 Sandra Mendoza-Elizalde1,2,3
Sandra Mendoza-Elizalde1,2,3 Nancy K. Arteaga-Resendiz1,4
Nancy K. Arteaga-Resendiz1,4 Pedro Valencia-Mayoral5Raúl C. Luna6Sarbelio Moreno-Espinosa1Francisco Arenas-Huertero7Gerardo Zúñiga3*
Pedro Valencia-Mayoral5Raúl C. Luna6Sarbelio Moreno-Espinosa1Francisco Arenas-Huertero7Gerardo Zúñiga3* Norma Velázquez-Guadarrama1*
Norma Velázquez-Guadarrama1*- 1Laboratorio de Infectología, Departamento de Infectologia, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico
- 2Posgrado en Ciencias Químicobiológicas, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Ciudad de México, Mexico
- 3Laboratorio de Variación Biológica y Evolución, Departamento de Zoología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Ciudad de México, Mexico
- 4Posgrado en Ciencias en Biomedicina y Biotecnología Molecular, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Ciudad de México, Mexico
- 5Dirección de Planeación, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico
- 6Bioterio, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico
- 7Laboratorio de Investigación en Patología Experimental, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico
The bacterium Helicobacter pylori exhibits great genetic diversity, and the pathogenic roles of its virulence factors have been widely studied. However, the evolutionary dynamics of H. pylori strains during stomach colonization are not well-characterized. Here, we analyzed the microevolutionary dynamics of the toxigenic strain vacAs1m1, the non-toxigenic strain vacAs2m2, and a combination of both strains in an animal model over time. Meriones unguiculatus were inoculated with the following bacteria: group 1-toxigenic strain vacAs1m1/cagA+/cagE+/babA2+; ST181, group 2-non-toxigenic strain vacAs2m2/cagA+/cagE+/babA2+; ST2901, and group 3-both strains. The gerbils were euthanized at different time points (3, 6, 12, and 18 months). In group 1, genetic alterations were observed at 6 and 12 months. With the combination of both strains, group 3 also exhibited genetic alterations at 3 and 18 months; moreover, a chimera, vacA m1-m2, was detected. Additionally, four new sequence types (STs) were reported in the PubMLST database for H. pylori. Synonymous and non-synonymous mutations were analyzed and associated with alterations in amino acids. Microevolutionary analysis of the STs (PHYLOViZ) identified in each group revealed many mutational changes in the toxigenic (vacAs1m1) and non-toxigenic (vacAs2m2) strains. Phylogenetic assessments (eBURST) did not reveal clonal complexes. Our findings indicate that the toxigenic strain, vacAs1m1, and a combination of toxigenic and non-toxigenic strains acquired genetic material by recombination. The allelic combination, vacAs2m1, displayed the best adaptation in the animal model over time, and a chimera, m1-m2, was also identified, which confirmed previous reports.
Introduction
Helicobacter pylori, a well-known member of the human microbiota, has a global distribution that is related to the migration of Homo sapiens over the past 60,000 years (Morelli et al., 2010; Moodley et al., 2012). H. pylori is a Gram-negative spiral bacterium that is associated with the development of peptic ulcers as well as some types of gastric lymphomas and gastric adenocarcinomas in humans (Mbulaiteye et al., 2009; Testerman and Morris, 2014).
The evolution of distinct genetic prototypes in H. pylori is linked to different human ethnic groups worldwide, supporting the presence of genetic mechanisms that have permitted rapid adaptation in human populations (Linz et al., 2007). High mutation rates and frequent inter-strain exchanges of genetic material that occur during infection are responsible for the extreme variation and genetic diversity among H. pylori strains (Blaser and Atherton, 2004; Suerbaum and Josenhans, 2007). Additionally, transmission mainly occurs through direct human-to-human contact, and single or multiple strains of H. pylori can colonize and recolonize a host to increase its variability (Brown, 2000; Frenck and Clemens, 2003). Consequently, mutations, inter-strain genetic exchange and the mode of transmission appear to account for the capacity of H. pylori to colonize different habitats in the stomach, and its indirect and direct interactions with the human host trigger different selective pressures that regulate the presence of strains in this changing habitat (Raymond et al., 2004; Kivi et al., 2007; Costa et al., 2009; Secka et al., 2011). Researchers have hypothesized that selective pressures that determine the presence of H. pylori in the stomach operate on three types of bacterial genes (e.g., genes that affect intrabacterial mutations, DNA uptake, repair and recombination; genes that favor bacteria–bacteria interactions; and genes that influence bacterial properties, such as adherence and immune responses that modulate interactions with the host) (Gangwer et al., 2010; Tanih et al., 2011).
The dynamics of H. pylori genotypes during stomach colonization are unknown because the successful establishment of these strains is an inadvertent process. A model that potentially explains the genotypic evolution of H. pylori in its human host assumes that strains with genotype vacA+/cagA+/babA+ are at a higher “fitness peak” (Montecucco and Rappuoli, 2001). These virulence genes encode proteins (i.e., VacA, CagA, and BabA) that help the bacteria to adhere and persist in the gastric epithelium by modifying and altering apical and cell junctions (Wroblewski et al., 2010); i.e., the vacAs1m1 allelic combination is capable of producing the VacA toxin (which induces vacuolation of gastric epithelial cells), whereas the vacAs2m2 allelic combination produces low amounts or none of the VacA toxin (Atherton et al., 1995; Letley and Atherton, 2000). Consequently, inactivation of any of these factors can shift the fitness of the strains. H. pylori can live in the stomach of an individual for many years, so it is possible that strains may emerge with vacA+/-. cag+/-, or babA+/- genotypes, and strains with the genotype cag- or cag+ can be isolated from the same patient. However, these strains likely cannot survive long because of the high recombination rates observed for this bacterium. Thus, only those bacteria that are efficient over long durations and engage in person-to-person transmission are thought to govern the evolution of H. pylori (Montecucco and Rappuoli, 2001; Prouzet-Mauleon et al., 2005).
Genetic alterations that are produced during the microevolution of H. pylori have not been studied because they can only be detected during the transition phase, i.e., after the passage of an in vitro strain (culture) to an in vivo (animal) setting or during colonization of a host that is not infected (Ferrero and Jenks, 2001). Animals that are infected (e.g., Rhesus monkeys, mice, and gerbils) with strains of known genotypes provide experimental models (Peek, 2008; Behrens et al., 2013; Linz et al., 2014) that can be used to follow the evolution of these strains in vivo, from the initial inoculation until the definitive establishment of the strain (Morelli et al., 2010; Linz et al., 2014). In the present study, we analyzed the evolutionary dynamics of the toxigenic strain, vacAs1m1, and the non-toxigenic strain, vacAs2m2, separately and together in an animal model over time.
Materials and Methods
Animal Model
The 8-week-old Mongolian gerbils (Meriones unguiculatus Hsd:MON, Harlan Teklad, Madison, WI, USA) used in this study were housed under specific pathogen-free conditions in plastic metabolic cages to prevent coprophagy under standard laboratory conditions (i.e., room temperature, 23 ± 2°C; relative humidity 40–60%; and a 12-h light–dark cycle). Free access to a standard diet (special rodent food; Harlan Teklad, Madison, WI, USA) and sterilized tap water were provided. The Ethics, Biosafety and Scientific committees at the Health Institute approved the experiment.
