 Yanzhi Mao1†
Yanzhi Mao1† Xuhong Sun1†Jianguo Shen2
Xuhong Sun1†Jianguo Shen2 Fangluan Gao3*Guangwei Qiu1
Fangluan Gao3*Guangwei Qiu1 Teng Wang1
Teng Wang1 Xianzhou Nie4Wei Zhang1Yanling Gao1Yanju Bai1*
Xianzhou Nie4Wei Zhang1Yanling Gao1Yanju Bai1*- 1Heilongjiang Academy of Agricultural Sciences, Harbin, China
- 2Inspection and Quarantine Technology Center, Fujian Exit-Entry, Inspection and Quarantine Bureau, Fuzhou, China
- 3Institute of Plant Virology, Fujian Agriculture and Forestry University, Fuzhou, China
- 4Fredericton Research and Development Centre, Agriculture and Agri-Food Canada, Fredericton, NB, Canada
Potato virus Y (PVY) is an important plant pathogen infecting solanaceous crops, causing significant losses to global potato and tobacco production. Some aspects of the plant pathology and molecular biology of PVY have been studied intensively, but the evolutionary dynamics of this virus are poorly understood. Here, we performed a comprehensive set of rigorous evolutionary analyses using 177 nucleotide sequences of the viral genome linked protein (VPg) gene, which interacts with the plant eukaryotic translation initiation factor 4E (eIF4E). Our Bayesian analysis reveals that the VPg gene of PVY has been evolving at a rate of 5.60 × 10–4 subs/site/year (95% credibility interval 3.35 × 10–4–8.17 × 10–4), which is equivalent to those of other plant-infecting RNA viruses. We identified different evolutionary constraints on the two clades of PVY, clade N and clade O, whose diverge time were estimated at the year 1861 CE (95% credibility interval 1750–1948 CE). We also found that genetic variations were correlated with geographic regions, suggesting that the evolution of this pathogen is strongly affected by geographical associated factors. Taken together, the results of our study have potential implications for the control strategies of PVY.
Introduction
Potato virus Y (PVY), the type member of the genus Potyvirus (Lefkowitz et al., 2018), is one of the most destructive plant pathogens causing considerable economic losses in Solanaceae crops. It has become one of the most prevalent viruses of potato and tobacco productions worldwide (Karasev and Gray, 2013; Quenouille et al., 2013). PVY exists as different strain groups, including the conventional PVYO and PVYN strains and the newly emerged recombinant strains such as European (Eu)-PVYNTN, PVYN:O, PVYN–Wi, and PVYNTN–NW (Karasev and Gray, 2013; Bai et al., 2019). The classic strains (i.e., PVYO and PVYN) are defined according to their reactions in potato cultivars bearing different N genes and in common tobacco (Singh et al., 2008). PVYO and PVYC induce hypersensitive responses (HR) in cultivars bearing the Ny and Nc genes, respectively, whereas PVYN does not induce HR in cultivars harboring these genes. In contrast, PVYN elicits the classic veinal necrosis on common tobacco plants whereas PVYO and PVYC only cause mosaic symptoms on the plants (Singh et al., 2008; Karasev and Gray, 2013). Most of the newly emerged strains were derivatives of PVYO and PVYN via genome recombination (Singh et al., 2008; Karasev and Gray, 2013; Nie et al., 2013). These strains possess one to four recombinant events (RE) (Green et al., 2017). For instance, in PVYN:O, one RE located at ca. ∼nt 2,400 nt (N to O) can be found; whereas in Eu-PVYNTN, three REs at ∼nt 2,400 nt (N to O), ∼nt 5,820 (O to N) and ∼nt 9,180 (N to O) can be found (Nie et al., 2004). PVY induces various foliar symptoms, ranging from mosaic, mottling, lesion to stunting and necrosis on potato. The severity of symptoms depends on the potato varieties, the type of virus strain and environment conditions. It is particularly noteworthy that PVYNTN not only induces symptoms on foliage, but also causes a severe tuber condition termed potato tuber necrotic ringspot disease in sensitive cultivars (Nie et al., 2012, 2013). In nature, PVY is mainly transmitted horizontally from plants to plants by a number of aphid species in a non-persistent manner, and vertically through infected potato seed tubers (Lacomme et al., 2017). Mechanical transmission caused by management practices (e.g., tractor movement during pesticide applications) has also been reported (MacKenzie et al., 2018).
PVY consists of a positive-strand RNA genome ∼9.7 kb in size and encodes a polyprotein of 3061 amino acids (Lefkowitz et al., 2018). The polyprotein is cleaved by three virus-encoded proteinases to yield up to 10 mature proteins (Adams et al., 2005), in which the viral protein genome-linked (VPg) is an intrinsically disorder protein (Grzela et al., 2008) and has been shown to interact with a variety of partners, including itself, allowing it to participate in RNA replication, virus movement, translation, RNA-silencing suppression, and phloem loading of the virus (Quenouille et al., 2013). The free VPg is linked covalently to the 5′-terminus of the genomic RNA and is required for potyvirus infectivity during the virus life cycle (Eskelin et al., 2011; Ivanov et al., 2014). Interestingly, a central intrinsically disordered region (IDR) localized between the amino acid positions 95 and 120 in VPg is a key determinant of PVY adaptation to the eukaryotic translation initiation factor 4E (eIF4E)-based recessive resistance against the virus (Moury et al., 2004, 2014). The contribution of intrinsic disorder to VPg-mediated PVY adaptation has recently been evaluated (Charon et al., 2018).
As one of the top 10 most famous plant viruses (Scholthof et al., 2011), some aspects of the plant pathology, molecular biology and population genetics of PVY have been studied intensively (Lorenzen et al., 2006; Chikh-Ali et al., 2013; Kim et al., 2016; Gao et al., 2017; Bai et al., 2019). However, few studies have focused on the evolutionary dynamics and timescale of PVY. A previous review suggested that PVY group of viruses primarily diverged in the Americas approximately 2400 years ago (Gibbs and Ohshima, 2010). Using Bayesian molecular dating the parental strains of PVY were dated to the period of the first introduction of potatoes to Europe (Visser et al., 2012). This is consistent with the recent origins inferred for various plant viruses (Gibbs et al., 2010). However, studies of the more recent evolutionary dynamics of PVY remain limited, especially at the global level, although such information is vital for designing strategies and management schemes. In this study, we use a Bayesian phylodynamics framework to infer the evolutionary rate and the time to recent common ancestor (tMRCA) based on the VPg gene sequences of PVY isolates collected from the main producing regions in China, combined with all published sequences in GenBank. We also investigated the correlation between the genetic variation and geography during PVY evolution.
