 Ana P. B. Nascimento1
Ana P. B. Nascimento1 Mauro F. Ortiz1
Mauro F. Ortiz1 Willames M. B. S. Martins2
Willames M. B. S. Martins2 Guilherme L. Morais1
Guilherme L. Morais1 Lorena C. C. Fehlberg2
Lorena C. C. Fehlberg2 Luiz G. P. Almeida1
Luiz G. P. Almeida1 Luciane P. Ciapina1
Luciane P. Ciapina1 Ana C. Gales2*
Ana C. Gales2* Ana T. R. Vasconcelos1*
Ana T. R. Vasconcelos1*- 1Laboratório de Bioinformática, Laboratório Nacional de Computação Científica, Petrópolis, Brazil
- 2Laboratório Alerta, Division of Infectious Diseases, Department of Internal Medicine, Escola Paulista de Medicina, Universidade Federal de São Paulo, São Paulo, Brazil
Carbapenems represent the mainstay therapy for the treatment of serious P. aeruginosa infections. However, the emergence of carbapenem resistance has jeopardized the clinical use of this important class of compounds. The production of SPM-1 metallo-β-lactamase has been the most common mechanism of carbapenem resistance identified in P. aeruginosa isolated from Brazilian medical centers. Interestingly, a single SPM-1-producing P. aeruginosa clone belonging to the ST277 has been widely spread within the Brazilian territory. In the current study, we performed a next-generation sequencing of six SPM-1-producing P. aeruginosa ST277 isolates. The core genome contains 5899 coding genes relative to the reference strain P. aeruginosa PAO1. A total of 26 genomic islands were detected in these isolates. We identified remarkable elements inside these genomic islands, such as copies of the blaSPM−1 gene conferring resistance to carbapenems and a type I-C CRISPR-Cas system, which is involved in protection of the chromosome against foreign DNA. In addition, we identified single nucleotide polymorphisms causing amino acid changes in antimicrobial resistance and virulence-related genes. Together, these factors could contribute to the marked resistance and persistence of the SPM-1-producing P. aeruginosa ST277 clone. A comparison of the SPM-1-producing P. aeruginosa ST277 genomes showed that their core genome has a high level nucleotide similarity and synteny conservation. The variability observed was mainly due to acquisition of genomic islands carrying several antibiotic resistance genes.
Introduction
Pseudomonas aeruginosa is a ubiquitous microorganism present in many diverse ecological niches, including water, soil, plants, animals, and humans. The ability of P. aeruginosa to survive on minimal nutritional requirements and to tolerate a variety of physical conditions has allowed this organism to persist in environmental and hospital settings (Pier and Ramphal, 2010). Carbapenems represent the main therapy for the treatment of serious P. aeruginosa infections. However, the emergence of carbapenem resistance has jeopardized the clinical use of this important class of compounds (Papp-Wallace et al., 2011). Among P. aeruginosa, hyperproduction of AmpC and/or metallo-β-lactamases coupled with alteration in the outer membrane permeability represent the main mechanism of carbapenem resistance (Lister et al., 2009; Papp-Wallace et al., 2011). In Brazil, P. aeruginosa is an important pathogen in the nosocomial environment. According to the latest report of the Brazilian Health Surveillance Agency1, P. aeruginosa ranked as the fifth most common pathogen causing catheter-related bloodstream infections in adult patients hospitalized at Brazilian intensive care units. Among the 2480 P. aeruginosa reported, nearly 42% were resistant to carbapenems. To date, the production of SPM-1, São Paulo metallo-β-lactamase, has been the most common mechanism of carbapenem resistance identified in P. aeruginosa isolated from Brazilian medical centers (Toleman et al., 2002; Scheffer et al., 2010; Rossi, 2011). However, unlike other carbapenemases such as NDM, IMP, and KPC, SPM-1 has been only reported in P. aeruginosa isolates. Previous studies have shown the presence of a SPM-1-producing P. aeruginosa clone belonging to the ST277, clone SP, widely spread within the Brazilian territory (Gales et al., 2003; Scheffer et al., 2010; Silva et al., 2011; Silveira et al., 2014).
This study was undertaken to determine the possible presence of genetic factors associated with the resistance and persistence of this clone within Brazilian institutions In addition, we aimed to compare the genome of the SPM-1-producing P. aeruginosa isolates collected from a single intensive care unit over a 9-year period to evaluate whether this clone had suffered any temporal changes compared with the index isolate.This is the first study to date to comprehensively evaluate and compare the complete genome of the SPM-1-producing P. aeruginosa ST277 isolates, as the genome of few SPM-1-producing P. aeruginosa strains have only been partially analyzed (Boyle et al., 2012; Silveira et al., 2014; van Belkum et al., 2015).
Methods
Bacterial Strains, Culture Conditions, and DNA Isolation
We studied six SPM-1-producing P. aeruginosa isolates, including the index isolate PA1088 (previously named 48-1997A), which was the first reported clinical isolate to carry blaSPM−1 (Toleman et al., 2002). The remaining five isolates were recovered from distinct patients admitted to a single intensive care unit between the years 2003 and 2012 (Table 1). All isolates were collected from the same tertiary teaching hospital located in the city of São Paulo, Brazil. The presence of blaSPM−1 was initially confirmed by PCR and DNA sequencing (BigDye Terminator Cycle Sequencing, Applied Biosystems, Foster City, USA) using primers previously described (Mendes et al., 2004). For whole genome sequencing, the bacteria were grown overnight in LB broth (Oxoid, Basingstoke, England) at 37°C. Total DNA was extracted using the Qiamp DNA Stool Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The DNA concentration was measured in a NanoVue digital spectrophotometer (GE Healthcare Life Sciences, New Jersey, USA) and submitted to the Unidade de Genômica Computacional Darcy Fontoura de Almeida (UGCDFA) of Laboratório Nacional de Computação Científica (LNCC) for further analysis.
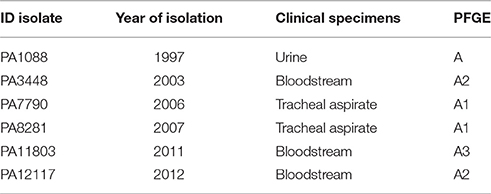
Table 1. Bacterial isolates sequenced in this work.
DNA Sequencing, Genome Assembly, Genome Annotation, and Comparative Genomics
Six whole genome sequencing libraries were generated using the Illumina TruSeq DNA PCR-free sample preparation kit with a median insert size of 550 bp according to the manufacturer's protocols. Briefly, 2 μg of genomic DNA was sheared using a Covaris M220 Focused-ultrasonicator, end-repaired, A-tailed, and adapter ligated. Library quantification was carried out by real-time PCR. Libraries were pooled together in equimolar amounts and sequenced by an Illumina MiSeq instrument in one 2 × 300 bp paired-end run. Genome assembly was performed using Newbler version 3.0. In addition, Celera assembler version 8.2 was used to close eventual gaps. Gaps within scaffolds resulting from repetitive sequences were resolved by in silico gap filling. We achieved mean sequence coverage of 170-fold for each of the six genomes. Mauve-based alignment of contigs revealed extensive synteny between the genomes of the six isolates and the reference genome of P. aeruginosa PAO1. However, two contigs of 49 kb did not align with chromosomal sequences. Notably, the mean sequence coverage for these putative extrachromosomal contigs was 3-fold higher than that observed for the syntenic contigs. Moreover, the two contigs showed different start points in different assemblies, indicating a circular sequence. The System for Automated Bacterial Integrated Annotation (SABIA) pipeline was used for gene prediction and automatic annotation followed by manual validation (Almeida et al., 2004). After annotation, the genomes were analyzed by the Bidirectional Best-Hits (BBH) clustering method (Overbeek et al., 1999), which compares different genomes with each other using the BLAST program (Altschul et al., 1997) to identify pairs of corresponding genes (clusters) and to recognize the best hit in other genomes. The parameters applied were 90% coverage, 90% of similarity and an e < 10−5. The GView Server (Petkau et al., 2010) was used to obtain the sequence of pan, core and unique genome of the six isolates using the P. aeruginosa PAO1 strain as a reference when necessary, with a minimum identity of 90% and an e < 10−5. The genomes of the six SPM-1-producing P. aeruginosa isolates were deposited in Genbank repository under the accession numbers: CP015001 (PA1088); LVWC01000000 (PA3448 contigs and plasmid); CP014999 (PA7790); CP015000 (PA7790 plasmid); CP015002 (PA8281); CP015003 (PA11803) and LVXB01000000 (PA12117).
Phylogenetic Analysis and Multilocus Sequence Typing
We used all conserved open reading frames (ORFs) among our six strains, the reference genome PAO1 (Stover et al., 2000), 11 ST277 strains with genome available:19BR (GCA_000223945.2), 213BR (GCA_000223965.2), 9BR (GCA_000223925.2), BWHPSA041 (GCA_000520375.1), AZPAE12409 (GCA_000797005.1), AZPAE14819 (GCA_000795205.1), AZPAE14821 (GCA_000795235.1), AZPAE14822 (GCA_000795265.1), AZPAE14853 (GCA_000789905.1), AZPAE14923 (GCA_000791205.1), CCBH4851 (GCA_000763245.1) and a single MLST locus variant strain: BWHPSA007 (GCA_000481565.1) to reconstruct the phylogeny. A total of 5 042 protein sequences were concatenated applying neighbor joining, minimum evolution, UPGMA and maximum parsimony methods for reconstruct initial trees using Poisson method, uniform rates among sites, complete deletion treatment to gaps and bootstrap 100 with MEGA software (Kumar et al., 2016). The tree was visualized using the TreeView tool (Page, 1996).
The seven housekeeping genes acs, aro, gua, mut, nuo, pps, and trp were selected according to the multilocus sequence typing (MLST) scheme for P. aeruginosa2 to confirm the allelic profiles of the six isolates.
Genomic Islands and Insertion Sequences Identification
Genomic islands (GIs) are segments of DNA mostly acquired by horizontal gene transfer. IslandViewer 3 (Dhillon et al., 2015) and PIPS (Soares et al., 2012) software were applied to detect genomic islands. IslandViewer 3 is based on sequence composition and comparative genomic methods, which may result in a prediction of false positives GIs, whereas PIPS includes the detection of virulence factors, hypothetical proteins, and flanking tRNAs in its analysis, which may exclude regions that do not meet these parameters. Both outputs were validated manually by observing the following criteria: (i) atypical G+C content; (ii) presence of mobile elements; (iii) adjacency to tRNA genes; (iv) size above 5 000 bp; and (v) comparison of the boundaries to the genomic context of the PAO1 reference strain. All criteria should be fulfilled, except for items (ii) and (iii), which were not mandatory to characterize a genomic island. We also used the IS Finder database and tools (Siguier et al., 2006) to identify the insertion sequences (ISs) from P. aeruginosa genomes. We considered full elements or fragments with e < 10−6 in BLASTn searches (Altschul et al., 1997). We also incorporated ORF regions sharing similarities with transposases into genome annotation with the SABIA platform (Almeida et al., 2004).