The three groups of gerbils included five animals each one, ensuring that three animal would present H. pylori infection (Velazquez-Guadarrama et al., 2007). Gerbils were inoculated intragastrically with 500 mL NaHCO3 (0.2 M), and 1 h later with a bacterial suspension of different genotypes of H. pylori [6 × 108 colony forming units (CFUs)/mL]. For 1 week, group 1 was inoculated with the toxigenic strain, vacAs1m1/cagA+/cagE+/babA2+; group 2 received the non-toxigenic strain, vacAs2m2/cagA+/cagE+/babA2+; and group 3 received both the toxigenic strain, vacAs1m1, and the non-toxigenic strain, vacAs2m2. The gerbils were fasted 18 h prior to the first inoculation until the end of the fifth inoculation. The H. pylori strains used in this study included reference strain 26695 (positive control for the vacAs1m1 genotype) and clinical strain 174F2 (positive control for the vacAs2m2 genotype). Control animals received saline alone. The gerbils were euthanized at 3, 6, 12, and 18 months by cervical dislocation under anesthesia to harvest the stomach. Assuming that the H. pylori generation time is at least 3 h (Jiang and Doyle, 2000; Joo et al., 2010), the generation numbers (G) achieved at these months were 654 G, 1309 G, 2617 G, and 3926 G, respectively. The stomach was dissected along the greater curvature and washed with phosphate-buffered saline (PBS pH 7.4, 0.01 M). It was then divided longitudinally into parts and macerated with Brucella broth (BD BBL) in a final volume of 200 μL. Next, 10 μL was used for a urease test, and 10 μL (STOCK) was inoculated in Casman agar plates (BD BBL, Sparks, MD, USA) with or without antibiotics (3 mg/mL vancomycin, 5 mg/mL trimeptoprim, and 2 mg/mL amphotericin B). Additionally, 170 μL was used for serial dilutions (1:10, 1:100, 1:1,000, and 1:10,000) on Casman agar plates with and without antibiotics. The plates were grown under microaerophilic conditions (5% O2, 5% CO2, 85% N2, and 10% humidity) at 37°C for 7–14 days. The control group was compared with the infected groups.
Isolation and Identification of H. pylori
Bacterial isolation was performed using 10 presumptive colonies of H. pylori for each dilution of the different generations. Bacterial identification was based on colony morphology, Gram staining, and tests for urease, catalase, and oxidase. H. pylori was stored at -70°C in 1.5 mL Brucella broth (BD BBL) supplemented with 10% fetal bovine serum and 25% glycerol.
Detection of Virulence Genes by PCR
Genomic DNA was extracted from a section of the stomach of gerbils, and colonies of H. pylori were isolated using a Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions with slight modifications of the incubation times. The DNA was quantified in an Epoch Microplate Spectrophotometer (BioTek, software Gen5TM, Winooski, VT, USA), and the DNA integrity was evaluated by electrophoresis using 1% agarose gels. H. pylori was identified in gastric tissue and isolates based on the presence of the glmM gene (Smith et al., 2004). The vacA (s1, s2, m1, and m2), cagA, cagE, and babA2 genes were amplified by PCR using the conditions described by Atherton et al. (1995), Mizushima et al. (2001), and Kauser et al. (2004). Amplification was performed in a reaction volume of 25 μL Master Mix (Promega) containing 100 ng bacterial DNA, 2.5 mM MgCl2, 10 mM dNTPs, 2 U Taq DNA polymerase, 20 pmol each primer and nuclease-free water in a Thermo Hybrid thermal cycler (PCR Express, Emeryville, CA, USA). The PCR products were separated by electrophoresis using 1% agarose gels at 80 V, followed by staining with ethidium bromide and imaging under UV illumination (ChemiDoc Transilluminator, Bio-Rad, Hercules, CA, USA). DNA from reference strain 26695 was included as a positive control.
Multilocus Sequence Typing (MLST)
Internal fragments were amplified and sequenced in both directions for seven housekeeping genes [mutY, HP0142, specific adenine glycosylase A/G; ureI, HP0071, urea transporter; atpA, HP1134, ATP synthase F1 α subunit; efp, HP0177, elongation factor P (EF-P); ppa, HP0620, inorganic pyrophosphatase; trpC, HP1279, indole-3-glycerol phosphate synthase; and yphC, HP0834, GTPase], as reported in previous studies of H. pylori (Lundin et al., 2005; Kivi et al., 2007). The PCR conditions were as follows: 35 cycles of 94°C for 15 s, 55–62°C for 30 s, and 72°C for 1.5 min and a final extension at 72°C for 5 min (Achtman et al., 1999). The PCR products were purified using ExoSAP-IT® (Affymetrix, Cleveland, OH, USA) according to the manufacturer’s recommendations. The purified products were sequenced using a BigDye Terminator v3.1 Cycle Sequencing kit with an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Bioinformatics and Phylogenetic Analyses
The sequences of the seven loci were aligned using ClustalX v2 (Larkin et al., 2007), edited with Seaview v4.2.5 (Gouy et al., 2010) and FinchTV V.1.4.0 Software (Geospiza, Inc.), and compared with those of all known alleles from H. pylori deposited in the PubMLST database1. Each strain was defined based on the presence of alleles of the seven genes (allelic profile); every allelic profile was defined as a sequence type (ST) (Feil et al., 2004; Vázquez and Berrón, 2004). To establish the open reading frame of each protein, the nucleotide sequences of the different STs from each housekeeping gene were translated into amino acid sequences using the translate tool in ExPASy2. To determine the presence of synonymous and non-synonymous mutations in different positions of the seven housekeeping genes, we used DnaSP v5.103 (Librado and Rozas, 2009).
The clonal relationship among strains of H. pylori was determined using the PHYLOViZ platform4. PHYLOViZ infers evolutionary descent patterns among allelic profiles using the goeBURST algorithm and a full Minimal Spanning Tree (MST)-like approach (Francisco et al., 2009). The phylogenetic relationships among the strains was determined using the eBURST algorithm5, which subdivided large multilocus sequence typing (MLST) datasets into non-overlapping groups of related STs or clonal complexes to discern the location of the most parsimonious isolates within groups or clonal complexes from the predicted founder (Feil et al., 2004). In addition, the eBURST algorithm explores the diversification of strains and can provide evidence for the emergence of clones of particular clinical relevance.
The nucleotide sequences (alleles) and STs found in this study were deposited in the PubMLST database for H. pylori1 (Jolley et al., 2004). The accession numbers for each gene were as follows: atpA 2358, 2470; efp 2228, 2354; mutY 2391; ppa 2252; trpC 2503, 2512; ureI 2474; and yhpC 2583, and for STs: ST2901, ST2902, ST2903, ST2904, and ST2905.
Results
Helicobacter pylori was identified in all infected groups by endpoint PCR. However, H. pylori strains were isolated from groups 1 and 3. The genotypes derived from the toxigenic strain (vacAs1m1/cagA+/cagE+/ babA2+; ST181) in group 1 exhibited genetic alterations at 6 and 12 months (1309 and 2617 G, respectively), and we also observed the emergence of new clones [vacAs1m1/cagA+/cagE-/babA2-; ST2902 (1309 G) and vacAs1m1/cagA-/cagE+/babA2-; ST2903 (2617 G)] (Figure 1). Among the seven housekeeping genes that were analyzed, five genes in ST2902 and ST2093, atpA. efp. mutY. ppa and trpC, had more synonymous mutations. However, the proportions of each gene at 6 and 12 months were similar. The trpC gene exhibited the most variation (Figure 2).