Materials and Methods
Sampling and VPg Gene Sequencing
A total of 44 PVY isolates from potato (Solanum tuberosum) across the main potato producing regions in China including Heilongjian, Hunan, and Hebei provinces between 2011 and 2017 were randomly collected (Supplementary Table S1). Leaf samples infected with PVY were stored at −80°C for later use. The PVY-infected tuber samples were kept in a greenhouse as described in Han et al. (2017), and the infected in vitro plantlets were maintained in a tissue culture room.
Total RNA was extracted from leaf tissue using Trizol reagent and was reverse transcribed according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, United States). The VPg gene was amplified using two primers designed from highly conserved regions of PVY genomes, as descripted by Gao et al. (2014). PCR amplifications were conducted in a total volume of 50 μL containing 2 μL of template cDNA, 10 μL 5 × PrimeSTAR Buffer (Mg2+ Plus), 4 μL of dNTP Mixture (2.5 mM each), 1.5 μL of forward primer (5′-GGYTCTGCATGYGAGGARAAT-3′) and 1.5 μL of reverse primer (5′-CAAACTGTTTGRGCAATTGGRTT-3′), 0.5 μL of PrimeSTAR HS DNA Polymerase (2.5 U/μl), and 30.5 μL of ddH2O. The PCR program conditions were as follows: after initial denaturation step at 94°C for 3 min; 35 cycles were performed consisting of three steps: denaturation at 94°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 50 s. The final elongation step was performed at 72°C for 5 min. PCR products were electrophoresed on 1.0% agarose gels in Tris–acetate-EDTA (TAE) buffer and visualized under UV illumination after staining with ethidium bromide (0.5 μg/mL). PCR products were purified using an QiAquick Gel Extraction kit (TransGen, Beijing), ligated into the pMD18-T vector (TaKaRa, Dalian), and transformed into Escherichia coli strain DH5α cells. The recombinant plasmids were purified and at least three cDNA clones were sequenced to ensure consensus in both directions by Sangon Biotech (Shanghai) Co., Ltd.
Data Set
Host-driven adaptation could affect the diversification of viral isolates (Cuevas et al., 2012b). Only PVY sequences derived from potato were included in this study. In addition to 44 novel sequences, 133 published VPg sequences derived from potato were retrieved from GenBank database to give a total data set consisting of 177 VPg sequences (Supplementary Table S1). The isolates were collected from 15 countries between 1963 and 2017 with known sampling dates and geographic regions (Supplementary Figure S1). The sampling regions were Africa (n = 11), Asia (n = 74), Europe (n = 17), North America (n = 73), and South America (n = 2). We constructed a codon-based alignment for the sequences using the MAFFT algorithm (Katoh and Standley, 2013) within TranslatorX online server with the default settings (Abascal et al., 2010).
Recombination Analysis
To investigate the role of recombination in PVY evolution, we constructed recombination analyses using two different approaches to identify potential recombination events in VPg sequences. Firstly, we performed a split-decomposition network analysis and calculated the pairwise homoplasy index using the neighbor-net algorithm in SplitsTree 4.13.1 (Huson, 1998). We subsequently identified potential recombinant and parental sequences using seven different algorithms, including RDP, GENECONV, BOOTSCAN, MAXCHI, CHIMAERA, SISCAN, and 3SEQ, implemented in the RDP 4.95 packages (Martin et al., 2015). Default settings were used throughout, except that the highest acceptable p-value cut-off was set to 0.01 with a standard Bonferroni correction. To avoid misidentification of recombination, recombination events were only considered to be significant when they were simultaneously supported by at least four of the seven algorithms with p-value < 10–6.
Tests for Temporal Signal
The standard Bayesian dating permutation test for temporal signal can be seriously misled for data sets in which temporal and genetic structures are confounded (Murray et al., 2016). To exclude this possible effect on molecular dating, we first performed a Mantel test of the correlation between pairwise genetic distances and differences in sampling dates and calculated the p-value of this test with 1000 permutations. We found evidence of confounding of temporal and genetic structure in the data (P = 0.001, Supplementary Figure S2A), so we applied the clustered permutation test introduced by Duchêne et al. (2015), which was recommended by Murray et al. (2016), to confirm the presence of temporal signal. In this test, a data set has sufficient temporal signal when the mean rate estimate from the original data set falls outside the 95% credibility intervals of the rate estimates from 10 clustered date-randomized replicates.
Bayesian Phylogenetic Analysis
To analyze the evolutionary rate and time scale of PVY, we applied a Bayesian framework for inference as in BEAST 1.8.4 (Drummond et al., 2012). The sequences were analyzed using the HKY + G4 substitution model, which was selected using ModelFinder implemented in IQ-Tree 1.5.5 (Nguyen et al., 2014), based on Bayesian Information Criterion (BIC). We further confirmed the adequacy of the selected substitution model for our data set using PhyloMAd (Duchêne et al., 2018; Supplementary Figure S3). The best-fit clock model (the strict and relaxed clocks) and the best-fit tree prior (among the constant size, exponential-growth, and Bayesian skyline coalescent), were selected using marginal likelihoods estimated by path sampling (Baele et al., 2012). An uncorrelated lognormal relaxed clock and Bayesian skyline coalescent tree prior provided the best fit for our data set (Supplementary Table S2). Isolation time of viral sequences were used to calibrate the molecular clock. Four independent Markov Chain Monte Carlo (MCMC) analyses were run simultaneously for 100 million generations, with samples drawn every 10,000 generations. Convergence of all parameters was verified visually using Tracer 1.7 (Rambaut et al., 2018).
In addition to Bayesian rate estimate, we employed an approximate maximum likelihood approach implemented in TreeTime (Sagulenko et al., 2018) to infer the evolutionary rate of the VPg gene using a regression of phylogenetic root-to-tip distances against sampling date.
Phylogeny-Geography Association and Population Structure Analyses
To assess the association between phylogeny and the pattern of the geographical structure of PVY, we used the software BaTS 2.0 (Parker et al., 2008) to calculate values of association index (AI), parsimony score (PS), and monophyletic clade (MC) size statistics from the posterior sample of trees produced by BEAST, as described above. This method accounts for phylogenetic uncertainty in investigating phylogeny-trait correlations, with 1000 random permutations of tip locations to estimate a null distribution for each statistic. Low AI index and PS and high MC scores suggest a strong phylogeny–geography association and low spatial admixture. The ratio of the observed to expected mean association index was also computed, as a measure of the strength of the association between the geography and the phylogeny. This ratio ranges from 0, indicating complete population subdivision, to 1, suggesting an unstructured population. Since the newly collected 44 PVY isolates were sampled within a relatively narrow time window (2011–2017), we used a bootstrapping approach to standardize sample sizes and performed analyses of 10 replicate subsamples to minimize the influence of sampling biases in our data set. For each bootstrap replicate, we randomly sampled 11 sequences without replacement from each geographic region.
