 Solomon A. Yimer1,2*
Solomon A. Yimer1,2* Alemayehu G. Birhanu2,3
Alemayehu G. Birhanu2,3 Shewit Kalayou1
Shewit Kalayou1 Tahira Riaz2
Tahira Riaz2 Ephrem D. Zegeye4
Ephrem D. Zegeye4 Getachew T. Beyene1
Getachew T. Beyene1 Carol Holm-Hansen5
Carol Holm-Hansen5 Gunnstein Norheim5
Gunnstein Norheim5 Markos Abebe6
Markos Abebe6 Abraham Aseffa6
Abraham Aseffa6 Tone Tønjum1,2
Tone Tønjum1,2- 1Department of Microbiology, Oslo University Hospital, Oslo, Norway
- 2Department of Microbiology, University of Oslo, Oslo, Norway
- 3Department of Medical Biotechnology, Institute of Biotechnology, Addis Ababa University, Addis Ababa, Ethiopia
- 4Centre for Applied Biotechnology, Uni Research Environment, Bergen, Norway
- 5Infection Control and Environmental Health, Norwegian Institute of Public Health, Oslo, Norway
- 6Department of Research and Innovation, Armauer Hansen Research Institute, Addis Ababa, Ethiopia
In order to decipher the nature of the slowly growing Mycobacterium tuberculosis (M.tuberculosis) lineage 7, the differentially abundant proteins in strains of M. tuberculosis lineage 7 and lineage 4 were defined. Comparative proteomic analysis by mass spectrometry was employed to identify, quantitate and compare the protein profiles of strains from the two M. tuberculosis lineages. Label-free peptide quantification of whole cells from M. tuberculosis lineage 7 and 4 yielded the identification of 2825 and 2541 proteins, respectively. A combined total of 2867 protein groups covering 71% of the predicted M. tuberculosis proteome were identified. The abundance of 125 proteins in M. tuberculosis lineage 7 and 4 strains was significantly altered. Notably, the analysis showed that a number of M. tuberculosis proteins involved in growth and virulence were less abundant in lineage 7 strains compared to lineage 4. Five ABC transporter proteins, three phosphate binding proteins essential for inorganic phosphate uptake, and six components of the type 7 secretion system ESX-3 involved in iron acquisition were less abundant in M. tuberculosis lineage 7. This proteogenomic analysis provided an insight into the lineage 7-specific protein profile which may provide clues to understanding the differential properties of lineage 7 strains in terms of slow growth, survival fitness, and pathogenesis.
Introduction
Tuberculosis (TB) has claimed an uncountable number of lives over centuries. One third of the global population is infected with the causative agent Mycobacterium tuberculosis (M. tuberculosis), which is the main cause of TB. Each year, ~10.4 million people contract TB and 1.8 million die from the disease (WHO, 2016). In line with the World Health Organization (WHO) vision, the world is united in the quest to eliminate TB by 2050 (WHO, 2016). A holistic approach to understand host, environmental and bacterial factors to TB susceptibility and disease is crucial to achieve the global TB elimination target. There is a large body of evidence on host and environmental attributes for TB (Rieder, 1999). However, given the potential implications for severity of illness and transmission, there is limited knowledge regarding the basic mechanisms underlying the physiology and pathogenesis of the different M. tuberculosis lineages.
Large sequence polymorphisms classify M. tuberculosis into 7 main lineages. These are lineage 1 (Indo-Oceanic), lineage 2 (East Asian including “Beijing”), lineage 3 (CAS/Delhi), lineage 4 (Euro-American including Latin American Mediterranean (LAM), Haarlem, X type and T families), lineage 5 and lineage 6 (West African 1 and 2, respectively), and lineage 7 (Comas et al., 2013). Lineage 7 was recently detected in Ethiopia and among Ethiopian immigrants in Djibouti (Blouin et al., 2012; Firdessa et al., 2013; Yimer et al., 2013, 2015). We have shown that cells of the M. tuberculosis lineage 7 grow slowly in vitro and that lineage 7 infections are associated with prolonged delay in seeking health care among patients compared to other lineages (Yimer et al., 2015). Whole genome sequencing (WGS) demonstrated that M. tuberculosis lineage 7 cells host a high number of mutations in genes involved in carbohydrate transport and metabolism, transcription, energy production and conversion, all of which contribute to the slow-growth phenotype (Yimer et al., 2016). Former WGS studies have described characteristics and attributes of the other M. tuberculosis lineages that may play important roles in the pathogenesis of TB (Coscolla and Gagneux, 2014). For example, the Beijing genotype (lineage 2) is associated with high bacillary load in acid-fast bacillus (AFB) smears and frequently acquires drug resistance (Coscolla and Gagneux, 2014). Lineages 2, 3, and 4 exhibited a lower early inflammatory response compared to lineage 1 and lineage 6 (Chacon-Salinas et al., 2005). Lineage 3 showed a higher anti-inflammatory phenotype compared to Lineage 4 (Portevin et al., 2011). Mycobacterium africanum (lineage 6) acquires drug resistance at a lower rate than the Euro-American (lineage 4) in Ghana (Albanna et al., 2011). This shows that even though they are genetically closely related, different strains of M. tuberculosis present very diverse clinical phenotypes in terms of virulence. We therefore hypothesized that there may be a mechanism underlying the variation in pathogenicity observed between strains of M. tuberculosis detectable at the proteomic level. Global proteomic characterization by use of mass spectrometry represents a powerful tool and is an important supplement to genomics in defining the protein expression patterns (indicating which genes are expressed and down- or upregulated; Peirs et al., 2005; Kelkar et al., 2011; Schubert et al., 2015; Bespyatykh et al., 2016; Jhingan et al., 2016; Peters et al., 2016). To date, no studies have addressed the relationship between the M. tuberculosis lineage 7 slow-growth phenotype and its proteomic signatures.
The objective of this study was to characterize the differentially abundant protein profile of M. tuberculosis lineage 7 (L7-35 and L7-28) and lineage 4 H37Rv strains and define the proteomic profiles relevant for growth and pathogenicity. This proteomic study generated novel insight into the differentially abundant (DA) proteins in M. tuberculosis lineage 7 vs. lineage 4 strains. A total of 2,867 proteins covering 71% of the predicted M. tuberculosis proteome were identified. The analysis of DA proteins by pathway clustering provided an overview on the lineage-specific protein interaction profile. This information may be used to explain the differential behavior of lineage 7 strains in terms of slow growth, survival fitness, and pathogenesis.
Materials and Methods
Mycobacterial Strains and Growth Conditions
Whole genome sequenced M. tuberculosis lineage 7 strains (L7-35 and L7-28) collected form the Amhara Region of Ethiopia and lineage 4 strain H37Rv were streaked onto Middlebrook 7H10 plates in triplicates from freezer stocks, and incubated in a humidified 37°C, 5% CO2 incubator. After 32 days the cells were carefully scrapped off the agar plates and put into 50 mL Falcon® tubes. The cell pellets were gently resuspended in 30 mL PBS, pH 7.4, and centrifuged at 3,900 rpm for 20 min at 4°C. The cell pellets were subsequently transferred into 2 mL screw capped tubes (Sarstedt, Nümbrecht, Germany) and resuspended in 1 mL PBS, and heat inactivated at 80°C for 90 min. Culturing and processing of the M. tuberculosis samples up until the heat inactivation step were conducted in a biosafety level 3 facility at Oslo University Hospital, Norway. The heat-inactivated M. tuberculosis samples were stored at −80°C until lysed for mass spectrometry analysis.
Proteomic Analysis
Cell lysis
The heat-inactivated cell pellets were resuspended in lysis buffer containing 2% SDS, 10 mM Tris-HCl (pH 7.5), 1 tablet per 50 mL EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich, Cleveland, US) and 1 tablet per 10 mL PhosSTOP Phosphatase Inhibitor Cocktail (Roche). The samples were subsequently transferred into Lysing Matrix B tubes (Roche) and disrupted mechanically by bead beating using MagNa Lyser (Roche Diognostics, GmbH, Mannheim, Germany) for 90 s, speed 6.0. The lysis procedure followed by 1 min cooling on ice was repeated six times. The lysate was clarified by centrifugation (15,000 × g for 15 min) at 21°C, and the supernatant containing the whole cell lysate proteins was transferred in to new 2 mL screw cap micro tubes (Sarstedt, Nümbrecht, Germany).