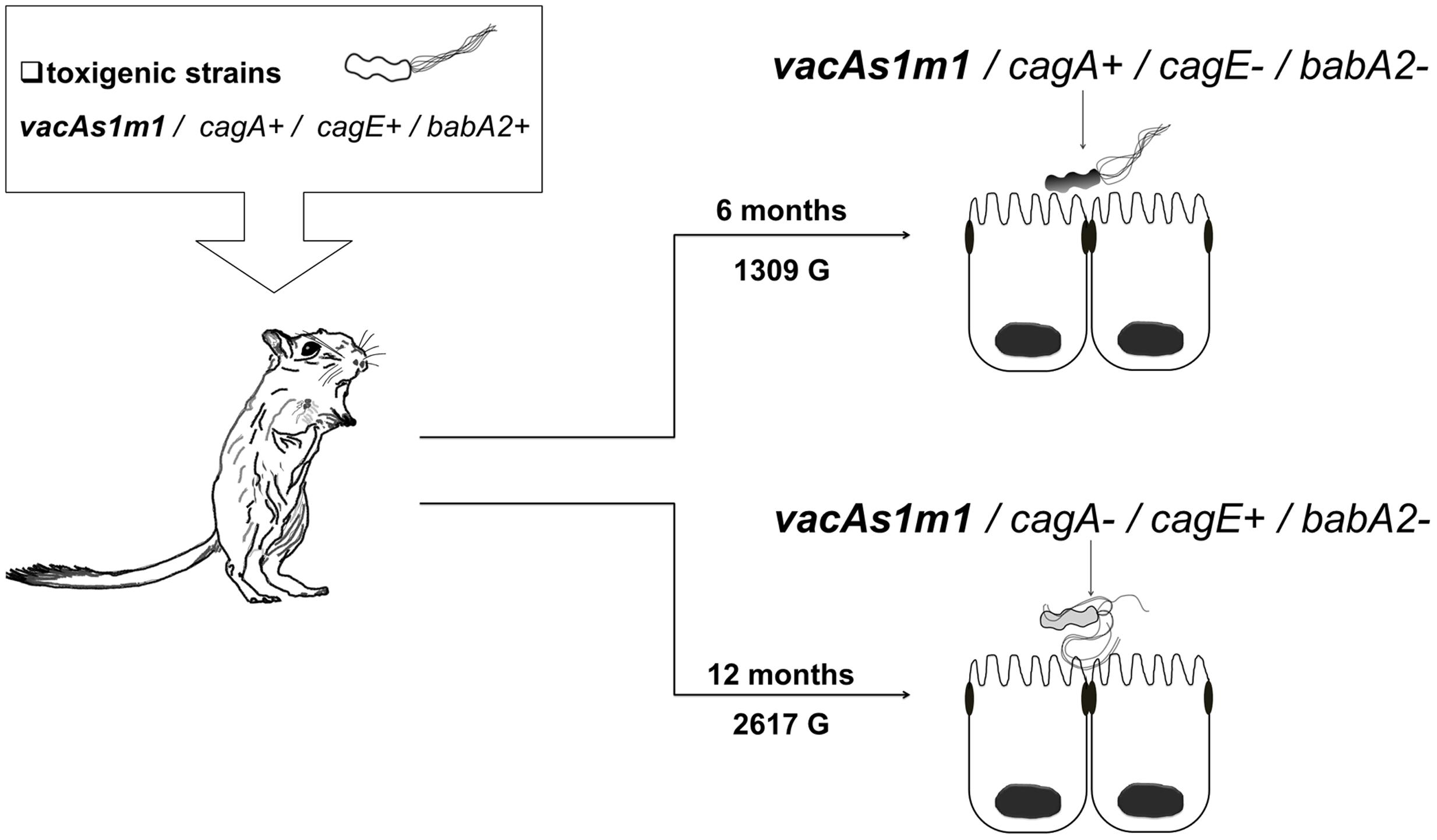
FIGURE 1. Helicobacter pylori genotypes identified with the group 1 toxigenic strain (vacAs1m1) in the animal model, Meriones unguiculatus. Alterations in the cag-PAI and babA2 genes occurred at 6 (1309 G) and 12 (2617 G) months.
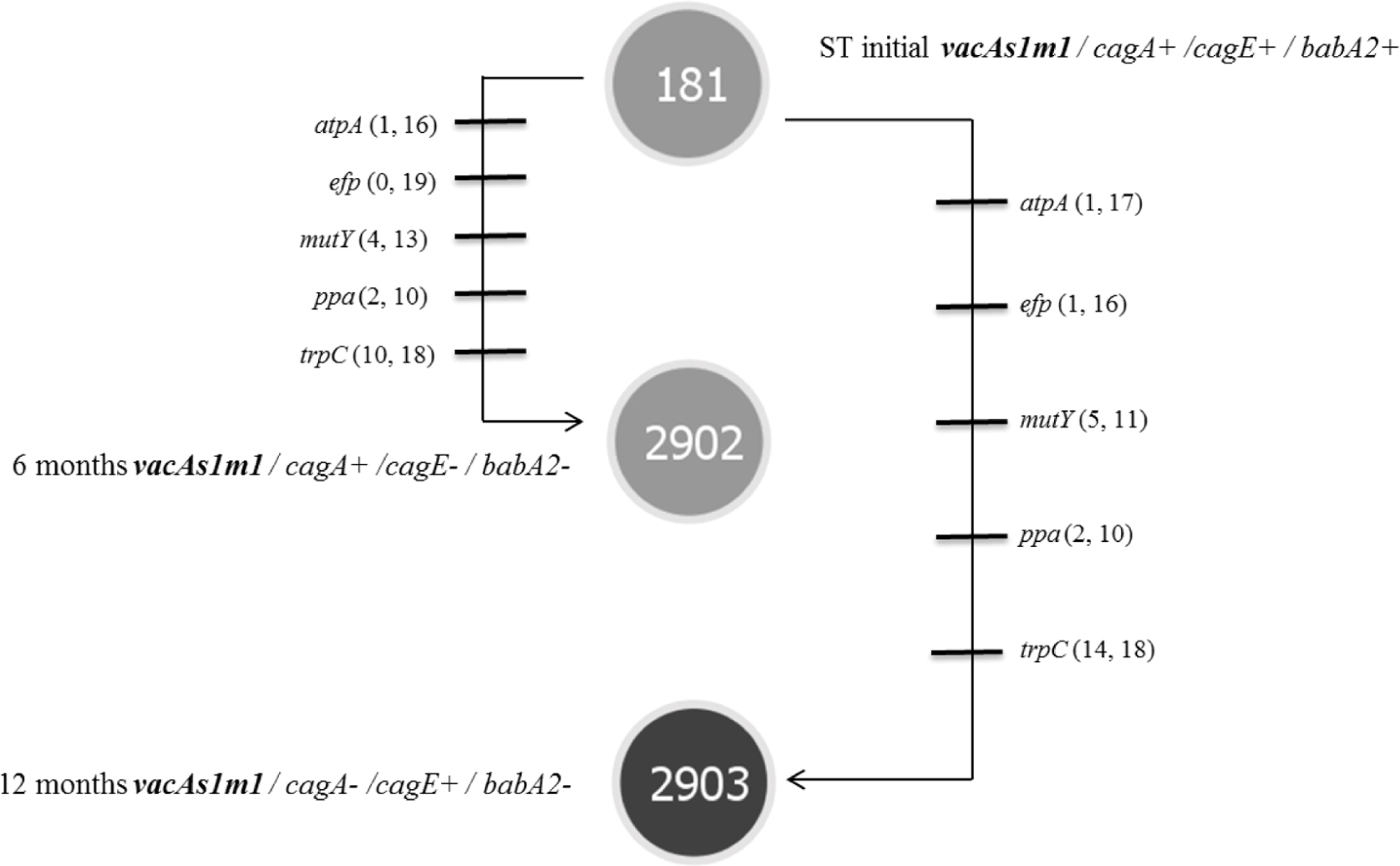
FIGURE 2. Microevolution of STs identified in group 1 (toxigenic strain vacAs1m1), as defined by PHYLOViZ (goeBURST algorithm) for strains of H. pylori isolated from the M. unguiculatus animal model. STs: ST181 corresponds to the reference strain 26695 of H. pylori (vacAs1m1 toxigenic strain). Each line represents a different allele with mutational changes. The numbers of non-synonymous and synonymous mutations are indicated in parentheses. ST2902 and ST2903 were identified at 6 and 12 months and exhibited 5 changes in alleles compared with the initial strain ST181.