We also used the package adegenet implemented in R 3.5.1 to investigate the genetic population structure present among geographic regions using a discriminant analysis of principal components (DAPC), which does not rely on assumptions of Hardy Weinberg Equilibrium and panmixia (Jombart et al., 2010). The PVY population of South America was excluded from analyses due to an inadequate sample size.
Analysis of Selection Pressures
The inference of selection was performed using the branch-specific models (Yang and Nielsen, 1998, 2002) of the CODEML algorithm (Yang, 2007) implemented in EasyCodeML (Gao et al., 2019). For the branch model, the ratio of non-synonymous vs. synonymous substitutions (ω = dN/dS) under two a priori assumptions: one-ratio model assuming that the entire tree has been evolving at the same rate and two-ratio model allowing foreground branch to evolve under a different rate. We used likelihood ratio test to verify which of the models best fit the data by comparing twice the difference in log-likelihood values between pairs of the models using a x2-distribution, with the degrees of freedom equal to the differences in the number of parameters between the models (Yang, 1998). We also applied the false discovery rate (FDR) method to performed multiple testing correction (Storey and Tibshirani, 2003) implemented in R. Only a p-value of less than 0.05 in the likelihood ratio test was considered to be significant. As an additional check on the sensitives test for the branch-specific model, we estimated pairwise dN/dS ratios using the yn00 program in PAML (Yang and Nielsen, 2000). For more-inclusive analysis, the site models were used to detect signatures of positive selection in VPg using EasyCodeML (Gao et al., 2019). We performed likelihood-ratio test to compare the fit of three nested models, including M3 vs. M0, M2a vs. M1a, and M8 vs. M7. When these tests yielded a significant result (p < 0.01), we then used the Bayes empirical Bayes method to identify individual positively selected codon sites (posterior probability > 0.95).
Results
Sequencing of PVY in China
The VPg gene sequences obtained in this study have been deposited in GenBank with accession numbers MK144421-MK144464. Using the SplitsTree program, we obtained a split network that showed reticulate topologies, indicating several potential recombination events among the PVY isolates (Figure 1). The pairwise homoplasy index also consistently identified a significant signal of recombination in our data set (p = 7.82 × 10–4). Using the RDP4 suites, one likely recombination event in the PVY-(accession number: MK144459) was identified as recombinant at high level significance by four algorithms (GENECONV, p ≤ 4.26 × 10–9; MAXCHI, p ≤ 2.31 × 10–8; CHIMAERA, p ≤ 7.68 × 10–8; and 3SEQ, p ≤ 2.9 × 10–12). Since including recombinants in evolutionary analysis may result in incorrect estimates of the rate of evolution (Sironi et al., 2015), this recombinant was removed. All recombination-free VPg sequence data from PVY isolates, even those that have combined recombination structure based on whole genome, were used for the subsequent phylogenetic dating and phylodynamic analyses.
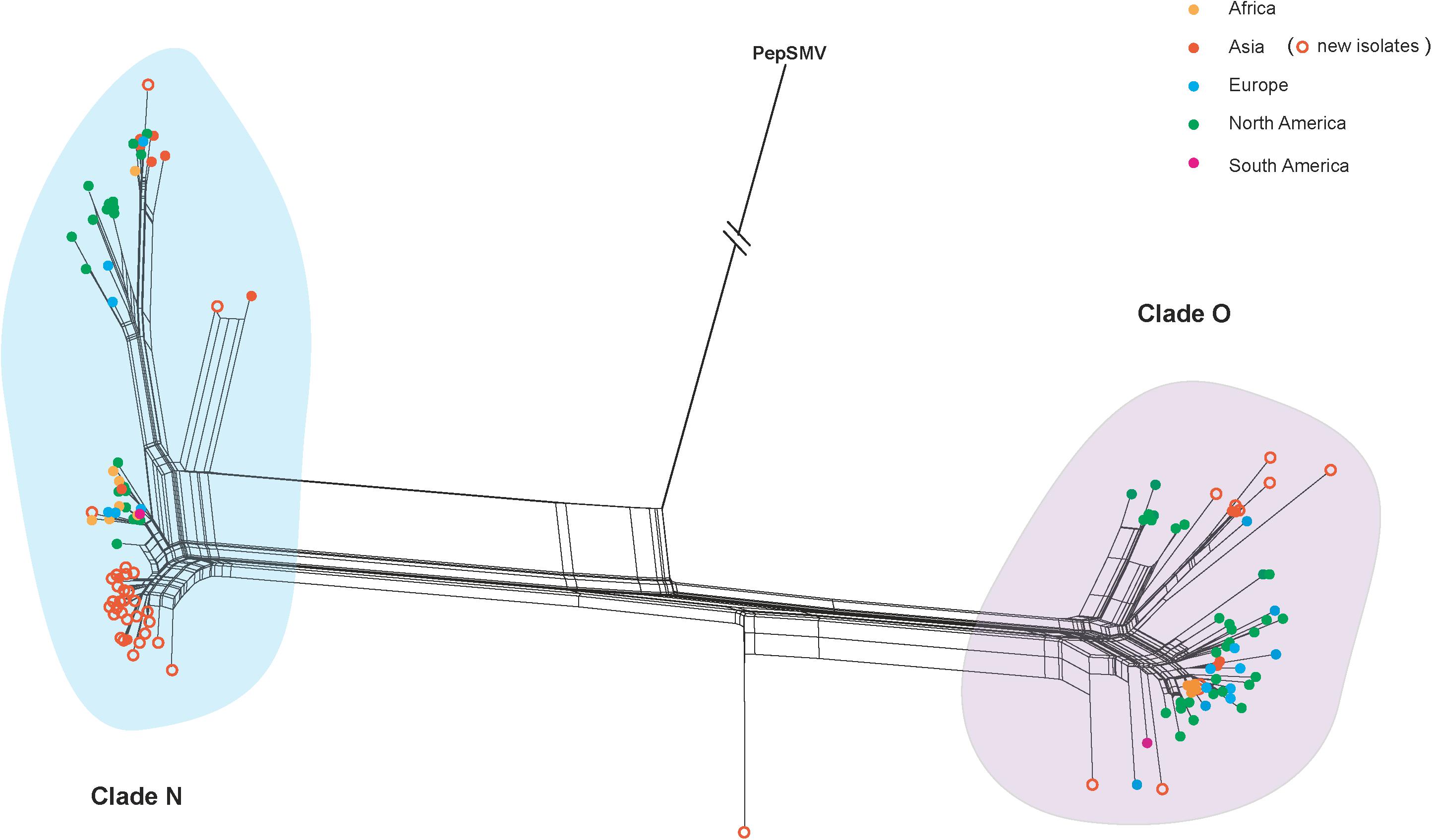
Figure 1. Splits network analysis of the VPg gene from 177 potato virus Y isolates from different geographic regions. An isolate of pepper severe mosaic virus (accession number: NC_008393) was used as an outgroup. Colors indicate isolates from different geographic regions. The scale bar is the genetic distances. PVY isolates newly sequenced in this study are marked with empty circle in orange.