In-Gel Trypsin Digestion of Cellular Proteins
Hundred μg of protein sample dissolved in NuPAGE LDS sample buffer (4x) and NuPAGE Sample Reducing Agent (10X) (Life Technologies, USA) were incubated for 10 min at 70°C and pre-fractionated by 1.0 mm, 4–12% NuPAGE Novex Bis-Tris SDS-PAGE gel (Life Technologies), at 80 V for 5 min followed by 20 min at 200 V. Gels were Coomassie-stained using a Colloidal Blue Staining kit for NuPAGE as per manufacturer's instructions. After staining, each gel lane was divided into 6 fractions, and each fraction was subjected to in-gel reduction, alkylation, and tryptic digestion (Shevchenko et al., 2006). In brief, proteins were reduced using 10 mM DTT for 1 h at 56°C and alkylated with 55 mM iodoacetamide for 1 h at room temperature (Sigma-Aldrich, Cleveland, US). The reduced and alkylated peptides were digested with sequencing-grade trypsin (Promega, WI, USA, 1:100; w/w) for 16 h at 37°C in 50 mM NH4HCO3. The trypsin-digested protein samples were extracted from the gel using sequential (50 and 100%) acetonitrile (ACN), dried by SpeedVac concentrator (Eppendorf, concentrator 5301) and re-suspended using 0.05% trifluoroaceticacid (TFA). For desalting, the peptide samples were loaded on to C18 stage tips activated and equilibrated with 95% ACN/0.1% FA and 0.1% formic acid (FA), respectively. The loaded samples were washed with 0.05% TFA and eluted with 95% ACN/0.1% FA. The eluent was dried using a SpeedVac concentrator, re-suspended in 0.1% FA, transferred to auto-sampler nano-liquid chromatography (LC) vials and stored at −20°C. The proteomic work flow is depicted in Supplementary file 1.
Mass spectrometry (MS) analysis
Peptide identification and quantitation were performed by label-free quantification (LFQ) LC-MS/MS using a Q Exactive hybrid quadropole-orbitrap instrument interfaced with an EASY 1000-nano-LC electrospray ion source (Thermo-Fisher Scientific, Biberach, Germany). Peptides were injected in triplicates into a pre-analytic column (Acclaim PepMap 100, 75 μm × 2 cm, nanoviper, C18, 3 μm, 100 Å, Thermo Fisher Scientific) and separated on an analytical column (PepMap RSLC, C18, 2 μm, 100 Å, 50 μm × 15 cm, Thermo Fisher Scientific) with a 75 min solvent gradient and flow rate of 0.3 μL/min at 60°C. The gradient used was from 2 to 30% solvent B for 30 min followed by 30–75% solvent B from 30 to 35 min and 75 to 90% solvent B from 35 to 70 min. Thereafter the gradient was kept at 90% solvent B from 70 to 75 min, using 0.1% formic acid (FA) in 3% acetonitrile (ACN) as solvent A and 0.1% FA in 97% ACN as solvent B (FA: LC-MS grade, Fluka; ACN: LC-MS grade, Merck Laboratories). The MS instrument was operated in the data-dependent acquisition mode with automatic switching between MS and MS/MS scans. The full MS scans were acquired at 70 K resolution, with automatic gain control target of 1 × 106 ions, maximum injection time of 200 ms and MS scan range 300–1,800 m/z. Higher energy collision dissociation (HCD) was used for peptide fragmentation with normalized collision energy set to 28. The MS/MS scans were performed using a data-dependent top10 method at a resolution of 17.5 K with an automatic gain control target of 5 × 104 ions at maximum injection time of 100 ms and isolation window of 2.0 m/z units. An under fill ratio of 10% and dynamic exclusion duration of 30 s were applied. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository [1] with the dataset identifier PXD006117.
Database search
The MS/MS data analysis was performed using the MaxQuant (MQ) software package (version 1.4.0.5; Cox and Matthias, 2008) for analyzing large MS/MS data sets, employing its integrated Andromeda search algorithms (Cox et al., 2011). The raw spectral data were searched against the M. tuberculosis H37Rv reference proteome UP000001584 (UniProt-proteome) using reverse decoy databases and a selection of known contaminants provided by MQ. The following parameters were applied for the database search: Enzyme specificity was set as Trypsin/P, and a maximum of two missed cleavages and a mass tolerance of 0.5 Da for fragment ion were applied. The “re-quantify” and “match between runs” options were utilized with a retention time alignment window of 3 min. Carbamidomethylation of cysteine was set as a fixed modification and acetylation of the protein N-terminus, conversion of N-terminal glutamine and glutamic acid to pyroglutamic acid and oxidation of methionine were set as variable modifications for database searches. The first search for precursor ions was performed with a mass tolerance of 20 ppm for calibration, while 6 ppm was applied for the main search. For protein identification, at least 1 unique peptide was required per protein group (Cox and Matthias, 2008; Zhao and Lin, 2010). Minimum peptide length of 7 amino acids was required for identification. The maximum false discovery rate (FDR) cutoff of 0.01 (1%) was set at both the peptide spectra matches and the protein group levels. For all other parameters, the default setting was applied. Following protein identification by a database search, validation for multiple comparisons was corrected using Benjamini-Hochberg correction (Benjamini et al., 2001). To aid in the control of false positives, the database was supplemented with additional sequences for common contaminants and reversed sequences of each entry.
Bioinformatics analysis
Bioinformatics analysis was performed using the Perseus software (version 1.5.1.6) as previously described (Tyanova et al., 2016). The protein groups output from MQ was used as the basis for all the subsequent statistical and ontology enrichment analysis. LFQ intensities were used to assess differences in the abundance of proteins between the two M. tuberculosis lineages. Abundance estimation of the proteins identified was performed using intensity-based absolute quantification (iBAQ) values. Briefly, the protein groups output was filtered by removing matches to the reverse database, matches only identified by site, and common contaminants. Subsequently, LFQ intensities were transformed to log2. For quantitative comparisons, three technical replicates of each biological experiment (n = 3) were averaged based on their median values. An LFQ intensity category was then created that consisted of the intensities from two clinical isolates (M. tuberculosis L728 and L735) of lineage 7. This merger served as a core proteome for lineage 7, which was later used to compare against the H37Rv lineage 4 reference strain. For statistical analysis, at least two valid LFQ intensities out of the three biological experiments were required. Signals that originally were zero (missing values) were imputed with random numbers from a normal distribution. The mean and standard deviation were chosen to best simulate low abundance values below the noise level (width = 0.3; shift = 1.8; Hubner et al., 2010). A two-tailed unpaired t-test with an FDR value of 0.05 and S0 = 2 was applied to identify proteins for which the abundance was significantly changed between the two M. tuberculosis lineages (Tusher et al., 2001). The resulting t-test -significant proteins for each M. tuberculosis lineage were analyzed for annotation enrichments. A two-tailed Fisher's Exact Test was used to assess the significance of enrichment terms. Proteins assigned to enriched term categories (p < 0.05) were grouped according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) classification. The GO and KEGG categories of proteins identified were added using the Uniprot annotation for the M. tuberculosis reference proteome database (Gene ontology1).
Protein Interaction and Network Analysis
The Search Tool for the Retrieval of Interacting Genes version 10.0 (Franceschini et al., 2013; STRING, http://string-db.org/) was used to interpret the biological significance of DA proteins in terms of predicted protein-protein interaction networks. The required minimum interaction score of at least 0.4 was used as the cut-off point criterion. The Cytoscape software (Shannon et al., 2003) was used to visualize the interaction network predicted. A list of DA proteins, the official gene identifier, and the corresponding relative abundance value were separately uploaded to Cytoscape (http://www.cytoscape.org/). Properties of the network including node degree and edge attributes were then analyzed. Nodes represent proteins and edges represent the interactions/connections between the proteins. The degree represents the number of interactions associated with the protein. Proteins with a large degree are known as hub proteins (Azuaje et al., 2010) and are considered to be the essential or key proteins in the network (Ideker and Sharan, 2008). The Network Analyzer option in Cytoscape 3.4.1 was used to compute the degree and between-ness centrality of the network (Assenov et al., 2008). The MCODE program was used to identify the most inter-connected nodes (Bader and Hogue, 2003).
Ethics Approval
The study obtained ethics approval from the Regional Committee for Medical Research Ethics in Eastern Norway (REK Øst) and the Ethiopian Science and Technology Ministry in Addis Ababa, Ethiopia. Written informed consent was obtained from the study participants before the study was conducted.
Results
Comprehensive Proteome Analysis of M. tuberculosis Lineage 7 and Lineage 4 Strains
A total of 2867 M. tuberculosis proteins were identified with 99% confidence at the peptide and protein levels (Supplemental file 2) representing 71% protein coverage of the predicted M. tuberculosis proteome. The total number of proteins identified in M. tuberculosis lineage 7 (L7-35 and L7-28) and in lineage 4 were 2,825 and 2,541, respectively. Among the 2,825 identified proteins in lineage 7 strains, 2499 (87%) proteins were shared in all the biological experiments (Figures 1A–C). Mutual exclusivity analysis revealed 326 and 42 strain-specific protein groups in lineage 7 and lineage 4, respectively (Supplemental file 2), and the main DA component pathways are addressed below. The overlap in protein identification in the different biological replicates and M. tuberculosis lineages is shown in the Venn diagram (Figures 1A–C). Of the annotated components, 1,783 (62%) have an assigned molecular function, 1,110 (38.7%) are involved in known biological processes, 948 (33%) are assigned by cellular compartment, and 829 (28.9%) have an assigned KEGG function. The complete list of protein groups is presented in Supplemental file 3.
