
- 1 Department of Biochemistry, Microbiology and Immunology, Faculty of Medicine, Ottawa Institute of Systems Biology, University of Ottawa, Ottawa, ON, Canada
- 2 Department of Physiological Sciences, College of Veterinary Medicine, Oklahoma State University, Stillwater, OK, USA
- 3 College of Veterinary Medicine, Oregon State University, Corvallis, OR, USA
Despite the prevalence of Campylobacter jejuni as an important food borne pathogen, the microbial factors governing its infection process are poorly characterized. In this study, we developed a novel rabbit soft tissue chamber model to investigate C. jejuni interactions with its host. The in vivo transcriptome profile of C. jejuni was monitored as a function of time post-infection by competitive microarray hybridization with cDNA obtained from C. jejuni grown in vitro. Genome-wide expression analysis identified 449 genes expressed at significantly different levels in vivo. Genes implicated to play important roles in early colonization of C. jejuni within the tissue chamber include up-regulation of genes involved in ribosomal protein synthesis and modification, heat shock response, and primary adaptation to the host environment (DccSR regulon). Genes encoding proteins involved in the TCA cycle and flagella related components were found to be significantly down-regulated during early colonization. Oxidative stress defense and stringent response genes were found to be maximally induced during the acute infectious phase. Overall, these findings reveal possible mechanisms involved in adaptation of Campylobacter to the host.
Introduction
Campylobacter jejuni infection results in human gastroenteritis and in rare circumstances triggers Guillain–Barré syndrome, which is the primary cause of acute neuromuscular paralysis (Mead et al., 1999; Skirrow and Blaser, 2000; Butzler, 2004; Moore et al., 2005). Transmission occurs primarily through consumption of contaminated food and is most frequently associated with consumption of undercooked poultry products (Humphrey et al., 2007). While most Campylobacter infections are self-limiting, antimicrobial therapies are recommended to treat both severe cases and immuno-compromised patients. Macrolides and fluoroquinolones are the drugs of choice for treatment (Engberg et al., 2001; Bos et al., 2006). However, resistance to these two classes of antibiotics has drastically increased during the last decade and this increased resistance may compromise future treatments (Engberg et al., 2001; Bos et al., 2006). Despite years of research and millions of Campylobacter cases annually, the mechanisms involved in C. jejuni pathogenicity remain obscure, preventing the development of new therapeutics and prevention approaches. While many colonization determinants have been identified, such as flagella, iron acquisition, host cell adherence and invasion, the stringent and heat shock responses, toxin production, capsule, and lipooligosaccharide, very little is known about precisely how this organism causes disease (Young et al., 2007; Poly and Guerry, 2008; Dasti et al., 2010).
Clearly, defining genes that are differentially expressed by C. jejuni during host colonization and interaction will help contribute to a better understanding of Campylobacter pathogenicity. To the best of our knowledge, only two genome-wide transcriptional studies have been performed to characterize the C. jejuni transcriptome in vivo during host colonization. Woodall et al. (2005) have evaluated the transcriptome of C. jejuni during colonization of the chick cecum. This study indicated the in vivo expression of specific electron transport and metabolic pathways which might enable successful colonization of the chick’s gastrointestinal tract. We have also previously reported the genome-wide expression profiling of C. jejuni during host colonization and pathogenic development using the rabbit ileal loop model which mimics human gastroenteritis (Stintzi et al., 2005). Our study indicated a remodeling of the C. jejuni envelope in vivo by altering the expression of genes encoding membrane proteins and proteins involved in peptidoglycan biosynthesis and glycosylation pathways. The transcriptional profile of genes involved in metabolic processes were also differentially expressed in vivo as compared to in vitro growth, reflecting an oxygen-limited, nutrient poor, and hyperosmotic niche. Although these in vivo studies have generated valuable insights into the potential mechanisms of gut colonization, limitations associated with their experimental design prevented the full characterization of the transcriptional events leading to a successful adaptation to the host. The study from Woodall et al. (2005) was restricted to the evaluation of C. jejuni transcriptome 12 h following chicks’ inoculation, thus representing the early colonization phase. In contrast, the transcriptome of C. jejuni growing in the rabbit intestine was obtained 48-h post-inoculation, thus reflecting gene expression during the acute phase of infection (Stintzi et al., 2005).
In order to gain new insights into the mechanisms of host adaptation, we developed a novel animal model of C. jejuni infection which enables longitudinal study of C. jejuni transcriptional responses to the host from the early colonization to the acute phases of infection. To note, the use of the terms “early colonization” and “acute phases of infection” refer to time points post-inoculation of the tissue chambers. These terms are used to provide time point references that correspond to the events that occur within the intestine during colonization and/or infection. This model is based on tissue chambers which are subcutaneously implanted into the dorsolumbar regions of New Zealand white rabbits. These chambers become vascularized and accumulate tissue fluid after implantation. The chambers constitute a convenient model to study microorganisms in vivo as their relatively large volume allows repetitive sampling. Tissue chambers have been extensively used to investigate the antimicrobial efficacy of antibiotics, to study bacterial growth characteristics in vivo, and to characterize the pathophysiological mechanisms of inflammation (Clarke, 1989). Although subcutaneous chambers cannot be expected to mimic the intestinal environment, they provide models to investigate the interactions between microorganisms and the major host defense components (e.g., neutrophils). Here, we have used these tissue chambers in conjunction with genome-wide expression profiling to characterize the time course of C. jejuni transcriptional alterations during host adaptation and interaction. This report validates the use of tissue chambers to study the mechanisms of C. jejuni pathogenesis and improves our understanding of C. jejuni interactions with the host.
Materials and Methods
Bacterial Strain and Growth Conditions
The C. jejuni NCTC 11168 strain was obtained from the National Collection of Type Culture (NCTC, England), and routinely cultured on Mueller-Hinton (MH) broth or agar plates at 37°C in a microaerophilic chamber (Don Whiteley, West Yorkshire, England) containing 84% N2, 5% O2, and 11% CO2.
Rabbit Tissue Chamber Model (RTC) And RNA Extraction
Round tissue chamber discs (1-cm thick, 3.5 cm in diameter) were implanted subcutaneously in the dorsolumbar region of four New Zealand white rabbits (+4 kg, male or female) under general anesthesia. The chambers used were constructed using Delrin thermoplastic and were fitted with 12.0 μm polycarbonate membranes. The skin was incised (4-cm incision), the subcutaneous tissues lateral and ventral to the incision were undermined, the chambers were inserted in the subcutaneous space, and the skin incisions were closed using cruciate, interrupted sutures. The sterility of the tissue chamber fluid was monitored for 20 days after implantation by aerobic and anaerobic culture of an aspirate of chamber fluid (collected aseptically by percutaneous aspiration). Forty five days after implantation, the fur over the chambers was clipped, the skin aseptically prepped with Betadine® and 70% isopropyl alcohol, and approximately 3 × 1010 cfu of C. jejuni NCTC 11168 (in vitro grown mid-log phase bacteria resuspended in 0.5 ml PBS buffer) was inoculated into each chamber using a 21–23 ga. needle and 1 cc syringe. The chamber fluid was aseptically sampled at different times post-infection by needle aspiration of the serum/exudate from the chamber using 21–23 ga. needle and 3 cc syringe. For one rabbit, a 1 ml aliquot of the chamber fluid was collected at 2-, 6-, 24-, and 48-h post-infection. For the three other rabbits, the chamber fluid (approximately 5 ml) was collected at 48-h post-infection. Chamber fluids were immediately mixed with 10% v/v solution of a cold RNA degradation stop solution (10% buffer-saturated phenol in ethanol), which has been previously shown to prevent degradation of the bacterial transcriptome. This suspension was centrifuged at 4°C at low speed to remove epithelial cells. Finally, the total bacterial RNA was purified using a hot-phenol chloroform protocol as previously described (Palyada et al., 2004). In addition, aliquots of the chamber fluids were diluted in PBS buffer and plated on MH plates for bacterial counts. Rabbits were humanely euthanized after the last sample was collected. All studies and procedures were approved by the Animal Care and Use committee at Oklahoma State University with the protocol number VM-01-41.