Identification of Evolutionary Constraints
Our time-scaled maximum clade credibility (MCC) tree indicated that PVY isolates could be clustered into two distinct clades, called clade N and clade O (Figure 2). In despite of a certain degree diversity in sampling regions, most PVY isolates tended to cluster according to their geographic origin, either within clade N or clade O. The overall ω estimates were 0.031 for the clade N and 0.001 for the clade O, respectively (Supplementary Table S3 and Figure 2). This result indicates that clade N evolved under reduced purifying selection compared with clade O. Furthermore, similar results obtained from pair-wise analyses showed that there are differences between the distribution of dN/dS values between clade N and clade O (Supplementary Figure S4). Using the site model, purifying selection was detected at the majority of polymorphic sites (Supplementary Figure S5). Positive selection was also detected at amino acid positions 96 and 164, but without significant (>0.95) posterior probability (Supplementary Figure S5).
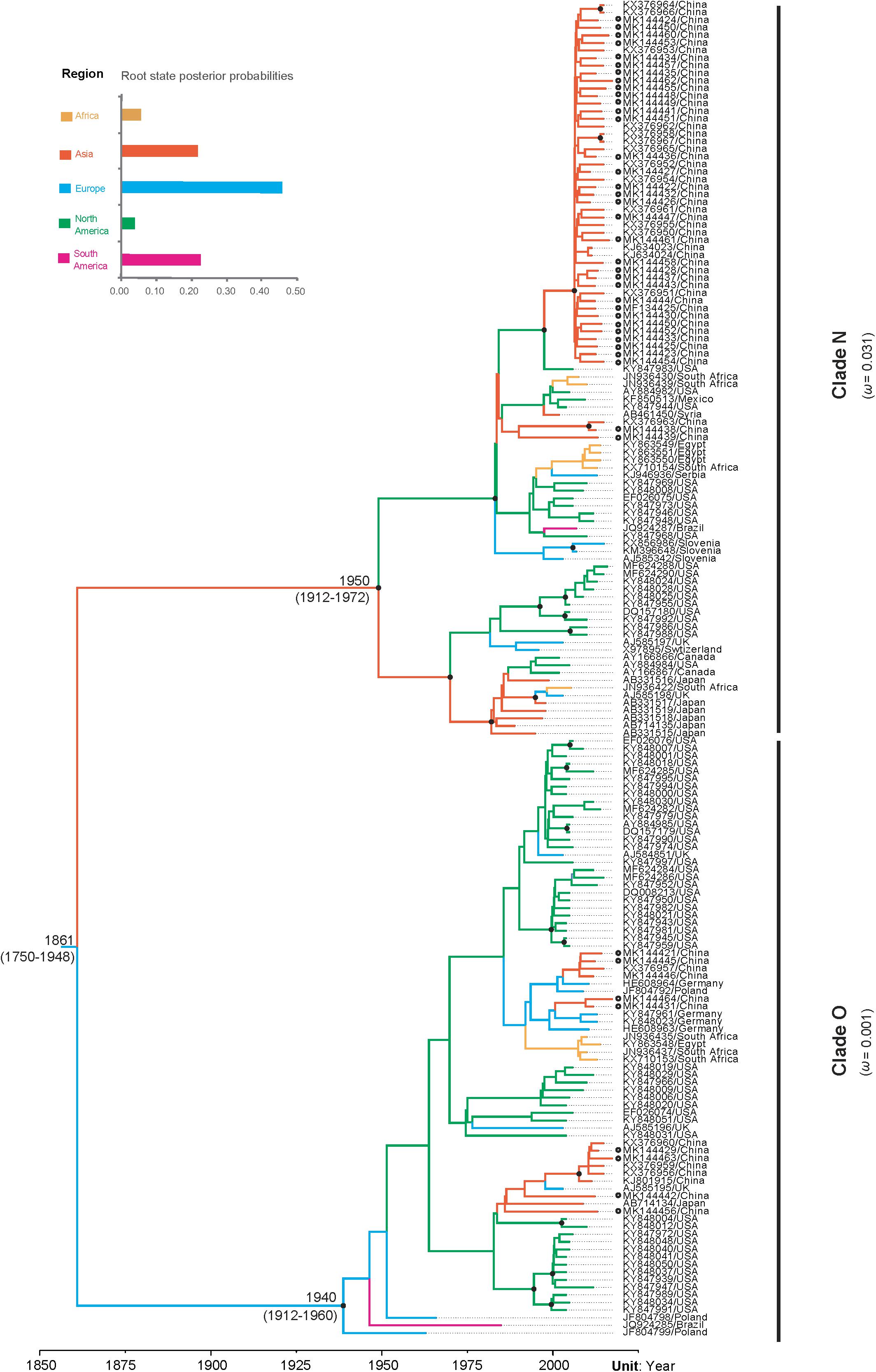
Figure 2. The maximum clade credibility tree inferred by Bayesian analysis of sequences of the VPg gene of potato virus Y and comparison of dN/dS values between the two clades. Bayesian estimates of divergence time, upper and lower limits of the 95% highest posterior density (HPD) estimates are shown for major nodes. Black dots indicate strong node support (posterior probability ≥ 95%). Branch lengths are scaled in units of time, as indicated by the time axis. Branch colors denote inferred location states, as shown in the color key. The root state posterior probabilities estimated for each geographic region are shown in the inset panel. PVY isolates newly sequenced in this study are marked with empty circle.