Single Nucleotide Polymorphisms Analysis
To identify possible polymorphisms in the six P. aeruginosa samples, we performed a single nucleotide polymorphism (SNP) calling using the PAO1 genome (NC_002516) as a reference. Briefly, the FASTA genome and GTF gene coordinates files were retrieved from NCBI. The deep sequencing libraries files were quality checked with the FastQC tool3 and trimmed with the FASTX_Toolkit4. The trimmed reads from the six samples were mapped separately against the PAO1 genome with the Bowtie 2 (Langmead and Salzberg, 2012) mapper with one mismatch per seed region (20 nucleotides in length), using three different seed regions for each read with repetitive regions and trying to extend 20 nucleotide after mapped seed region (very sensitive preset). The resulting mapping files were treated with the SAMtools program (Li et al., 2009); only mapping reads with a map quality (mapQ) greater than 30 were kept. The Picard mark duplicates tool5 was used to flag putative sequencing artifacts, such as optical duplicates. The Genome Analysis Toolkit (GATK) (McKenna et al., 2010) was used to call the variants using default parameters. The SNPs were annotated with the SnpEff tool (Cingolani et al., 2012) and custom Python scripts. Only SNPs with coverage larger than 10 reads were considered in further analyses. To find SNPs among the six P. aeruginosa isolates, the PA1088 genome was used as a reference, and the SNP call was performed as previously mentioned. In addition to the SNP call analysis, we performed a BLASTp search (Altschul et al., 1997), using the protein sequences of the six isolates against the PAO1 encoded proteins. This approach enabled us to find any polymorphism that was not detected by the previous method.
Susceptibility Testing, Pulsed-Field Gel Electrophoresis, and qRT-PCR
Antimicrobial susceptibility testing was performed by broth microdilution, and the results were interpreted according to the criteria of the European Committee on Antimicrobial Susceptibility Testing6. P. aeruginosa ATCC 29853 and Escherichia coli ATCC 25922 were tested as quality control strains. The genetic relatedness was initially determined by pulsed-field gel electrophoresis (PFGE) using SpeI enzyme (200 V [6 V/cm]; 13°C; switch time initial 5.0 and final, 60.0; 23 h) (Pfaller et al., 1992), and the results (Figure S1) were interpreted as previously recommended (Tenover et al., 1995). Two copies of blaSPM−1 were identified in the isolates PA3448 and PA8281. To confirm whether the blaSPM−1 multiple copies had led to an increase in transcriptional levels, qRT-PCR experiments were carried out. Total RNA was collected from SPM-1-producing P. aeruginosa isolates using the RNeasy Mini Kit (Qiagen, Hilden, Germany) with addition of RNase-free DNase (Qiagen, Hilden, Germany). Reverse transcription of the extracted RNA was performed using the High Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA, USA). The pair of primers used for the amplification of the blaSPM−1 and 16S rDNA genes were as previously reported (Mendes et al., 2007). Transcripts were quantified in triplicate using SYBR® Green PCR Master Mix (Life Technologies, Carlsbad, CA, USA) and the 7500 Real Time system (Life Technologies, Carlsbad, CA, USA). The 16S rDNA gene was used as a reference to normalize the relative amount of mRNA. The blaSPM−1 transcriptional levels were compared using PA1088 as the reference strain because it has been known to carry a single copy of blaSPM−1. The transcriptional level of genes encoding efflux pumps (mexB, mexD, mexF, and mexY), and the OprD porin (oprD) was also studied but using the PAO1 as the reference strain. Mean values (± standard deviations) of mRNA levels obtained in triplicate were calculated. Strains showing mRNA values of >5-fold for mexD, mexF, or mexY or >2-fold for mexB were considered to overexpress these genes (Cabot et al., 2011).
Results
General Genomic Features
A summary of the genomic features of the six newly sequenced genomes of P. aeruginosa isolates is provided in Table 2. The whole genome size ranged from 6,643,783 to 7,018,690 bp, with only the PA3448 and PA7790 isolates observed to carry a plasmid. Although the isolates had similar G+C contents, it was possible to note some differences in the total number of coding sequences (CDSs) according to the variation in the chromosome size. An overview of the whole genome homology among the P. aeruginosa isolates, core genome and unique regions is presented in Figure 1.
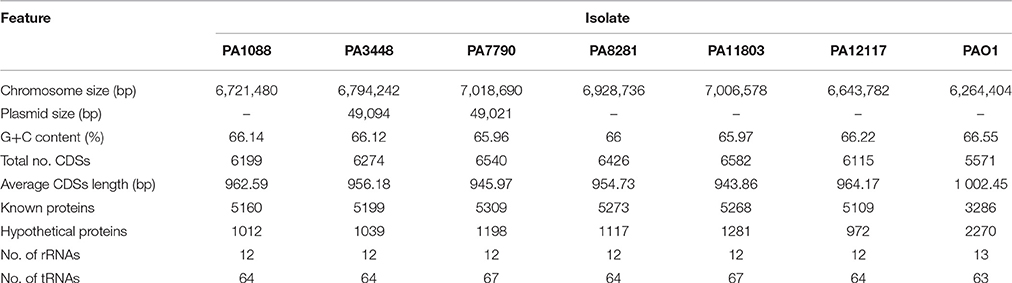
Table 2. General features of the genomes of SPM-1-producing P. aeruginosa ST277 clinical isolates relative to the reference strain P. aeruginosa PAO1.
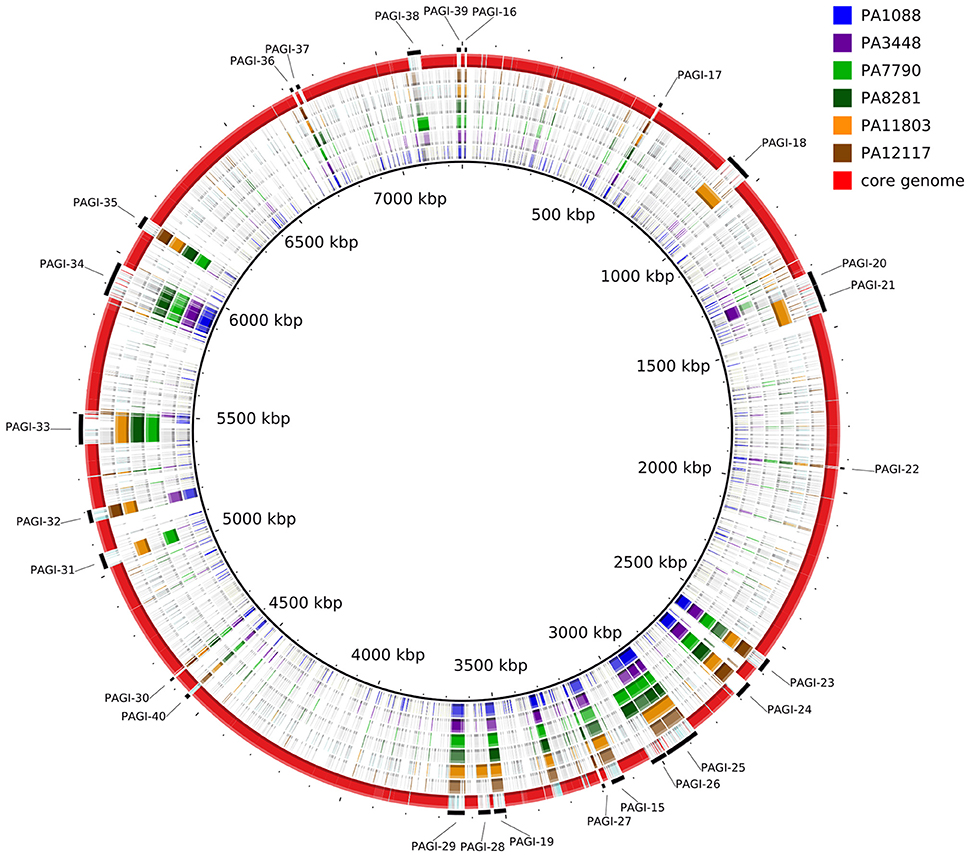
Figure 1. Circular map depicting the unique regions of each SPM-1-producing P. aeruginosa isolate relative to the reference strain PAO1. The red ring represents the core genome shared by all isolates. The outermost, interspaced ring represents the localization of the predicted genomic islands found in each isolate.
A functional classification based on KEGG analysis assigned the CDSs into the 19 main categories was performed (Table S1). All isolates showed a conserved distribution of CDSs among the categories relative to the reference strain PAO1; one exception was the replication and repair category, in which 10 additional CDS were found (Table S2), such as the subunits A and B of excinuclease ABC, a single-stranded DNA-binding (SSB) protein and a DNA methyltransferase present in all six isolates. Interestingly, these additional CDSs involved in replication and repair were located in the genomic islands found among the six isolates.
Plasmids
The isolates PA3448 and PA7790 carried a plasmid with a size of approximately 49 Kb and a G+C content of 58.8%. A major portion of this plasmid (89%) shared 96% of its identity with a chromosomal region of P. aeruginosa PSE305 (Wright et al., 2015). The majority of ORFs were predicted to encode hypothetical proteins, except for those encoding proteins that could be related to the type II secretion system (T2SS) (1 ORF), plasmid stabilization and mobilization (4 ORFs), DNA replication and repair (3 ORFs), and other functions assigned by homology Table S3). The plasmids were identical except for one CDS present only in PA3448's plasmid, ORF 44, which encodes a putative adhesin (Figure S2).
Phylogenetic Analysis and Allelic Profiles
The MLST allelic profiles confirmed the expected relationship showing that all isolates were grouped under the same sequence type, ST277. The concatenated ORFs were used to build a phylogenetic tree representing approximately 70% of the size of each genome. Our phylogenic reconstruction showed that the six P. aeruginosa isolates formed a monophyletic group with other ST277 strains, which supports the high similarity observed among these isolates (Figure 2). All methods used showed the same result, corroborating the validity of the groups. Among the ST277 strains were found two main groups, one including the PA3448 and PA12117 isolates, and another with PA11803, PA7790, and PA8281 isolates. Only the PA1088 isolate showed an unclear relationship due to its low branch support.
