 Zhanna A. Ktsoyan1 Natalia V. Beloborodova2 Anahit M. Sedrakyan1 George A. Osipov3 Zaruhi A. Khachatryan1 Denise Kelly4 Gayane P. Manukyan1 Karine A. Arakelova1 Alvard I. Hovhannisyan1 Andrey Y. Olenin2 Arsen A. Arakelyan1 Karine A. Ghazaryan1 Rustam I. Aminov4*
Zhanna A. Ktsoyan1 Natalia V. Beloborodova2 Anahit M. Sedrakyan1 George A. Osipov3 Zaruhi A. Khachatryan1 Denise Kelly4 Gayane P. Manukyan1 Karine A. Arakelova1 Alvard I. Hovhannisyan1 Andrey Y. Olenin2 Arsen A. Arakelyan1 Karine A. Ghazaryan1 Rustam I. Aminov4*
- 1 Institute of Molecular Biology, National Academy of Sciences of Republic of Armenia, Yerevan, Republic of Armenia
- 2 Research Institute of General Reanimatology, Russian Academy of Medical Sciences, Moscow, Russian Federation
- 3 Scientific Center of Cardiovascular Surgery, Russian Academy of Medical Sciences, Moscow, Russian Federation
- 4 Rowett Institute of Nutrition and Health, University of Aberdeen, Aberdeen, UK
The human gastrointestinal tract is inhabited by a diverse and dense symbiotic microbiota, the composition of which is the result of host–microbe co-evolution and co-adaptation. This tight integration creates intense cross-talk and signaling between the host and microbiota at the cellular and metabolic levels. In many genetic or infectious diseases the balance between host and microbiota may be compromised resulting in erroneous communication. Consequently, the composition of the human metabolome, which includes the gut metabolome, may be different in health and disease states in terms of microbial products and metabolites entering systemic circulation. To test this hypothesis, we measured the level of hydroxy, branched, cyclopropyl and unsaturated fatty acids, aldehydes, and phenyl derivatives in blood of patients with a hereditary autoinflammatory disorder, familial Mediterranean fever (FMF), and in patients with peptic ulceration (PU) resulting from Helicobacter pylori infection. Discriminant function analysis of a data matrix consisting of 94 cases as statistical units (37 FMF patients, 14 PU patients, and 43 healthy controls) and the concentration of 35 microbial products in the blood as statistical variables revealed a high accuracy of the proposed model (all cases were correctly classified). This suggests that the profile of microbial products and metabolites in the human metabolome is specific for a given disease and may potentially serve as a biomarker for disease.
Introduction
The human gastrointestinal tract (GIT) represents the main site where intensive cross-talk takes place between the host and the immense, diverse commensal microbiota. In this host–microbe dialog (Bäckhed et al., 2005; Sartor, 2006) microbial components and metabolites are important biomolecules, contributing to the structural and functional stability in the complex GIT environment (Zoetendal et al., 2008). Many disorders in humans are due to a restructured microbiota, which fails to “communicate” properly with its host thus leading to aberrant host responses and contributing to the disease state (Farthing, 2004; Stecher and Hardt, 2008).
Much less, however, is known about the metabolic events along the gut microbiota–metabolite–host signal chain associated with the risk of inflammation and subsequent host disease. There is evidence suggesting that gut microbiota alterations correlate with a wide spectrum of human diseases, both within and outside the GIT (Frank et al., 2007; Adams et al., 2008; Jia et al., 2008). Even if a disease results from a genetic predisposition, the normal gut microbiota may be transformed into a “pathobiota” inflicting the disease on a genetically resistant host (Garrett et al., 2007). Structural changes in the gut microbiota in disease are predictably accompanied by changes in the gut metabolome (Jansson et al., 2009) but the role of these metabolites in disease remains elusive.
We demonstrated recently that in an autoinflammatory disorder, familial Mediterranean fever (FMF, MIM249100), the commensal gut microbiota composition is specifically restructured and that the observed structural changes are determined by the host genotype (Khachatryan et al., 2008). This disease, which is caused by mutations in the MEFV gene, is characterized by periodic attacks of acute inflammation and fever, followed by periods of remission without any clinical sign of disease (The French FMF Consortium, 1997; The International FMF Consortium, 1997). However, the elevated levels of cytokines and C-reactive protein in remission suggest the persisting subclinical inflammation (Manukyan et al., 2008a). In the present work, we attempted to extend the investigation beyond structural analysis of the gut microbial community and establish if the restructured gut microbiota affects the human systemic metabolome.
Patients with peptic ulcer (PU) due to Helicobacter pylori infection were chosen as a comparative disease model for this metabolomic study because it is a GIT inflammatory disease of known infectious nature. Although there is a contributory role of host genetics in the disease outcome (Snaith and El-Omar, 2008) the infection itself is a major driving force in development of the disease (Makola et al., 2007). Cultivation-independent studies revealed a much higher microbial diversity in the healthy stomach than previously thought (Bik et al., 2006). Infection by this well-adapted pathogen drastically reduces microbial diversity and it becomes the dominant species in this ecological niche (Andersson et al., 2008). The H. pylori colonization induces a chronic inflammatory response with the risk of a broad range of the upper GIT disorders, including gastritis, PU, gastric cancer, and mucosa-associated lymphoid tissue lymphoma (Israel and Peek, 2006; Amieva and El-Omar, 2008). Using this disease model, it is possible to discover if the changes in the metabolome are attributable to the products of a single infective agent or if the situation is more complex. Whilst the role of H. pylori as an initiator of inflammation, as well as a modifier of the microbial environment is evident, the influence of its colonization of the gut on the host metabolome remains largely uncharacterized.
The rapidly developing field of metabolomics is an essential part of understanding the functionality of complex biological systems. The application of highly automated sequencing methodologies has tremendously changed the landscape of biological and biomedical research. However, in the post-genomic era the wealth of genotype information has to be complemented by phenotype characterization because it is only at this level that it is possible to assess the outcome of genotype–environment interaction. In gut microbiology, for example, a wealth of information is accumulated about the structure of microbial communities and the corresponding gene repertoires in health and disease. Much less is known, though, about the outcome of such genotypic differences at a mechanistic level. Metabolomics attempts to comprehensively analyze small molecules characterizing intact biological systems and in this regard mass spectrometry (MS) is the method of choice for metabolomic studies because it allows highly robust, reproducible, and sensitive qualitative or quantitative analysis (Koal and Deigner, 2010).