Analysis of Campylobacter Gene Expression Using an in-House Microarray
To serve as an in vitro comparison, total RNA was extracted from C. jejuni grown in vitro to mid-log phase in MH broth. Both the in vivo and in vitro RNA samples were processed for microarray hybridization as previously described (Palyada et al., 2004; Stintzi et al., 2005). Briefly, each RNA sample (∼16 μg) was converted to cDNA using reverse transcriptase Superscript II in presence of random hexamers and aminoallyl-dUTP. Next, the aminoallyl cDNA was fluorescently labeled by coupling with monoreactive succinimidyl ester fluor dyes (either Cy3 or Cy5 dye). C. jejuni microarrays consisting of PCR products of each predicted open-reading frame of NCTC 11168 were constructed as previously described (Stintzi, 2003). The microarray slides were prehybridized in 5× SSC buffer, 25% formamide, 0.1% SDS, and 1% bovine serum albumin at 42°C for 45 min. Labeled Cy3 and Cy5-cDNAs were pooled, purified using a Qiaquick PCR spin column, resuspended in 36 μl of a hybridization solution (5× SSC buffer, 25% formamide, 0.1% SDS, and 25 μg salmon sperm DNA), denatured at 99°C for 2 min, and hybridized on the microarray slide overnight under a glass coverslip at 42°C using a humidified chamber (ArrayIt). The microarray slides were subsequently washed for 5 min with 2 × SSC and 0.1% SDS solution at 42°C. This was followed by a 10 min wash in 0.1 × SSC, 0.1% SDS at room temperature and four more 1 min washes in 0.1 × SSC before a brief final wash with distilled water. The microarray slides were scanned with a PerkinElmer ScanArray Express scanner at 10-μm resolution and the data collected using GenePix Pro 4.0 (Axon Instrument). The obtained data were processed using the MIDAS software (Saeed et al., 2003) (available from TIGR; http://www.tigr.org/software/) and OriginPro7 software (OriginLab corporation) as previously described (Palyada et al., 2004; Stintzi et al., 2005). For the temporal gene expression study, total RNA from each time point (2, 6, 24, and 48 h) was hybridized once to the microarray slides, yielding up to two measurements per gene (given that each gene is spotted on duplicate on each slide). For the 48-h time point, three biological replicates were performed and each one was hybridized to one microarray slide, yielding up to six measurements per gene. The gene expression ratios were statistically analyzed using a statistical test based on analysis of variance and a Bayesian prior (Long et al., 2001) (by using the web interface of CyberT accessible at http://molgen.biol.rug.nl/cybert/index.shtml). Finally, genes were identified as differentially expressed if the fold change in transcript abundance was higher than 2 and the Bayesian P value was below 10−3 in at least one of the time points. A hierarchical clustering analysis was performed using the Genesis software (Sturn et al., 2002) (version 1.0.2; Graz University of Technology; http://genome.tugraz.at).
Results
Growth of C. Jejuni in the Rabbit Soft Tissue Chamber
Campylobacter jejuni, a human pathogen, primarily colonizes the gastrointestinal tract of warm blooded animals and causes gastroenteritis in humans (Skirrow and Blaser, 2000; Butzler, 2004; Humphrey et al., 2007). Although several animal models have been developed to study the ability of Campylobacter to colonize the intestine and induce disease (Dasti et al., 2010; Haddad et al., 2010), to date, none of these models are suitable to perform genome-wide expression studies in vivo over the course of C. jejuni–host interactions within the same animal. Consequently, to study C. jejuni gene expression in vivo as a function of time, we developed a rabbit tissue chamber (RTC) model. The tissue chambers consist of shallow perforated cups covered with a silicone rubber diaphragm and a semipermeable membrane of 12-μm pore size. The tissue chambers were implanted subcutaneously in the dorsolumbar region of New Zealand White rabbits. Over the course of several weeks after implantation, the chamber becomes vascularized and filled with approximately 5 ml of tissue fluid (Clarke, 1989). This fluid can be inoculated or sampled aseptically by inserting a needle through the skin and the covering diaphragm. Although the chambers are freely accessible to the host defense components, C. jejuni introduced into the chamber remain confined to the inoculated chamber as confirmed by the absence of cross-contamination between two chambers implanted into the same rabbit. Control experiments confirmed that when one of the two chambers was inoculated with C. jejuni, the other chamber remained sterile (data not shown).
To study C. jejuni growth in the tissue chambers, we first performed a pilot study in which two chambers were inoculated with 5 × 107 cfu of C. jejuni NCTC 11168 (yielding ∼107 cfu/ml of tissue fluid). The growth of C. jejuni in the tissue chambers is characterized by an exponential increase in bacterial numbers reaching 5 × 107 and 1 × 108 cfu/ml of fluid at 8- and 24-h post-inoculation respectively. Considering that approximately 109 cfu of C. jejuni are required to yield sufficient RNA to perform microarray hybridization, it was not possible to monitor the C. jejuni transcriptome during its growth in the tissue chamber. Consequently, we decided to inoculate the tissue chamber with 3 × 1010 cfu (corresponding to approximately 5 × 109 cfu/ml of tissue fluid) and study the immediate response of C. jejuni to the host at 2-, 6-, 24-, and 48-h post-inoculation by collecting 1 ml of tissue fluid at each time point. Under these experimental conditions the bacterial counts remained at 4 × 109 ± 1 × 109 cfu/ml for the entire 48-h period post-inoculation. Polymorphonuclear cell counts (especially neutrophils) in the chamber increased from approximately 104 cells per chamber at the time of inoculation, to 105 cells per chamber at 8-h post-infection. The polymorphonuclear levels remained stable at 105 cells per chamber for the next 40 h of the experiment.