In group 3, which was inoculated with the toxigenic (vacAs1m1/cagA+/cagE+/babA2+; ST181) and non-toxigenic (vacAs2m2/cagA+/cagE+/ babA2+; ST2901) strains, we also observed genetic alterations at 3 and 18 months (654 G and 3926 G). The strains in this group gave rise to three new clones [vacAs2m1-2/cagA+/cagE+/babA2+; ST2901 (654 G), vacAs2m1-2/cagA+/cagE+/babA2+; ST2904 and vacAs2m1/cagA+/cagE+/babA2; ST2095 (3926 G)] (Figure 3). Notably, the genotype identified at 3 months exhibited an alteration in the middle region of the vacA gene (m1-m2). The nucleotide sequences of the chimera showed 99% and 97% identity with the allelic sequences of vacAs1m1 and vacAs2m2, respectively, from GenBank6. In addition, alignment of these chimera sequences with those of other chimeras reported in other studies yielded similar identity values (Figure 4).
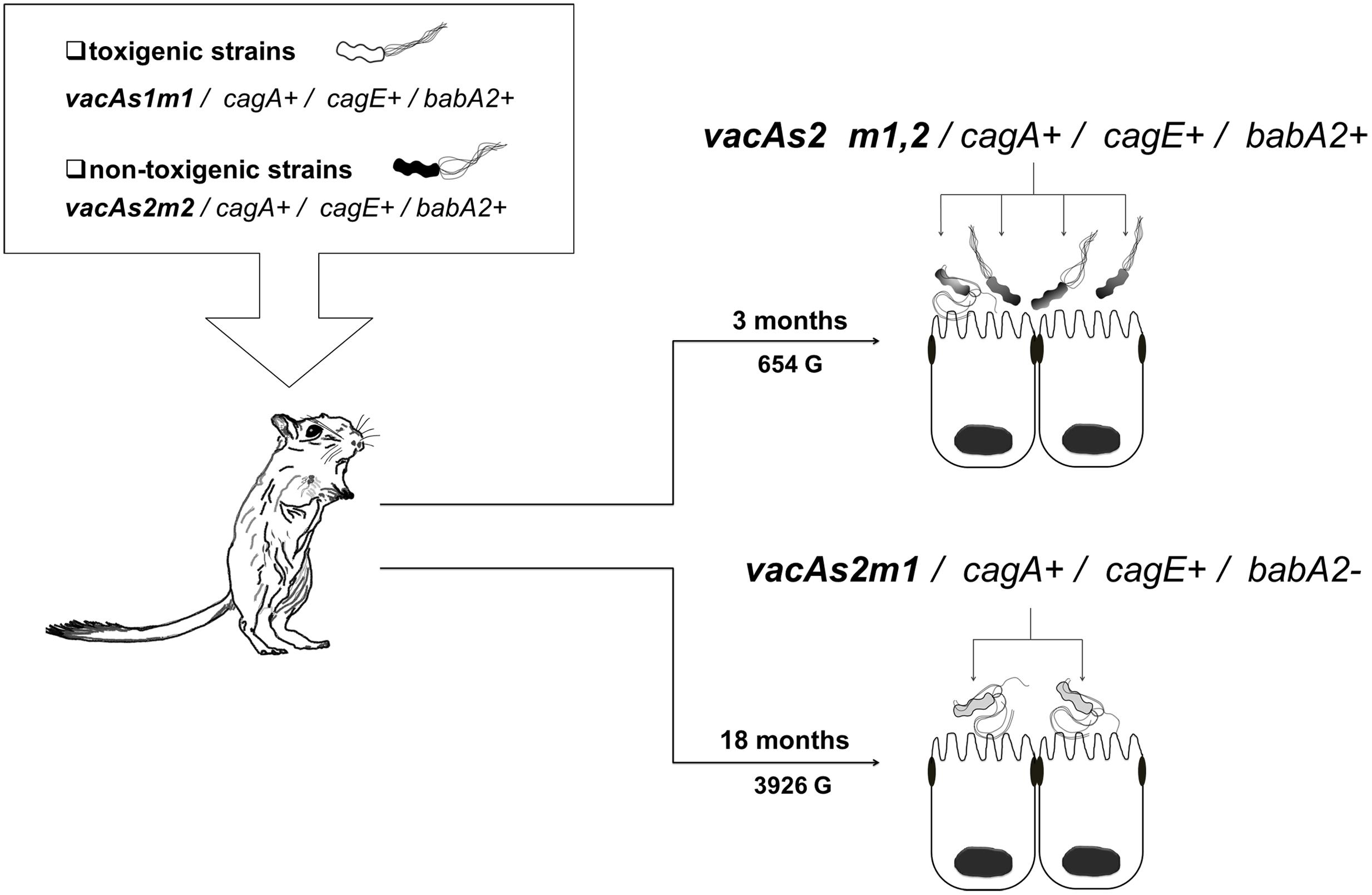
FIGURE 3. Helicobacter pylori genotypes identified in the group 3 toxigenic (vacAs1m1) and non-toxigenic (vacAs2m2) strains in the Meriones unguiculatus animal model. At 3 months (654 G), both alleles in the middle region of vacA m1 and m2 were present (∗chimera). At 3 (654 G) and 18 (3926 G) months, alterations in the babA2 gene were noted.

FIGURE 4. Alignment of the nucleotide sequences of the middle region of the vacA gene of H. pylori. The middle region corresponding to nucleotides (nt) 2308–4400 of the m1 allele of the reference strain, NCTC11638 (Cover et al., 1994), is presented. Asterisks indicate nucleotides that are identical between the analyzed alleles. Comparisons of m1-m2 chimeric alleles of H. pylori strains obtained from the animal model (strains 3A, 3B, 3C, and 3D), m2 allele of strain 87–203 (Cover et al., 1994), m2 allele of strain 95–54 (Pagliaccia et al., 1998), m1–m2 chimeric alleles of strain ch2 (Ji et al., 2000), m1-m2 chimeric alleles of strain R10A (Pan et al., 1998), m1-m2 chimeric alleles of strain India100 (Mukhopadhyay et al., 2000), m1b allele of strain R13A (Pan et al., 1998), and m1 allele of the reference strain, NCTC11638 (Cover et al., 1994). Black stripes identify the portion of the m1, m2, and m1-m2 chimeric alleles (14 nt) in which recombination would have generated chimeric alleles m1-m2, as proposed by Pan et al. (1998). The region marked by an arrow identifies the segment that is absent in the m1 allele but is present in the m2 allele. The GenBank accession numbers for the presented sequences are U07145 (NCTC11638), U05677 (87–203), U95971 (95–54), AF191639 (ch2), AF035609 (R10A), AF220120 (India100), and AF035610 (R13A).
The housekeeping genes of the STs in group 3 exhibited many mutational changes in both the toxigenic and the non-toxigenic strains (Figure 5). The number of mutated genes and the number of mutations within each gene were different in the STs. However, the genotype identified at 3 months did not exhibit mutational changes in the housekeeping genes, i.e., the ST was similar to that of the non-toxigenic strain, and the number of synonymous and non-synonymous mutations was similar to that of the toxigenic strain (ST181). All non-synonymous mutations occurred outside of the active site and other functionally important sites of the proteins (e.g., signature motif and substrate-binding domains); some synonymous mutations (15.29%) were identified in these regions.