In this work, two disease states, FMF and PU, were studied using the gas chromatography/mass spectrometry (GC/MS) technique to analyze microbial products and metabolites encountered in systemic circulation. The profile of hydroxy, branched, cyclopropyl and unsaturated fatty acids, aldehydes, and phenyl derivatives in these two diseases was distinct from the healthy state and was also disease-specific.
Materials and Methods
Subjects
A total of 37 FMF patients (13 in remission and 24 in attack periods), 14 PU patients in exacerbation period, and 43 healthy controls participated in the study. All FMF cases were diagnosed based on Tel-Hashomer criteria with further genetic confirmation of MEFV mutations. All PU patients were diagnosed based on clinical symptoms (pain or discomfort in the upper abdomen, dyspepsia, bloating, an early sense of fullness after eating, acid reflux, lack of appetite, nausea, water brash, vomiting, blood in stool), and were H. pylori-positive by H. pylori breath test. The control group comprised of healthy volunteers without family history of FMF or any previously diagnosed PU disease. All study participants gave their informed consent to the protocol, which was approved by the local ethical committee at IMB NAS RA.
Sample Preparation
Blood samples for GC/MS analysis were collected into sterile heparinized vials and immediately frozen at −20°C until analysis. Defrosted blood samples were air dried at 80°C and subjected to acid methanolysis using 1 M HCl in methanol. As a result, fatty acids and aldehydes were extracted from complex lipids of microorganisms in the form of methyl esters and aldehyde dimethyl acetals. Internal standard, deuteromethyl ether of tridecane acid, prepared in hexane, was added to the cooled reaction mixture at 300 ng. The fraction of methyl esters of fatty acids with other lipid components was extracted twice from the reaction mixture with 200 μl of hexane. Phenyl-carbonic acids were extracted from the acidified whole blood (pH 2.0) with diethyl ester. The hexane and ester extracts were dried at 40°C, and the dry residue was treated with 20 μl of N,O-bis-(trimethylsilyl)trifluoroacetamide for 15 min at 80°C to form trimethylsilyl derivatives of hydroxy acids (Beloborodova and Osipov, 2000).
GC/MS Analysis
A GC/MS system (Agilent AT-5973, Agilent Technologies) with a cross-linked methyl silicone capillary column (DB-5 ms EVDX; dimensions: 0.2 mm × 25 m × 0.33 mkm, Agilent Technologies) was used to identify microbial components and metabolites in blood. Two microliters of the samples derived were injected into the capillary column. Fatty acids and other lipid components separated in the GC column were analyzed in the selected ion-monitoring (SIM) mode (multi-ion mass-fragmentography regimen). The substances eluted were identified by measuring the specific retention times and the ratio of chromatographic peak areas for selected ions were measured primarily in automatic mode by using standard library search program (NIST 02 MS library), with subsequent manual checking of peaks. Correlation between the products identified and the corresponding source microorganisms was carried out as described before (Osipov and Turova, 1997; Osipov et al., 2009) using the Sherlock® microbial identification system (MIS; www.midi-inc.com).
Statistical Analysis
Statistical analysis was performed using the SPSS package (SPSS Inc., Chicago, IL, USA). Mann–Whitney U-test was applied to determine statistical significance among the mean values of studied groups. p-Values below 0.05 were considered statistically significant. Discriminant function analysis (DA) was used to classify cases into groups. The investigated groups of subjects were taken as dependent variables of DA, and microbial substances and metabolites in blood were taken as predictors.
Results
The GC/MS approach was used in this study to detect and evaluate the concentration of products of microbial origin in blood of FMF (remission and attack) and PU patients compared to healthy control subjects (Table 1). For convenience, the chemically related structural compounds of microorganisms are placed together and presented as groups of hydroxy, branched, cyclopropyl and unsaturated acids, and aldehydes. Phenolic compounds, which are mostly the products of microbial metabolism, are presented as a separate group. Putative microbial sources of particular compounds are also shown.
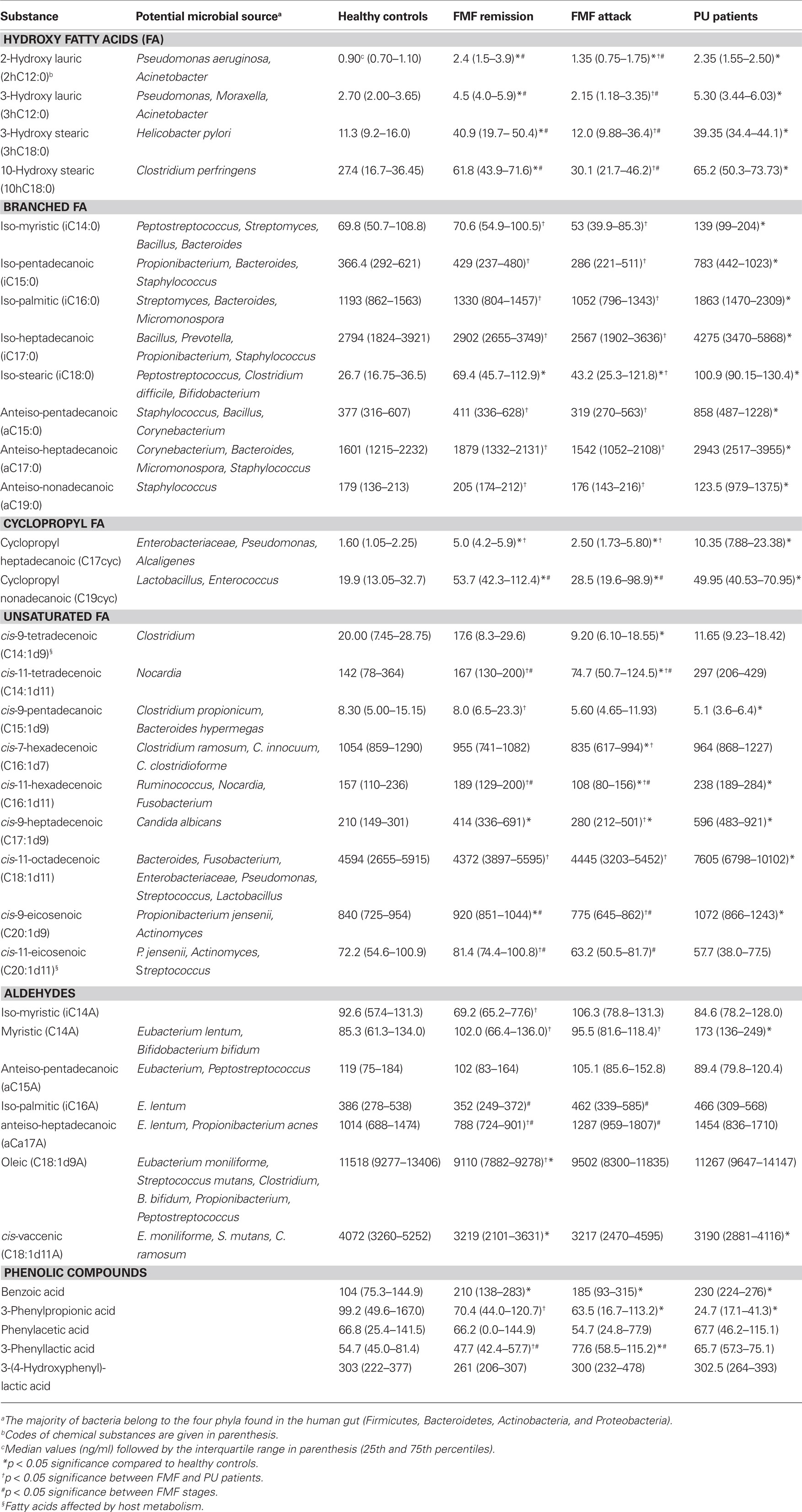
Table 1. Concentration of compounds and their potential microbial source in blood of FMF and PU patients and healthy controls.