Identification of C. Jejuni genes Expressed In Vivo Over the Course of its Interaction with the Host
The aim of this work was to follow, as a function of time post-infection, the alterations of the C. jejuni transcriptome in response to its interaction with the host environment. The experimental approach consisted of inoculating three RTCs (implanted in three different rabbits) with 3 × 1010 cfu of C. jejuni. A fourth chamber was kept non-inoculated and served as a control. One rabbit was selected for longitudinal sampling and 1 ml aliquots of the chamber fluid were collected at 2-, 6-, 24- and 48-h post-inoculation and the C. jejuni total RNA was extracted. The bacterial RNA from the chamber fluid of the two other rabbits was extracted at 48-h post-inoculation. Approximately 22–30 μg of total RNA was recovered per sample. The control chamber was processed identically to the inoculated chamber; however, no RNA was recovered, validating our experimental protocol to monitor chamber-grown C. jejuni gene expression. Additionally, C. jejuni was not recovered from tissues surrounding the chambers or from rabbit blood samples. In order to directly compare the transcriptome of C. jejuni grown in the tissue chamber with the previously described transcriptome of C. jejuni grown in the ligated rabbit ileal loop (Stintzi et al., 2005), the relative level of transcripts obtained from chamber-grown C. jejuni was monitored by competitive hybridization with RNA extracted from bacteria grown in vitro to mid-log phase. The extraction and preparation of RNA from the in vitro grown C. jejuni was performed as described previously (Stintzi et al., 2005). Following microarray data analysis as described in the “Materials and Methods” section, genes were considered to be differentially expressed if their change in transcript level was ≥2-fold in at least one of the conditions tested with a Bayesian p value ≤ 10−3. To note, analysis and validation of gene expression by RT-PCR was not possible due to the low amount of isolated RNA, a problem especially challenging for in vivo experiments. Nevertheless, the same microarray platform and protocols were previously shown to produce data with high level of concordance with quantitative RT-PCR (Stintzi, 2003; Palyada et al., 2004; Stintzi et al., 2005; Reid et al., 2008a,b; Palyada et al., 2009). Overall, it was found that 449 genes were differentially expressed in vivo as compared to in vitro conditions in at least one of the experimental time points (2, 6, 24, or 48 h) using the RTC model (Figures 1 and 2). Of these 449 genes, 213 were identified to be down-regulated, while 236 genes had an increase in their transcript abundance. Mapping the differentially expressed genes on the C. jejuni chromosome highlights the co-regulation of genes belonging to the same operon and supports the validity of our data (Figure 1).
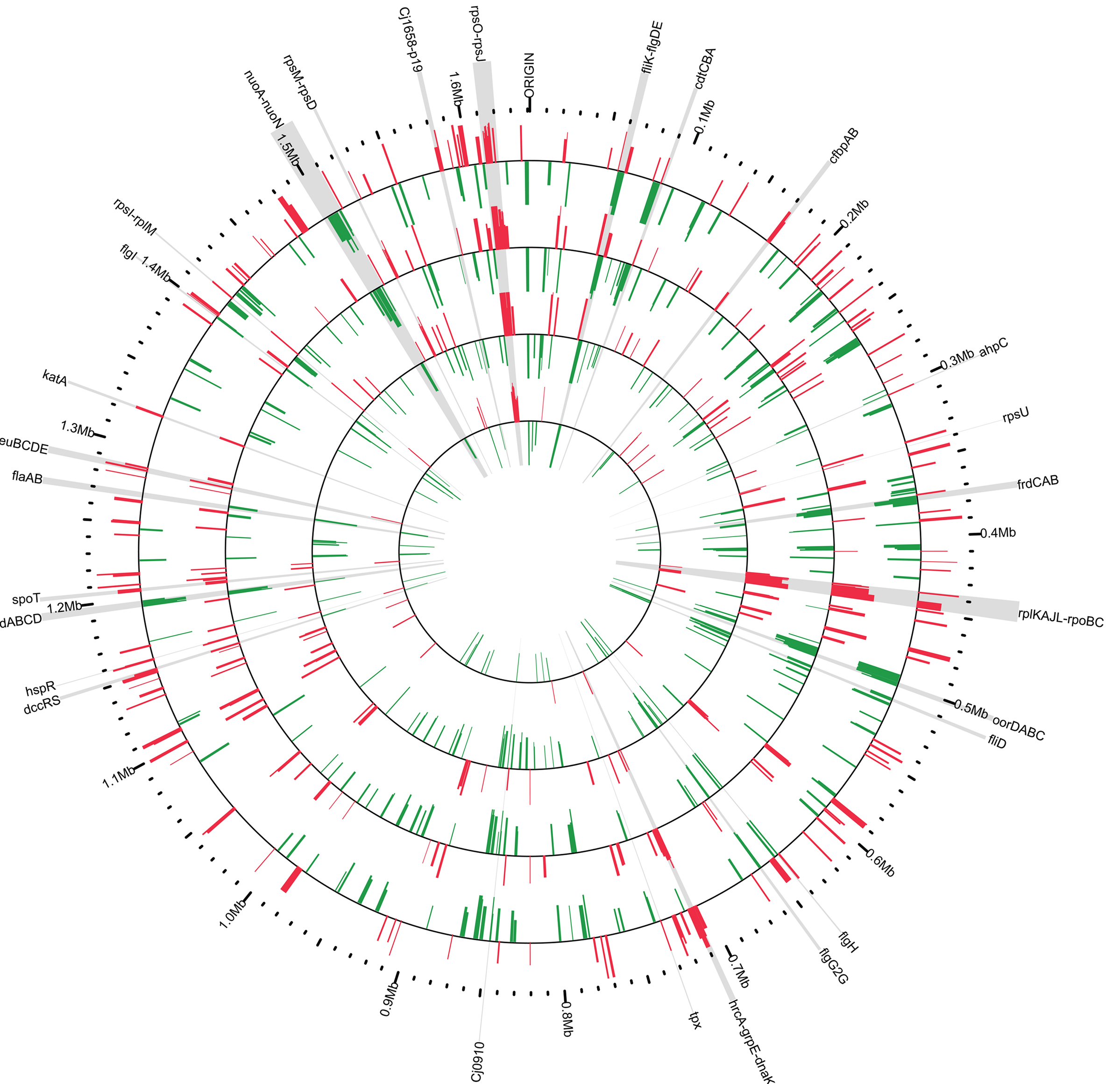
Figure 1. Chromosomal mapping of Campylobacter jejuni transcriptomes. Genes found to be differentially expressed (fold change ≥2, p value <10−3) were mapped onto the C. jejuni NCTC11168 genome. Red signifies up-regulation in the tissue chamber while green represents down-regulation. The tracks are scaled from <0.25- to >4-fold transcript change with respect to in vitro cultures. The tracks are arranged temporally with the earliest time point closest to the center and represent the C. jejuni transcriptomes at 2 (track 1), 6 (track 2), 28 (track 3), and 48 h (track 4) post-inoculation of the tissue chambers. Key genes mentioned in the text are highlighted in gray and their approximate genomic location is enumerated on the outermost ring. Figure was created using Circos v0.52 (Krzywinski et al., 2009).

Figure 2. Hierarchical cluster analysis of genes differentially expressed using the rabbit tissue chamber model. Each row represents one gene. Columns 1–4 represent the expression profile for one biological replicate at fixed time points; column 1, 2 h; 2, 6 h; 3, 28 h; 4, 48 h. Column 5 represents the differential gene expression at 48 h for three biological replicates. Columns 6–10 represent changes in the transcriptome for five different rabbits using the rabbit ileal loop model (as reported previously Stintzi et al., 2005) and are shown for comparative purposes between the two different host interaction models. The color intensity is proportional to the fold change as represented by the scale. Gray colors represent missing data. Genes in boldface are further discussed in the text.