The Phylodynamic Patterns and Demographic History of PVY
Our data set passed the date-randomization test (DRT) that showed no overlaps between the true estimate of evolutionary rate and 95% credibility interval generated from 10 replicates of randomized data sets (Supplementary Figure S2B). This suggests that the data set has adequate temporal signal for reliable Bayesian tip-dated analyses.
The results of our Bayesian phylogenetic analysis suggested that PVY probably emerged in Europe (root posterior probability = 0.46), with a recent common ancestor in 1861 CE (95% credibility interval 1750–1948 CE). The most recent common ancestor of the PVYN strain was located at approximately 1940 CE (95% credibility interval: 1912–1972 CE), whereas the most recent common ancestor of PVYO strains was 1950 CE (95% credibility interval: 1912–1960 CE) (Figure 3). Our Bayesian estimated mean substitution rate was 5.60 × 10–4 subs/site/year (95% credibility interval 3.35 × 10–4–8.17 × 10–4), whereas the root-to-tip regression method obtained an estimate of 8.73 × 10–4 subs/site/year.
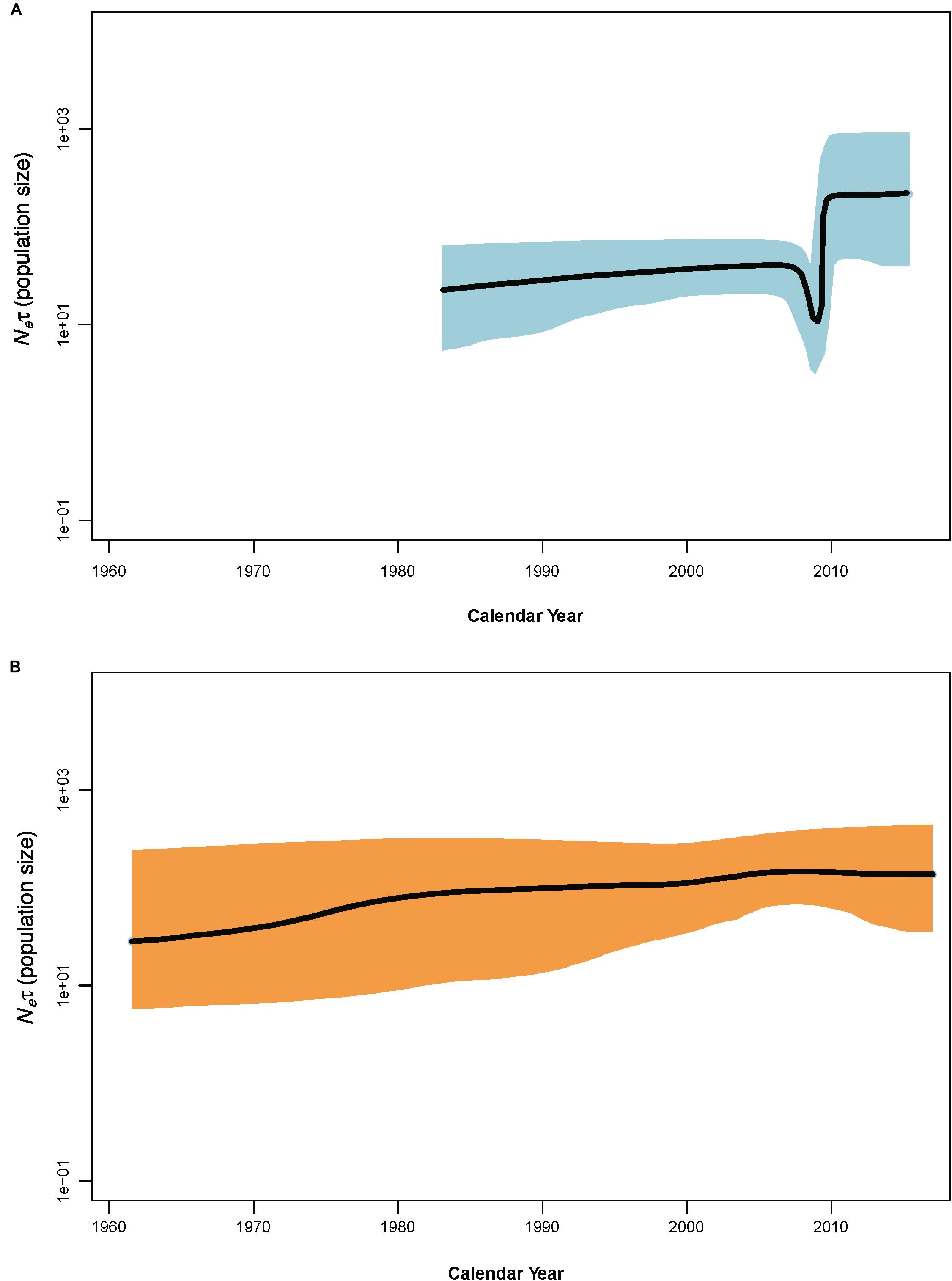
Figure 3. Bayesian skyline plots of population dynamics for clade N (A) and clade O (B) of potato virus Y. The x–axis gives units of years, and y–axis is a measure of relative genetic diversity. The shade areas in light blue are within the 95% highest posterior density interval. The dotted vertical lines indicate important population-size changes for potato virus Y. The shorter timescales of the clade N skyline plots are due to the shorter sampling period.
Coalescence-based Bayesian skyline plot (BSP) revealed an explicit demographic history for the PVY populations of clade N and clade O (Figure 3) – showing that the population size of PVY from clade N suffered from a decline in size in 2006–2007 prior to a period of slight increase and then experienced a sudden population expansion approximately in 2009, followed by a period of stability from 2010 to the last sampling year, whereas the population size of PVY from clade O remained relatively constant throughout the past decades.
The Patterns of Geographic Structure of PVY
Although the MCC tree for the VPg gene seem to not show a clear geographic structure (Figure 2), the global trait association tests of phylogeographic structure rejected the null hypothesis of no association between the geographic and phylogenetic relationship (AI, p < 0.001 and PS, p < 0.001, Table 1). Significant population subdivision was observed for four geographic regions either in original data set or among the 10 replicate subsamples when the MC statistic was shown (Table 1 and Supplementary Table S4). This indicates strong geographic structure in PVY, suggesting the diversification of the virus is explained by geography-driven adaptation. We also found that the index ratios of the observed values to those expected were between 0 and 1 among geographic regions, with association indices of 0.12 (95% credibility interval: 0.07–0.20) and parsimony scores of 0.33 (95% credibility interval: 0.27–0.39), suggesting the evolution of PVY is accounted for a geographic structure but not homogeneous.