Hydroxy Fatty Acids
Significantly higher levels of hydroxy acids were detected in blood of PU patients and FMF patients in remission compared with healthy controls, whereas in an FMF attack it was found to be comparable with the controls, except for 2-hydroxy lauric acid. Further comparison among the diseased cohorts revealed a significant reduction of these substances in acute FMF patients compared to either FMF patients in remission or in PU patients. The corresponding values between FMF patients in remission and in PU patients were not significantly different. Surprisingly, no difference in concentration of H. pylori marker, 3-hydroxy stearic acid, was found between the FMF remission and PU diseases, whilst these values exceeded the normal controls by up to four-fold. A similar tendency, with a two-fold increase, was also observed in the case of 10-hydroxy stearic acid, a reference marker for Clostridium perfringens. It is important to note that 2-hydroxy lauric and 3-hydroxy lauric acids are the signature compounds of non-fermentative Gram-negative bacteria (NFB; Table 1).
Cyclopropyl Fatty Acids
In all three disease cohorts the concentration of cyclopropyl heptadecanoic acid (17cyc) was significantly higher compared to the norm (Table 1). It is also a signature compound of NFB further supporting the overrepresentation of this group in these diseased states. Cyclopropyl nonadecanoic acid, which is associated with Lactobacillus and Enterococcus showed a similar tendency for elevation in all diseases studied.
Branched Fatty Acids
Unidirectional inter-cohort alterations were observed for the branched fatty acids studied, which are mostly the components of commensal microbiota. In particular, the average levels of these acids were not significantly different between FMF remission and the normal controls, and tended to be less represented in blood of acute FMF patients compared to healthy subjects; whereas high concentrations of these compounds were associated with PU disorder. The correlation of these acids with their potential microbial source is evidence of elevated content of such bacteria as Peptostreptococcus, Streptomyces, Bacillus, Bacteroides, Propionibacterium, and Micromonospora in PU disorders. The only exceptions to this tendency were observed in the case of iso-stearic (i18) and anteiso-nonadecanoic (a19) acids. The latter was detected in significantly lower concentrations in PU patients compared to other groups. Concentrations of iso-stearic acid were found to be higher in samples from both FMF and PU patients compared to controls.
Unsaturated Fatty Acids
In contrast to iso- and anteiso-fatty acids, the levels of unsaturated fatty acids were highly variable among the groups studied. The highest concentration of unsaturated fatty acids was detected mainly in PU patients suggesting an overrepresentation of the associated microorganisms in these patients, with the exception of C15:1d9, which was lower compared to all other groups (Table 1). The values for FMF remission patients and healthy controls were approximately equal. In the FMF attack state, levels of unsaturated acids were reduced compared to other groups (or differences were not statistically significant) and only cis-9-heptadecenoic acid, an indicator of Candida albicans, was increased compared to the controls.
Aldehydes
Measurement of this group of compounds in the human metabolome revealed a significantly higher concentration of myristic aldehyde, an indicator of Eubacterium lentum and Bifidobacterium, in PU patients compared to other groups studied (Table 1). Elevated concentrations of iso-myristic, oleic, and anteiso-heptadecanoic aldehydes were found in blood of PU patients compared to FMF patients in remission. The latter substance along with iso-palmitic aldehyde was also detected at higher concentrations in patients in the acute phase of FMF. Both of these markers are indicators of Eubacterium and Propionibacterium. In all disease cohorts the concentration of cis-vaccenic aldehyde was uniformly reduced compared to control.
Phenolic Compounds
These are products of dietary aromatic amino acid transformation by intestinal microbiota. A number of significant differences were also observed in profiles of these compounds, particularly in benzoic, 3-phenylpropionic, and 3-phenyllactic acids (Table 1). Benzoic acid increased in both FMF disease states compared to the healthy control group, with considerable elevation of levels in blood of PU patients. In contrast, the level of 3-phenylpropionic acid was reduced in the diseased cohorts compared to the normal controls, reaching a four-fold reduction in PU patients. In particular, 3-phenyllactic acid levels significantly increased in the FMF attack state compared to normal controls or FMF remission. In the latter group, the level of 3-phenyllactic acid was significantly lower than in PU patients.
Discriminant Function Analysis
Although the pair-wise comparisons revealed significant changes in the human metabolome due to disease, it is not clear if these changes are disease-specific or not. Evidence that some groups of microorganisms may be over- or under-represented in these diseases provides indirect support for disease-specific changes (Table 1). If true, then the level of specific microbial products should correlate with those originating from the same group of microorganisms. In addition, we have demonstrated previously that in the case of FMF patients the gut microbiota is restructured (Khachatryan et al., 2008), which possibly leads to the specific shifts observed in the range of microbial compounds and metabolites produced.
