnoySCombesODeckerJMet alCombinatorial optimization of a CD4-mimetic miniprotein and cocrystal structures with HIV-1 gp120 envelope glycoprotein.J Mol Biol. (2008) 382:510–24. 10.1016/j.jmb.2008.06.069
69.
Van HerrewegeYMorellatoLDescoursAAertsLMichielsJHeyndrickxLet alCD4 mimetic miniproteins: potent anti-HIV compounds with promising activity as microbicides.J Antimicrobial Chemother. (2008) 61:818–26. 10.1093/jac/dkn042
70.
ChoiYHRhoWSKimNDParkSJShinDHKimJWet alShort peptides with induced β-turn inhibit the interaction between HIV-1 gp120 and CD4.J Med Chem. (2001) 44:1356–63. 10.1021/jm000403
71.
BiornACCocklinSMadaniNSiZIvanovicTSamanenJet alMode of action for linear peptide inhibitors of HIV-1 gp120 interactions.Biochemistry. (2004) 43:1928–38. 10.1021/bi035088i
72.
AndrianovAMKashynIATuzikovAV. Computational identification of novel entry inhibitor scaffolds mimicking primary receptor CD4 of HIV-1 gp120.J Mol Model. (2017) 23:18. 10.1007/s00894-016-3189-4
73.
AndrianovAMKashynIATuzikovAV. Computational discovery of novel HIV-1 entry inhibitors based on potent and broad neutralizing antibody VRC01.J Mol Graph Modell. (2015) 61:262–71. 10.1016/j.jmgm.2015.08.003
74.
KortemmeTBakerD. A simple physical model for binding energy hot spots in protein–protein complexes.Proc Natl Acad Sci USA. (2002) 99:14116–21. 10.1073/pnas.202485799
75.
KyteJDoolittleRF. A simple method for displaying the hydropathic character of a protein.J Mol Biol. (1982) 157:105–32. 10.1016/0022-2836(82)90515-0
76.
ChangKYYangJ-R. Analysis and prediction of highly effective antiviral peptides based on random forests.PLoS One. (2013) 8:e70166. 10.1371/journal.pone.0070166
77.
ScharfLWestAPGaoHLeeTScheidJFNussenzweigMCet alStructural basis for HIV-1 gp120 recognition by a germ-line version of a broadly neutralizing antibody.Proc Natl Acad Sci USA. (2013) 110:6049–54. 10.1073/pnas.1303682110
78.
LiaoH-XLynchRZhouTGaoFAlamSMBoydSDet alCo-evolution of a broadly neutralizing HIV-1 antibody and founder virus.Nature. (2013) 496:469–76. 10.1038/nature12053
Summary
Keywords
HIV-1, peptide therapeutics, CD4-binding site, neutralizing peptides, molecular dynamics simulation
Citation
Vivekanandan S, Vetrivel U and Hanna LE (2022) Design of human immunodeficiency virus-1 neutralizing peptides targeting CD4-binding site: An integrative computational biologics approach. Front. Med. 9:1036874. doi: 10.3389/fmed.2022.1036874
Received
05 September 2022
Accepted
26 October 2022
Published
18 November 2022
Volume
9 - 2022
Edited by
Balu Kamaraj, Imam Abdulrahman Bin Faisal University, Saudi Arabia
Reviewed by
Qingbing Zheng, Xiamen University, China; E. Srinivasan, Indian Institute of Science (IISc), India; Karthick Vasudevan, Reva University, India
Updates
Copyright
© 2022 Vivekanandan, Vetrivel and Hanna.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luke Elizabeth Hanna, hannatrc@yahoo.comUmashankar Vetrivel, umashankar.v@icmr.gov.in
†These authors have contributed equally to this work
This article was submitted to Precision Medicine, a section of the journal Frontiers in Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.