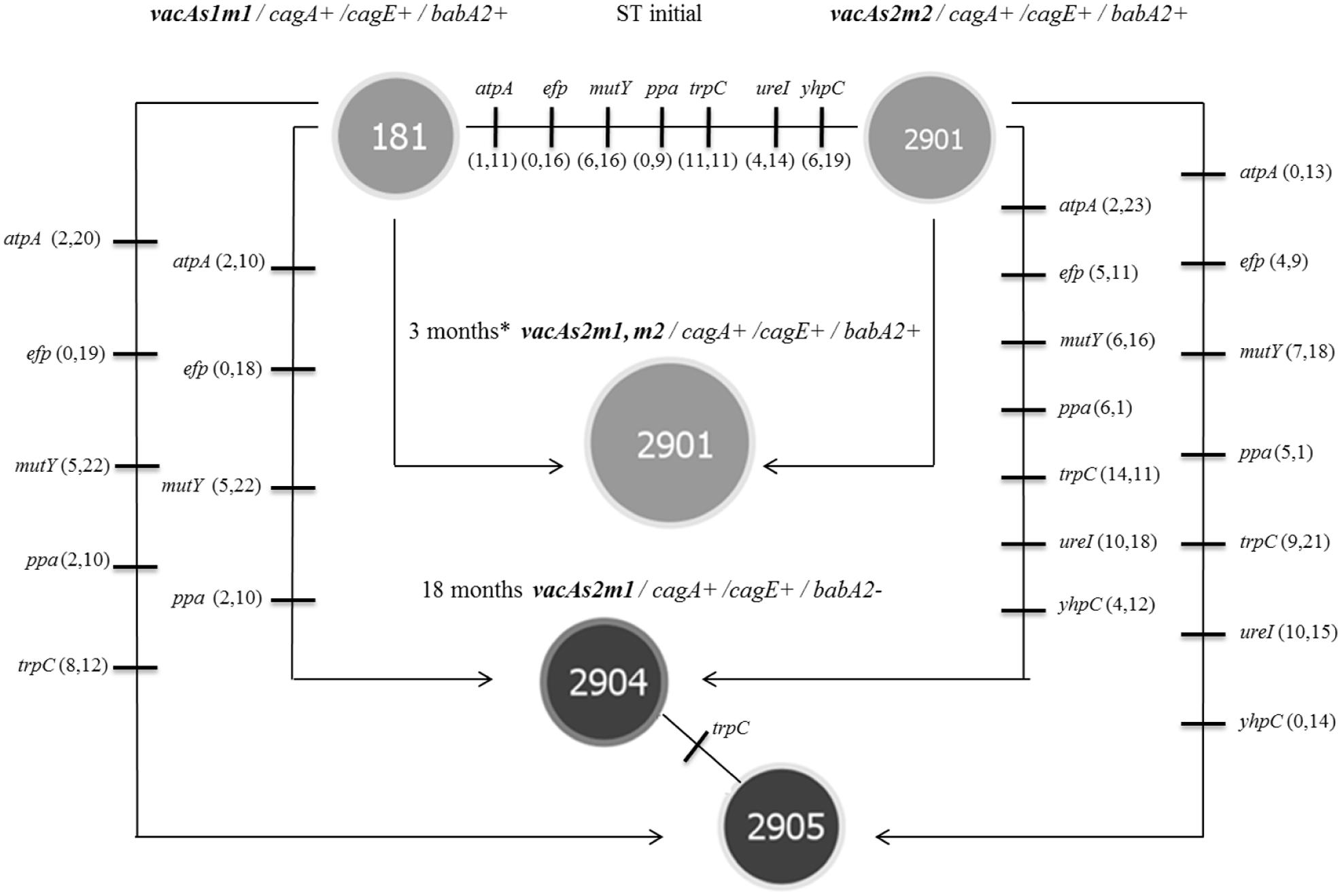
FIGURE 5. Microevolution of STs was identified in group 3 (toxigenic strain vacAs1m1 and non-toxigenic strain vacAs2m2), as defined by PHYLOViZ (goeBURST algorithm) for strains of H. pylori isolated from the M. unguiculatus animal model. ST181 and ST2901 correspond to the reference 26695 (vacAs1m1 toxigenic strain) and clinical 172F2 (vacAs2m2 non-toxigenic strain) strains of H. pylori, respectively. Each line represents a different allele with mutational changes. The number of non-synonymous and synonymous mutations are indicated in parentheses. ST2901 was identified at 3 months∗ (chimera, Figure 3); the data are presented for both original strains. The reference strain 26695 (ST181) donated the vacAm1 allele, which originated the chimera and clinical strain 174F2 (ST2901) alleles of the housekeeping genes. The trpC gene (ST2904 and ST2905) was the only housekeeping gene with mutational change.
The phylogenetic analysis of 1993 isolates deposited in the PubMLST database for H. pylori along with those isolates of H. pylori obtained from the animal model in the present study exhibited an overview of clonal complexes (Figure 6). Clusters of related isolates and individual unlinked STs are shown as a single-tree eBURST, establishing the definition of category zero for seven shared alleles. ST2904 and ST2905 displayed a ‘double-link’ that featured six alleles in common. Moreover, ST2901, ST2902, and ST2903 are individual unlinked STs.
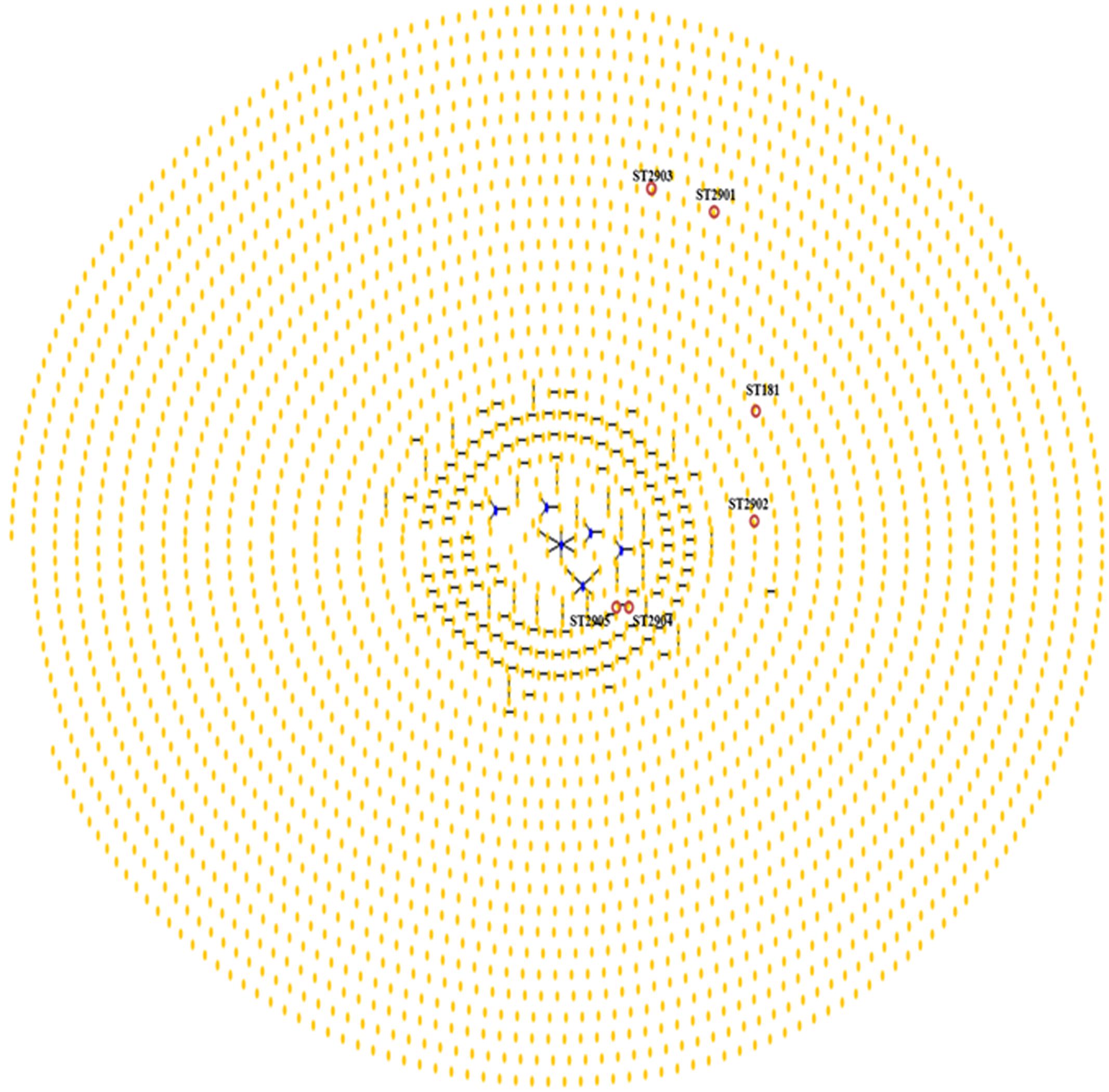
FIGURE 6. Population “snapshot” of H. pylori and STs found in strains of H. pylori that were isolated from the M. unguiculatus animal model. Clusters of related isolates and individual unlinked STs found in the MLST database for H. pylori are presented as a single-tree eBURST to define category zero of seven shared alleles. Unions link isolates that correspond to clonal complexes. Primary founders (blue) are located in the center of the group, and founders of subgroups are shown in yellow, as shown for ST2904 and ST2905 (red circle). ST181, ST2901, ST2902, and ST2903 are marked; the labels for the other STs (http://pubmlst.org//helicobacter/) have been removed for clarity.