To test this hypothesis, the raw data were subjected to multivariate DA to determine the distribution of low-weight microbial patterns from the complex datasets obtained from each group studied, and the resulting scatterplot of DA calculations is shown in Figure 1. DA of a data matrix consisting of 94 cases as statistical units (13 cases of FMF remission, 24 – acute FMF, 14 – PU, and 43 – healthy controls) and the concentration of 35 microbial products in peripheral blood (Table 1) as statistical variables revealed a 100% accuracy of disease classification (Table 2).
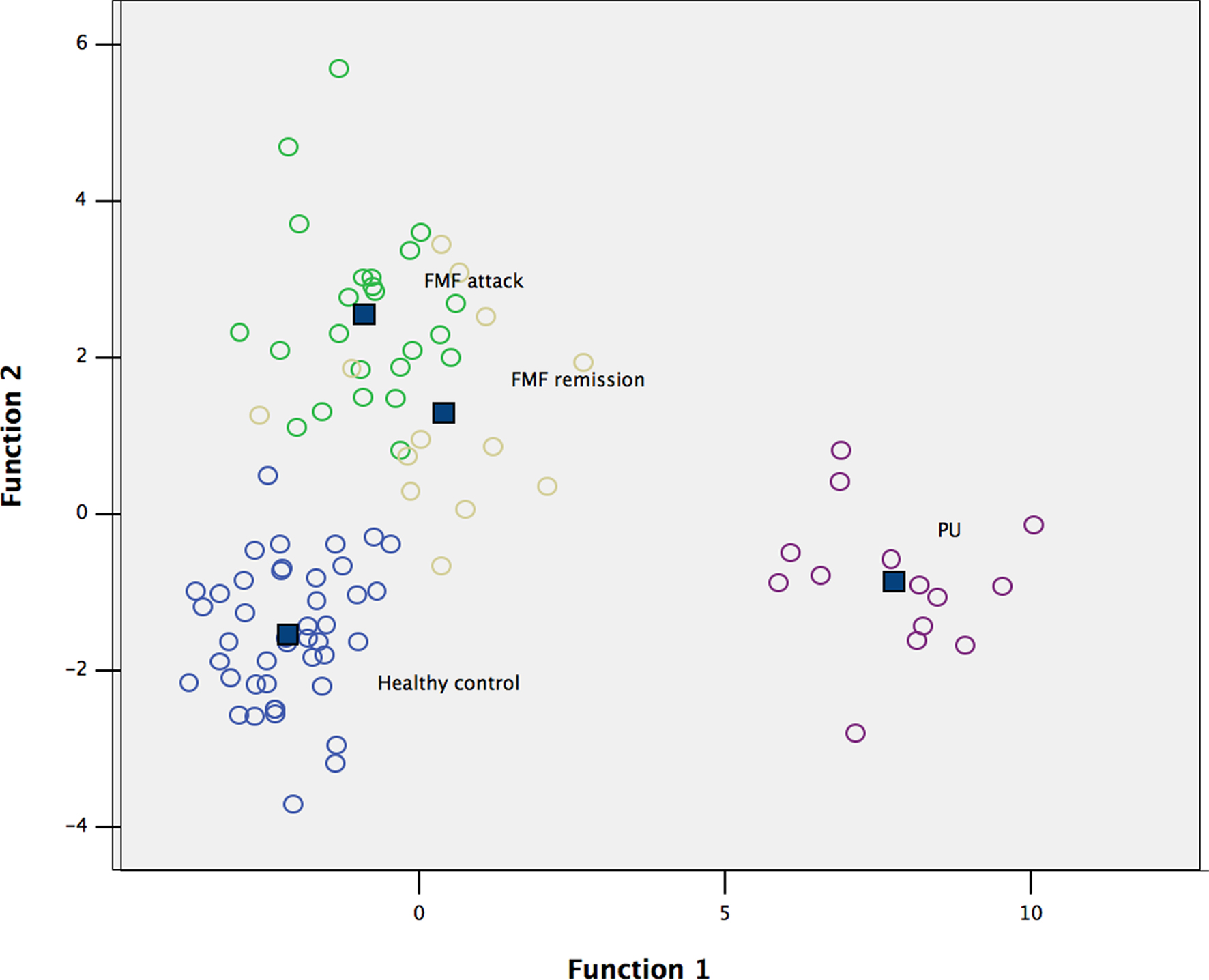
Figure 1. Scatterplot of DA model based on concentration of fatty acids and phenolics in blood. Number of variables in model: 35; grouping – 4 groups. Root 1, 2 – discriminant functions 1 and 2 (first and second canonical roots). Healthy controls are shown in blue circles; FMF attack – in green circles; FMF remission – in yellow circles; and PU – in purple circles. Class centers are shown in dark blue squares.
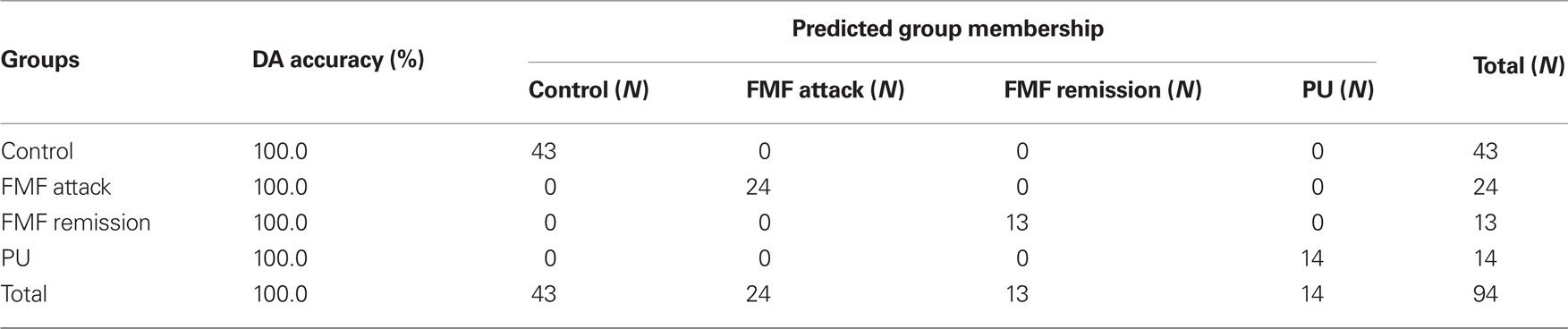
Table 2. Classification table of correct estimates based on 35 variables set.