Global In Vivo Transcript Profiling
To visually investigate the temporal change of C. jejuni’s transcriptome during host adaptation, the 449 differentially expressed genes were subjected to hierarchical clustering analysis, revealing 6 major clusters (A, B, C, D, E, and F) as shown in Figure 2. Clusters A, B, and C represent the genes that have a decrease in transcript abundance. Cluster A consists of genes that are down-regulated early on during host adaptation (2-h post-infection); whereas cluster C represents the genes that decrease in transcript level at later time points during the acute phases of infection (24- to 48-h post-infection). Clusters D, E, and F consist of genes that are up-regulated during early adaptation (cluster D) to genes that are induced after 24–48 h, representing the gene expression profile during acute infection (cluster F).
Cluster A consists of genes whose transcript level dropped sharply and early on during host adaptation. These genes remained significantly down-regulated during the entire course of the experiment. Only 12 genes segregate to this cluster and are almost exclusively involved in flagella biogenesis (flgDEE2GG2HI and fliDK).
Cluster B consists of genes that were relatively unchanged at 2- and 6-h post-inoculation, but become significantly down-regulated at later time points. This cluster is composed of 29 genes that are primarily involved in small molecule metabolism. Cluster C is the largest cluster of genes whose expression was down-regulated during the course of these experiments. These genes had their expression levels slowly decrease during the sampling time course, but not to the same extent as genes in cluster B. Cluster C consists of 172 genes and similarly to cluster B, these genes are primarily involved in small molecule metabolism and energy metabolism. Representative genes from clusters B and C include the fumarate reductase (frdCAB), NADH dehydrogenase I (nuoBCGIJKLN) and α-ketoglutarate dehydrogenase (oorABC).
Cluster D contains 30 transcripts that were up-regulated early on during the time course of the experiment and continued to remain over-expressed 48-h post-inoculation. Most of the genes belonging to this cluster encode proteins involved in ribosome biogenesis and modification. These genes include rplABEFMNOPQR, rpmC, and rpsBEHJNQ, which are 50S ribosomal proteins and 30S ribosomal proteins respectively. Another interesting set of genes from this cluster encodes proteins involved in the heat shock response, including the ATP-dependent CLP protease (clpB) and the heat shock protein GrpE (grpE). ClpB, in association with the heat protein DnaK, helps resolubilize aggregated proteins formed in response to various stresses (Lund, 2001; Chow and Baneyx, 2005; Reid et al., 2008a,b). GrpE, a nucleotide exchange factor, releases ADP from the ATPase domain on DnaK enhancing substrate release and recycling of DnaK (Lund, 2001). To note, dnaK belongs to cluster F and its transcript abundance was also found to increase in vivo, in concordance with several other genes encoding heat shock proteins (groEL, groES, hrcA, cbpA, hspR, dnaJ, lon, Cj1034c, and htpJ).
The next cluster of induced gene transcripts, cluster E, is comprised of 80 transcripts that are most highly induced in the RTC between 6- and 24-h post-inoculation. Within this cluster, the 50S ribosomal and 30S ribosomal genes rplCDJKLTVWX, rpmA and rpsCDGKLMOSU were induced. Two genes that function in iron transport and binding, Cj1658 and ceuE were also found to up-regulated in vivo. Cj1658 encodes for a putative iron permease, whereas ceuE is an enterobactin uptake periplasmic binding protein, a component of the ferric-enterobactin ABC transporter system ceuBCDE (Palyada et al., 2004) (To note, ceuC belongs to cluster C, and no data was obtained for either ceuB or ceuD). Another gene of interest within cluster E is tpx. Tpx is a thiol peroxidase involved in the detoxification of hydrogen peroxide and peroxides within the cell (Atack et al., 2008). Peb3 (encoding major antigenic peptide), dnaJ (a chaperone involved in heat shock response), and htpG (hsp90 family heat shock protein) were also found to be induced in vivo.
Cluster F contains genes that were induced during acute infection (24- to 48-h post-infection) and consists of 126 genes. C. jejuni catalase (katA) and alkyl hydroperoxide reductase (ahpC), two genes involved in oxidative stress defense (Palyada et al., 2009), were found to be significantly up-regulated. KatA and AhpC are both involved in the direct detoxification of hydrogen peroxide within the cell (Palyada et al., 2009). In addition, the primary peroxide regulator perR, also had a significant increase in transcript abundance. As previously mentioned, genes involved in the heat shock response and protein degradation, dnaK, groES, groEL, and lon were also found to be induced. DnaK encodes for a chaperonin, that along with DnaJ and GrpE, refolds misfolded proteins under stressful conditions (Lund, 2001). GroES is another chaperonin that binds to GroEL to suppress the ATPase activity of GroEL, while GroEL promotes refolding of misfolded proteins (Lund, 2001). The ATP-dependent protease Lon is involved in the hydrolysis of regulatory or misfolded proteins (Ingmer and Brondsted, 2009). The transcriptional regulators of these heat shock proteins hspR and hrcA were also found to be induced. Other genes of interest include iron uptake proteins such as the periplasmic iron binding protein p19 (Chan et al., 2010), the enterobactin transporter cfrA (Palyada et al., 2004), and both cfbpA and cfbpB which have been reported to be involved in transferrin/lactoferrin transport (Miller et al., 2008). Additionally both the stringent response regulator spoT and the active component of the cytolethal distending toxin (CDT) protein cdtB, were induced. Finally, several genes involved in the biosynthesis of cofactors were also found to be induced including bioB (involved in biotin biosynthesis), folP (involved in folic acid biosynthesis), moaD (involved in molybdopterin biosynthesis), panB (involved in pantothenate biosynthesis), Cj1613c and pdxA (involved in pyridoxine biosynthesis), thiS (involved in thiamine biosynthesis), and ribF ((involved in riboflavin biosynthesis).
Correlation of C. Jejuni Genes Expressed in the Rabbit Soft Tissue Chamber and in the Ligated Ileal Loop
Our lab has previously used a rabbit ileal loop model to study C. jejuni gene expression during infection of the host (Stintzi et al., 2005). However, these experiments revealed weak correlations (r2 ∼0.49) for gene expression profiles between different rabbits. This is most likely due to the inherent complexities of the rabbit intestinal tract and the endogenous microflora that result in variations in both the colonization ability of C. jejuni and its intestinal environment. Importantly, very few genes were reported to be antagonistically regulated between rabbits. Indeed, the trend of differential expression remained essentially the same, with only the amplitude of change in transcript level varying. In contrast, and as shown Figure 3, the gene expression profiles of the three biological replicates obtained at 48-h post-inoculation of the tissue chambers correlated relatively well. We obtained r2 values corresponding to 0.66, 0.67, and 0.72 when comparing the three 48-h biological replicates. In order to validate the usefulness of the tissue chamber model to study Campylobacter pathogenesis, we compared the 48-h transcriptome of chamber-grown C. jejuni to the transcriptome of C. jejuni recovered from rabbit ileal loop. C. jejuni colonization of the rabbit ileal loop is shown in Figure 2 (columns 6–10). Genes that were found to be differentially expressed in the RTC at 48 h were directly compared to the genes that were differentially expressed at 48 h in each of the five rabbits from the ileal loop model. Approximately 84% of the genes exhibited similar expression profile (up- or down-regulation independently of the absolute extent of fold change) between the two animal models. While we recognize that the tissue chamber model has limitations, this result validates our model as an infectious model and indicates that the genes responsive to mammalian host-specific signals should be identified in this study.