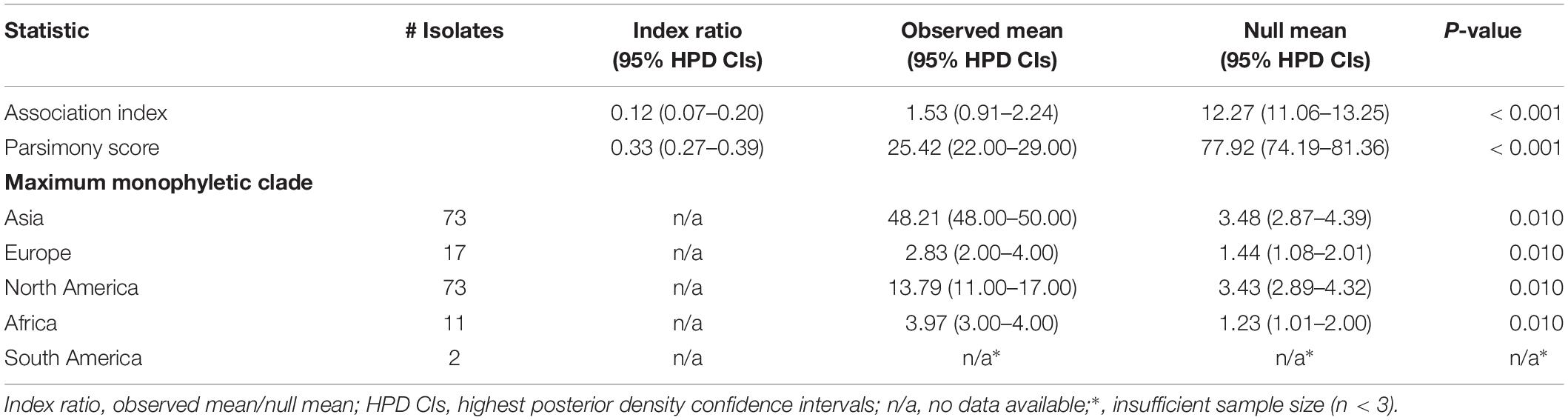
Table 1. phylogeny–trait association analysis for the phylogeographic structure of potato virus Y using Bayesian tip-association significance testing.
Consistent with the results from the phylogeny- geography association analysis (Table 1), DAPC scatter plots also indicated that the Asia population were relatively distinct from the other populations along the first discriminant function axis, while the Africa population exhibited more subtle structure along the third discriminant function axis (Figure 4). This suggests that geography contributes to population differentiation among PVY populations.
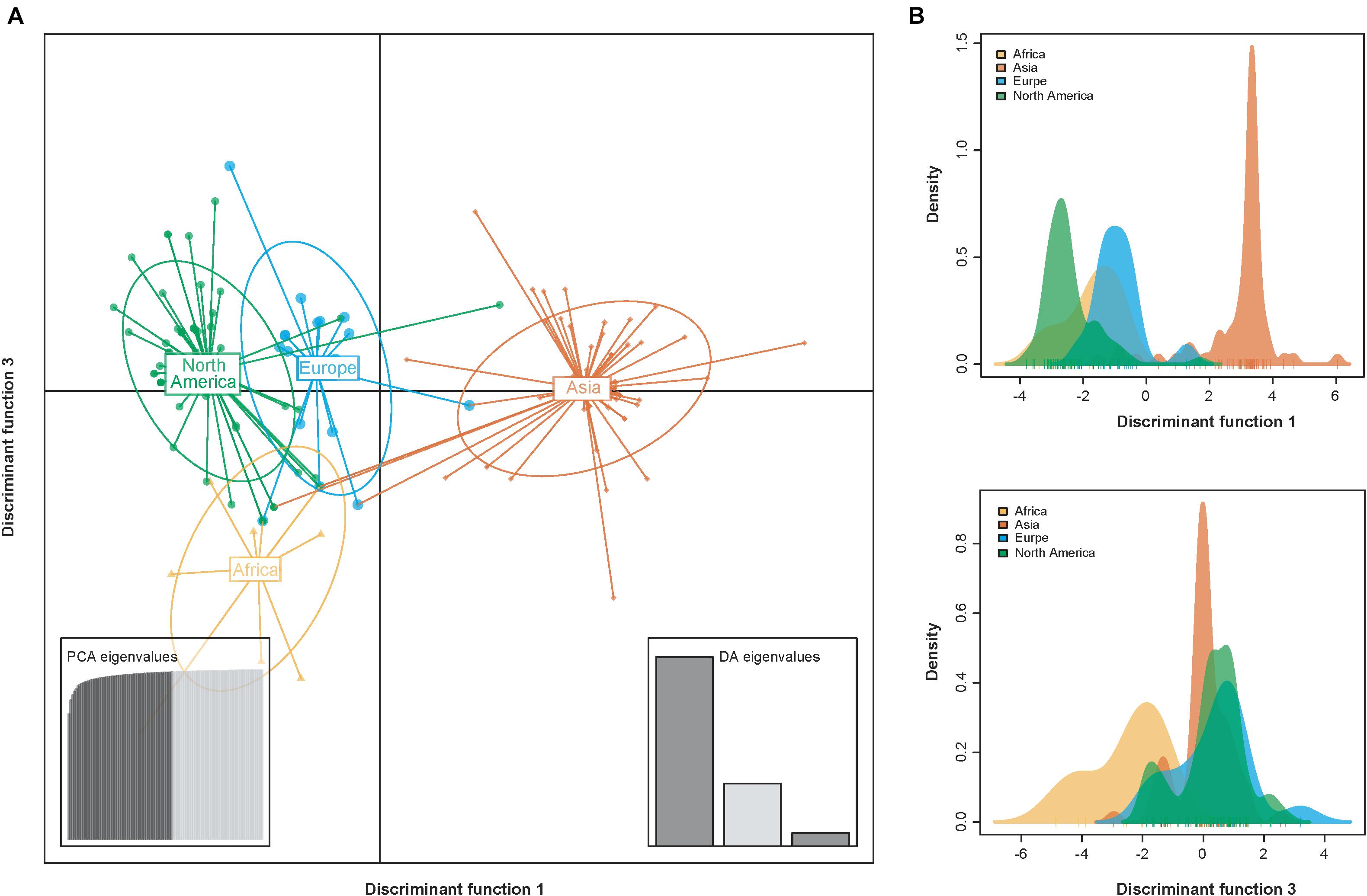
Figure 4. Scatterplots resulting from the discriminant analysis of principal components (DAPC). (A) Individual isolates from the same geographic regions are depicted as unique color shapes and surrounded by 95% inertia ellipses. The PCA and DA eigenvalues inset panels show the overall variability among individuals and the relative capture of variance for each discriminant function, respectively. The y- and x-axes respectively indicate the first and third discriminant principal components, which best summarize the differences between clusters while neglecting within-cluster variation. (B) The discriminant functions for separating the clusters.