The statistical significance of the DA model was also supported by the Wilks’ lambda test (Table 3).
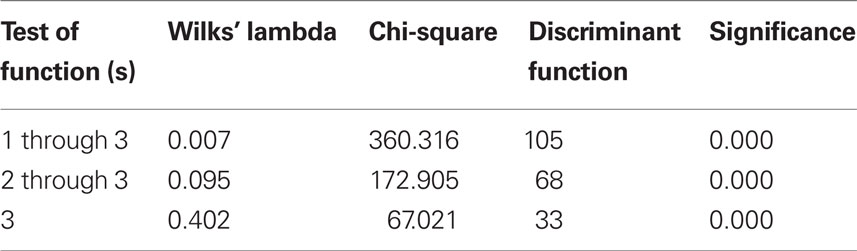
Table 3. Wilks’ lambda test of class center separation.
The DA model, which is based on the set of markers of microbial origin, has good predictive accuracy resulting in excellent separation of the diseased and healthy cohorts studied. This statistical analysis also supports the hypothesis that changes in the human metabolome are disease-specific.
Discussion
In this work, we have investigated the influence of human gut microbiota on the human metabolome through a pioneering approach involving the quantification of bacterial structural components, long chain fatty acids (LCFAs), present in human systemic circulation. The concentration of 30 molecules comprising hydroxy, branched, cyclopropyl and unsaturated fatty acids and aldehydes was assessed using GS/MS analysis of peripheral blood from healthy control subjects and was compared with the corresponding values in patients with a hereditary autoinflammatory disorder (FMF) and in patients with PU caused by H. pylori infection (94 subjects in total). The concentration of five phenolic compounds was also measured in these cohorts. Statistically significant differences were observed in the level of LCFAs and phenolics in healthy and disease states. Moreover, DA analysis confirmed that this set of variables is sufficient for accurate classification of disease state and suggests that metabolomic profiles based on bacterial structural components and metabolites are disease-specific.
Previously, we established that the gut microbial community is restructured in FMF (Khachatryan et al., 2008). The present work complements this finding and suggests that the structural changes observed are accompanied by corresponding changes in the human systemic metabolome. In particular, we note a statistically significant increase of the γ-proteobacteria-related SSU rRNA sequences in the gut community of FMF patients in remission, compared to control patients (Khachatryan et al., 2008). In the present work, the concentration of specific markers for this group of bacteria (2hC12:0, 3hC12:0, and C17cyc in Table 1) also increased significantly in the metabolome of FMF patients. A similar trend was observed for PU patients (Table 1). The markers 2hC12:0 and 3hC12:0 are precursors of lipid A and subsequently lipopolysaccharide (LPS) biosynthesis in one of the representatives of γ-proteobacteria, Pseudomonas aeruginosa (Goldman et al., 1988). However, the monomers themselves are only weak inducers of inflammation and the pro-inflammatory effect. For example, the effect of 3hC12:0 can only be seen at much higher concentrations than detected in this work (Soydan et al., 2006). It may be important to note that the monomers detected are released by the acid catalyzed methanolysis used during the analysis procedure, and the original compound in blood may well be LPS, which is an extremely potent inducer of the inflammatory cascade even at low concentrations (Beutler and Rietschel, 2003). The microbial community structure shifts observed in disease may not only result in microbial compounds that are presently considered as neutral to the host but also in products potentially contributing to the diseased state. This may, in part, explain how the normal gut microbiota can acquire pathogenic properties through the selection and restructuring of microbial community structure in the diseased host (Garrett et al., 2007, 2010).
The concentration of phenolic compounds in the normal human gut is usually high (Jenner et al., 2005) and these are mainly the products of degradation of aromatic amino acids in the large intestine. Phenylpropionate, phenylacetate, and phenyllactate that we measured in systemic circulation in this study are the products of phenylalanine catabolism by gut bacteria (Macfarlane and Cummings, 1991). The pool of benzoic acid, in contrast, arises from several sources, including direct dietary intake (berries and food preservatives) and as a byproduct of dietary phenylalanine and polyphenols metabolism in gut bacteria. The only hydroxyphenyl-containing molecule measured, hydroxyphenyllactate, is the product of endogenous tyrosine metabolism, ingested as a dietary component or obtained by hydroxylation of phenylalanine. The level of hydroxyphenyllactic acid is elevated in patients with inherited deficiency of the enzyme p-hydroxyphenylpyruvate oxidase (Wishart et al., 2009) and we observed no difference in the level of this compound among our cohorts. Phenolic compounds may influence a range of cellular functions including differentiation, apoptosis, mitochondrial function, and inflammation (Li et al., 2004; Karlsson et al., 2005; Fedotcheva et al., 2008).
We found that the plasma concentrations of phenylpropionate in both PU and FMF attack state patients were significantly reduced (Table 1). This compound inhibits the TNF-α-stimulated induction of cyclooxygenase-2 (COX-2) protein in HT29 cells leading to the suppression of COX-2-catalyzed prostaglandin biosynthesis involved in inflammation (Karlsson et al., 2005). Thus the lower concentration of phenylpropionate found in PU and FMF patients may be a factor contributing to the maintenance of chronic inflammation in these two diseases. Interestingly, despite that these phenyl-containing compounds are all derived from the catabolism of the same amino acid, phenylalanine, the level of resulting products is very different between cohorts. This may reflect the structural differences in microbiota that possess different metabolic routes or catabolic efficiencies in transformation of phenyl-containing compounds.