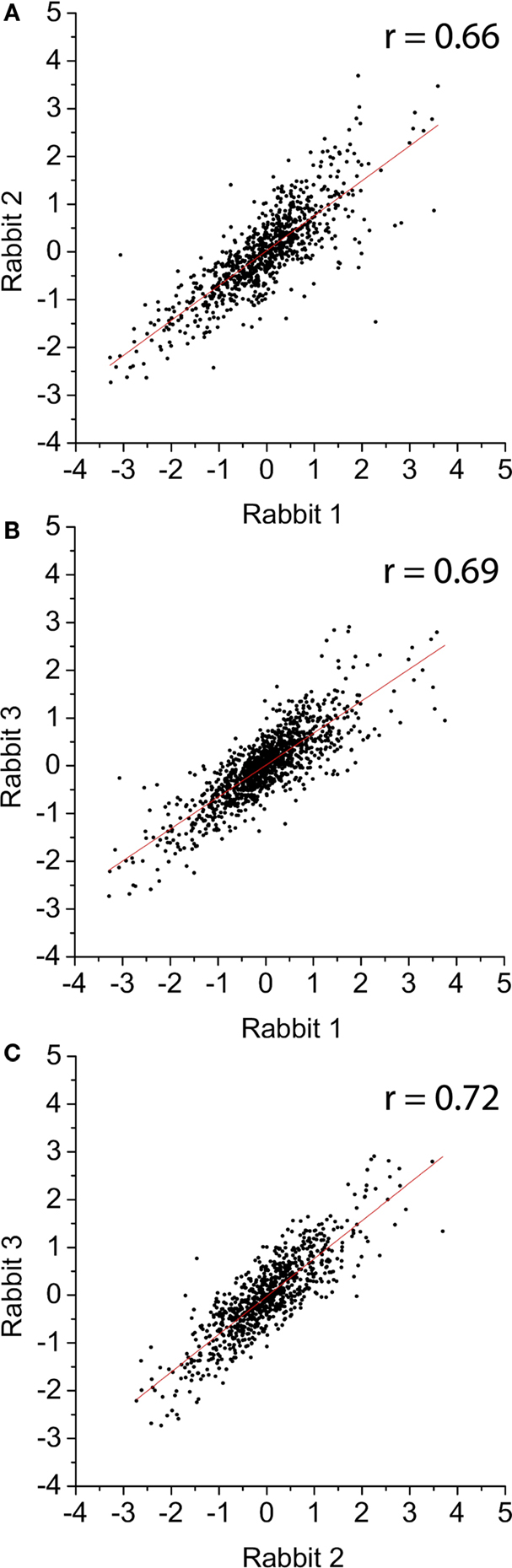
Figure 3. Correlation between log2 values of the gene expression ratio obtained from microarray experiments using bacterial cDNA from three different rabbits: (A) rabbit 1 vs. rabbit 2; (B) rabbit 1 vs. rabbit 3; and (C) rabbit 2 vs. rabbit 3. The red line represents the linear regression fit of the data. r, correlation coefficient.
Discussion
In order to gain a better understanding of C. jejuni factors involved in host interaction and niche adaptation, we analyzed the transcriptome profiles of C. jejuni during in vivo infection from early stage colonization to acute infection by development and use of a novel RTC model. Although the tissue chambers cannot duplicate host defenses specific to specialized organs such as the gastrointestinal tract, they do provide an in vivo model that represents important protective functions of the inflammatory reactions observed in most tissues (Clarke, 1989). In addition, the ease of sampling and accessibility to neutrophil-mediated and humoral components of the host defense response makes the tissue chamber model well suited to study C. jejuni gene expression as a function of time post-infection. The transcriptome of C. jejuni was assessed at 2, 6, 24, and 48 h following the chambers’ inoculation. Comparison of the transcriptomes of C. jejuni grown in the tissue chambers to in vitro growth conditions revealed the alteration in abundance of 449 transcripts. This study represents the first temporal assessment of C. jejuni gene induction during in vivo adaptation and survival. The genes identified from this study will help provide insight into pathways and mechanisms that play an important role in C. jejuni colonization, immune system evasion, and survival within the host. In addition, this study provides valuable information on the expression of surface proteins in vivo, which might guide future vaccine development.
Growth of C. jejuni within the tissue chambers led to a rapid increase in abundance of transcripts associated with ribosomal protein synthesis and modification, rplA, rplB, rplE, rplF, rplM, rplN, rplO, rplP, rplQ, rplR, rpmC and rpsB, rpsE, rpsH, rpsI, rpsJ, rpsN, rpsQ. These genes remain highly up-regulated throughout the 48 h survival within the tissue chambers. In E. coli, ribosome biogenesis occurs during periods of rapid growth in the presence of an adequate nutrient supply (Kaczanowska and Ryden-Aulin, 2007). The up-regulation of these genes suggests that the tissue chambers provide a suitable environment for the survival and multiplication of C. jejuni. Our observations that C. jejuni was able to grow in these tissue chambers also supports this fact. This is somewhat counterintuitive, given that tissue fluids are relatively poor in nutrients as hosts sequester key nutrients, such as iron, as a defense mechanism against pathogen invasion (Weinberg, 2009). The presence of this sequestration is evident as revealed by the up-regulation of genes involved in amino-acid biosynthesis and iron acquisition. Nevertheless, genes encoding ribosomal proteins were also found to be up-regulated in the rabbit intestine (Stintzi et al., 2005). This suggests that C. jejuni is able to acquire the nutrients it needs for growth and additionally suggests that the current in vitro growth conditions employed to cultivate C. jejuni are sub-optimal as compared to actual in vivo conditions.
The in vivo transcriptome profile is suggestive of major metabolic reprogramming from a nutrient-rich microaerophilic growth condition to an anaerobic environment deficient in specific cofactors such as biotin, thiamine, and riboflavin. The transcript abundance of genes encoding proteins involved in small metabolite metabolism was found to either gradually increase or decrease from 2- to 48-h post-inoculation. Thus, these genes are likely to be specific for the adaptation to the new in vivo environment. In particular, genes encoding proteins involved in the tricarboxylic acid cycle (TCA), which is highly active under aerobic conditions, were among the most highly down-regulated genes in vivo. These genes code for components of fumarate reductase (frdABC), succinyl-coA synthetase (sucCD), malate dehydrogenase (mdh), citrate synthase (gltA), aconitate hydratase (acnB), isocitrate dehydrogenase (icd), andα-ketoglutarate dehydrogenase (oorABCD). In C. jejuni, the TCA cycle is linked to the respiratory chain via flavodoxin (Weerakoon and Olson, 2008), which is the electron acceptor for the enzyme α-ketoglutarate dehydrogenase (oorABCD). In agreement with a decrease in TCA cycle activity, the genes encoding the NADH:ubiquinone oxidoreductase (nuoBCGHIJKLN), which transfers electrons from flavodoxin to the menaquinone pool, was also found to be down-regulated. Altogether, the down-regulation of these enzymes indicates an abrupt shift from aerobic to anaerobic metabolism in vivo. In agreement with this statement, the genes encoding formate dehydrogenase (fdhABCD), an enzyme of anaerobic metabolism, were found to be up-regulated or equally expressed in vivo as compared to in vitro growth conditions. In turn, formate dehydrogenase might allow C. jejuni to carry out anaerobic respiration in vivo.