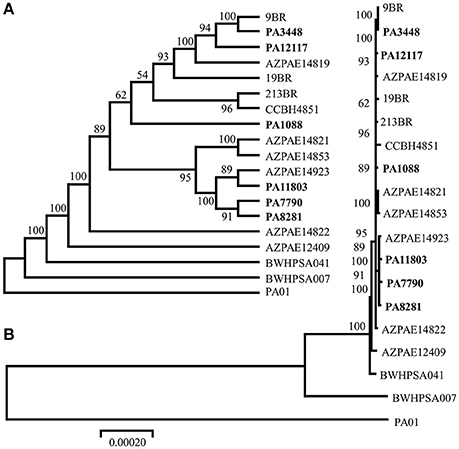
Figure 2. Phylogenic tree of the six sequenced P. aeruginosa isolates, the reference genome PAO1, the 11 ST277 strains: 19BR (GCA_000223945.2), 213BR (GCA_000223965.2), 9BR (GCA_000223925.2), BWHPSA041 (GCA_000520375.1), AZPAE12409 (GCA_000797005.1), AZPAE14819 (GCA_000795205.1), AZPAE14821 (GCA_000795235.1), AZPAE14822 (GCA_000795265.1), AZPAE14853 (GCA_000789905.1), AZPAE14923 (GCA_000791205.1), CCBH4851 (GCA_000763245.1), and a single MLST locus variant strain: BWHPSA007 (GCA_000481565.1). The numbers indicate the bootstrap value associated with the nodes. (A) Consensus tree with neighbor joining method and (B) best tree drawing on scale.
Comparative Genomics
The overall chromosome organization of the P. aeruginosa isolates was compared with that of the reference strain PAO1 using Mauve software (Darling et al., 2004). This analysis revealed a conserved structure among the chromosomes of the SPM-1-producing P. aeruginosa isolates. The multiple alignments showed the existence of 11 conserved blocks; however, it was observed that unique regions were also present (Figure 3). Compared with the PAO1 strain, a major rearrangement was observed in all six P. aeruginosa clones. This rearrangement is an inversion that could be a result of a homologous recombination between genes encoding a 23S ribosomal RNA (PA0668.4 and PA4280.2 relative to PAO1 strain), which was orientated in opposite directions, and share 99% identity.
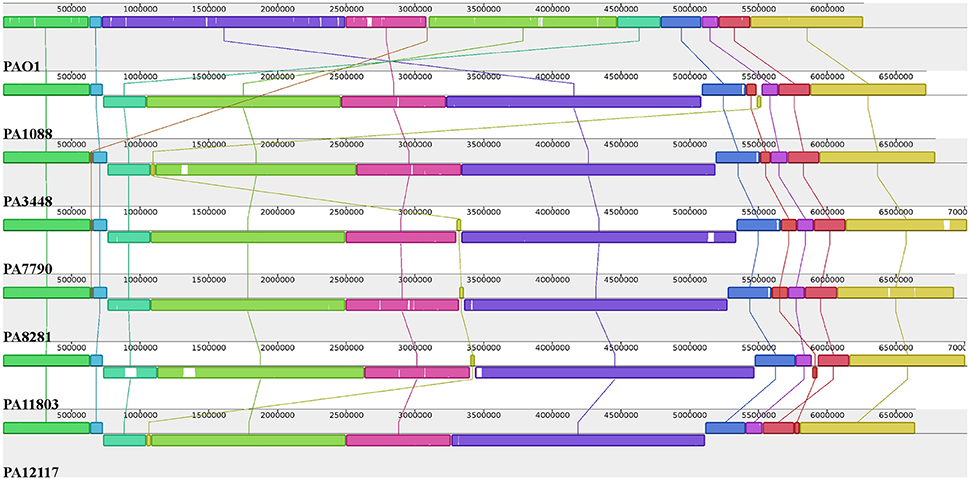
Figure 3. Pairwise alignment between the P. aeruginosa chromosomes. Colors indicate conserved and highly related genomic regions, and white areas identify unique or low-identity regions. Blocks shifted below the horizontal axis indicate segments that align in the reverse orientation relative to the reference strain PAO1.
Comparison of Genetic Repertoire with P. aeruginosa PAO1
The genomes of the six SPM-1-producing P. aeruginosa were compared with the genome of the reference strain PAO1 using the BBH method to evaluate the absence or partial homology of CDSs. All six isolates lacked 102 PAO1 ORFs encoding pyocins, phage elements, regulators, transporters, several hypothetical proteins and others. Among these pyocins, two, S2 and S4, were completely absent. In addition, 18 PAO1 ORFs shared only partial homology with predicted ORFs of SPM-1 isolates, including two porins encoded by PA0958 and PA2213 loci, and the transcriptional regulator encoded by PA2020 (Table S4). All six SPM-1-producing P. aeruginosa isolates showed a 2 bp deletion of 380 and 381 nucleotides in PA0958 (oprD), changing the reading frame and causing a gain of a premature stop codon. The porin encoded by the PA2213 gene also gained a premature stop codon because of a nucleotide change in position 193. The sequence of PA2020 (mexZ) lost 19 bp in all isolates, leading to a gain of a premature stop codon.
Comparison of the Unique and Shared Genes among the SPM-1-producing P. aeruginosa Isolates
A core genome containing 5899 coding genes was identified by the BBH method, representing 89–96% of the total number of CDSs of each clone. Genes conserved among all genomes encode proteins contributing mainly to fundamental housekeeping functions. The set of unique genes encountered for each isolate represents between 0.4 and 5% of the total number of CDSs: 38 for PA1088, 56 for PA3448, 111 for PA7790, 75 for PA8281, 334 for PA11803, and 27 for the PA12117 isolate. The majority of unique genes were annotated as hypotheticals because no function could have been attributed. Other genes were mostly associated with phages, transposases and integrases (Table S5). Among the unique genes, we identified PAO1 partial genes, such as kinB (encoding a two component system sensor protein) in the PA1088 isolate, radC (DNA repair protein) in the PA8281 isolate and lasR (transcriptional regulator) in the PA12117 isolate.
Genomic Islands and Insertion Sequences
We identified 26 genomic islands in the six studied P. aeruginosa isolates. They were named PAGI (P. aeruginosa genomic island) and numbered accordingly from 15 to 40, i.e., PAGI-15 to PAGI-40. The size of the smallest region found was 5 914 bp, whereas the largest was 132,631 bp. A total of 14 islands were common to all six isolates, whereas other 6 were unique, 3 of which were present only in PA11803 (Figure 4; Table S6). Most CDSs observed in PAGIs were predicted to encode hypothetical proteins in addition to transposases, integrases and phage-related proteins (Table S7). Genes conferring antibiotic resistance were mainly located in PAGI-15 and -25, such as blaSPM−1, blaOXA−56, rmtD, cmx, and sul1. Genes homologous to those encoding proteins involved in the cell response to a stress condition, such as hicAB, hipAB, and higAB, were identified in PAGI-17, -24, and -31, respectively. PA1088, PA34448, PA7790, and PA8281 isolates acquired a gene predicted to encode a pyocin, a potent bacteriocin implicated in intraspecific microbial competition, homologous to the S5 type. This gene was carried by PAGI-34. Additionally, PAGI-20, -21, -32, -33, and -38 harbor genes homologous to prtN, ptrB, and prtR, which are involved in the regulation of pyocin production. Interestingly, PAGI-34 was 63% similar to a previously described island, PAPI-1; this island, in contrast to PAGI-34, did not carry a CRISPR-Cas system. This system, which possesses cas3, cas5, cas8c, cas7, cas4, cas1, and cas2 genes (type I-C), was present in four (PA1088, PA3448, PA7790, PA8281) of the six sequenced isolates, but it was absent in the remaining two isolates. Whereas the CRISPR-Cas system was intact in the PA1088 and PA3448 isolates, it was interrupted by the insertion of another island (PAGI-35) in the PA7790 and PA8281 isolates (Figure S3). PAGI-34 also shared a higher coverage (99%) and similarity (99%) to other mobile elements, the pKLC102-like ICEs, which have been previously reported to carry a type I-C CRISPR-Cas system (van Belkum et al., 2015). PAGI-28, an island present only in PA11803, carried a gene locus predicted to encode proteins homologous to type I-E and type I-F anti-CRISPR systems, namely JBD5-gp34, -gp35, -gp36, and -gp37 phage proteins (Bondy-Denomy et al., 2013; Pawluk et al., 2014).
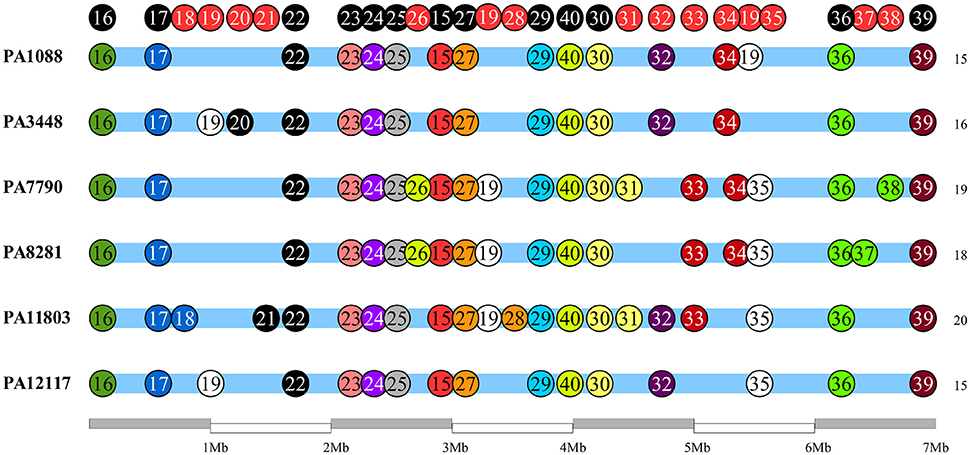
Figure 4. Schematic overview of genomic island (PAGI) distribution in the six SPM-1 isolates. Each circle represents a PAGI throughout the bacterial chromosomes. There are a total of 26 PAGIs, and each isolate carries between 16 and 21 PAGI (number at right). The circles at the top represent conserved sites (black) and variable sites (red).