Benzoic acid is detoxified as a conjugate with glycine and excreted through the hepatic and renal routes (Tremblay and Qureshi, 1993). The rate of conjugation of benzoic acid with glycine varies among subjects but it is normally distributed (Temellini et al., 1993). We found a statistically significant increase in systemic benzoate concentration in all disease cohorts compared to control (Table 1). Due to its comparatively larger mass the liver is the main route of benzoylglycine disposal, but, there is no indication that liver function is compromised in these two diseases, which may result in a higher residual benzoate level. Moreover, the limiting step in the formation of hippurate is availability of glycine (Beliveau and Brusilow, 1987) and there is no literature data suggesting any abnormality of glycine metabolism in these two diseases. Therefore, the statistically significant increase in systemic benzoic acid concentration observed in PU and FMF patients may be due to a restructured gut microbiota, with a higher proportion of gut bacteria that are more efficient at degradation of dietary plant polyphenols.
The human metabolome results from the combination of genetic and environmental components such as diet, ethnicity and geographical location, disease, and other factors (Rezzi et al., 2007; Holmes et al., 2008; Li et al., 2008). Being tightly integrated within the host in a symbiotic relationship, the gut microbiome is one of the major contributors to a diverse range of endogenous metabolites found in the human metabolome. The best example of metabolites produced by the gut microbiota are probably short chain fatty acids produced in large quantities, part of which such as butyrate is metabolized in situ by colonic epithelium and the remainder including acetate and propionate enter systemic circulation to serve the energy and biosynthetic needs of the host (Cummings, 1994). The role of these end products of bacterial metabolism is not limited to supplying the catabolic and anabolic needs of the host but may also have clinical implications. The effect of decreased availability of butyrate, for example, may result in a reduction of their protective effect toward bowel cancer in inflammatory bowel diseases (Thibault et al., 2010).
The range of systemic bacterial metabolites under investigation is constantly increasing in order to find suitable biomarkers for evaluation of various aspects of human health. A novel aspect of this study is that, in addition to the products of bacterial metabolism such as phenolic compounds, we also attempted to quantify a broad range of other molecules in the human metabolome, which are not the bacterial metabolites per se but are structural components of microbial cells. Because of the extremely potent effects on the host that have been discovered more than a century ago, the best-studied example among the bacterial structural components is LPS. It is a major component of the outer membrane in Gram-negative bacteria and consists of lipid A (or endotoxin), the core oligosaccharide, and the O antigen (Raetz and Whitfield, 2002). In addition to the more obvious cases of infectious disease, plasma LPS concentration is responsive to diet suggesting that diet-related shifts in the population structure of gut microbiota may be responsible for this phenomenon because dietary LPS intake was similar between the diets (Ghanim et al. 2009). We also found that LPS-related monomer levels increased in plasma of FMF and PU patients. Restructured microbiota with overrepresentation of γ-proteobacteria (Khachatryan et al., 2008) and impaired gut barrier function due to chronic inflammation in these diseases may lead to an elevated level of these bacterial structural components in the human metabolome. We established earlier that in FMF the level of systemic antibodies toward antigens of gut commensal bacteria is elevated suggesting that the gut barrier function may be compromised in this disease (Manukyan et al., 2008b).
Very little is known about the concentration of other bacterial structural compounds entering into, and circulating in, the human metabolome. In this respect, LCFAs represent good candidates for measuring the impact of changes in the gut microbiota on the human metabolome. The repertoire of endogenous LCFAs in humans (and eukaryotes in general) is limited because the biosynthetic needs are served by a single type I fatty acid synthase (Lomakin et al., 2007; Maier et al., 2008), whilst bacteria synthesize a wide diversity of different types of fatty acids through type II fatty acid synthase systems (Lu et al., 2004). The LCFAs profile of a given bacterium is highly specific and GC/MS may serve as a rapid and sensitive method for identification or detection of bacteria (Li et al., 2010). Bacterial ecosystems may, therefore, be characterized by unique sets of LCFAs “fingerprints” specific for given microbial communities. In this work, although not measured directly in the gut microbiota, the bacterial LCFAs fingerprints observed in the human metabolome were highly specific for the cohorts studied (Figure 1). This observation correlates well with our previous analyses of microbial community structure performed using different SSU rRNA-based molecular ecology approaches (Khachatryan et al., 2008). Interestingly, the LCFAs profile in the metabolome of PU patients suggests extensive structural changes in the gut microbiota involving other bacterial populations in the lower parts of the GIT rather than a local effect of a single infectious agent, H. pylori, in the stomach. This suggests that further investigations of community structure in lower parts of the GIT in this disease using molecular ecology approaches may be useful.
Need to note, however, that not all fatty acids measured in the metabolome are exclusively of microbial origin. Some unsaturated fatty acids such as myristoleic acid (cis-9-tetradecenoic acid) may also be formed endogenously from the dietary myristic acid by the enzyme stearoyl-CoA desaturase (delta-9 desaturase; Wishart et al., 2009). This enzyme is a key regulator of lipid metabolism and plays an important role in human metabolic disease (Sampath and Ntambi, 2008). The high-saturated as well as monounsaturated-fat diets can increase its expression and the contribution of microbial fatty acids on this highly regulated enzyme has to be elucidated. Another unsaturated fatty acid, cis-11-eicosenoic acid, is found in a variety of plant oils (Wishart et al., 2009) and part of it in the human metabolome may be of dietary origin. Increased concentration of eicosenoic acid in red blood cells is associated with autism in children (Bu et al., 2006). Thus the microbially produced fatty acids may intercalate into the endogenous fatty acid metabolism affecting the human health.
In conclusion, in this study we have established that significant concentrations of microbial LCFAs present in the human metabolome and that the profile of these LCFAs is specific for the healthy and diseased states. The LCFA profile therefore may potentially serve as a biomarker for disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the International Science and Technology Centre (project A-1055). Rustam I. Aminov is supported by the Scottish Government Rural and Environment Research and Analysis Directorate. We thank Dr. A. J. Travis for proofreading the manuscript.
References
Adams, R. J., Heazlewood, S. P., Gilshenan, K. S., O’Brien, M., McGuckin, M. A., and Florin, T. H. (2008). IgG antibodies against common gut bacteria are more diagnostic for Crohn’s disease than IgG against mannan or flagellin. Am. J. Gastroenterol. 103, 386–396.
Amieva, M. R., and El-Omar, E. M. (2008). Host–bacterial interactions in Helicobacter pylori infection. Gastroenterology 134, 306–323.