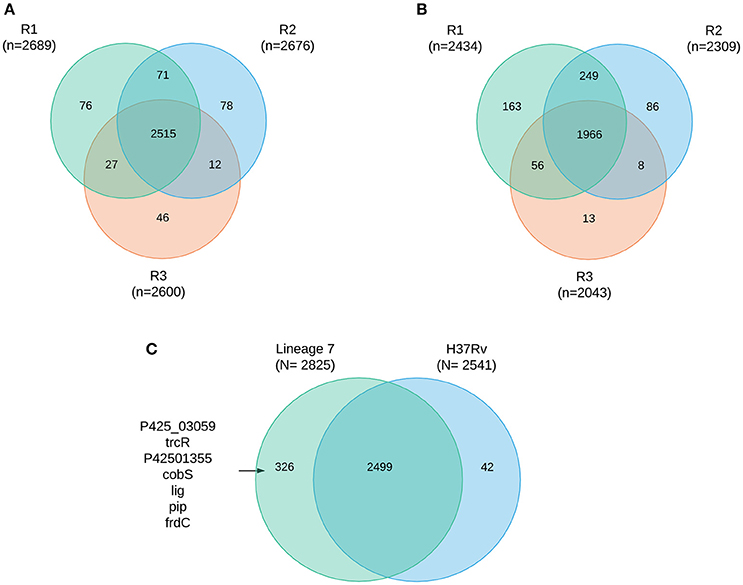
Figure 1. Protein coverage Venn plot illustrating protein identification overlaps among the three biological replicates (R1–R3) in M. tuberculosis lineage 7 (A) and in lineage 4 (B) isolates, (C) overlap of protein identification between lineages and some of the mutually exclusively identified proteins.
Protein Profile of M. tuberculosis Lineage 7 (L7-35 and L7-28) and Lineage 4 Strains
The abundance of the M. tuberculosis proteins identified was quantified by iBAQ. This technique takes into account the normalization and summation of MS/MS signals in relation to peptide size, length, and number of theoretical peptides considered acceptable for all the proteins that are defined in a specific proteome run. The 10 most abundant protein classes identified in lineage 7 strain were chaperones, hydrolase isomerase, ligase, oxidoreductase, transfer carrier protein, and nucleic acid-binding proteins. The least abundant proteins identified include the ESAT-6-like protein (EsxT), MCE-family protein (Mce2F), PE-PGRS family protein and PE family protein. The iBAQ intensity in the aggregate proteome covered a dynamic range of six orders of magnitude between the most abundant and least abundant proteins (Figures 2A,B; Supplemental file 3).
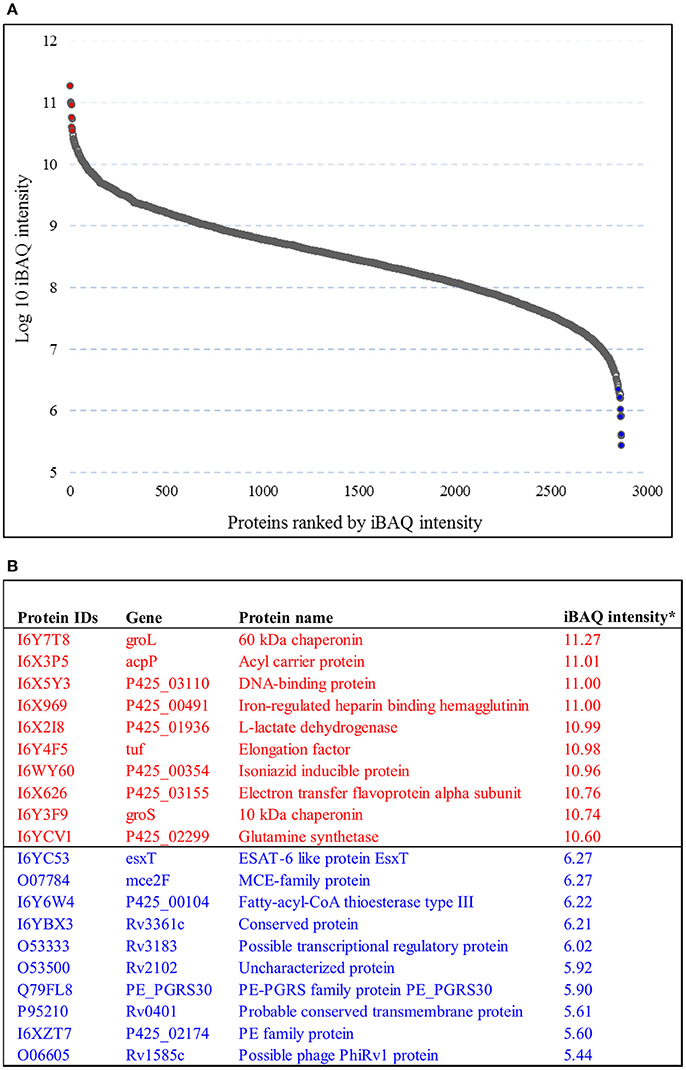
Figure 2. Protein dynamic range estimation. (A) Combined intensity based absolute quantification (iBAQ) values for the 2,825 proteins were plotted with log10 iBAQ intensity on the y axis, and proteins were ranked by iBAQ intensity on the x axis. The plot shows a dynamic range of 6 orders of magnitude. (B) List of the ten most (colored red) and 10 least (colored blue) abundant proteins based on iBAQ intensity.
After identifying the 2,867 proteins in the composite M. tuberculosis proteome, the reproducibility of our label-free quantification workflow was assessed (Supplemental file 1). The Pearson correlations (R-values) of biological replicates using normalized protein LFQ intensities were computed. The analysis showed that the R-value between normalized intensities was high (Figures 3A,B, Supplemental file 4) and thus was suitable for accurate comparisons of protein abundance differences.
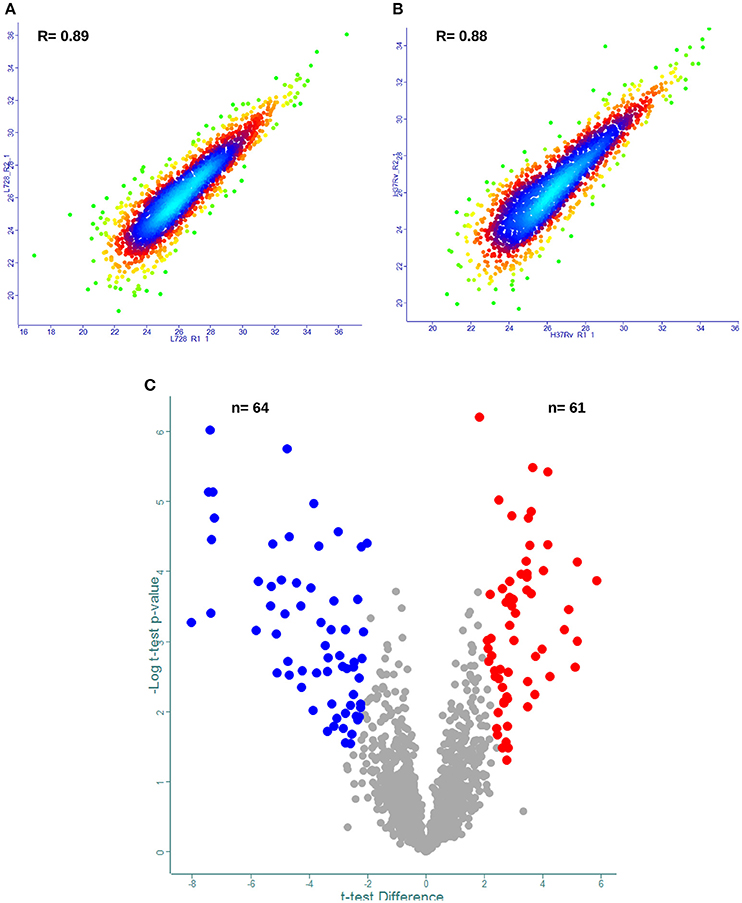
Figure 3. Label free proteome quantification of Mycobacterium tuberculosis lineages. Quantitative analysis was performed using the MaxQuant and Perseus software environments as described in the method section. Reproducibility of the analytic workflow for biological replicates was assessed by Pearson correlation coefficients (R-values). (A,B) Represent density scatter plot of peptide intensities for biological replicates of M. tuberculosis lineage 7 and lineage 4, respectively. (C) volcano plot of protein abundance differences as a function of statistical significance (t-test p ≤ 0.05 and fold change cutoff point ±2) between lineage 7 and lineage 4 isolates. Y-axis indicates p-value (−log10). X-axis shows protein ratio (x-axis) in lineage 7 vs. lineage 4 stains. The color code indicates upregulation (red) and downregulation (blue). Proteins with no statistically significant difference in abundances between the two lineages are shown in gray.