Discussion
Overall, we performed a large-scale phylodynamic analysis of the global population of PVY using newly sequenced and curated sequence data retrieved from GenBank. Previous works have found evidence that recombination is an important driving force in the evolution and divergence of many plant viruses, including potyviruses (Nagy, 2008). However, only one recombinant was identified in our analysis. One plausible explanation for this observation is strong selective pressure against survival of new PVY recombinants. Consistent with this possibility, our selective analyses suggested that the VPgs were under strong purifying selection within both clades (0 < ω < 1, Figure 2). Another explanation is that the genomic segment encoding VPg is not a hot spot for recombination. This explanation is consistent the results obtained by Green et al. (2017), which show that recombinant junctions are not frequently seen in the genomic segment corresponding to VPg.
Although PVY exists as complex strain groups, most potato-infecting PVYs are thought to be recombinants of PVYO and PVYN. Consistent with this, our Bayesian phylogenetic analysis resolved VPgs into two large clades, with VPgs from well-characterized PVYO (such as the representative isolate of PVYO Oz, accession number EF026074) and PVYN strains (such as the N605 isolate, accession number X97895) in either of the two clades. Like that observed in by Cuevas et al. (2012a), we also found that VPgs in clade O (mostly likely derived from PVYO strain) was subjected to stronger purifying selection than those in clade N (mostly likely derived from PVYN strain). A possible explanation for the difference is that genes evolving under relaxed selective constraints may more readily adopt new forms of biased expression during the evolution of alternate phenotypes (Hunt et al., 2011). As expected, PVYO strains can induce severe leaf mosaic symptoms on potato, whereas PVYN strains are reported to induce mild mosaic or mottle in leaves but not severe symptoms in potato plants (Quenouille et al., 2013). Cuevas et al. (2012b) detected a positive selection site in the central region of VPg. This site was not found in our study. However, we did find two positive selection sites in the vicinity of this site, although both of which were not statistically significant (with posterior probabilities < 80%). The fact that two independent studies, using different datasets and with different methods, detected positive selection in the central region of VPg is intriguing. A hypothesis that deserve further studies is that the central region of VPg may be involved in its physical interactions with eIF4E.
Our Bayesian phylogenetic analysis placed the root of the tree in Europe with strong supports (Figure 2). This coincides with previous studies that PVY was probably introduced to Europe by potato tubers taken from South America that subsequently seeded other regions of the world after the second half of the nineteenth century. However, there is no evidence that it is transmitted by sexually produced seed (Visser et al., 2012; Gibbs et al., 2017). In addition, our analysis dated the N and O clade in the year 1861 CE (95% credibility interval 1750–1948 CE). This timeframe is similar to that estimated by Visser et al. (2012), but older than that reported by Gibbs et al. (2017), which placed the most recent common ancestor around 1000 CE. These discrepancies might be due to the result of sampling differences, in which we excluded the early Chilean isolates that were first to diverge during the PVY evolution (Quenouille et al., 2013).
The estimated substitution rate of the VPg gene from our Bayesian analysis is 5.60 × 10–4 subs/site/year (95% credibility interval 3.35 × 10–4–8.17 × 10–4). This is similar to our estimate of 8.73 × 10–4 subs/site/year from a regression of root-to-tip genetic distances against the sampling dates. Several previous studies have shown that the estimates of substitution rates might be unreliable if there is strong population structure (Navascues and Emerson, 2009). In that case, the clustered permutation approach of Duchêne et al. (2015) can still give reliable results, regardless of whether it is applied to linear regression or Bayesian dating (Murray et al., 2016). In combination with the results of the clustered permutation test (Supplementary Figure S2B) in this study, this provides support for the substitution rate estimated from our Bayesian analysis to be reliable, although confounding of temporal and genetic structure is present in our data set (Supplementary Figure S2A).
Our demographic analyses found that PVY experienced obvious population expansions during the past 30–40 years. However, this expansion was interrupted approximately in the year 2006 (Figure 3). Such a demographic history of PVY coincides well with the history of potato production in the word. Based on FAO data1, the cultivation area of potato increased from ∼20.91 million ha in 1985 to ∼23.63 million ha in 2005, and from ∼23.89 million ha in 2009 to ∼24.62 in 2016. However, the period from 2006 to 2008 saw a slight decrease in potato cultivation (from ∼23.63 million ha in 2005 to ∼22.01 million ha in 2006). This coincidence suggested a direct impact of potato cultivation on the population size of PVY. Further studies aiming to understand how potato cultivation area influences the population size of PVY will be interesting.
Genetic variability of many potyviruses is related to the geographical origin of the viral isolates, including chili veinal mottle virus (Gao et al., 2016), papaya ringspot virus (Gibbs et al., 2008) and telosma mosaic virus (Xie et al., 2017). Similar observations have been found in PVY, where significant spatial structure in the differentiation of PVY population, on either a global scale or national scale, has been observed when using the CP sequences (Cuevas et al., 2012a; Gao et al., 2017). No surprisingly, the results obtained in this study have also shown a clear correlation between the phylogeny and the geographical origins (Table 1 and Figure 4), providing further evidence that geographically driven adaptation is an important determinant of PVY differentiation.
Conclusion
In summary, the results obtained in our study shows that phylodynamic analysis of the VPg gene data can shed light on the temporal dynamics of PVY. We found that genetic variations of this pathogen were correlated with geographic regions. It will also be important to study human-mediated dispersal in the global population of PVY, which will need to be further analyzed, which will lead to a more comprehensive picture of its evolution.
Data Availability
The datasets generated for this study can be found in NCBI, MK144421-MK144464.
Author Contributions
YB and XN conceived the study. YM, XS, GQ, TW, WZ, and YG performed the experiments. YB, FG, JS, and XN analyzed the data and interpreted the results. FG and YB led the writing of the manuscript. All authors contributed to the manuscript and agreed on the manuscript before review.