Discussion
The success of any infection in a host depends on a delicate balance between the host and the pathogen. For bacterial pathogens, the host appears to impose a selective pressure that drives variation within the bacterium (Thompson et al., 2004). In the present study, we infected M. unguiculatus with the following H. pylori strains with known virulence genotypes and STs: reference strain 26695 (vacAs1m1/cagA+cagE+bab2A+; ST181) and clinical strain 172F2 (vacAs2m2/cagA+cagE+bab2A+; ST2901). Our findings revealed genetic alterations of the introduced genotypes throughout the course of infection in an animal model. Group 1 exhibited genetic alterations in the cagA, cagE, and babA2 genes (Figure 1). Several previous studies have observed similar changes or alterations. Solnick et al. (2004) studied genetic alterations in H. pylori J166 and found replacements of babA with babB, suggesting that babA undergoes selective pressure early during the course of infection and that changes in babA might represent a crucial adaptation to the host stomach environment. In addition, Linz et al. (2014) reported that alterations within the babA promoter could occur as early as 1 week post-infection, also they reported mutations in the cag-PAI genes in J166 strain after 2 and 6 months in a macaque model, where the function of cag-PAI was apparently abolished, implying adaptation by mutation and recombination during early stages of the infection.
More than a decade ago, several authors described the potential for genetic rearrangements and recombination events among H. pylori strains during chronic infections in different parts of the stomach (Blaser and Berg, 2001; Blaser and Atherton, 2004). We identified different genotypes at 6 and 12 months post-infection in the group 1 (1309 and 2617 G, respectively) that exhibited alterations in the cagA, cagE, and babA2 genes. In these genes, the incorporation of genetic material by recombination between strains (e.g., cagA- cagE- and babA2-) was evident. The presence of these genotypes in group 1 after 6 and 12 months strongly suggested that mechanisms of recombination or mutation had occurred over long periods of colonization in our animal model, resulting in the acquisition of this genetic material (Figure 1). The same authors suggested that such genetic variation can lead to the development of different strains, genotypes or subclones within the same host. Our data documented mutational changes in housekeeping genes and virulence genotypes; thereby, we identified the following new STs: ST2902, ST2903, ST2904, and ST2905 derived from ST181 (group 1) and ST181 and ST2901 (group 3). In group 2, which was infected with strain ST2901 alone, the infection was identified at different periods of time by PCR. However, isolation any H. pylori colonies was not possible, and we still do not know why this was the case. Generally, genotype vacAs2m2 is reported less frequently in infected patients, probably because of its need for more nutrients for its own development in vitro or might suggest to be a genotype unsuitable to infect, it can enter to a viable but non-culturable stage (coccoid forms). The coccoid forms of H. pylori are less virulent, less likely to colonize and induce inflammation (Mazaheri Assadi et al., 2015). Meanwhile, Pagliaccia et al. (1998) observed m1 and m2 alleles of the VacA cytotoxin, which can recognize different receptors on gastric epithelial cells in humans; however, similar findings have not been previously published for gerbils.
Global studies have shown that high degrees of polymorphism in housekeeping genes are associated with changes in the third nucleotide position of codons, and many of the observed variation in STs are synonymous changes (Achtman et al., 1999). We also observed a greater number of synonymous mutations, in accord with other studies (Achtman et al., 1999; Morelli et al., 2010; Secka et al., 2011). In our study, the trpC gene (ST2903) was the most variable and uniquely introduced stop codon. However, these changes were unlikely to be deleterious for the micro-organisms because they were isolated of animal model. Martincorena et al. (2012) have suggested an evolutionary optimization of the mutation rate to reduce the risk of deleterious mutations.
Phylogenetic studies have indicated a clear separation between sequences of the middle region; the m2 sequence contains an insertion of 21–25 amino acids that was not present in the m1 sequence (van Doorn et al., 1998; Gangwer et al., 2007). In the present study, we observed that group 3 (toxigenic strain vacAs1m1; ST181 and non-toxigenic strain vacAs2m2; ST2901) (Figure 3) had a vacAs2m1-m2/cagA+cagE+babA2+ genotype with the presence of a natural chimera of the middle region of vacA at 3 months (654 G). To confirm the presence of the m1-m2 alleles of vacA in the chimera, each allele present in the four strains was sequenced. The identity percentages were 99% (m1) and 97% (m2), which were similar to those described by Tanih et al. (2011) who reported identity percentages of 87–99 and 89–98% for m1 and m2, respectively, in their chimeras. Our chimeras were aligned with other chimeras that have been previously reported (Figure 4), revealing the region proposed by Pan et al. (1998), in which recombination occurs between the m1 and m2 alleles in a region with limited homology (14 nt). Natural chimeras in strains of H. pylori have rarely been reported, suggesting that H. pylori strains with intact m1 or m2 of vacA provide favorable functional properties and, therefore, exhibit a selective advantage compared with strains containing m1-m2 chimera sequences (Ji et al., 2000). In the present work, we observed instability of the vacA m1-m2 chimera; moreover, the allelic combination, vacAs2m1, exhibited the best adaptation in the animal model over time. Our results demonstrate the combination of genotypes among the vacAs1m1 and vacAs2m2 strains in an animal model. Studies have shown that allelic combination of vacAs2m1 causes less damage to the host (Atherton et al., 1995). Once H. pylori is established in the stomach, it may or may not evolve to a vacAs1m1 genotype; however, this genotype is the most frequently noted in adult patients and is associated with duodenal and gastric ulcers and gastric cancer (Atherton et al., 1995, 1997; Miehlke et al., 2000).
The high rate of mutations between STs results in high genetic diversity that reflects a long evolutionary history of various strains of H. pylori. In this study, ST2904 and ST2905 exhibited seven different housekeeping genes derived from the initial ST2901. Thus, we identified four new STs that were reported in the database PubMLST of H. pylori. Among these STs, three had alleles that had been previously reported in the database: ST2901 with the mutY1504 allele reported in Ireland, and ST2904 and ST2905 each with an atpA1708 allele reported in Brazil. The phylogenetic analysis conducted with 1993 isolates (PubMLST H. pylori; Figure 6) provided results that were consistent with previous studies (Suerbaum et al., 1998; Feil and Spratt, 2001; Hanage et al., 2006; Turner et al., 2007), indicating that H. pylori forms a non-clonal population, presents a high mutation rate that generates a large number of alleles and that there is a high rate of recombination among these alleles.
The microevolutionary history of H. pylori infection in humans reveals remarkable genetic diversity within this bacterium, which is mainly generated by point mutations and recombination (intragenic or intergenic). The high variability of H. pylori is thought to maximize its ability to adapt to the changing environment of the host gastric habitat, consequently facilitating chronic colonization. This study provides evidence for processes of recombination between genotypes, the emergence of new clones and patterns of evolutionary non-clonal descent among H. pylori strains obtained from an animal model. Our findings suggest that the recombination process in H. pylori in the host results from the adaptation of the bacterium to the host.
Author Contributions
SM-El, GZ, and NV-G conceived and planned the study. SM-El and NA-R performed experiments and generated the database. RL maintained and provided care for the animal model. SM-El, GZ, and NV-G analyzed and interpreted data. PV-M helped with the animal model. SM-Es and FA-H reviewed and corrected the manuscript. The manuscript was prepared by SM-El, GZ, and NV-G. All authors revised and agreed on the final version of the manuscript.