For DA protein comparisons, criteria were set that fulfill two valid LFQ intensity values from each biological triplicate. This resulted in a total of 1,946 proteins. Using a set of statistical criteria, T-test p-value 0.05, S0 = 2 and fold change cutoff point ±2, we found the abundances of 125 proteins to be significantly changed (Figure 3C, Supplemental file 5).
The Proteomes of the M. tuberculosis Lineage 7 Strains Are Significantly Different from Lineage 4
Among the 125 differentially regulated proteins, 64 were downregulated and 61 proteins were upregulated (Figure 3C, Supplemental file 5). The 125 DA protein groups were further subjected to unsupervised hierarchical cluster analysis. The resulting cluster-gram is shown in Figure 4A. KEGG analysis was then conducted to investigate whether these proteins were enriched for any particular pathway. The pathway sub-clusters that were significantly enriched are shown in Figures 4B–D (Supplementary file 6). The DA proteins were also categorized into their functional categories as defined by TubercuList. A majority of the proteins detected belong to categories of intermediary metabolism and respiration (39.2%), lipid metabolism (20%), cell wall- and cell processes-related (24.8%), conserved hypotheticals (2.4%) and unknowns (0.8%) (Supplemental file 7). As shown in Figure 5, the number of less abundant proteins involved in cell wall and cell processes is greater in lineage 7 strains than in lineage 4. In addition, the number of more abundant proteins involved in intermediary metabolism and respiration is greater in lineage 7 strains than in lineage 4.
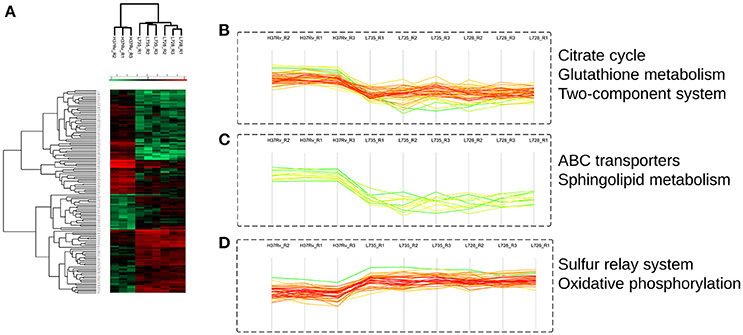
Figure 4. Quantitative proteomics analysis reveals distinct pattern of M. tuberculosis differentially abundant proteins that are involved in several important pathways. (A) Unsupervised hierarchical clustering representing the T-test significant proteins (n = 125). Color code indicates the normalized median abundance of the proteins belonging to the category (red more abundant; green less abundant). (B–D) Representative clusters of significantly enriched KEGG pathways.
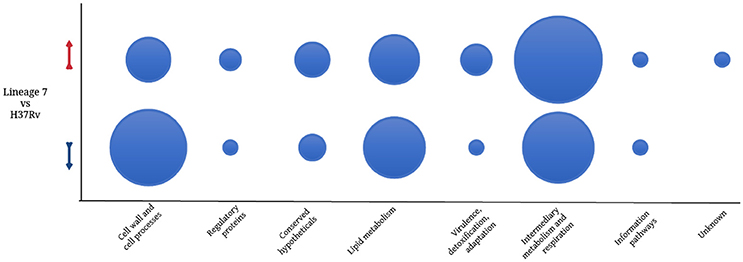
Figure 5. Bubble plot comparing the number of M. tuberculosis differentially abundant proteins in each functional category. Red arrow denotes upregulation and the blue arrow indicates downregulation. Functional categorization was performed according to TubercuList v 2.6 (http://tuberculist.epfl.ch/).
To visualize the functional and molecular interaction network, a total of 125 DA proteins were searched into the online STRING protein query database. Eighty eight of the 125 proteins were present in recognized and predicted networks with total interaction edges of 174 interaction networks. Figure 6 suggests that LpdA, FrdA, EccB3, EccC3, and PstB2 represent a significant protein hub. Three network sub clusters of proteins were highly correlated to functions of Pst system, Esx-3 secretion machinery, energy metabolism, and oxidative stress responses (Figure 6).
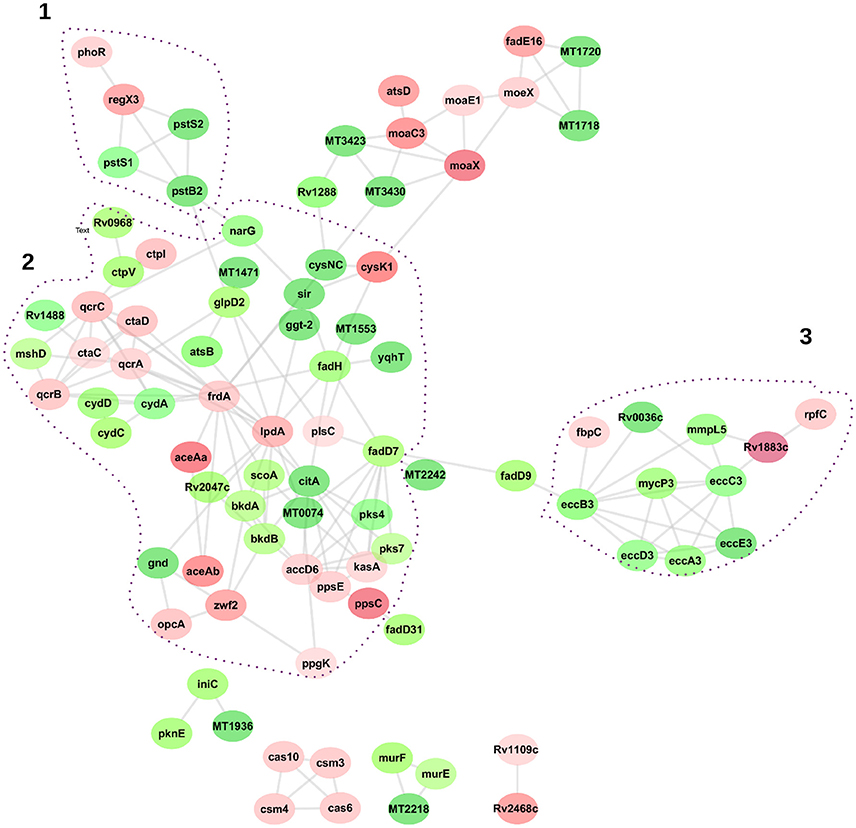
Figure 6. Network interaction analysis of differentially abundant proteins related to M. tuberculosis lineages 7 and 4. Graphical representation of the interaction was generated by the Cytoscape software version 3.4. Proteins are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). The intensity of the node color indicates the increased (red) or decreased (green) abundance according to fold changes. Dot-lined circles indicate sub-networks of proteins linked to specific KEGG pathways.
Cluster 1 in Figure 6 shows interactions of ABC transporter proteins Rv0932c (Pst1), (PstS2), Rv0933 (PstB), and Rv0292 which are essential for the high-affinity capture of periplasmic inorganic phosphate (Pi) in M. tuberculosis. These proteins were significantly downregulated with a fold change ranging from 31 to 262. In addition, Rv0491 (RegX3) and Rv0758 (PhoR) part of the two-component regulatory system were upregulated (Table 1). The Lineage 7 strains also exhibited downregulation of Rv3881c (EspB), Rv1196 (PPE18), Rv1743 (PknE), and Rv1980c (Mpt64), which play important roles in the ESX-1 secretion system and apoptosis (Table 2).
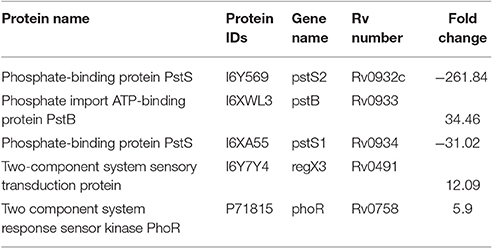
Table 1. List of the differentially abundant proteins involved in the Pst system in M. tuberculosis lineage 7 (L7-35 and L7-28) vs. lineage 4 (H37Rv) strains.
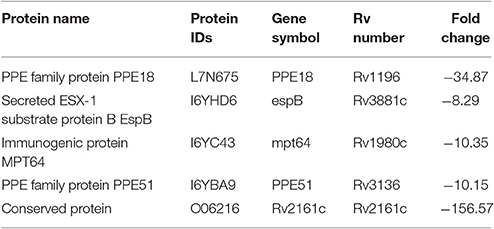
Table 2. List of the M. tuberculosis differentially abundant PPE proteins in M. tuberculosis lineage 7 (L7-35 and L7-28) vs. lineage 4 (H37Rv) strains.