Andersson, A. F., Lindberg, M., Jakobsson, H., Bäckhed, F., Nyrén, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3, e2836. doi: 10.1371/journal.pone.0002836
Bäckhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A., and Gordon, J. I. (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920.
Beliveau, G. P., and Brusilow, S. W. (1987). Glycine availability limits maximum hippurate synthesis in growing rats. J. Nutr. 117, 36–41.
Beloborodova, N. V., and Osipov, G. A. (2000). Small molecules originating from microbes (SMOM) and their role in microbes–host relationship. Microb. Ecol. Health Dis. 12, 12–21.
Beutler, B., and Rietschel, E. T. (2003). Innate immune sensing and its roots: the story of endotoxin. Nat. Rev. Immunol. 3, 169–176.
Bik, E. M., Eckburg, P. B., Gill, S. R., Nelson, K. E., Purdom, E. A., Francois, F., Perez-Perez, G., Blaser, M. J., and Relman, D. A. (2006). Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. U.S.A. 103, 732–737.
Bu, B., Ashwood, P., Harvey, D., King, I. B., Water, J. V., and Jin, L. W. (2006). Fatty acid compositions of red blood cell phospholipids in children with autism. Prostaglandins Leukot. Essent. Fatty Acids 74, 215–221.
Cummings, J. (1994). “Quantitative short chain fatty acid production in humans,” in Short-chain Fatty Acids, eds J. Cummings, H. J. Binder, and K. Soergel (Boston: Kluwer Academic Publisher), 11–19.
Farthing, M. J. (2004). Bugs and the gut: an unstable marriage. Best Pract. Res. Clin. Gastroenterol. 18, 233–239.
Fedotcheva, N. I., Kazakov, R. E., Kondrashova, M. N., and Beloborodova, N. V. (2008). Toxic effects of microbial phenolic acids on the function of mitochondria. Toxicol. Lett. 180, 182–188.
Frank, D. N., Amand, A. L., Feldman, R. A., Boedeker, E. C., Harpaz, N., and Pace, N. R. (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. U.S.A. 104, 13780–13785.
Garrett, W. S., Gallini, C. A., Yatsunenko, T., Michaud, M., DuBois, A., Delaney, M. L., Punit, S., Karlsson, M., Bry, L., Glickman, J. N., Gordon, J. I., Onderdonk, A. B., and Glimcher, L. H. (2010). Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300.
Garrett, W. S., Lord, G. M., Punit, S., Lugo-Villarino, G., Mazmanian, S. K., Ito, S., Glickman, J. N., and Glimcher, L. H. (2007). Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45.
Ghanim, H., Abuaysheh, S., Sia, C. L., Korzeniewski, K., Chaudhuri, A., Fernandez-Real, J. M., and Dandona, P. (2009). Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care 32, 2281–2287.
Goldman, R. C., Doran, C. C., Kadam, S. K., and Capobianco, J. O. (1988). Lipid A precursor from Pseudomonas aeruginosa is completely acylated prior to addition of 3-deoxy-d- manno-octulosonate. J. Biol. Chem. 263, 5217–5223.
Holmes, E., Loo, R. L., Stamler, J., Bictash, M., Yap, I. K., Chan, Q., Ebbels, T., De Iorio, M., Brown, I. J., Veselkov, K. A., Daviglus, M. L., Kesteloot, H., Ueshima, H., Zhao, L., Nicholson, J. K., and Elliott, P. (2008). Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 453, 396–400.
Israel, D. A., and Peek, R. M. (2006). The role of persistence in Helicobacter pylori pathogenesis. Curr. Opin. Gastroenterol. 22, 3–7.
Jansson, J., Willing, B., Lucio, M., Fekete, A., Dicksved, J., Halfvarson, J., Tysk, C., and Schmitt-Kopplin, P. (2009). Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS ONE 4, e6386. doi: 10.1371/journal.pone.0006386
Jenner, A. M., Rafter, J., and Halliwell, B. (2005). Human fecal water content of phenolics: the extent of colonic exposure to aromatic compounds. Free Radic. Biol. Med. 38, 763–772.
Jia, W., Li, H., Zhao, L., and Nicholson, J. K. (2008). Gut microbiota: a potential new territory for drug targeting. Nat. Rev. Drug Discov. 7, 123–129.
Karlsson, P. C., Huss, U., Jenner, A., Halliwell, B., Bohlin, L., and Rafter, J. J. (2005). Human fecal water inhibits COX-2 in colonic HT-29 Cellc: role of phenolic compounds. J. Nutr. 135, 2343–2349.
Khachatryan, Z. A., Ktsoyan, Z. A., Manukyan, G. P., Kelly, D., Ghazaryan, K. A., and Aminov, R. I. (2008). Predominant role of host genetics in controlling the composition of gut microbiota. PLoS ONE 3, e3064. doi: 10.1371/journal.pone.0003064
Koal, T., and Deigner, H. P. (2010). Challenges in mass spectrometry based targeted metabolomics. Curr. Mol. Med. 10, 216–226.
Li, M., Wang, B., Zhang, M., Rantalainen, M., Wang, S., Zhou, H., Zhang, Y., Shen, J., Pang, X., Zhang, M., Wei, H., Chen, Y., Lu, H., Zuo, J., Su, M., Qiu, Y., Jia, W., Xiao, C., Smith, L. M., Yang, S., Holmes, E., Tang, H., Zhao, G., Nicholson, J. K., Li, L., and Zhao, L. (2008). Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. U.S.A. 105, 2117–2122.
Li, X. N., Parikh, S., Shu, Q., Jung, H. L., Chow, C. W., Perlaky, L., Leung, H. C., Su, J., Blaney, S., and Lau, C. C. (2004). Phenylbutyrate and phenylacetate induce differentiation and inhibit proliferation on human medulloblastoma cells. Clin. Cancer Res. 10, 1150–1159.
Li, Y., Wu, S., Wang, L., Li, Y., Shi, F., and Wang, X. (2010). Differentiation of bacteria using fatty acid profiles from gas chromatography-tandem mass spectrometry. J. Sci. Food Agric. 90, 1380–1383.
Lomakin, I. B., Xiong, Y., and Steitz T. A. (2007). The crystal structure of yeast fatty acid synthase, a cellular machine with eight active sites working together. Cell 129, 319–332.