We identified 20 different types of insertion sequences in the six P. aeruginosa isolates; some ISs were found more than one time in the genome, such as IS222, CR4, TPAse5. The overall distribution of ISs was quite similar among the SPM-1 isolates (25–32) (Figure 5). We observed 21 IS sites conserved between all six isolates. Regarding the variable sites, mobile elements such as TPAse4 were observed at different locations in the genome inserted within PAGI-19, which was present in all six isolates at different chromosomal positions (Figure 5; Table S6). Other variable sites comprised the CR4 elements. Two of them were located inside of PAGI-15 but suffered duplication in PA3448 and PA8281 isolates, resulting in an additional copy of the blaSPM−1 gene. Additional copies of CR4 elements were found in PAGI-25; in PA1088, PA11803, and PA12117, we observed these copies occur next to one copy of the sul1 gene (absent in PA3448) and another next to the rmtD gene (absent in PA3448, PA7790, and PA8281) (Figure 6). The complete list of IS elements identified is presented in Table S8.
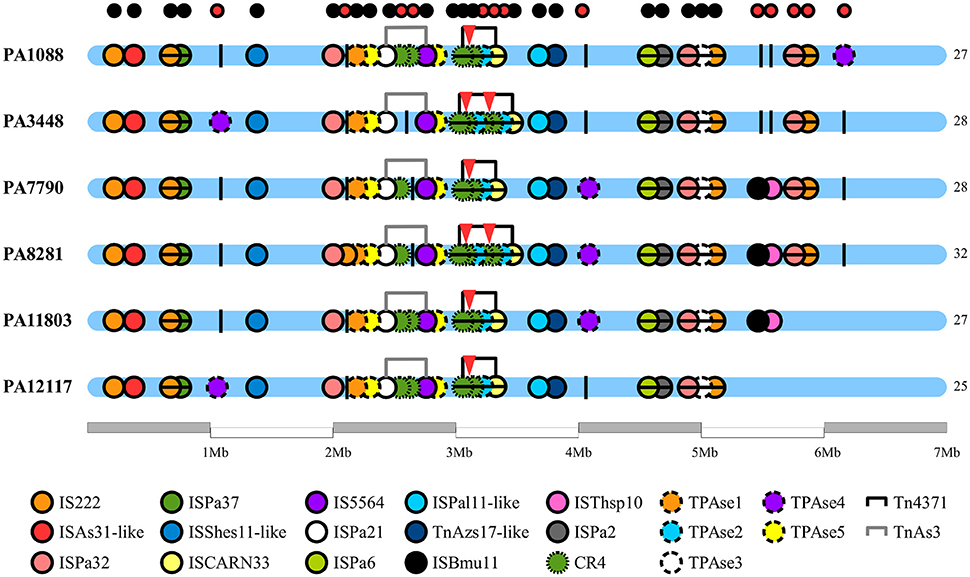
Figure 5. Chromosome distribution of insertion sequences of P. aeruginosa genomes. Each circle represents an insertion sequence (IS) throughout the bacterial chromosome. In the bottom of the figure, the legend differentiates the various ISs by color. The black vertical lines are the empty sites. The small circles at the top of the figure indicate the conservative (black) and variable (red) ISs sites. The red triangles indicate the presence of spm-1 gene copies.
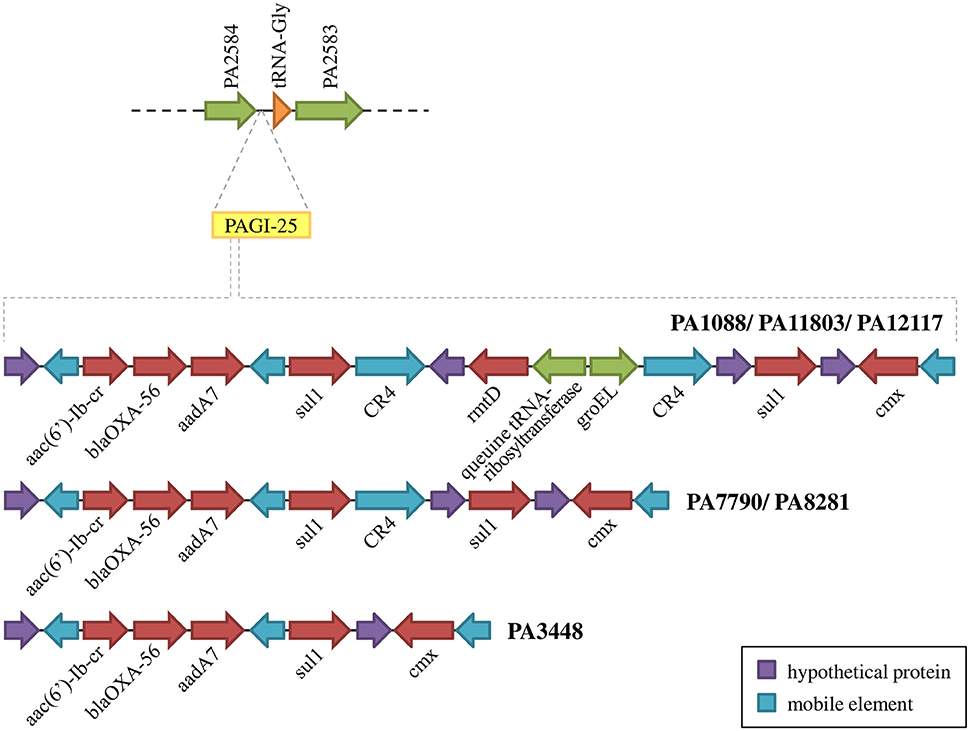
Figure 6. Schematic overview of PAGI-25 highlighting the region harboring genes conferring acquired antibiotic resistance.
SNPs Analysis
To find SNPs in the six P. aeruginosa isolates, the complete genome of P. aeruginosa PAO1 was used as a reference to perform an SNP call. The same strategy was used to find SNPs among the six isolates using the PA1088 genome as reference.
The overall number of SNPs was very similar among the six P. aeruginosa isolates in comparison to PAO1, demonstrating that most SNPs were commonly shared by all SPM-1-producing isolates (25%), except by PA11803, which exhibited approximately 400 more SNPs than the other five isolates (37%) (Table S9). The SNPs found in PA11803 were not homogeneously dispersed in the genome; instead, they were concentrated in a region located between 2,484,263 and 2,737,996 bp, a SNP hot spot measuring approximately 250 Kb in length (Figure 7). It is not uncommon to find these hot spot characteristics, as previously reported for other P. aeruginosa strains when using PAO1 as a reference genome (Bezuidt et al., 2013). Approximately 63% of SNPs in this region were classified as synonymous coding, followed by 22% classified as non-synonymous coding and 15% classified as intergenic. Most non-synonymous coding SNPs were found in genes predicted to encode hypothetical proteins (45%), but some were detected in the putative operon ambB-E and the pvd gene cluster (Table S10).
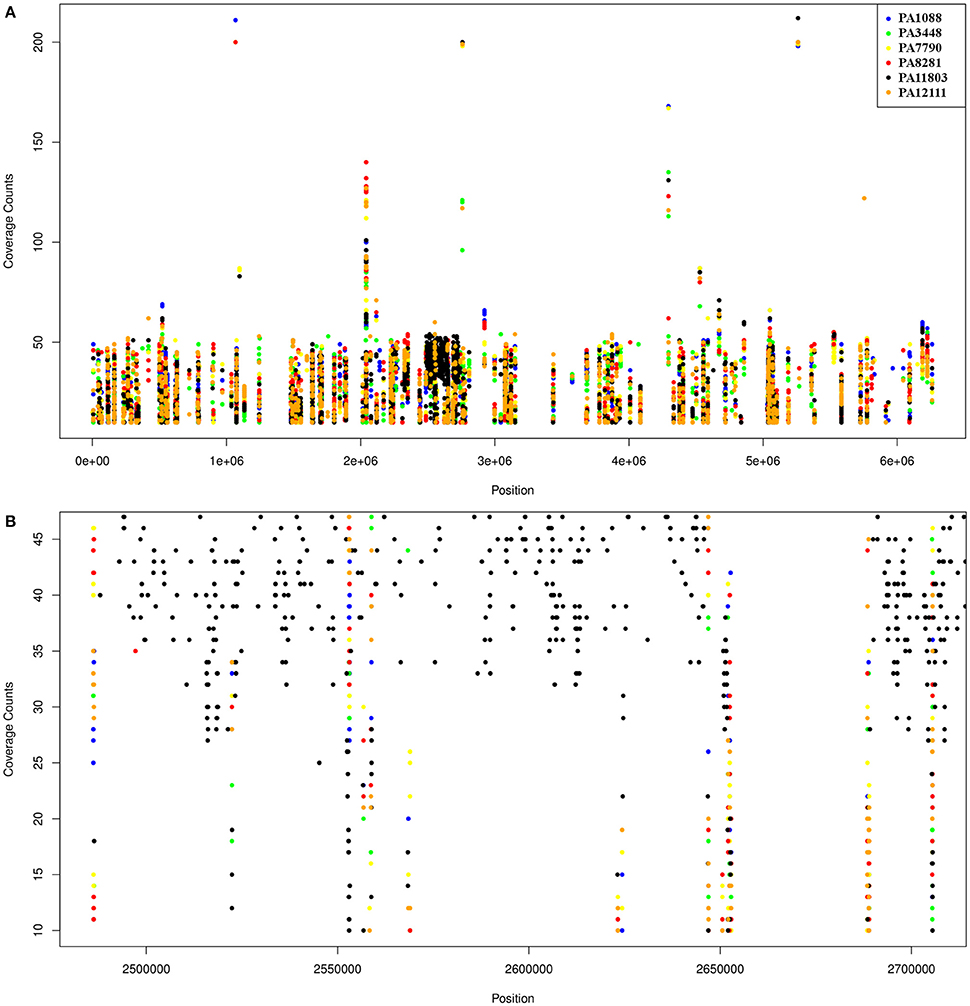
Figure 7. Schematic representation of SNPs distribution in the six P. aeruginosa isolates using PAO1 as a reference genome. Each dot represents an SNP. The coverage counts show how many mapped reads support the SNP. (A) SNPs distribution of the six isolates relative to PAO1; the black spot refers to the SNP enriched region in PA11803. (B) A higher resolution representation of the PA11803 SNP enriched region.
Among the SNPs shared by all P. aeruginosa isolates, the majority were located in the intergenic regions, followed by SNPs in CDSs, but without an amino acid change (synonymous coding). SNPs causing amino acid changes were also found in all six P. aeruginosa isolates (Table S9). We focused on this class of SNPs that could induce changes in phenotype. The remaining SNP classes were mostly found in hypothetical genes, except for a hydrolase, an amidase and an ABC transporter gene that harbored a lost stop codon (Table S10).
The SNP call performed using the PA1088 genome as a reference showed a number of SNPs lower than that detected in the previous analysis because of the high similarity among the isolates. Most of the non-synonymous coding SNPs found in genomic islands were located in genes predicted to encode hypothetical proteins, except for those encoding an ATP-dependent CLP protease found in PAGI-23 (PA3448), a zonula occludens toxin gene found in PAGI-29 (PA3448, PA7790, PA11803) and a lytic enzyme found in PAGI-32 (PA7790, PA8281) (Table S11).