Cluster 3 in Figure 6 depicts interactions of the type 7 secretion system (Esx-3) components that are crucial for iron acquisition in M. tuberculosis. The ESX-3 secretion machinery proteins (Rv2083 Rv0282 (EccA3), (EccB3), Rv0284 (EccC3), (Rv0290/EccD3), and Rv0292 (EccE3) were downregulated with a fold change ranging from 19 to 55. In addition, Rv0291 (MycP3), Rv0676c (MmpL5), and Rv0036 proteins were downregulated in lineage 7 strains. Rv1884c (RpfC), a resuscitation-promoting factor protein, was upregulated in lineage 7 strains compared to lineage 4 (Table 3).
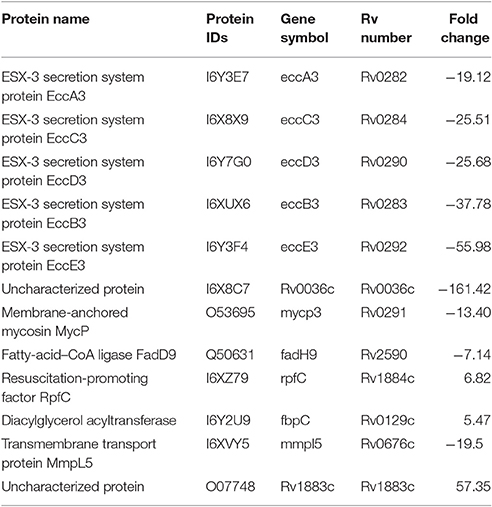
Table 3. List of the M. tuberculosis differentially abundant proteins and corresponding fold changes involved in type 7 secretion system.
Several proteins, Rv2952 (AhpD), Rv0969 (CtpV), Rv0968 (MshD), Rv1161 (NarG), Rv2394 (GgtB), CysD, CysN, CysH, and SirA that are involved in counteracting the effect of reactive oxygen intermediates (ROI) and/or reactive nitrogen intermediates (RNI) were less abundant in lineage 7 (L7-35 and L7-28) compared to lineage 4. Lineage 7 strains also showed downregulation of ABC transporter proteins Rv1620c (CydC) and Rv1621c (CydD). In contrast, a number of proteins that counteract the detrimental effects of ROI and RNI were upregulated in lineage 7 (L7-35 and L7-28) isolates compared to lineage 4. These included GnD1, ZwF1, Rv3119 (MoaE1), RV3324C (MoaC), RV3323C, and (MoaX) proteins (Table 4).
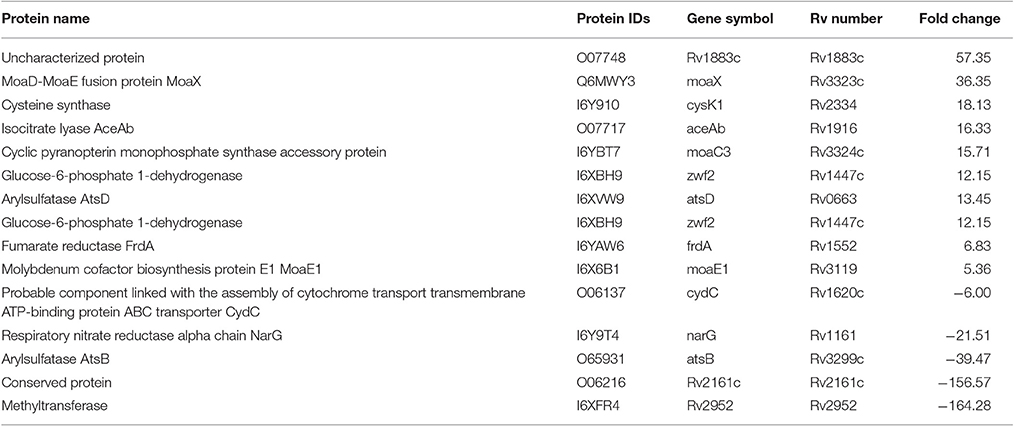
Table 4. List of the differentially abundant proteins involved in ROI and RNI stress exposure responses in M. tuberculosis lineage 7 (L7-35 and L7-28) vs. lineage 4 (H37Rv) strains.
The DA proteins CitA, BkdB BkdA, FrdA, and LpdA play important roles in the citrate cycle (TCA) pathway were differentially regulated. While CitA, BkdB, BkdA proteins were less abundant, the FrdA and Rv3303c (LpdA) proteins were upregulated in lineage 7 strains compared to lineage 4. Rv3043c (CtaD), Rv2200c (CtaC), QcrA (Rv1446c), QcrC (Rv2194), QcrB (Rv2196), which are the major respiratory route in mycobacteria, were also upregulated. In addition, the Rv1915 (AceaA) and Rv1916 (AceaB) GnD1 and PpgK proteins were upregulated in lineage 7 strains (Table 5).
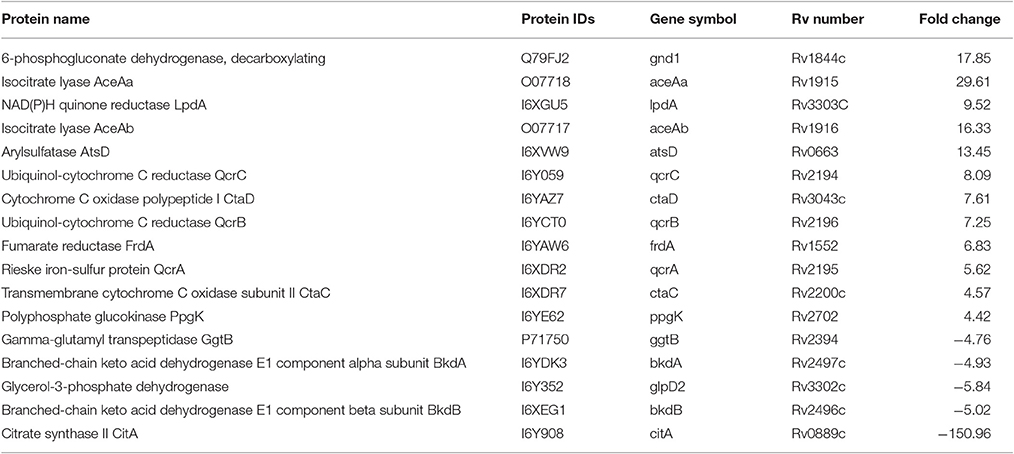
Table 5. List of the differentially abundant proteins involved in energy metabolism in M. tuberculosis lineage 7 (L7-35 and L7-28) vs. lineage 4 (H37Rv) strains.
Several proteins involved in cell wall/lipid biosynthesis were less abundant in lineage 7 than in lineage 4. The RV2952 was markedly downregulated with a 164-fold change in lineage 7 strains. Furthermore, the MmaA3, IniC, Pks4, MurE, and MurF proteins were less abundant in lineage 7 strains. The PpsE PpsC, AccD6, and KasA proteins were upregulated in lineage 7 than in lineage 4 (Table 6).
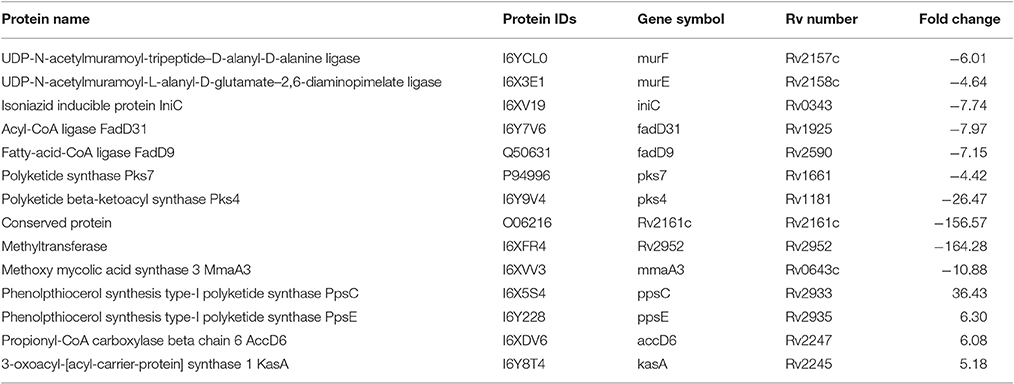
Table 6. List of the differentially abundant proteins involved in cell wall/lipid biosynthesis in M. tuberculosis lineage 7 (L7-35 and L7-28) vs. lineage 4 (H37Rv) strains.
Discussion
This is the first study to generate information on the DA proteomic profile of M. tuberculosis lineage 7 vs. lineage 4 strains. We compared the proteomes in the two lineages and obtained 2867 protein groups that cover 71% of the predicted M. tuberculosis proteome (n = 4023). Former studies have documented even higher protein coverages with maximal ranges of 77–82% of the predicted M. tuberculosis proteome (Kelkar et al., 2011; Peters et al., 2016). Compared to recent publications that reported total protein coverages of 46% (Bespyatykh et al., 2016), 54% (Jhingan et al., 2016) and 62% (Schubert et al., 2015), the number of proteins identified in our study is relatively high.