Genes encoding proteins involved in small molecule metabolism that were found to be up-regulated in vivo include components of the ATP synthase (atpBDEF) and genes encoding enzymes involved in the biosynthesis of amino acids (glnA, argC, dapBD, and metA). Purine (purBHL) and the cofactor biosynthesis, including thiamine, riboflavin, pyridoxine, pantothenate, molybdopterin, folic acid and biotin (thiS, ribDF, pdxA, panB, moaD, pabB, folP, and bioB) transcripts were also induced. The up-regulation of these genes in vivo indicates that the tissue chamber environment is deficient in several required amino acids and cofactors.
Several genes highly expressed in the tissue chambers are known or thought to be involved in stress responses, host colonization, and/or virulence. For example, the genes encoding components of the heat shock response, iron stimulon, and stringent response were found to be up-regulated in vivo. Heat shock proteins have been previously shown to have a significant role in C. jejuni colonization of the chick and rabbit intestines (Konkel et al., 1998; Stintzi et al., 2005). In C. jejuni, the heat stress response is negatively controlled though the regulators HrcA and HspR (Holmes et al., 2010). The HrcA and HspR repressors act at the transcriptional level by binding to conserved sequences known as cis-acting regulatory element (CIRCE) and HspR-associated inverted repeat (HAIR) respectively (Holmes et al., 2010). Following exposure to heat shock or other stresses, these repressors dissociate from their operators, leading to the transcriptional expression of the heat shock genes. Members of the HspR regulon have been previously characterized and include the genes encoding the heat shock proteins HspR, HrcA, GroES, GroEL, ClpB, GrpE, DnaK, and CbpA (Andersen et al., 2005; Holmes et al., 2010). The transcript abundance of every member of the HspR regulon was found to gradually increase in vivo suggesting a hostile environment and coincide with the recruitment of neutrophils within the chambers. In addition to its role in the control of the heat shock response, HspR has also been reported to modulate the expression of genes involved in the formation of the flagellum apparatus (Andersen et al., 2005). Previous transcriptome analysis of a ΔhspR mutant revealed a group of eight flagellum genes with lower transcript abundance as compared to the wild-type strain, and corroborated the requirement of HspR for normal motility and chemotaxis (Andersen et al., 2005). Interestingly, most of the flagellar genes were also found to be down-regulated in the tissue chambers, corroborating the induction of the HspR regulon. In fact, almost all the flagella biosynthesis genes were down-regulated in the tissue chambers. The apparent antagonistic regulation of proteins involved in the heat shock response and flagellum biogenesis is unclear and requires further investigation. Flagella are important virulence factors for C. jejuni and unflagellated C. jejuni mutants are unable to colonize the intestinal tract of chickens or cause disease in pathogenic models (Guerry, 2007; Poly and Guerry, 2008). However, flagella may only be required for the initial colonization of the intestinal tract and for penetrating the thick layer of mucus that separates the mucosal epithelium from the intestinal luminal space. After this initial penetration, the flagellar apparatus may not be required for replication within the mucosal layer and it has been suggested that the flagellum is not required for persistence in the chicken cecum (Wassenaar et al., 1993; Wosten et al., 2004). Moreover flagella components are key antigenic factors that are easily recognized by the host’s immune machinery (Nachamkin and Hart, 1985). Thus in contact with the host environment, C. jejuni may down-regulate its flagellar apparatus post-colonization as a way to evade the host immune defenses.
We also found that many genes encoding iron acquisition proteins were up-regulated in vivo, suggesting an iron limited environment. These genes include Cj1658 (putative iron permease), ceuE (enterobactin uptake periplasmic binding protein), p19 (periplasmic iron transporter), cfrA (ferric-enterobactin uptake receptor), cfbpAB (ABC transport system reported to be involved in tranferrin/lactoferrin transport). Genes involved in iron acquisition have been shown to be essential for successful colonization of C. jejuni in the gastrointestinal tracts of chicks and rabbit (Palyada et al., 2004). Induction of these iron acquisition genes highlights the importance of scavenging iron from the surrounding environment for C. jejuni to survive and multiply in the RTC.
Transcript abundance for genes involved in oxidative stress defense increased at the greatest levels at 24- and 48-h post-inoculation of the tissue chambers. These genes are maximally induced at these time points, which coincides with maximal numbers of neutrophils within the chambers and subsequently the inflammatory response (neutrophils were observed to increase in number up until 8-h post-inoculation and then remained constant through 48 h). Neutrophils are involved in protecting the host against invading bacteria by producing hydrogen peroxide, which can damage DNA, proteins and lipids of surrounding living cells (Dahlgren and Karlsson, 1999). Up-regulation of tpx, katA, and ahpC in C. jejuni suggests that these enzymes are involved in detoxification of oxidants present in the tissue chambers during acute infection.
A number of other genes associated with C. jejuni colonization and virulence were found to be up-regulated in the tissue chambers. These genes include spoT, dccS, cbrR, and cdtB. In C. jejuni, spoT encodes a bifunctional (p)ppGpp synthetase/hydrolase and regulates the stringent response required to survive several specific stresses, including nutrient deprivation, stationary phase growth conditions, rifampicin exposure, and low CO2/high O2 conditions (Gaynor et al., 2005). Interestingly, spoT was shown to be required for the virulence-related phenotypes of adherence, invasion, and intracellular survival within human epithelial cells and for colonization of the rabbit gastrointestinal tract (Gaynor et al., 2005; Stintzi et al., 2005). The gene dccS encodes the sensor of a two-component signal transduction system designated DccRS (MacKichan et al., 2004; Wosten et al., 2010). DccS has been shown to selectively phosphorylate the cognate response regulator DccR (Wosten et al., 2010). The DccRS regulon in C. jejuni NCTC 11168 consists of 6 genes, Cj0606, which encodes a putative secretion protein HlyD, Cj0200c, Cj1004, Cj1356c, Cj1626c, and Cj1723c, which are annotated as putative periplasmic or membrane proteins (MacKichan et al., 2004; Wosten et al., 2010). Interestingly, the transcript abundance of Cj0200c, Cj1356c, and Cj1626c was significantly up-regulated in the tissue chambers at 2- and 6-h post-inoculation. Cj0606 and Cj1723c exhibited a similar expression profile (1.5- and 1.9-fold up-regulation at 2- and/or 6-h post-inoculation) but were not selected by our data analysis threshold (>2-fold differential expression). The increased abundance of five out of six members from the DccSR regulon during the first few hours following inoculation is in agreement with the speculated role of these genes in the primary adaptation of C. jejuni to the host environment rather than being required for survival during persistence or acute infection (Wosten et al., 2010). DccR was shown to be required for early colonization of C. jejuni in 7-day-old chicks (Wosten et al., 2010). The gene cbrR encodes a response regulator of another two-component signal transduction system but with no known cognate histidine kinase sensor (Raphael et al., 2005). The transcript abundance of cbrR was also found to increase in the tissue chambers as compared to in vitro conditions. CbrR has been shown to modulate C. jejuni’s resistance to bile and its ability to colonize chickens (Raphael et al., 2005). Given that bile is unlikely to be present within tissue chambers, our data suggest an additional role for CbrR in vivo. Finally, the transcript level of cdtB, which encodes a component of the CDT, was found to increase at 24- and 48-h post-inoculation, coinciding with neutrophil influx within the chambers. In C. jejuni CDT is encoded by 3 genes, cdtA, cdtB, and cdtC. CdtB is the active subunit of CDT. The product of cdtA and cdtC form a tripartite complex with CdtB and are both required for the delivery and translocation of CdtB into the cell (Ge et al., 2008). While the exact function of CDT in Campylobacter pathogenesis remains unclear, a ΔcdtB mutant was found to be impaired in its ability to persistently colonize immunocompetent C57BL/129 mice (Fox et al., 2004), suggesting that CDT may contribute to evade the host immune surveillance. While the expression of cdtB was up-regulated in vivo after 24 and 48 h of inoculation, the expression of cdtA was unchanged and the expression of cdtC was slightly increased (but was below our selection threshold of >2-fold). Considering CdtB is the only component of the holotoxin that enters the cells, one possible explanation for the cdtABC expression pattern is that CdtA and CdtC are recycled. Therefore, cdtB would be the only gene from the cdtABC cluster that would need to be up-regulated.