Microbiological Characterization of Bacterial Isolates and Multidrug-Resistance Mechanisms Analysis
The six SPM-1-producing P. aeruginosa isolates were fully susceptible to polymyxin B (MICs, 0.25–0.5 μg/mL) but were resistant to ciprofloxacin (MICs, >32 μg/mL) and all β-lactams tested, except for aztreonam (Table 3). P. aeruginosa isolates exhibited intermediate susceptibility to this compound, which is not surprising because aztreonam is not recognized as a substrate by β-lactamases such as SPM-1, AmpC, OXA-50, and OXA-56 (Toleman et al., 2002; Lister et al., 2009; Leonard et al., 2013). All P. aeruginosa isolates were resistant to both amikacin and gentamicin, except PA3448. This isolate was susceptible to amikacin but resistant to gentamicin.
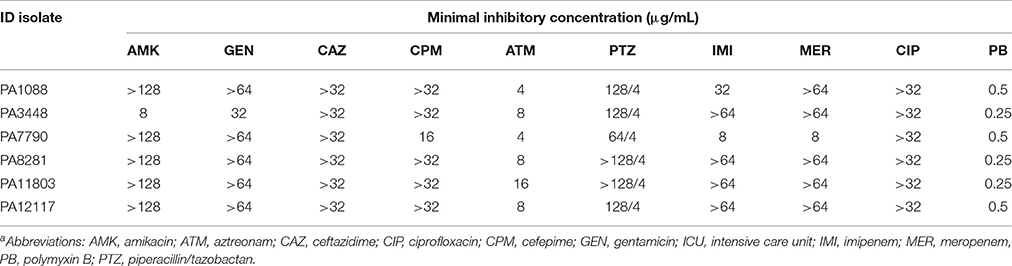
Table 3. Microbiological characteristics of the six SPM-1-producing P. aeruginosa ST277 clinical isolatesa.
The SPM-1 encoding gene, blaSPM−1, was found in all six P. aeruginosa isolates within two distinct genetic contexts (Figure 8). The blaSPM−1 gene was carried by a transposon with two CR4 elements in PAGI-15. The isolates PA1088, PA7790, PA11803, and PA12117 showed a duplication of a 4.2 Kb flanking sequence with two directly oriented copies of a region carrying one gene coding a hypothetical protein, one traR, one bcr1, and one virD2 genes. This context is similar to that described for ICETn43716061 (Fonseca et al., 2015), except that the transcription direction was inversely oriented since PAGI-15 is located in a genomic region that suffered a major chromosomal inversion. In the remaining two SPM-1-producing P. aeruginosa, PA3448, and PA8281, we observed a duplication of a region measuring approximately 10 Kb possibly caused by recombination of the directly oriented repeats aforementioned, between which blaSPM−1 was inserted, resulting in one additional copy of this gene (Wozniak and Waldor, 2010; Reams et al., 2012). The blaSPM−1 transcriptional levels were 2.6 and 1.6 times higher in the PA3448 and PA8281 isolates, respectively, compared with the transcription level of PA1088, suggesting that both blaSPM−1 copies were expressed (Figure 9A).
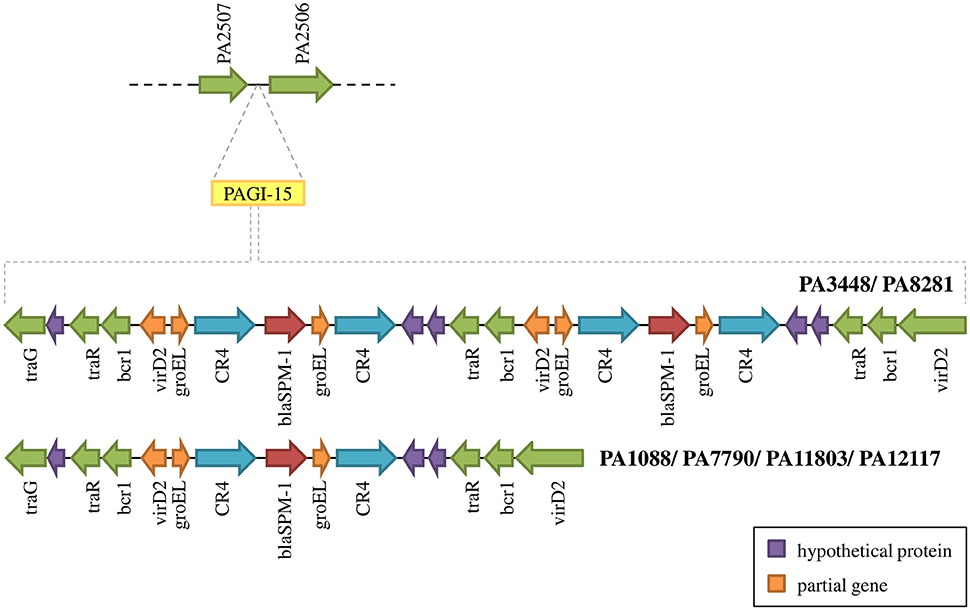
Figure 8. Schematic overview of PAGI-15 region harboring the spm-1 gene. PA3448 and PA8281 carry two copies of this gene as a result of a duplication inside the island.
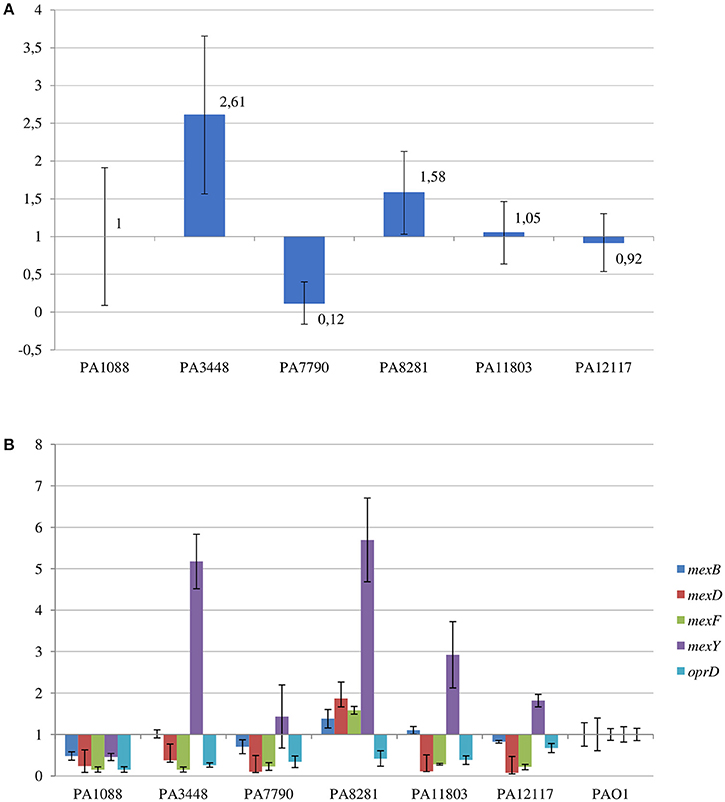
Figure 9. Relative transcriptional levels by qRT-PCR of (A) blaSPM−1 gene in the SPM-1-producing P. aeruginosa isolates compared to PA1088 strain; and (B) mexB, mexD, mexF, mexY, and oprD genes in the SPM-1-producing P. aeruginosa isolates compared to PAO1 strain.
To verify any difference in genes involved in multidrug resistance mechanisms in all six isolates relative to the PAO1 reference strain, we performed BLASTp searches using default parameters. The chromosomal β-lactamases blaOXA−50h and the cephalosporinase AmpC were detected among all six P. aeruginosa isolates. AmpC carried substitutions at R79Q and T105A that were identical to those described in the PDC-5 variant previously described (Rodríguez-Martínez et al., 2009). In addition, substitutions in AmpC regulators such as DacB (PBP4; A394P) and AmpD (G148A and S175L) were also observed in our study. However, no alterations in AmpD homologous proteins AmpDh2 and ApmDh3 were detected. Among the six P. aeruginosa isolates evaluated in this study, we also observed a substitution (A104P) in PbpC, a penicillin binding protein (PBP3A), which was detected by our SNP call analysis but only in the PA11803 isolate.
Other antimicrobial resistance genes were detected among P. aeruginosa isolates. The β-lactamase blaOXA−56, a narrow-spectrum oxacilinase; the aminoglycoside-modifying enzymes (AMEs) aadA7 and aac(6′)-Ib-cr; the sulphonamide resistance gene sul1; and the chloramphenicol-related resistance gene cmx were present in three distinct mobile genetic contexts carried by a TnAs3 transposon, which were inserted into the PAGI-25 (Figure 6). aac(6′)-Ib-cr, blaOXA−56, and aadA7 were harbored as gene cassettes of In163, a class 1 integron. These mobile genetic elements were integrated into the chromosome of all six P. aeruginosa isolates evaluated. We also observed two copies of sul1 in all isolates, except in PA3448.
The six P. aeruginosa isolates showed a deletion in oprD (coding the main porin for the uptake of carbapenems), as outlined before, and an important reduction in the oprD transcriptional levels by qRT-PCR when compared to that of PAO1 strain (Figure 9B).
Substitutions at quinolone resistance determining regions of GyrA (T83I) and ParC (S87L) were observed, justifying the ciprofloxacin resistance exhibited by all P. aeruginosa isolates. In addition, an unknown substitution H262Q in ParC was observed.
Among the RND-type efflux systems present in P. aeruginosa, a Q25L substitution in the MexR was only observed in the PA1088 isolate suggesting that MexAB-OprM was not overexpressed in most isolates. In all six clones, the mexZ sequence was incomplete likely leading to the overexpression of MexXY-OprM system. In contrast, substitutions (F172I, I341F and D345E for MexT; D249N for MexS) not related to date to the MexEF-OprN system overexpression were observed in both transcriptional regulators, MexT and MexS, in all six isolates. By qRT-PCR, comparing the results to those obtained for PAO1 strain, increased mexY transcriptional levels were observed for PA8281 (5.7-fold), PA3448 (5.2-fold), PA11803 (2.9-fold), PA12117 (1.8-fold), and PA7790 (1.6-fold). On the other hand, a not significant increase in the mexB transcriptional levels was observed only for PA8281 (1.4-fold, Figure 9B).