This study showed considerable differences in the levels of protein abundance between M. tuberculosis lineage 7 and lineage 4 strains. Transporter proteins of the Pst system Rv0932 (PstS2), RV0933 (PstB), and RV0934 (PstS1) were less abundant in the two lineage 7 strains. PstA, PstC, PstS, and PstB form an ABC transporter essential for the capture of periplasmic inorganic phosphate (Pi) in M. tuberculosis (Rifat et al., 2009). While PstS binds Pi with high affinity, PstB provides the energy required for Pi transport from the periplasm to the cytosol. A former study demonstrated that M. tuberculosis strains with a disruption in genes encoding the pst system were deficient in phosphate uptake and exhibited decreased virulence and attenuated growth, and this was attributed to the absence of the PstS2 protein (Peirs et al., 2005). In our study, PstS2 in lineage 7 strains was 261-fold lower than that in lineage 4. The phosphate starvation response (PSR) is an important mechanism for M. tuberculosis survival under phosphate-depleted conditions (Rifat et al., 2009). In this regard, the M. tuberculosis genes regX3 and phoR are known to encode components that are essential for bacillary survival during phosphate limitation and for regulation of PSR (Rifat et al., 2009, 2014). In the two lineage 7 strains, we observed upregulation of the RegX3 and PhoR proteins that may suggest enhanced expression of PSR. Pi is an essential component of DNA, RNA, ATP, phospholipids, and proteins, and is crucial for energy transfer, protein activation, and carbon and amino acid metabolic processes (Tischler et al., 2013). The significant downregulation of essential proteins for Pi uptake in lineage 7 (L7-35 and L7-28) strains suggests that these proteins may contribute to the relatively slow in vitro growth described in our former study (Yimer et al., 2015).
This study showed downregulation of proteins involved in the type 7 secretion systems ESX-3 and ESX-1 in lineage 7 (L7-35 and L7-28) strains. ESX-1, ESX-3, and ESX-5 have been shown to be crucial for virulence or viability in M. tuberculosis (van Winden et al., 2016). The ESX-3 secretion machinery is encoded by an operon, including the genes eccA3, eccB3, eccC3, eccD3, rv0291, and eccE3 (Table 1). All proteins encoded by these genes were downregulated in lineage 7 strains. The Esx-3 system is essential for siderophore-mediated iron acquisition, and is critical for in vitro growth of M. tuberculosis (Serafini et al., 2013). M. tuberculosis uses siderophore molecules (mycobactin and carboxymycobactin) to acquire iron. MmpS4/MmpL4 and MmpS5/MmpL5 proteins are essential for the biosynthesis and transport of siderophores in M. tuberculosis (Wells et al., 2013). In the current study, MmpL5 was downregulated in lineage 7 strains. A non-synonymous mutation in the mmpL4 gene was identified in our recent WGS study (Yimer et al., 2016). The Rv0291 (MycP3) protein downregulated in lineage 7 strains is an essential protein for M. tuberculosis growth in vitro (Sassetti et al., 2003). The downregulation of several components of the essential type 7 secretion system ESX-3 may expose lineage 7 strains to iron starvation that potentially affects its growth in vitro.
A number of proteins that are involved in counteracting the effect of ROI and RNI were downregulated in the two lineage 7 strains compared to lineage 4 (Table 3). The protein CtpV is responsible for copper transport in M. tuberculosis and copper levels increase during hypoxia (White et al., 2009; Ward et al., 2010). Excess copper is toxic and must be regulated in M. tuberculosis. Several proteins, including CysD, CysN, CysH, and SirA involved in cysteine biosynthesis and sulfur metabolism, were downregulated in lineage 7. Cysteine is a precursor for glutathione biosynthesis (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/). The MshD and GgtB proteins are involved in mycothiol biosynthesis (Buchmeier et al., 2006) and glutathione metabolism (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/), respectively. Lineage 7 strains showed downregulation of ABC transporter proteins Rv1620c (CydC) and Rv1621c (CydD) that are involved in cytochrome biosynthesis (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/). CydDC contributes to nitric oxide tolerance (Holyoake et al., 2016). In contrast, several proteins that counteract the lethal effects of ROI and RNI in phagocytic cells were upregulated in the two lineage 7 strains (Table 3). GnD1 and ZwF1 proteins are required for glutathione metabolic process (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/). Rv3119 (MoaE1), Rv3324c (MoaC), and Rv3323c (MoaX) proteins are important in sulfur relay system (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/) and redox reaction in various compounds and are crucial for molybdenum cofactor (MoCo) biosynthesis (Williams et al., 2014). Molybdenum enzymes are involved in the sulfur, carbon, and nitrogen metabolism (Williams et al., 2014). In general, the downregulation of several proteins that counteract the effect of ROI and RNI highlights the exposure of lineage 7 cells to hostile environments. Conversely, the upregulation of specific proteins points to a compensatory mechanism adapted by lineage 7 strains to escape from the detrimental effect of ROI and RNI stress exposures.
Lineage 7 (L7-35 and L7-28) strains expressed various proteins involved in metabolic pathways that generate energy for survival. A number of proteins that play an important role in the TCA pathway were differentially expressed. While CitA, BkdB, and BkdA were downregulated, FrdA and LpdA proteins were upregulated in lineage 7 strains compared to lineage 4. The TCA is an aerobic core metabolic pathway which is crucial for the final steps of the oxidation of carbohydrates and fatty acids (Wikipedia2). The TCA pathway supplies NADH for use in oxidative phosphorylation and other metabolic processes. One study showed that in dormant cultivatable M. tuberculosis, the TCA was suppressed (Converse et al., 2009). When M. tuberculosis experiences hypoxia due to oxygen depletion, increased levels of NADH and inability to close the respiratory cycle, the bacteria shifts the direction of TCA enzymes from an oxidative direction to a reductive direction and starts to actively secrete succinic acid to complete the respiratory cycle. This process is facilitated by an enzyme called fumarate reductase (FrdA). We found an upregulation of FrdA that may signal a shift in metabolic pathway by lineage 7 strains. A former study showed that the FrdA protein was upregulated under hypoxic conditions in M. tuberculosis (Watanabe et al., 2011). Lineage 7 strains exhibited upregulation of LpdA protein; LpdA has quinone reductase activity and catalyzes the formation of NADH, which is important for energy production under anaerobic conditions (Zheng et al., 2008). As shown in Figure 6, FrdA and LpdA exhibit interactions with a number of proteins indicating the central role of these enzymes in energy metabolism in lineage 7 strains.
We also found upregulation of the CtaD, CtaC, QcrA, QcrC, and QcrB proteins that are components of the bc1-aa3 pathway. The bc1-aa3 cytochrome pathway is essential for growth in vitro and is the major respiratory route in mycobacteria (Matsoso et al., 2005). These proteins are also upregulated under hypoxia and poor-energy environments (Cook et al., 2014). In addition, the AceaA and AceaB enzymes involved in glyoxylate cycle that serves as an alternative to the TCA cycle, were upregulated in lineage 7. The GnD1 protein upregulated in lineage 7 strains functions in the pentose phosphate pathway as the main generator of cellular NADPH (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/). In addition, the PpgK protein involved in glucose phosphorylation (Kyoto Encyclopedia of Genes and Genomes; http://www.genome.jp/kegg/) was upregulated in lineage 7 strains.
M. tuberculosis is adapted to inhabit a wide range of intracellular and extracellular environments. A typical feature of this adaptation is the ability to respire and regenerate ATP via various energy-generating metabolic pathways (Cook et al., 2014). We observed downregulation of essential enzymes involved in core metabolic pathway of the TCA cycle. Therefore, the upregulation of the proteins specified above, which are involved in aerobic and anaerobic respiration in lineage 7 strains, may be considered as a compensatory mechanism for generating energy to maintain basic physiological functions M. tuberculosis lineage 7 cells.
Several proteins involved in cell wall/lipid biosynthesis were differentially expressed between the two lineage 7 strains and lineage 4. The Rv2952 protein, which is required for the biosynthesis of phenolglycolipid (PGL) and production of dimycocerosates of phthiocerol (DIM), was markedly downregulated in lineage 7 cells. Rv2952 is a methyltransferase that catalyzes the transfer of a methyl group for the production of DIM and PGL (Pérez et al., 2004). A study indicated that a Pks12 mutant strain was deficient in the synthesis of DIM, and that that the growth and virulence of this strain were reduced (Sirakova et al., 2003). Our recent study exhibited non-synonymous mutations in the pkS12 gene (Yimer et al., 2016), suggesting a possible effect of this gene on the efficiency of PDIM synthesis. Similarly, the IniC protein downregulated in lineage 7 strains is suggested to participate in the regulation of cell wall growth (Alland et al., 2000). The downregulated MurE and MurF proteins are key enzymes of peptidoglycan biosynthetic pathway (Munshi et al., 2013). AccD6, KasA, PpsE, PpsC proteins were upregulated in lineage 7 strains compared to lineage 4. AccD6 and KasA proteins are key components in the mycolate biosynthesis (Bhatt et al., 2007; Pawelczyk et al., 2011). Mycolic acids are considered major virulence factors. PpsE and PpsC proteins are involved in phenolpthiocerol and phthiocerol dimycocerosate and important for virulence in M. tuberculosis (Bisson et al., 2012). The integrity of cell wall envelope is crucial for protection of M. tuberculosis against environmental stress factors. Therefore, the downregulation of a number of proteins involved in cell wall/lipid biosynthesis may have an effect on the growth and survival of M. tuberculosis lineage 7 cells.