Concluding Remarks
In this work we assessed the dynamics of gene expression during C. jejuni adaptation to its host using a novel RTC model. We monitored the C. jejuni transcriptome over time from early colonization to acute infection. This study clearly demonstrates that bacterial gene expression varies as a function of time in vivo, with a group of genes expressed during the early stage of adaptation to the host and another group of genes expressed during acute infection. For example, the DccSR regulon appears to be important for early adaptation, while the stress responses appear to be required later on during acute infection. In addition to genes of known or annotated function, several genes of unknown function were also found to be transiently expressed and might be specifically required for host adaptation. The genes involved in oxidative stress defense, heat shock response, stringent response, and toxin production were up-regulated after 24 h and their expression profiles coincided with the entry of neutrophils into the chambers, suggesting that they are necessary for counteracting the host immune defenses. Approximately 84% of the genes found to be differentially expressed at 48-h post-inoculation in the RTC model were also similarly differentially expressed in the rabbit ileal loop model (Stintzi et al., 2005). This result is somewhat surprising, giving that the intestinal and tissue chamber environments might be significantly different. For example, the intestine is colonized by a complex microbial flora which should result in competitive processes. In contrast, the chambers are sterile. While it is possible that the dynamics of gene expression might be different during the early stages of colonization between these two models, similar transcriptomes during acute infection suggests a steady stage adaptation to the host and validate this model for the study of Campylobacter pathogenesis. Moreover, this model might be very valuable for screening and efficacy testing of novel anti-Campylobacter drugs and/or vaccines.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Canadian Institutes of Health Research (CIHR) grant (to Alain Stintzi) and National Institutes of Health (NIH) grant RO1-055612 (to Alain Stintzi).
References
Andersen, M. T., Brondsted, L., Pearson, B. M., Mulholland, F., Parker, M., Pin, C., Wells, J. M., and Ingmer, H. (2005). Diverse roles for HspR in Campylobacter jejuni revealed by the proteome, transcriptome and phenotypic characterization of an hspR mutant. Microbiology 151(Pt 3), 905–915.
Atack, J. M., Harvey, P., Jones, M. A., and Kelly, D. J. (2008). The Campylobacter jejuni thiol peroxidases Tpx and Bcp both contribute to aerotolerance and peroxide-mediated stress resistance but have distinct substrate specificities. J. Bacteriol. 190, 5279–5290.
Bos, J. C., Schultsz, C., Vandenbroucke-Grauls, C. M., Speelman, P., and Prins, J. M. (2006). Optimising antibiotic policies in the Netherlands. IX. SWAB guidelines for antimicrobial therapy in adults with acute infectious diarrhoea. Ned. Tijdschr. Geneeskd. 150, 1116–1122.
Butzler, J. P. (2004). Campylobacter, from obscurity to celebrity. Clin. Microbiol. Infect. 10, 868–876.
Chan, A. C., Doukov, T. I., Scofield, M., Tom-Yew, S. A., Ramin, A. B., Mackichan, J. K., Gaynor, E. C., and Murphy, M. E. (2010). Structure and function of P19, a high-affinity iron transporter of the human pathogen Campylobacter jejuni. J. Mol. Biol. 401, 590–604.
Chow, I. T., and Baneyx, F. (2005). Coordinated synthesis of the two ClpB isoforms improves the ability of Escherichia coli to survive thermal stress. FEBS Lett. 579, 4235–4241.
Clarke, C. R. (1989). Tissue-chamber modeling systems – applications in veterinary medicine. J. Vet. Pharmacol. Ther. 12, 349–368.
Dahlgren, C., and Karlsson, A. (1999). Respiratory burst in human neutrophils. J. Immunol. Methods 232, 3–14.
Dasti, J. I., Tareen, A. M., Lugert, R., Zautner, A. E., and Gross, U. (2010). Campylobacter jejuni: a brief overview on pathogenicity-associated factors and disease-mediating mechanisms. Int. J. Med. Microbiol. 300, 205–211.
Engberg, J., Aarestrup, F. M., Taylor, D. E., Gerner-Smidt, P., and Nachamkin, I. (2001). Quinolone and macrolide resistance in Campylobacter jejuni and C. coli: resistance mechanisms and trends in human isolates. Emerg. Infect. Dis. 7, 24–34.
Fox, J. G., Rogers, A. B., Whary, M. T., Ge, Z., Taylor, N. S., Xu, S., Horwitz, B. H., and Erdman, S. E. (2004). Gastroenteritis in NF-kappaB-deficient mice is produced with wild-type Camplyobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect. Immun. 72, 1116–1125.
Gaynor, E. C., Wells, D. H., MacKichan, J. K., and Falkow, S. (2005). The Campylobacter jejuni stringent response controls specific stress survival and virulence-associated phenotypes. Mol. Microbiol. 56, 8–27.
Ge, Z., Schauer, D. B., and Fox, J. G. (2008). In vivo virulence properties of bacterial cytolethal-distending toxin. Cell Microbiol. 10, 1599–1607.
Haddad, N., Marce, C., Magras, C., and Cappelier, J. M. (2010). An overview of methods used to clarify pathogenesis mechanisms of Campylobacter jejuni. J. Food Prot. 73, 786–802.
Holmes, C. W., Penn, C. W., and Lund, P. A. (2010). The hrcA and hspR regulons of Campylobacter jejuni. Microbiology 156(Pt 1), 158–166.
Humphrey, T., O’Brien, S., and Madsen, M. (2007). Campylobacters as zoonotic pathogens: a food production perspective. Int. J. Food Microbiol. 117, 237–257.
Ingmer, H., and Brondsted, L. (2009). Proteases in bacterial pathogenesis. Res. Microbiol. 160, 704–710.