Virulence-Related Factors Analysis
A BLASTn search using the Virulence Factors of Pathogenic Bacteria (VFDB) database (Chen et al., 2016) to find homology between sequences of the genomic islands found in SPM-1-producing P. aeruginosa isolates showed no significant results. Moreover, the comparative analysis did not yield any results related to genes encoding the main known virulence-related factors (Table S4).
The six SPM-1-producing P. aeruginosa showed several SNPs in genes encoding virulence factors (Table S12). We found 19, 20, and 19 non-synonymous SNPs in cupC1, 2, and 3, respectively. These genes participate in the chaperone-usher pathway involved in biofilm formation (Vallet et al., 2004). Moreover, the S → Y SNP, previously reported in CupC2 (Bezuidt et al., 2013), was also present in our SNP call analysis only in PA3448. Our analysis also identified polymorphisms in genes related to virulence, such as clpV3, encoding a protease related to type VI secretion system (T6SS). We observed a non-synonymous SNP in clpV3 that has not been related until now; however, it is known that mutations in this gene can cause inactivation of T6SS (Hachani et al., 2011). In addition, the PA11803 isolate has one ClpV3 SNP (I → T) that is different from another isolates (L → F), but it is also not clear whether this SNP could affect T6SS. The vgrG1 gene is also related to T6SS, and it is responsible for encoding a protein that acts as a puncturing device (Hachani et al., 2014). Four isolates (PA3448, PA8281, PA11803, and PA12117) showed a non-synonymous coding SNP (N → D) that was previously identified (Bezuidt et al., 2013). The pvd operon plays a role in the pyoverdine pathway and is related to iron acquisition. Mutations in this operon could decrease the virulence of P. aeruginosa (Lehoux et al., 2000). Our analysis revealed polymorphisms in pvdA, pvdQ, pvdR, and pvdT, but only pvdR and pvdT showed non-synonymous SNPs, in contrast to a previous report (Bezuidt et al., 2013). The ptxR gene is another gene related to the virulence process and is involved in quorum sensing in P. aeruginosa (Carty et al., 2006). We found one non-synonymous coding SNP in this gene in PA11803 (S → G). The pilY1 gene is involved in pilus assembly, twitching motility and adhesion to host cells. All six isolates showed a non-synonymous SNP (F → Y) in PilY1 that was previously described (Bezuidt et al., 2013).
We found SNPs in the amb operon, which produces l-2-amino-4-methoxy-trans-3-butenoic acid (AMB), a potent antibiotic and toxin (Lee et al., 2010). PA11803 showed one non-synonymous SNP in ambB and ambD, three in ambC and eight in the ambE. It has been shown that some site mutations abolish AMB production (Lee et al., 2010), but in our case, more studies are necessary to elucidate whether these SNPs could cease the toxin production.
We identified polymorphisms among the six isolates located in genomic islands when using PA1088 as a reference genome. Our analysis identified a non-synonymous coding SNP (V → D) in a gene encoding a protein homologous to zonula occludens in PA3448, PA7790 and PA11803. The zonula occludens is an enterotoxin elaborated by Vibrio cholerae that increases intestinal permeability by interacting with a mammalian cell receptor with subsequent activation of intracellular signaling leading to the disassembly of intercellular tight junctions (Di Pierro et al., 2001).
Discussion
P. aeruginosa is an important pathogen in the nosocomial environment (Buhl et al., 2015). In this study, we characterized the full genome of six SPM-1-producing P. aeruginosa ST277 isolated from a Brazilian teaching hospital. This clone is widely spread within Brazilian hospitals and, although less frequently distributed than ST111 and ST235, has been recognized as a multidrug-resistant P. aeruginosa global clone (Kos et al., 2015; Oliver et al., 2015; van Belkum et al., 2015).
Most of the genomic features described are consistent with those reported for previously sequenced genomes of other Pseudomonas (Silby et al., 2011). The genomes had a size between 6.6 and 7 Mb with a minor variation in chromosome size and CDS numbers caused mainly by the acquisition of genomic islands. Despite these insertions, the overall functional classification of the CDSs was quite similar when comparing our six isolates with PAO1, except for additional CDSs grouped in the replication and repair category (Table S2). All isolates were observed in three additional CDSs predicted to encode an SSB protein. These proteins protect single-stranded DNA from degradation and are reported to play a role in the mobilization of other proteins in the process of DNA replication, recombination or repair (Shereda et al., 2008). The ABC excinuclease subunits A and B, involved in the recognition and removal of damaged DNA, were also present in all six isolates (Verhoeven et al., 2002). The presence of these replication- and repair-related proteins suggests a reinforcement of mechanisms for maintaining DNA integrity. In addition to these mechanisms, we identified a type I-C CRISPR-Cas system in PAGI-34. The CRISPR-Cas genes constitute a bacterial adaptive genetic immune system that plays a major role in controlling horizontal transfer of elements such as phages and plasmids, avoiding the insertion of mobile elements that could cause gene or operons interruptions or genetic rearrangements. The observation of the type I-C CRISPR-Cas systems in multidrug-resistant P. aeruginosa group ST277 carried by an acquired mobile element is a rare event and was recently described (van Belkum et al., 2015). Moreover, the presence of a type I-C CRISPR-Cas system has been correlated with resistance to amikacin and the presence of the rmtD, aad7 and blaOXA-encoding resistance genes (van Belkum et al., 2015). Thus, the CRISPR-Cas system could be in part responsible for the genomic stability of the six P. aeruginosa isolates. Based on our phylogenetic analysis, GI and IS detection (Figures 2, 5, 6, respectively), we hypothesized that this system would have been recently interrupted in the PA7790 and PA8281 isolates, and lost in PA11803 and PA12117 isolates. However, prior to these events, the type I-C CRISPR-Cas system could have been fully functional, playing its role in the genomic plasticity of these isolates in past years. This hypothesis and the protective effect of the type I-C CRISPR-Cas system need to be further investigated.
The presence of IS elements is related to horizontal gene transfer and genomic rearrangement (Kung et al., 2010; Al-Nayyef et al., 2015). The overall number of ISs in the SPM-1 isolates was quite similar. Slight differences can be observed between them according to the rearrangement or presence of specific genomic islands when the genomes are compared.
The SPM-1-producing P. aeruginosa clones evaluated in this study were fully resistant to all β-lactams, including carbapenems. The production of metallo-β-lactamases such as SPM-1 has been recognized as the main mechanism of carbapenem resistance among P. aeruginosa (Toleman et al., 2002; Lister et al., 2009; Kos et al., 2015). In addition, other mechanisms of β-lactam resistance were identified in these isolates, including loss of OprD, production of intrinsic and acquired β-lactamases, and overexpression of efflux systems. OprD serves as the preferred portal of entry for the carbapenems into the bacterial cell, and the loss of OprD significantly decreases the susceptibility of P. aeruginosa to available carbapenems, especially imipenem (Lister et al., 2009). OprD loss has been commonly reported as a mechanism of resistance to carbapenems, in P. aeruginosa isolated from Brazilian medical centers (Xavier et al., 2010; Fehlberg et al., 2012; Ocampo-Sosa et al., 2012; Cavalcanti et al., 2015; Kos et al., 2015).
Genes encoding intrinsic β-lactamases such as AmpC and OXA-50 h were also encountered in the evaluated genomes. Wild-type strains of P. aeruginosa produce only low basal levels of AmpC and are susceptible to antipseudomonal penicillins, penicillin-inhibitor combinations, cephalosporins, and carbapenems. When AmpC production is significantly increased, P. aeruginosa develops resistance to all β-lactams, with the exception of the carbapenems (Lister et al., 2009). However, it has been demonstrated that some AmpC variants are also able to hydrolyse carbapenems. The ampC carried by all SPM-1-producing P. aeruginosa isolates was identical to the PDC-5 variant and might have contributed to a decrease in susceptibility to oxyiminocephalosporins and imipenem (Rodríguez-Martínez et al., 2009). In addition, AmpC could be overexpressed by the ST277 clone because mutations were observed in ampD, a negative regulator of AmpC, and dacB, which codifies a non-essential low-molecular-weight PBP4 gene that was previously identified as an important component of ampC regulation (Moya et al., 2009). The blaSPM−1 gene was located in PAGI-15, and it was duplicated in two SPM-1 producers. PAGI-15 without the blaSPM−1 duplication can be found in at least five different P. aeruginosa ST277 strains previously sequenced (CCBH4851, PS106, 9BR, 19BR, and 213BR). The island without any copy of blaSPM−1 appears to be widely spread among other bacteria, including Pseudomonas and other genera, such as Ralstonia oxalatica. Together, these data suggest the recent insertion of this transposon and the acquisition of blaSPM−1 gene by P. aeruginosa.
Mechanisms of aminoglycoside resistance, such as the production of AMEs and rRNA methylases, were detected among the SPM-1-producing P. aeruginosa isolates (Doi et al., 2007a). The aadA7 and aac(6′)-Ib-cr genes were found in the six P. aeruginosa isolates. Whereas the aadA7 gene codifies an AME capable of modifying the molecular structure of streptomycin and spectinomycin by adenylation, aac(6′)-Ib-cr codifies an acetyltransferase, AAC(6′)-Ib-cr, that acetylates not only the molecular structure of kanamycin, tobramycin and amikacin but also that of ciprofloxacin (Robicsek et al., 2006; Ramirez and Tolmasky, 2010). In addition, three P. aeruginosa isolates also carried rmtD, a gene encoding RmtD, a rRNA methylase. The methylation of the 16S rRNA of the A site of the 30S ribosomal subunit interferes with aminoglycoside binding and promotes high-level resistance to all clinically available aminoglycosides (Doi and Arakawa, 2007). P. aeruginosa co-producing RmtD and SPM-1 have been frequently reported among Brazilian isolates (Doi et al., 2007b; Lincopan et al., 2010). At least three distinct mechanisms could be related to fluoroquinolone resistance profile observed among the SPM-1-producing P. aeruginosa isolates: the known gyrA and parC mutations, the presence of aac(6′)-Ib-cr, and possible overexpression of MexXY-OprM efflux systems.
Treatment of infections caused by SPM-1-producing P. aeruginosa is currently problematic because only polymyxins remain active. The combination of old antibiotics such as chloramphenicol or bicyclomycin may not be a valid strategy for the treatment of such infections because genes encoding mechanisms of resistance to chloramphenicol (cmx-1) and bicyclomycin (bcr1) were presented in the genome of all sequenced P. aeruginosa isolates.