This study addresses in detail the DA proteomic profile of M. tuberculosis lineage 7 (L7-35 and L7-28) vs. lineage 4 (H37Rv) strains. The analysis provides new insight into the lineage-specific protein profile variations that may explain the particular character of lineage 7 cells in terms of growth/survival and pathogenesis as compared to lineage 4. A number of proteins involved in M. tuberculosis growth and virulence fitness are less abundant in the two lineage 7 isolates in contrast to lineage 4. This may suggest that the in vitro slow-growth of M. tuberculosis lineage 7 bacilli and delayed health seeking among patients infected with lineage 7 strains observed in our earlier study may be phonotypic characteristics of lineage 7 cells. However, the most preferred method of linking phenotypic characteristics with a particular protein is to disrupt the gene encoding that protein and assess phenotypic alterations that the unavailability of that protein vests on the phenotype in vitro and in animal models. Therefore, further study by knocking out genes encoding the pst system and ESX-3 secretion system proteins, and observing phenotypic changes that the absence the corresponding proteins confers on the phenotype of lineage 7 vs. other lineages in in vitro and in vivo models, are warranted. In addition, further work examining the immune response of patients infected with lineage 7 vs. other lineages is imperative.
Author Contributions
TT and SY conceived the study and study design. SY collected the lineage 7 isolates. SY and EZ performed specimen handling and cultivation. AB, TR, and SK performed the MS analysis. SK, SY, and AB performed the bioinformatics analysis. SY, SK, AB, and TT evaluated and interpreted the data and drafted the paper. All authors edited and approved the final manuscript.
Funding
Funding was received from the Research Council of Norway (RCN) FRIMEDBIO project 204747 and RCN GLOBVAC projects 234506 to TT and 192468 to CH, and Norwegian South-Eastern Health Authority project 2013080 to SY and TT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patients for consenting to participate in the study. We also thank the selected health care facilities in the Amhara Region, Ethiopia, for facilitating the study. We are grateful to the Armauer Hansen Research Institute (AHRI), Addis Ababa, Ethiopia, and the Norwegian Institute of Public Health for facilitating the transfer of lineage 7 strains for WGS at Oslo University Hospital.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00795/full#supplementary-material
Footnotes
References
Albanna, A. S., Reed, M. B., Kotar, K. V., Fallow, A., McIntosh, F. A., Behr, M. A., et al. (2011). Reduced transmissibility of East African Indian strains of Mtb. PLoS ONE 6:e25075. doi: 10.1371/journal.pone.0025075
Alland, D., Steyn, A. J., Weisbrod, T., Aldrich, K., and Jacobs, W. R. (2000). Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J. Bacteriol. 182, 1802–1811. doi: 10.1128/JB.182.7.1802-1811.2000
Assenov, Y., Ramírez, F., Schelhorn, S. E., Lengauer, T., and Albrecht, M. (2008). Computing topological parameters of biological networks. Bioinformatics 24, 282–284. doi: 10.1093/bioinformatics/btm554
Azuaje, F., Devaux, Y., and Wagner, D. R. (2010). Coordinated modular functionality and prognostic potential of a heart failure biomarker-driven interaction network. BMC Syst. Biol. 4:60. doi: 10.1186/1752-0509-4-60.
Bader, G. D., and Hogue, C. W. (2003). An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4:2. doi: 10.1186/1471-2105-4-2
Benjamini, Y., Drai, D., Elmer, G., Kafkafi, N., and Golani, I. (2001). Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 125, 279–284. doi: 10.1016/S0166-4328(01)00297-2
Bespyatykh, J., Shitikov, E., Butenko, I., Altukhov, I., Alexeev, D., and Mokrousov, I. (2016). Proteome analysis of the Mycobacterium tuberculosis Beijing B0/W148 cluster. Sci. Rep. 6:28985. doi: 10.1038/srep28985
Bhatt, A., Molle, V., Besra, G. S., Jacobs, W. R. Jr., and Kremer, L. (2007). The Mycobacterium tuberculosis FAS-II condensing enzymes: their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 64, 1442–1454. doi: 10.1111/j.1365-2958.2007.05761.x
Bisson, G. P., Mehaffy, C., Broeckling, C., Prenni, J., Rifat, D., and Lun, D. (2012). Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, rpoB mutant Mycobacterium tuberculosis. J. Bacteriol. 194, 6441–6452. doi: 10.1128/JB.01013-12
Blouin, Y., Hauck, Y., Soler, C., Fabre, M., Vong, R., and Dehan, C. (2012). Significance of the identification in the Horn of Africa of an exceptionally deep branching Mycobacterium tuberculosis clade. PLoS ONE 7:12. doi: 10.1371/journal.pone.0052841
Buchmeier, N. A., Newton, G. L., and Fahey, R. C. (2006). A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J. Bacteriol. 188, 6245–6252. doi: 10.1128/JB.00393-06
Chacon-Salinas, R., Serafín-López, J., Ramos-Payán, R., Méndez-Aragón, P., Hernández-Pando, R., Van Soolingen, D., et al. (2005). Differential pattern of cytokine expression by macrophages infected in vitro with different Mycobacterium tuberculosis genotypes. Clin. Exp. Immunol. 140, 443–449. doi: 10.1111/j.1365-2249.2005.02797.x
Comas, I., Coscolla, M., Luo, T., Borrell, S., Holt, K. E., Kato-Maeda, M., et al. (2013). Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182. doi: 10.1038/ng.2744
Converse, P. J., Karakousis, P. C., Klinkenberg, L. G., Kesavan, A. K., Ly, L. H., and Allen, S. S. (2009). Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect. Immun. 77, 1230–1237. doi: 10.1128/IAI.01117-08
Cook, G. M., Hards, K., Vilchèze, C., Hartman, T., and Berney, M. (2014). Energetics of respiration and oxidative phosphorylation in mycobacteria. Microbiol. Spectr. 2:3. doi: 10.1128/microbiolspec.MGM2-0015-2013
Coscolla, M., and Gagneux, S. (2014). Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 26, 441–444. doi: 10.1016/j.smim.2014.09.012
Cox, J., and Matthias, M. (2008). MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi: 10.1038/nbt.1511
Cox, J., Neuhauser, N., Michalski, A., Scheltema, R. A., Olsen, J. V., and Mann, M. (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805. doi: 10.1021/pr101065j
Firdessa, R., Berg, S., Hailu, E., Schelling, E., Gumi, B., Erenso, G., et al. (2013). Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerging Infect. Dis. 19, 460–463. doi: 10.3201/eid1903.120256
Franceschini, A., Szklarczyk, D., Frankild, S., Kuhn, M., Simonovic, M., and Roth, A. (2013). STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815. doi: 10.1093/nar/gks1094
Holyoake, L. V., Hunt, S., Sanguinetti, G., Cook, G. M., Howard, M. J., Rowe, M. L., et al. (2016). CydDC-mediated reductant export in Escherichia coli controls the transcriptional wiring of energy metabolism and combats nitrosative stress. Biochem. J. 473, 693–701. doi: 10.1042/BJ20150536
Hubner, N. C., Bird, A. W., Cox, J., Splettstoesser, B., Bandilla, P., Poser, I., et al. (2010). Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J. Cell Biol. 17, 739–754. doi: 10.1083/jcb.200911091
Ideker, T., and Sharan, R. (2008). Protein networks in disease. Genome Res. 18, 644–652. doi: 10.1101/gr.071852.107
Jhingan, G. D., Kumari, S., Jamwal, S. V., Kalam, H., Arora, D., Jain, N., et al. (2016). Comparative proteomic analyses of avirulent, virulent, and clinical strains of Mycobacterium tuberculosis identify strain-specific patterns. J. Biol. Chem. 291, 14257–14273. doi: 10.1074/jbc.M115.666123
Kelkar, D. S., Kumar, D., Kumar, P., Balakrishnan, L., Muthusamy, B., Yadav, A. K., et al. (2011). Proteogenomic analysis of Mycobacterium tuberculosis by high resolution mass spectrometry. Mol. Cell. Proteomics. 10:M111.011627. doi: 10.1074/mcp.M111.011627
Matsoso, L. G., Kana, B. D., Crellin, P. K., Lea-Smith, D. J., Pelosi, A., Powell, D., et al. (2005). Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 187, 6300–6308. doi: 10.1128/JB.187.18.6300-6308.2005
Munshi, T., Gupta, A., Evangelopoulos, D., Guzman, J. D., Gibbons, S., Nicholas, H., et al. (2013). Characterization of ATP-Dependent Mur Ligases involved in the biogenesis of cell wall peptidoglycan in Mycobacterium tuberculosis. PLoS ONE 8:e60143. doi: 10.1371/journal.pone.0060143
Pawelczyk, J., Brzostek, A., Kremer, L., Dziadek, B., Rumijowska-Galewicz, A., Fiolka, M., et al. (2011). AccD6, a key carboxyltransferase essential for mycolic acid synthesis in Mycobacterium tuberculosis, is dispensable in a nonpathogenic strain. J. Bacteriol. 193, 6960–6972. doi: 10.1128/JB.05638-11
Peirs, P., Lefèvre, P., Boarbi, S., Wang X. M., Denis, O., Braibant, M., et al (2005). Mycobacterium tuberculosis with disruption in genes encoding the phosphate binding proteins PstS1 and PstS2 is deficient in phosphate uptake and demonstrates reduced in vivo virulence. Infect. Immun. 73, 1898–1902. doi: 10.1128/IAI.73.3.1898-1902.2005
Pérez, E., Constant, P., Laval, F., Lemassu, A., Lanéelle, M. A., Daffé, M., et al. (2004). Molecular dissection of the role of two methyltransferases in the biosynthesis of phenolglycolipids and phthiocerol dimycoserosate in the Mycobacterium tuberculosis complex. J. Biol. Chem. 279, 42584–42592. doi: 10.1074/jbc.M406134200
Peters, J. S., Calder, B., Gonnelli, G., Degroeve, S., Rajaonarifara, E., Mulder, N., et al. (2016). Identification of quantitative proteomic differences between Mycobacterium tuberculosis lineages with altered virulence. Front. Microbiol. 7:813. doi: 10.3389/fmicb.2016.00813
Portevin, D., Gagneux, S., Comas, I., and Young, D. (2011). Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog. 7:e1001307. doi: 10.1371/journal.ppat.1001307
Rieder, H. L. (1999). Epidemiologic Basis of Tuberculosis Control. Paris: International Union Against Tuberculosis and Lung Disease.