Funding
This project was supported by Federal Resources (HIM/2011/080 SSa. 1005) from SSA, Mexico.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank M. en C. Leonel Martínez Cristóbal and Technical Marco Antonio Ochoa Arias (Hospital Infantil de México Federico Gómez) for providing help in the care and handling of animals and Biol. Juan Carlos Vigueras for his technical assistance. This work was part of the Ph.D. dissertation of MES, a CONACyT (216177) fellow. NCAR was a CONACyT (254412) and PIFI-IPN (20161878) fellow.
Footnotes
- ^http://pubmlst.org/
- ^http://www.expasy.org
- ^http://www.softpedia.com/get/Science-CAD/DnaSP.shtml
- ^http://www.phyloviz.net/
- ^http://eburst.mlst.net/
- ^http://www.ncbi.nlm.nih.gov/genbank
References
Achtman, M., Azuma, T., Berg, D. E., Ito, Y., Morelli, G., Pan, Z. J., et al. (1999). Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol. Microbiol. 32, 459–470. doi: 10.1046/j.1365-2958.1999.01382.x
Atherton, J. C., Cao, P., Peek, R. M. Jr., Tummuru, M. K., Blaser, M. J., and Cover, T. L. (1995). Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori: association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 270, 17771–17777. doi: 10.1074/jbc.270.30.17771
Atherton, J. C., Peek, R. M. Jr., Tham, K. T., Cover, T. L., and Blaser, M. J. (1997). Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology 112, 92–99. doi: 10.1016/S0016-5085(97)70223-3
Behrens, W., Schweinitzer, T., Bal, J., Dorsch, M., Bleich, A., Kops, F., et al. (2013). Role of energy sensor TlpD of Helicobacter pylori in gerbil colonization and genome analyses after adaptation in the gerbil. Infect. Immun. 81, 3534–3551. doi: 10.1128/IAI.00750-13
Blaser, M. J., and Atherton, J. C. (2004). Helicobacter pylori persistence: biology and disease. J. Clin. Invest. 113, 321–333. doi: 10.1172/JCI20925
Blaser, M. J., and Berg, D. E. (2001). Helicobacter pylori genetic diversity and risk of human disease. J. Clin. Invest. 107, 767–773. doi: 10.1172/JCI12672
Brown, L. M. (2000). Helicobacter pylori: epidemiology and routes of transmission. Epidemiol. Rev. 22, 283–297. doi: 10.1093/oxfordjournals.epirev.a018040
Costa, A. C., Figueiredo, C., and Touati, E. (2009). Pathogenesis of Helicobacter pylori infection. Helicobacter 14, 15–20. doi: 10.1111/j.1523-5378.2009.00702.x
Cover, T. L., Tummuru, M. K. R., Cao, P., Thompson, S. A., and Blaser, M. J. (1994). Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem. 269, 10566–10573.
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Feil, E. J., and Spratt, B. G. (2001). Recombination and the population structures of bacterial pathogens. Annu. Rev. Microbiol. 55, 561–590. doi: 10.1146/annurev.micro.55.1.561
Ferrero, R. L., and Jenks, P. J. (2001). “In vivo adaptation to the host,” in Helicobacter pylori: physiology and genetics, eds H. L. T. Mobley, G. L. Mendz, and S. L. Hazell (Washington, DC: ASM Press).
Francisco, A. P., Bugalho, M., Ramirez, M., and Carriço, J. A. (2009). Global optimal eBURST analysis of multilocus typing data using a graphical matroid approach. BMC Bioinformatics 10:152. doi: 10.1186/1471-2105-10-152
Frenck, R. W. Jr., and Clemens, J. (2003). Helicobacter in the developing world. Microbes Infect. 5, 705–713. doi: 10.1016/S1286-4579(03)00112-6
Gangwer, K. A., Mushrush, D. J., Stauff, D. L., Spiller, B., McClain, M. S., Cover, T. L., et al. (2007). Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc. Natl. Acad. Sci. U.S.A.. 104, 16293–16298. doi: 10.1073/pnas.0707447104
Gangwer, K. A., Shaffer, C. L., Suerbaum, S., Lacy, D. B., Cover, T. L., and Bordenstein, S. R. (2010). Molecular evolution of the Helicobacter pylori vacuolating toxin gene vacA. J. Bacteriol. 192, 6126–6135. doi: 10.1128/JB.01081-10
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Hanage, W. P., Fraser, C., and Spratt, B. G. (2006). The impact of homologous recombination on the generation of diversity in bacteria. J. Theor. Biol. 239, 210–219. doi: 10.1016/j.jtbi.2005.08.035
Ji, X., Fernandez, T., Burroni, D., Pagliaccia, C., Atherton, J. C., Reyrat, J. M., et al. (2000). Cell specificity of Helicobacter pylori cytotoxin is determined by a short region in the polymorphic midregion. Infect. Immun. 68, 3754–3757. doi: 10.1128/IAI.68.6.3754-3757.2000
Jiang, X., and Doyle, M. P. (2000). Growth supplements for Helicobacter pylori. J. Clin. Microbiol. 38, 1984–1987.
Jolley, K. A., Chan, M. S., and Maiden, M. C. (2004). mlstdbNet - distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5:86. doi: 10.1186/1471-2105-5-86
Joo, J. S., Park, K. C., Song, J. Y., Kim, D. H., Lee, K. J., Kwon, Y. C., et al. (2010). A thin-layer liquid culture technique for the growth of Helicobacter pylori. Helicobacter 15, 295–302. doi: 10.1111/j.1523-5378.2010.00767.x
Kauser, F., Khan, A. A., Hussain, M. A., Carroll, I. M., Ahmad, N., Tiwari, S., et al. (2004). The cag pathogenicity island of Helicobacter pylori is disrupted in the majority of patient isolates from different human populations. J. Clin. Microbiol. 42, 5302–5308. doi: 10.1128/JCM.42.11.5302-5308.2004
Kivi, M., Rodin, S., Kupershmidt, I., Lundin, A., Tindberg, Y., Granstrom, M., et al. (2007). Helicobacter pylori genome variability in a framework of familial transmission. BMC Microbiol. 7:54. doi: 10.1186/1471-2180-7-1
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). ClustalW and ClustalX version 2. Bioinformatics. 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Letley, D. P., and Atherton, J. C. (2000). Natural diversity in the N terminus of the mature vacuolating cytotoxin of Helicobacter pylori determines cytotoxin activity. J. Bacteriol. 182, 3278–3280. doi: 10.1128/JB.182.11.3278-3280.2000
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Linz, B., Moodley, Y., Manica, A., Liu, H., Roumagnac, P., Falush, D., et al. (2007). An African origin for the intimate association between humans and Helicobacter pylori. Nature 445, 915–918. doi: 10.1038/nature05562
Linz, B., Windsor, H. M., McGraw, J. J., Hansen, L. M., Gajewski, J. P., Tomsho, L. P., et al. (2014). A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nat. Commun. 5, 4165. doi: 10.1038/ncomms5165
Lundin, A., Björkholm, B., Kupershmidt, I., Unemo, M., Nilsson, P., Andersson, D. I., et al. (2005). Slow genetic divergence of Helicobacter pylori strains during long-term colonization. Infect. Immun. 73, 4818–4822. doi: 10.1128/IAI.73.8.4818-4822.2005
Martincorena, I., Seshasayee, A. S., and Luscombe, N. M. (2012). Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature 485, 95–98. doi: 10.1038/nature10995