The pathogenicity of P. aeruginosa has been attributed to the production of several virulence factors, among them are pili, exotoxins, pyoverdin, secretion systems, biofilm formation, all of which tightly controlled by regulatory systems (Balasubramanian et al., 2013; Gellatly and Hancock, 2013). Although we found alterations in several genes encoding virulence factors such as: clpV3, gene components of the pvd cluster, cupC and pilY1, the impact of these findings on their transcription has not yet been studied and need to be further investigated. Surprisingly, the clpV3 SNP found in our analysis has never been described. We also found several possible new transcriptional regulators carried by the acquired genomic islands, such as genes sharing a homology with prtN, ptrB and prtR, which appears to have their expression affected when exposed to β-lactam stress (Matsui et al., 1993; Balasubramanian et al., 2012). These finding suggest additional players in P. aeruginosa regulatory network and, consequently, in the bacterial response to the antibiotic therapy.
Despite the slight variations observed among the SPM-1-producing P. aeruginosa isolates, our work demonstrated, by comparative genomics, IS and SNP analysis, that the six isolates did not present high genome plasticity over the 9-year period even after being exposed to an environment of antibiotic selective pressure. We attributed this finding to the presence of additional replication- and repair-related proteins and the type I-C CRISPR-Cas system in PAGI-34 because these factors could be responsible for modulating the shape of the P. aeruginosa genome. This comparative genomics report is an important way of determining strain features to enable the development of new therapies to combat infections and to avoid the occurrence of future outbreaks and the worldwide dissemination of the SPM-1-producing P. aeruginosa ST277 strains, which despite being widely spread only in Brazilian hospitals, have already been recognized as multidrug-resistant global clones.
Author Contributions
AN performed genome annotation, comparative genomics, interpreted the results and wrote the manuscript. MO performed insertion sequences and phylogeny, analysis and wrote the manuscript. WM performed microbiological characterization. GM performed the single-nucleotide polymorphisms analysis and wrote the manuscript. LF performed microbiological characterization. LA performed the genome assembly. LC performed genome analysis and revised the manuscript. AG and AV designed the experiment, supervised the research, revised the manuscript and served as corresponding authors.
Funding
This work was funded by National Counsel of Technological and Scientific Development (CNPq) (grant numbers: 305535/2014-5, 302768/2011-4, 312864/2015-9), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (grant number: E-26/202.903/2016) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the staff of LNCC for support.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01946/full#supplementary-material
Footnotes
1. ^ANVISA. “Relatório de resistência microbiana em infecções primárias de corrente sanguínea confirmadas laboratorialmente associadas a cateter venoso central em unidades de terapia intensiva (2014)”. Last modified December 31, 2015. http://www20.anvisa.gov.br/segurancadopaciente/index.php/publicacoes/item/12.
2. ^PubMLST. “Pseudomonas aeruginosa MLST”. Accessed May 15, 2015. http://pubmlst.org/paeruginosa.
3. ^Babraham Bioinformatics. “FastQC”. Last modified March 08, 2016. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
4. ^FASTX-Toolkit. “FASTQ/A short-reads pre-processing tools”. Last modified February 02, 2010. http://hannonlab.cshl.edu/fastx_toolkit/index.html.
5. ^Picard Tools by Broad Institute. “Picard”. Accessed December 02, 2015. http://broadinstitute.github.io/picard.
6. ^EUCAST. “Clinical breakpoints”. Accessed January 18, 2016. http://www.eucast.org/clinical_breakpoints.
References
Almeida, L. G., Paixão, R., Souza, R. C., Costa, G. C., Barrientos, F. J., Santos, M. T., et al. (2004). A system for automated bacterial (genome) integrated annotation–SABIA. Bioinformatics 20, 2832–2833. doi: 10.1093/bioinformatics/bth273
Al-Nayyef, H., Guyeux, C., Petitjean, M., Hocquet, D., and Bahi, J. (2015). “Relation between insertion sequences and genome rearrangements in Pseudomonas aeruginosa,” in Bioinformatics and Biomedical Engineering, eds F. Ortuño, and I. Rojas (Granada: Springer International Publishing), 426–437.
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Balasubramanian, D., Schneper, L., Kumari, H., and Mathee, K. (2013). A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res. 41, 1–20. doi: 10.1093/nar/gks1039
Balasubramanian, D., Schneper, L., Merighi, M., Smith, R., Narasimhan, G., Lory, S., et al. (2012). The regulatory repertoire of Pseudomonas aeruginosa AmpC β-lactamase regulator AmpR includes virulence genes. PLoS ONE 7:e34067. doi: 10.1371/journal.pone.0034067
Bezuidt, O. K., Klockgether, J., Elsen, S., Attree, I., Davenport, C. F., and Tümmler, B. (2013). Intraclonal genome diversity of Pseudomonas aeruginosa clones CHA and TB. BMC Genomics 14:416. doi: 10.1186/1471-2164-14-416
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L., and Davidson, A. R. (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. doi: 10.1038/nature11723
Boyle, B., Fernandez, L., Laroche, J., Kukavica-Ibrulj, I., Mendes, C. M., Hancock, R. W., et al. (2012). Complete genome sequences of three Pseudomonas aeruginosa isolates with phenotypes of polymyxin B adaptation and inducible resistance. J. Bacteriol. 194, 529–530. doi: 10.1128/JB.06246-11
Buhl, M., Peter, S., and Willmann, M. (2015). Prevalence and risk factors associated with colonization and infection of extensively drug-resistant Pseudomonas aeruginosa: a systematic review. Expert Rev. Anti Infect. Ther. 13, 1159–1170. doi: 10.1586/14787210.2015.1064310
Cabot, G., Ocampo-Sosa, A. A., Tubau, F., Macia, M. D., Rodríguez, C., Moya, B., et al. (2011). Overexpression of AmpC and efflux pumps in Pseudomonas aeruginosa isolates from bloodstream infections: prevalence and impact on resistance in a Spanish multicenter study. Antimicrob. Agents Chemother. 55, 1906–1911. doi: 10.1128/AAC.01645-10
Carty, N. L., Layland, N., Colmer-Hamood, J. A., Calfee, M. W., Pesci, E. C., and Hamood, A. N. (2006). PtxR modulates the expression of QS-controlled virulence factors in the Pseudomonas aeruginosa strain PAO1. Mol. Microbiol. 61, 782–794. doi: 10.1111/j.1365-2958.2006.05269.x
Cavalcanti, F. L., Mirones, C. R., Paucar, E. R., Montes, L., Leal-Balbino, T. C., Morais, M. M., et al. (2015). Mutational and acquired carbapenem resistance mechanisms in multidrug resistant Pseudomonas aeruginosa clinical isolates from Recife, Brazil. Mem. Inst. Oswaldo Cruz 110, 1003–1009. doi: 10.1590/0074-02760150233
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis-10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6, 80–92. doi: 10.4161/fly.19695
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dhillon, B. K., Laird, M. R., Shay, J. A., Winsor, G. L., Lo, R., Nizam, F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43, W104–W108. doi: 10.1093/nar/gkv401
Di Pierro, M., Lu, R., Uzzau, S., Wang, W., Margaretten, K., Pazzani, C., et al. (2001). Zonula occludens toxin structure-function analysis. Identification of the fragment biologically active on tight junctions and of the zonulin receptor binding domain. J. Biol. Chem. 276, 19160–19165. doi: 10.1074/jbc.M009674200
Doi, Y., and Arakawa, Y. (2007). 16S ribosomal RNA methylation: emerging resistance mechanism against aminoglycosides. Clin. Infect. Dis. 45, 88–94. doi: 10.1086/518605
Doi, Y., de Oliveira Garcia, D., Adams, J., and Paterson, D. L. (2007a). Coproduction of novel 16S rRNA methylase RmtD and metallo-β-lactamase SPM-1 in a panresistant Pseudomonas aeruginosa isolate from Brazil. Antimicrob. Agents Chemother. 51, 852–856. doi: 10.1128/AAC.01345-06
Doi, Y., Ghilardi, A. C., Adams, J., de Oliveira Garcia, D., and Paterson, D. L. (2007b). High prevalence of metallo-β-lactamase and 16S rRNA methylase coproduction among imipenem-resistant Pseudomonas aeruginosa isolates in Brazil. Antimicrob. Agents Chemother. 51, 3388–3390. doi: 10.1128/AAC.00443-07
Fehlberg, L. C., Xavier, D. E., Peraro, P. P., Marra, A. R., Edmond, M. B., and Gales, A. C. (2012). Beta-lactam resistance mechanisms in Pseudomonas aeruginosa strains causing bloodstream infections: comparative results between Brazilian and American isolates. Microb. Drug Resist. 18, 402–407. doi: 10.1089/mdr.2011.0174
Fonseca, E. L., Marin, M. A., Encinas, F., and Vicente, A. C. (2015). Full characterization of the integrative and conjugative element carrying the metallo-β-lactamase blaSPM-1 and bicyclomycin bcr1 resistance genes found in the pandemic Pseudomonas aeruginosa clone SP/ST277. J. Antimicrob. Chemother. 70, 2547–2550. doi: 10.1093/jac/dkv152
Gales, A. C., Menezes, L. C., Silbert, S., and Sader, H. S. (2003). Dissemination in distinct Brazilian regions of an epidemic carbapenem-resistant Pseudomonas aeruginosa producing SPM metallo-β-lactamase. J. Antimicrob. Chemother. 52, 699–702. doi: 10.1093/jac/dkg416
Gellatly, S. L., and Hancock, R. E. (2013). Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog. Dis. 67, 159–173. doi: 10.1111/2049-632X.12033
Hachani, A., Allsopp, L. P., Oduko, Y., and Filloux, A. (2014). The VgrG proteins are “à la carte” delivery systems for bacterial type VI effectors. J. Biol. Chem. 289, 17872–17884. doi: 10.1074/jbc.M114.563429
Hachani, A., Lossi, N. S., Hamilton, A., Jones, C., Bleves, S., Albesa-Jové, D., et al. (2011). Type VI secretion system in Pseudomonas aeruginosa: secretion and multimerization of VgrG proteins. J. Biol. Chem. 286, 12317–12327. doi: 10.1074/jbc.M110.193045