Rifat, D., Belchis, D. A., and Karakousis, P. C. (2014). senX3-independent contribution of regX3 to Mycobacterium tuberculosis virulence. BMC Microbiol. 14:265. doi: 10.1186/s12866-014-0265-8
Rifat, D., Bishai, W. R., and Karakousis, P. C. (2009). Phosphate depletion: a novel trigger for Mycobacterium tuberculosis persistence. J. Infect. Dis. 200, 1126–1135. doi: 10.1086/605700
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high-density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Schubert, O. T., Ludwig, C., Kogadeeva, M., Zimmermann, M., Rosenberger, G., Gengenbacher, M., et al. (2015). Absolute proteome composition and dynamics during dormancy and resuscitation of Mycobacterium tuberculosis. Cell Host Microbe 18, 96–108. doi: 10.1016/j.chom.2015.06.001
Serafini, A., Pisu, D., Palù, G., Rodriguez, G. M., and Manganelli, R. (2013). The ESX-3 secretion system is necessary for iron and zinc homeostasis in Mycobacterium tuberculosis. PLoS ONE 8:e78351. doi: 10.1371/journal.pone.0078351
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shevchenko, A., Tomas, H., Havli, J., Olsen, J. V., and Mann, M. (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860. doi: 10.1038/nprot.2006.468
Sirakova, T. D., Dubey, V. S., Kim, H. J., Cynamon, M. H., and Kolattukudy, P. E. (2003). The largest open reading frame (pks12) in the Mycobacterium tuberculosis genome is involved in pathogenesis and dimycocerosyl phthiocerol synthesis. Infect. Immun. 71, 3794–3801. doi: 10.1128/IAI.71.7.3794-3801.2003
Tischler, A. D., Leistikow, R. L., Kirksey, M. A., Voskuil, M. I., and McKinney, J. D. (2013). Mycobacterium tuberculosis requires phosphate-responsive gene regulation to resist host immunity. Infect. Immun. 81, 317–328. doi: 10.1128/IAI.01136-12
Tusher, V. G., Tibshirani, R., and Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121. doi: 10.1073/pnas.091062498
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016). The Perseus computational platform for comprehensive analysis of (prote) omics data. Nat. Methods 13, 731–740. doi: 10.1038/nmeth.3901
van Winden, V. J., Ummels, R., Piersma, S. R., Jiménez, C. R., Korotkov, K. V., Bitter, W., et al. (2016). Mycosins are required for the stabilization of the ESX-1 and ESX-5 Type VII secretion membrane complexes. MBio 7:e01471-16. doi: 10.1128/mbio.01471-16
Ward, S. K., Abomoelak, B., Hoye, E. A., Steinberg, H., and Talaat, A. M. (2010). CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol. Microbiol. 77, 1096–1110. doi: 10.1111/j.1365-2958.2010.07273.x
Watanabe, S., Zimmermann, M., Goodwin, M. B., Sauer, U., Barry, C. F., and Boshoff, H. I. (2011). Fumarate Reductase Activity Maintains an Energized Membrane in Anaerobic Mycobacterium tuberculosis. PLoS Pathog. 7:e1002287. doi: 10.1371/journal.ppat.1002287
Wells, R. M., Jones, C. M., Xi, Z., Speer, A., Danilchanka, O., Doornbos, K. S., et al. (2013). Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog. 9:e1003120. doi: 10.1371/journal.ppat.1003120
White, C., Lee, J., Kambe, T., Fritsche, K., and Petris, M. J. (2009). A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 284, 33949–33956. doi: 10.1074/jbc.M109.070201
WHO (2016). World Health Organization and Geneva Switzerland. Available online at: http://www.who.int/tb/publications/global_report/en/www.who.int/tb/publications/global_report/en/ (Accessed Dec 14, 2016).
Williams, M., Mizrahi, V., and Kana, B. D. (2014). Molybdenum cofactor: a key component of Mycobacterium tuberculosis pathogenesis? Crit. Rev. Microbiol. 40, 18–29. doi: 10.3109/1040841X.2012.749211
Yimer, S. A., Hailu, E., Derese, Y., Bjune, G. A., and Holm-Hansen, C. (2013). Spoligotyping of Mycobacterium tuberculosis isolates among pulmonary tuberculosis patients in Amhara Region, Ethiopia. APMIS. 121, 878–885. doi: 10.1111/apm.12046
Yimer, S. A., Namouchi, A., Zegeye, E. D., Holm-Hansen, C., Norheim, G., Abebe, M., et al. (2016). Deciphering the recent phylogenetic expansion of the originally deeply rooted Mycobacterium tuberculosis lineage 7. BMC Evol Biol. 16:146. doi: 10.1186/s12862-016-0715-z
Yimer, S. A., Norheim, G., Namouchi, A., Zegeye, E. D., Kinander, W., Tønjum, T., et al. (2015). Mycobacterium tuberculosis lineage 7 strains are associated with prolonged patient delay in seeking treatment for pulmonary tuberculosis in Amhara Region, Ethiopia. Clin. Microbiol. 53, 1301–1309. doi: 10.1128/JCM.03566-14
Zhao, Y., and Lin, Y. H. (2010). Whole-cell protein identification using the concept of unique peptides. Genomics Proteomics Bioinform. 8, 33–41. doi: 10.1016/S1672-0229(10)60004-6
Keywords: Mycobacterium tuberculosis, tuberculosis, lineage 7, proteomics, Ethiopia, mass spectrometry, type 7 secretion
Citation: Yimer SA, Birhanu AG, Kalayou S, Riaz T, Zegeye ED, Beyene GT, Holm-Hansen C, Norheim G, Abebe M, Aseffa A and Tønjum T (2017) Comparative Proteomic Analysis of Mycobacterium tuberculosis Lineage 7 and Lineage 4 Strains Reveals Differentially Abundant Proteins Linked to Slow Growth and Virulence. Front. Microbiol. 8:795. doi: 10.3389/fmicb.2017.00795
Received: 28 December 2016; Accepted: 18 April 2017;
Published: 09 May 2017.
Edited by:
Biswarup Mukhopadhyay, Virginia Tech, USAReviewed by:
Thomas Dick, Rutgers University, USANomakorinte Gcebe, Agricultural Research Council, South Africa
Jonathan M. Blackburn, University of Cape Town, South Africa
Copyright © 2017 Yimer, Birhanu, Kalayou, Riaz, Zegeye, Beyene, Holm-Hansen, Norheim, Abebe, Aseffa and Tønjum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Solomon A. Yimer, s.a.yimer@medisin.uio.no