Kaczanowska, M., and Ryden-Aulin, M. (2007). Ribosome biogenesis and the translation process in Escherichia coli. Microbiol. Mol. Biol. Rev. 71, 477–494.
Konkel, M. E., Kim, B. J., Klena, J. D., Young, C. R., and Ziprin, R. (1998). Characterization of the thermal stress response of Campylobacter jejuni. Infect. Immun. 66, 3666–3672.
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., Jones, S. J., and Marra, M. A. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645.
Long, A. D., Mangalam, H. J., Chan, B. Y., Tolleri, L., Hatfield, G. W., and Baldi, P. (2001). Improved statistical inference from DNA microarray data using analysis of variance and a Bayesian statistical framework. Analysis of global gene expression in Escherichia coli K12. J. Biol. Chem. 276, 19937–19944.
MacKichan, J. K., Gaynor, E. C., Chang, C., Cawthraw, S., Newell, D. G., Miller, J. F., and Falkow, S. (2004). The Campylobacter jejuni dccRS two-component system is required for optimal in vivo colonization but is dispensable for in vitro growth. Mol. Microbiol. 54, 1269–1286.
Mead, P. S., Slutsker, L., Dietz, V., McCaig, L. F., Bresee, J. S., Shapiro, C., Griffin, P. M., and Tauxe, R. V. (1999). Food-related illness and death in the United States. Emerg. Infect. Dis. 5, 607–625.
Miller, C. E., Rock, J. D., Ridley, K. A., Williams, P. H., and Ketley, J. M. (2008). Utilization of lactoferrin-bound and transferrin-bound iron by Campylobacter jejuni. J. Bacteriol. 190, 1900–1911.
Moore, J. E., Corcoran, D., Dooley, J. S., Fanning, S., Lucey, B., Matsuda, M., McDowell, D. A., Megraud, F., Millar, B. C., O’Mahony, R., O’Riordan, L., O’Rourke, M., Rao, J. R., Rooney, P. J., Sails, A., and Whyte, P. (2005). Campylobacter. Vet. Res. 36, 351–382.
Nachamkin, I., and Hart, A. M. (1985). Western blot analysis of the human antibody response to Campylobacter jejuni cellular antigens during gastrointestinal infection. J. Clin. Microbiol. 21, 33–38.
Palyada, K., Sun, Y. Q., Flint, A., Butcher, J., Naikare, H., and Stintzi, A. (2009). Characterization of the oxidative stress stimulon and PerR regulon of Campylobacter jejuni. BMC Genomics 10, 481.
Palyada, K., Threadgill, D., and Stintzi, A. (2004). Iron acquisition and regulation in Campylobacter jejuni. J. Bacteriol. 186, 4714–4729.
Poly, F., and Guerry, P. (2008). Pathogenesis of Campylobacter. Curr. Opin. Gastroenterol. 24, 27–31.
Raphael, B. H., Pereira, S., Flom, G. A., Zhang, Q., Ketley, J. M., and Konkel, M. E. (2005). The Campylobacter jejuni response regulator, CbrR, modulates sodium deoxycholate resistance and chicken colonization. J. Bacteriol. 187, 3662–3670.
Reid, A. N., Pandey, R., Palyada, K., Naikare, H., and Stintzi, A. (2008a). Identification of Campylobacter jejuni genes involved in the response to acidic pH and stomach transit. Appl. Environ. Microbiol. 74, 1583-97.
Reid, A. N., Pandey, R., Palyada, K., Whitworth, L., Doukhanine, E., and Stintzi, A. (2008b). Identification of Campylobacter jejuni genes contributing to acid adaptation by transcriptional profiling and genome-wide mutagenesis. Appl. Environ. Microbiol. 74, 1598–1612.
Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., Braisted, J., Klapa, M., Currier, T., Thiagarajan, M., Sturn, A., Snuffin, M., Rezantsev, A., Popov, D., Ryltsov, A., Kostukovich, E., Borisovsky, I., Liu, Z., Vinsavich, A., Trush, V., and Quackenbush, J. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378.
Skirrow, M. B., and Blaser, M. J. (2000). “Clinical aspects of Campylobacter infection,” in Campylobacter, 2nd Edn, eds I. Nachamkin and M. J. Blaser (Washington, DC: American Society for Microbiology), 69–88.
Stintzi, A. (2003). Gene expression profile of Campylobacter jejuni in response to growth temperature variation. J. Bacteriol. 185, 2009–2016.
Stintzi, A., Marlow, D., Palyada, K., Naikare, H., Panciera, R., Whitworth, L., and Clarke, C. (2005). Use of genome-wide expression profiling and mutagenesis to study the intestinal lifestyle of Campylobacter jejuni. Infect. Immun. 73, 1797–1810.
Sturn, A., Quackenbush, J., and Trajanoski, Z. (2002). Genesis: cluster analysis of microarray data. Bioinformatics 18, 207–208.
Wassenaar, T. M., van der Zeijst, B. A., Ayling, R., and Newell, D. G. (1993). Colonization of chicks by motility mutants of Campylobacter jejuni demonstrates the importance of flagellin A expression. J. Gen. Microbiol. 139(Pt 6), 1171–1175.
Weerakoon, D. R., and Olson, J. W. (2008). The Campylobacter jejuni NADH:ubiquinone oxidoreductase (complex I) utilizes flavodoxin rather than NADH. J. Bacteriol. 190, 915–925.
Woodall, C. A., Jones, M. A., Barrow, P. A., Hinds, J., Marsden, G. L., Kelly, D. J., Dorrell, N., Wren, B. W., and Maskell, D. J. (2005). Campylobacter jejuni gene expression in the chick cecum: evidence for adaptation to a low-oxygen environment. Infect. Immun. 73, 5278–5285.
Wosten, M. M., van Dijk, L., Parker, C. T., Guilhabert, M. R., van der Meer-Janssen, Y. P., Wagenaar, J. A., and van Putten, J. P. (2010). Growth phase-dependent activation of the DccRS regulon of Campylobacter jejuni. J. Bacteriol. 192, 2729–2736.
Wosten, M. M., Wagenaar, J. A., and van Putten, J. P. (2004). The FlgS/FlgR two-component signal transduction system regulates the fla regulon in Campylobacter jejuni. J. Biol. Chem. 279, 16214–16222.
Keywords: Campylobacter jejuni, animal model, host interactions, transcriptome
Citation: Flint A, Butcher J, Clarke C, Marlow D and Stintzi A (2010) Use of a rabbit soft tissue chamber model to investigate Campylobacter jejuni–host interactions. Front. Microbio. 1:126. doi: 10.3389/fmicb.2010.00126
Received: 13 August 2010;
Accepted: 25 October 2010;
Published online: 16 November 2010.
Edited by:
Rey Carabeo, Imperial College London, UKReviewed by:
Michael Konkel, Washington State University, USAStuart A. Thompson, Medical College of Georgia, USA
Paul Howard Everest, University of Glasgow, UK
Copyright: © 2010 Flint, Butcher, Clarke, Marlow and Stintzi. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Alain Stintzi, Department of Biochemistry, Microbiology and Immunology, Faculty of Medicine, Ottawa Institute of Systems Biology, University of Ottawa, 451 Smyth road, Ottawa, ON, Canada K1H 8M5. e-mail: astintzi@uottawa.ca