Mazaheri Assadi, M., Chamanrokh, P., Whitehouse, C. A., and Huq, A. (2015). Methods for detecting the environmental coccoid form of Helicobacter pylori. Front. Public Health 28:147. doi: 10.3389/fpubh.2015.00147
Mbulaiteye, S. M., Hisada, M., and El-Omar, E. M. (2009). Helicobacter pylori associated global gastric cancer burden. Front. Biosci. (Landmark Ed). 14:1490–1504. doi: 10.2741/3320
Miehlke, S., Kirsch, C., Agha-Amiri, K., Gunther, T., Lehn, N., Malfertheiner, P., et al. (2000). The Helicobacter pylori vacA s1, m1 genotype and cagA is associated with gastric carcinoma in germany. Int. J. Cancer 87, 322–327. doi: 10.1002/1097-0215(20000801)87:3<322::AID-IJC3>3.3.CO;2-D
Mizushima, T., Sugiyama, T., Komatsu, Y., Ishizuka, J., Kato, M., and Asaka, M. (2001). Clinical Relevance of the babA2 Genotype of Helicobacter pylori in Japanese clinical isolates. J. Clin. Microbiol. 39, 2463–2465. doi: 10.1128/JCM.39.7.2463-2465.2001
Montecucco, C., and Rappuoli, R. (2001). Living dangerously: how Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2, 457–466. doi: 10.1038/35073084
Moodley, Y., Linz, B., Bond, R. P., Nieuwoudt, M., Soodyall, H., Schlebusch, C. M., et al. (2012). Age of the association between Helicobacter pylori and man. PLoS. Pathog. 8:e1002693. doi: 10.1371/journal.ppat.1002693
Morelli, G., Didelot, X., Kusecek, B., Schwarz, S., Bahlawane, C., Falush, D., et al. (2010). Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 6:e1001036. doi: 10.1371/journal.pgen.1001036
Mukhopadhyay, A. K., Kersulyte, D., Jeong, J. Y., Datta, S., Ito, Y., Chowdhury, A., et al. (2000). Distinctiveness of genotypes of Helicobacter pylori in Calcutta, India. J. Bacteriol. 182, 3219–3227. doi: 10.1128/JB.182.11.3219-3227.2000
Pagliaccia, C., de Bernard, M., Lupetti, P., Ji, X., Burroni, D., Cover, T. L., et al. (1998). The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc. Natl. Acad. Sci. USA. 95, 10212–10217. doi: 10.1073/pnas.95.17.10212
Pan, Z. J., Berg, D. E., van der Hulst, R. W., Su, W. W., Raudonikiene, A., Xiao, S. D., et al. (1998). Prevalence of vacuolating cytotoxin production and distribution of distinct vacA alleles in Helicobacter pylori from China. J. Infect. Dis. 178, 220–226. doi: 10.1086/515601
Peek, R. M. (2008). Helicobacter pylori infection and disease: from humans to animal models. Dis. Model. Mech. 1, 50–55. doi: 10.1242/dmm.000364
Prouzet-Mauleon, V., Abid, M. H., Lamouliatte, H., Kauser, F., Megraud, F., and Ahmed, N. (2005). Pathogen evolution in vivo: genome dynamics of two isolates obtained 9 years apart from a duodenal ulcer patient infected with a single Helicobacter pylori strain. J. Clin. Microbiol. 43, 4237–4241. doi: 10.1128/JCM.43.8.4237-4241.2005
Raymond, J., Thiberg, J. M., Chevalier, C., Kalach, N., Bergeret, M., Labigne, A., et al. (2004). Genetic and transmission analysis of Helicobacter pylori strains within a family. Emerg. Infect. Dis. 10, 1816–1821. doi: 10.3201/eid1010.040042
Secka, O., Antonio, M., Berg, D. E., Tapgun, M., Bottomley, C., Thomas, V., et al. (2011). Mixed infection with cagA positive and cagA negative strains of Helicobacter pylori lowers disease burden in the gambia. PLoS ONE 6:e27954. doi: 10.1371/journal.pone.0027954
Smith, S. I., Oyedeji, K. S., Arigbabu, A. O., Cantet, F., Megraud, F., Ojo, O. O., et al. (2004). Comparison of three PCR methods for detection of Helicobacter pylori DNA and detection of cagA gene in gastric biopsy specimens. World. J. Gastroenterol. 10, 1958–1960. doi: 10.3748/wjg.v10.i13.1958
Solnick, J. V., Hansen, L. M., Salama, N. R., Boonjakuakul, J. K., and Syvanen, M. (2004). Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc. Natl. Acad. Sci. U.S.A. 101, 2106–2111. doi: 10.1073/pnas.0308573100
Suerbaum, S., and Josenhans, C. (2007). Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 5, 441–452. doi: 10.1038/nrmicro1658
Suerbaum, S., Smith, J. M., Bapumia, K., Morelli, G., Smith, N. H., Kunstmann, E., et al. (1998). Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. 95, 12619–12624. doi: 10.1073/pnas.95.21.12619
Tanih, N. F., Ndip, L. M., and Ndip, R. N. (2011). DNA sequence analysis of South African Helicobacter pylori vacuolating cytotoxin gene (vacA). Int. J. Mol. Sci. 12, 7459–7468. doi: 10.3390/ijms12117459
Testerman, T. L., and Morris, J. (2014). Beyond the stomach: an updated view of Helicobacter pyloripathogenesis, diagnosis, and treatment. World. J. Gastroenterol. 20, 12781–12808. doi: 10.3748/wjg.v20.i36.12781
Thompson, L. J., Danon, S. J., Wilson, J. E., O’Rourke, J. L., Salama, N. R., Falkow, S., et al. (2004). Chronic Helicobacter pylori infection with Sydney strain 1 and a newly identified mouse-adapted strain (Sydney strain 2000) in C57BL/6 and BALB/c mice. Infect. Immun. 72, 4668–4679. doi: 10.1128/IAI.72.8.4668-4679.2004
Turner, K. M., Hanage, W. P., Fraser, C., Connor, T. R., and Spratt, B. G. (2007). Assessing the reliability of eBURST using simulated populations with knownancestry. BMC Microbiol. 7:30. doi: 10.1186/1471-2180-7-30
van Doorn, L. J., Figueiredo, C., Sanna, R., Pena, S., Midolo, P., Ng, E. K., et al. (1998). Expanding allelic diversity of Helicobacter pylori vacA. J. Clin. Microbiol. 36, 2597–2603.
Vázquez, J. A., and Berrón, S. (2004). Multilocus sequence typing: the molecular marker of the internet era. Enferm. Infecc. Microbiol. Clin. 22, 113–120. doi: 10.1016/S0213-005X(04)73045-1
Velazquez-Guadarrama, N., Olivares, A., Valencia, P., De los Monteros, L., Madrigal-Santillán, E., and Madrigal-Bujaidar, E. (2007). Genotoxic and oxidative damage induced by Helicobacter pylori in Meriones unguiculatus. J. Environ. Pathol. Toxicol. Oncol. 26, 39–49. doi: 10.1615/JEnvironPatholToxicolOncol.v26.i1.50
Keywords: H. pylori, Meriones unguiculatus, animal model, diversification of genotypes, natural chimera, eBURST, PHYLOViZ
Citation: Mendoza-Elizalde S, Arteaga-Resendiz NK, Valencia-Mayoral P, Luna RC, Moreno-Espinosa S, Arenas-Huertero F, Zuñiga G and Velázquez-Guadarrama N (2016) Diversification of the vacAs1m1 and vacAs2m2 Strains of Helicobacter pylori in Meriones unguiculatus. Front. Microbiol. 7:1758. doi: 10.3389/fmicb.2016.01758
Received: 24 June 2016; Accepted: 19 October 2016;
Published: 08 November 2016.
Edited by:
John R. Battista, Louisiana State University, USAReviewed by:
Irina Pinchuk, University of Texas Medical Branch, USAFilipa F. Vale, Faculdade de Farmácia Universidade de Lisboa, Portugal
Copyright © 2016 Mendoza-Elizalde, Arteaga-Resendiz, Valencia-Mayoral, Luna, Moreno-Espinosa, Arenas-Huertero, Zuñiga and Velázquez-Guadarrama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Norma Velázquez-Guadarrama, normave@himfg.edu.mx Gerardo Zúñiga, capotezu@hotmail.com