Lu, Y. J., Zhang, Y. M., and Rock, C. O. (2004). Product diversity and regulation of type II fatty acid synthases. Biochem. Cell Biol. 82, 145–155.
Macfarlane, G. T., and Cummings, J. H. (1991). “The colonic flora, fermentation and large bowel digestive function,” in The Large Intestine: Physiology, Pathophysiology and Disease, eds S. F. Phillips, J. H. Pemberton, and R. G. Shorter (New York: Raven Press), 51–92.
Maier, T., Leibundgut, M., and Ban, N. (2008). The crystal structure of a mammalian fatty acid synthase. Science 321, 1315–1322.
Makola, D., Peura, D. A., and Crowe, S. E. J. (2007). Helicobacter pylori infection and related gastrointestinal diseases. J. Clin. Gastroenterol. 42, 548–558.
Manukyan, G. P., Ghazaryan, K. A., Ktsoyan, Z. A., Tatyan, M. V., Khachatryan, Z. A., Hakobyan, G. S., Mkrtchyan, V. A., Kelly, D., Coutts, A., and Aminov. R. I. (2008a). Cytokine profile of Armenian patients with familial Mediterranean fever. Clin. Biochem. 41, 920–922.
Manukyan, G. P., Ghazaryan, K. A., Ktsoyan, Z. A., Khachatryan, Z. A., Arakelova, K. A., Kelly, D., Grant, G., and Aminov, R. I. (2008b). Elevated systemic antibodies towards commensal gut microbiota in autoinflammatory condition. PLoS ONE 3, e3172. doi: 10.1371/journal.pone.0003172
Osipov, G. A., Boiko, N. B., Fedosova, N. F., Kasikhina, S. A., and Lyadov, K. V. (2009). Comparative gas chromatography-mass spectrometry study of the composition of microbial chemical markers in faeces. Microb. Ecol. Health Dis. 21, 159–171.
Osipov, G. A., and Turova, E. S. (1997). Studying species composition of microbial communities with the use of gas chromatography-mass spectrometry. Microbial community of kaolin. FEMS Microbiol. Rev. 20, 437–446.
Raetz, C. R., and Whitfield, C. (2002). Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–670.
Rezzi, S., Ramadan, Z., Martin, F. P., Fay, L. B., van Bladeren, P., Lindon, J. C., Nicholson, J. K., and Kochhar, S. (2007). Human metabolic phenotypes link directly to specific dietary preferences in healthy individuals. J. Proteome Res. 6, 4469–4477.
Sampath, H., and Ntambi, J. M. (2008). Role of stearoyl-CoA desaturase in human metabolic disease. Future Lipidol. 3, 163–173.
Sartor, R. B. (2006). Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 390–407.
Snaith, A., and El-Omar, E. M. (2008). Helicobacter pylori: host genetics and disease outcomes. Expert Rev. Gastroenterol. Hepatol. 2, 577–585.
Soydan, A. S., Dokmetas, H. S., Cetin, M., Koyuncu, A., Kaptanoglu, E., and Elden, H. (2006). The evaluation of the role of beta-hydroxy fatty acids on chronic inflammation and insulin resistance. Mediators Inflamm. 2006, 64980.
Stecher, B., and Hardt, W. D. (2008). The role of microbiota in infectious disease. Trends Microbiol. 16, 107–114.
Temellini, A., Mogavero, S., Giulianotti, P. C., Pietrabissa, A., Mosca, F., and Pacifici, G. M. (1993). Conjugation of benzoic acid with glycine in human liver and kidney: a study on the interindividual variability. Xenobiotica 12, 1427–1433.
The French FMF Consortium. (1997). A candidate gene for familial Mediterranean fever. Nat. Genet. 17, 25–31.
The International FMF Consortium. (1997). Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90, 797–807.
Thibault, R., Blachier, F., Darcy-Vrillon, B., de Coppet, P., Bourreille, A., and Segain, J. P. (2010). Butyrate utilization by the colonic mucosa in inflammatory bowel diseases: a transport deficiency. Inflamm. Bowel Dis. 16, 684–695.
Tremblay, G. C., and Qureshi, I. A. (1993). The biochemistry and toxicology of benzoic acid metabolism and its relationship to the elimination of waste nitrogen. Pharmacol. Ther. 60, 63–90.
Wishart, D. S., Knox, C., Guo, A. C., Eisner, R., Young, N., Gautam, B., Hau, D. D., Psychogios, N., Dong, E., Bouatra, S., Mandal, R., Sinelnikov, I., Xia, J., Jia, L., Cruz, J. A., Lim, E., Sobsey, C. A., Shrivastava, S., Huang, P., Liu, P., Fang, L., Peng, J., Fradette, R., Cheng, D., Tzur, D., Clements, M., Lewis, A., De Souza, A., Zuniga, A., Dawe, M., Xiong, Y., Clive, D., Greiner, R., Nazyrova, A., Shaykhutdinov, R., Li, L., Vogel, H. J., and Forsythe, I. (2009). HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 37, D603–D610.
Keywords: familial Mediterranean fever, peptic ulcer, metabolome, gas chromatography/mass spectrometry, microbial fatty acids, phenolic compounds, discriminant function analysis
Citation: Ktsoyan ZA, Beloborodova NV, Sedrakyan AM, Osipov GA, Khachatryan ZA, Kelly D, Manukyan GP, Arakelova KA, Hovhannisyan AI, Olenin AY, Arakelyan AA, Ghazaryan KA and Aminov RI (2011) Profiles of microbial fatty acids in the human metabolome are disease-specific. Front. Microbio. 1:148. doi: 10.3389/fmicb.2010.00148
Received: 06 November 2010;
Accepted: 27 December 2010;
Published online: 20 January 2011.
Edited by:
Murty V. Madiraju, University of Texas Health Center, USAReviewed by:
Jeong-Heon Cha, Yonsei University, South KoreaAlain Stintzi, Ottawa Institute of Systems Biology, Canada
Copyright: © 2011 Ktsoyan, Beloborodova, Sedrakyan, Osipov, Khachatryan, Kelly, Manukyan, Arakelova, Hovhannisyan, Olenin, Arakelyan, Ghazaryan and Aminov. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Rustam I. Aminov, Rowett Institute of Nutrition and Health, University of Aberdeen, Aberdeen AB21 9SB, UK. e-mail: r.aminov@abdn.ac.uk