Kos, V. N., Déraspe, M., McLaughlin, R. E., Whiteaker, J. D., Roy, P. H., Alm, R. A., et al. (2015). The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob. Agents Chemother. 59, 427–436. doi: 10.1128/AAC.03954-14
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kung, V. L., Ozer, E. A., and Hauser, A. R. (2010). The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 74, 621–641. doi: 10.1128/MMBR.00027-10
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, X., Fox, A., Sufrin, J., Henry, H., Majcherczyk, P., Haas, D., et al. (2010). Identification of the biosynthetic gene cluster for the Pseudomonas aeruginosa antimetabolite L-2-amino-4-methoxy-trans-3-butenoic acid. J. Bacteriol. 192, 4251–4255. doi: 10.1128/JB.00492-10
Lehoux, D. E., Sanschagrin, F., and Levesque, R. C. (2000). Genomics of the 35-kb pvd locus and analysis of novel pvdIJK genes implicated in pyoverdine biosynthesis in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 190, 141–146. doi: 10.1111/j.1574-6968.2000.tb09276.x
Leonard, D. A., Bonomo, R. A., and Powers, R. A. (2013). Class D β-lactamases: a reappraisal after five decades. Acc. Chem. Res. 46, 2407–2415. doi: 10.1021/ar300327a
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lincopan, N., Neves, P., Mamizuka, E. M., and Levy, C. E. (2010). Balanoposthitis caused by Pseudomonas aeruginosa co-producing metallo-β-lactamase and 16S rRNA methylase in children with hematological malignancies. Int. J. Infect. Dis. 14, e344–e347. doi: 10.1016/j.ijid.2009.04.016
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. doi: 10.1128/CMR.00040-09
Matsui, H., Sano, Y., Ishihara, H., and Shinomiya, T. (1993). Regulation of pyocin genes in Pseudomonas aeruginosa by positive (prtN) and negative (prtR) regulatory genes. J. Bacteriol. 175, 1257–1263. doi: 10.1128/jb.175.5.1257-1263.1993
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mendes, R. E., Kiyota, K. A., Monteiro, J., Castanheira, M., Andrade, S. S., Gales, A. C., et al. (2007). Rapid detection and identification of metallo-β-lactamase-encoding genes by multiplex real-time PCR assay and melt curve analysis. J. Clin. Microbiol. 45, 544–547. doi: 10.1128/JCM.01728-06
Mendes, R. E., Toleman, M. A., Ribeiro, J., Sader, H. S., Jones, R. N., and Walsh, T. R. (2004). Integron carrying a novel metallo-β-lactamase gene, blaIMP-16, and a fused form of aminoglycoside-resistant gene aac(6′)-30/aac(6′)-Ib′: report from the SENTRY Antimicrobial Surveillance Program. Antimicrob. Agents Chemother. 48, 4693–4702. doi: 10.1128/AAC.48.12.4693-4702.2004
Moya, B., Dötsch, A., Juan, C., Blázquez, J., Zamorano, L., Haussler, S., et al. (2009). Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 5:e1000353. doi: 10.1371/journal.ppat.1000353
Ocampo-Sosa, A. A., Cabot, G., Rodríguez, C., Roman, E., Tubau, F., Macia, M. D., et al. (2012). Alterations of OprD in carbapenem-intermediate and -susceptible strains of Pseudomonas aeruginosa isolated from patients with bacteremia in a Spanish multicenter study. Antimicrob. Agents Chemother. 56, 1703–1713. doi: 10.1128/AAC.05451-11
Oliver, A., Mulet, X., López-Causapé, C., and Juan, C. (2015). The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 21–22, 41–59. doi: 10.1016/j.drup.2015.08.002
Overbeek, R., Fonstein, M., D'souza, M., Pusch, G. D., and Maltsev, N. (1999). The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. U.S.A. 96, 2896–2901. doi: 10.1073/pnas.96.6.2896
Page, R. D. (1996). TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12, 357–358.
Papp-Wallace, K. M., Endimiani, A., Taracila, M. A., and Bonomo, R. A. (2011). Carbapenems: past, present, and future. Antimicrob. Agents Chemother. 55, 4943–4960. doi: 10.1128/AAC.00296-11
Pawluk, A., Bondy-Denomy, J., Cheung, V. H., Maxwell, K. L., and Davidson, A. R. (2014). A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio 5:e00896. doi: 10.1128/mBio.00896-14
Petkau, A., Stuart-Edwards, M., Stothard, P., and Van Domselaar, G. (2010). Interactive microbial genome visualization with GView. Bioinformatics 26, 3125–3126. doi: 10.1093/bioinformatics/btq588
Pfaller, M., Hollis, R., and Sader, H. (1992). “Molecular Biology - PFGE analysis of chromosomal restriction fragments,” in Clinical Microbiology Procedures Handbook, ed H. Isenberg (Washington, DC: ASM Press), 10.5.c.1–10.5.c.11.
Pier, G. B., and Ramphal, R. (2010). “Pseudomonas aeruginosa,” in Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases, 7th Edn., eds G. L. Mandell, J. E. Bennett, and R. Dolin (Philadelphia: Churchill Livingstone Elsevier), 2835–2860.
Ramirez, M. S., and Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Updat. 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Reams, A. B., Kofoid, E., Kugelberg, E., and Roth, J. R. (2012). Multiple pathways of duplication formation with and without recombination (RecA) in Salmonella enterica. Genetics 192, 397–415. doi: 10.1534/genetics.112.142570
Robicsek, A., Strahilevitz, J., Jacoby, G. A., Macielag, M., Abbanat, D., Park, C. H., et al. (2006). Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 12, 83–88. doi: 10.1038/nm1347
Rodríguez-Martínez, J. M., Poirel, L., and Nordmann, P. (2009). Extended-spectrum cephalosporinases in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 1766–1771. doi: 10.1128/AAC.01410-08
Rossi, F. (2011). The challenges of antimicrobial resistance in Brazil. Clin. Infect. Dis. 52, 1138–1143. doi: 10.1093/cid/cir120
Scheffer, M. C., Gales, A. C., Barth, A. L., Carmo Filho, J. R., and Dalla-Costa, L. M. (2010). Carbapenem-resistant Pseudomonas aeruginosa: clonal spread in southern Brazil and in the state of Goiás. Braz. J. Infect. Dis. 14, 508–509. doi: 10.1590/s1413-86702010000500014
Shereda, R. D., Kozlov, A. G., Lohman, T. M., Cox, M. M., and Keck, J. L. (2008). SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 43, 289–318. doi: 10.1080/10409230802341296
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Silby, M. W., Winstanley, C., Godfrey, S. A., Levy, S. B., and Jackson, R. W. (2011). Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 35, 652–680. doi: 10.1111/j.1574-6976.2011.00269.x
Silva, F. M., Carmo, M. S., Silbert, S., and Gales, A. C. (2011). SPM-1-producing Pseudomonas aeruginosa: analysis of the ancestor relationship using multilocus sequence typing, pulsed-field gel electrophoresis, and automated ribotyping. Microb. Drug Resist. 17, 215–220. doi: 10.1089/mdr.2010.0140
Silveira, M., Albano, R., Asensi, M., and Assef, A. P. (2014). The draft genome sequence of multidrug-resistant Pseudomonas aeruginosa strain CCBH4851, a nosocomial isolate belonging to clone SP (ST277) that is prevalent in Brazil. Mem. Inst. Oswaldo Cruz 109, 1086–1087. doi: 10.1590/0074-0276140336
Soares, S. C., Abreu, V. A., Ramos, R. T., Cerdeira, L., Silva, A., Baumbach, J., et al. (2012). PIPS: pathogenicity island prediction software. PLoS ONE 7:e30848. doi: 10.1371/journal.pone.0030848
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964. doi: 10.1038/35023079
Tenover, F. C., Arbeit, R. D., Goering, R. V., Mickelsen, P. A., Murray, B. E., Persing, D. H., et al. (1995). Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol. 33, 2233–2239.
Toleman, M. A., Simm, A. M., Murphy, T. A., Gales, A. C., Biedenbach, D. J., Jones, R. N., et al. (2002). Molecular characterization of SPM-1, a novel metallo-β-lactamase isolated in Latin America: report from the SENTRY antimicrobial surveillance programme. J. Antimicrob. Chemother. 50, 673–679. doi: 10.1093/jac/dkf210
Vallet, I., Diggle, S. P., Stacey, R. E., Cámara, M., Ventre, I., Lory, S., et al. (2004). Biofilm formation in Pseudomonas aeruginosa: fimbrial cup gene clusters are controlled by the transcriptional regulator MvaT. J. Bacteriol. 186, 2880–2890. doi: 10.1128/JB.186.9.2880-2890.2004
van Belkum, A., Soriaga, L. B., LaFave, M. C., Akella, S., Veyrieras, J. B., Barbu, E. M., et al. (2015). Phylogenetic distribution of CRISPR-Cas systems in antibiotic-resistant Pseudomonas aeruginosa. MBio 6:e01796-15. doi: 10.1128/mBio.01796-15
Verhoeven, E. E., Wyman, C., Moolenaar, G. F., and Goosen, N. (2002). The presence of two UvrB subunits in the UvrAB complex ensures damage detection in both DNA strands. EMBO J. 21, 4196–4205. doi: 10.1093/emboj/cdf396
Wozniak, R. A., and Waldor, M. K. (2010). Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8, 552–563. doi: 10.1038/nrmicro2382
Wright, E. A., Di Lorenzo, V., Trappetti, C., Liciardi, M., Orru, G., Viti, C., et al. (2015). Divergence of a strain of Pseudomonas aeruginosa during an outbreak of ovine mastitis. Vet. Microbiol. 175, 105–113. doi: 10.1016/j.vetmic.2014.11.011
Xavier, D. E., Picão, R. C., Girardello, R., Fehlberg, L. C., and Gales, A. C. (2010). Efflux pumps expression and its association with porin down-regulation and β-lactamase production among Pseudomonas aeruginosa causing bloodstream infections in Brazil. BMC Microbiol. 10:217. doi: 10.1186/1471-2180-10-217
Keywords: drug resistance, comparative genomics, pathogenic bacteria, antimicrobial resistance, carbapenemase, Gram-negative bacilli
Citation: Nascimento APB, Ortiz MF, Martins WMBS, Morais GL, Fehlberg LCC, Almeida LGP, Ciapina LP, Gales AC and Vasconcelos ATR (2016) Intraclonal Genome Stability of the Metallo-β-lactamase SPM-1-producing Pseudomonas aeruginosa ST277, an Endemic Clone Disseminated in Brazilian Hospitals. Front. Microbiol. 7:1946. doi: 10.3389/fmicb.2016.01946
Received: 23 September 2016; Accepted: 21 November 2016;
Published: 05 December 2016.
Edited by:
Teresa M. Coque, Instituto Ramón y Cajal de Investigación Sanitaria, SpainReviewed by:
Antonio Oliver, Hospital Universitario Son Dureta, SpainRaffaele Zarrilli, University of Naples Federico II, Italy
Copyright © 2016 Nascimento, Ortiz, Martins, Morais, Fehlberg, Almeida, Ciapina, Gales and Vasconcelos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana C. Gales, ana.gales@gmail.com
Ana T. R. Vasconcelos, atrv@lncc.br