Funding
This work was supported by the China Agriculture Research System (Grant No. CARS-09-16), the National Natural Science Foundation of China (Grant No. 31772103), and the Research Award Foundation of Heilongjiang Academy of Agricultural Sciences (Grant No. 2019KYJL008). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Zhenguo Du (Fujian Agriculture and Forestry University) for his comments and suggestions on the manuscript. We are especially grateful to Dr. Yingbo Zhang (Chinese Academy of Tropical Agricultural Science) and Dr. Jinlong Zhang (Kadoorie Farm and Botanic Garden) for their assistance during the preparation of Supplementary Figure S1.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01708/full#supplementary-material
FIGURE S1 | Map showing the localities of potato virus Y isolates included in this study. The ggplot2 and maps libraries in R 3.5.1were used to create the map. Novel viral isolates obtained in this study are indicated by black dot.
FIGURE S2 | Results of tests for the presence of temporal signal in the sequence data. (A) Mantel test of confounding of genetic and temporal distances. The y-axis shows differences in sampling years, and the x-axis indicates genetic distance. (B) Date-randomization test using clustered permutations. The y-axis indicates the substitution rate on a log10 scale, and the x-axis shows the actual data set and 10 different clustered permutations of the dates in the data set. The dashed red lines indicate the 95% credibility interval of the rate estimate from the actual data set.
FIGURE S3 | Test of substitution model adequacy for the HKY + G4 substitution model, as assessed using five test statistics available in PhyMad: (A) chi-squared statistic, (B) multinomial likelihood, (C) biochemical diversity, (D) consistency index, and (E) squared Mahalanobis distance based on the first four statistics. The histograms of the values of the test statistics are calculated from simulation data set, with the red line indicating the value for the empirical data set. The tail-area probability, empirical test statistic, and standard deviations of the predictive distribution (SDPD) are shown in the top-right of each panel.
FIGURE S4 | Comparison of dN/dS values between the two clades. Boxplots showing the dN/dS ratio of clade O and clade N for the VPg gene.
FIGURE S5 | Sliding window plot of dN/dS ratios for the VPg gene. Sites under neutral (dN/dS = 1) are indicated in red dotted line. The window size is 20 codons, and the offset between windows is one codon.
TABLE S1 | Isolates of potato virus Y used in this study.
TABLE S2 | Marginal likelihoods of different combinations of clock model and tree prior.
TABLE S3 | Selective constraint acting on the VPg gene.
TABLE S4 | Results of subsampling-Bayesian tip-association significance testing.
Footnotes
References
Abascal, F., Zardoya, R., and Telford, M. J. (2010). TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Resour. 38, W7–W13. doi: 10.1093/nar/gkq291
Adams, M. J., Antoniw, J. F., and Beaudoin, F. (2005). Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Mol. Plant Pathol. 6, 471–487. doi: 10.1111/j.1364-3703.2005.00296.x
Baele, G., Lemey, P., Bedford, T., Rambaut, A., Suchard, M. A., and Alekseyenko, A. V. (2012). Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 29, 2157–2167. doi: 10.1093/molbev/mss084
Bai, Y., Han, S., Gao, Y., Zhang, W., Fan, G., Qiu, C., et al. (2019). Genetic diversity of Potato virus Y (PVY) in potato production areas in Northeast China. Plant Dis. 103, 289–297. doi: 10.1094/PDIS-04-18-0687-RE
Charon, J., Barra, A., Walter, J., Millot, P., Hebrard, E., Moury, B., and Michon, T. (2018). First experimental assessment of protein intrinsic disorder involvement in an RNA virus natural adaptive process. Mol. Biol. Evol. 35, 38–49. doi: 10.1093/molbev/msx249
Chikh-Ali, M., Gray, S. M., and Karasev, A. V. (2013). An improved multiplex IC-RT-PCR assay distinguishes nine strains of Potato virus Y. Plant Dis. 97, 1370–1374. doi: 10.1094/PDIS-02-13-0161-SR
Cuevas, J. M., Delaunay, A., Rupar, M., Jacquot, E., and Elena, S. F. (2012a). Molecular evolution and phylogeography of Potato virus Y based on the CP gene. J. Gen. Virol. 93, 2496–2501. doi: 10.1099/vir.0.044347-44340
Cuevas, J. M., Delaunay, A., Visser, J. C., Bellstedt, D. U., Jacquot, E., and Elena, S. F. (2012b). Phylogeography and molecular evolution of Potato virus Y. PLoS One 7:e37853. doi: 10.1371/journal.pone.0037853
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with beauti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Duchêne, D. A., Duchêne, S., and Ho, S. Y. W. (2018). Phylomad: efficient assessment of phylogenomic model adequacy. Bioinformatics 34, 2300–2301. doi: 10.1093/bioinformatics/bty103
Duchêne, S., Duchêne, D., Holmes, E. C., and Ho, S. Y. W. (2015). The performance of the date-randomization test in phylogenetic analyses of time-structured virus data. Mol. Biol. Evol. 32, 1895–1906. doi: 10.1093/molbev/msv056
Eskelin, K., Hafren, A., Rantalainen, K. I., and Makinen, K. (2011). Potyviral VPg enhances viral RNA translation and inhibits reporter mRNA translation in planta. J. Virol. 85, 9210–9221. doi: 10.1128/jvi.00052-11
Gao, F., Chang, F., Shen, J., Shi, F., Xie, L., and Zhan, J. (2014). Complete genome analysis of a novel recombinant isolate of Potato virus Y from China. Arch. Virol. 159, 3439–3442. doi: 10.1007/s00705-014-2184-2182
Gao, F., Chen, C., Arab, D. A., Du, Z., He, Y., and Ho, S. Y. W. (2019). Easycodeml: a visual tool for analysis of selection using codeML. Ecol. Evol. 9, 3891–3898. doi: 10.1002/ece3.5015
Gao, F., Jin, J., Zou, W., Liao, F., and Shen, J. (2016). Geographically driven adaptation of chilli veinal mottle virus revealed by genetic diversity analysis of the coat protein gene. Arch. Virol. 161, 1329–1333. doi: 10.1007/s00705-016-2761-2767
Gao, F., Zou, W., Xie, L., and Zhan, J. (2017). Adaptive evolution and demographic history contribute to the divergent population genetic structure of Potato virus Y between China and Japan. Evol. Appl. 10, 379–390. doi: 10.1111/eva.12459
Gibbs, A., Fargette, D., Garcia-Arenal, F., and Gibbs, M. (2010). Time – the emerging dimension of plant virus studies. J. Gen. Virol. 91, 13–22. doi: 10.1099/vir.0.015925-15920
Gibbs, A., and Ohshima, K. (2010). Potyviruses and the digital revolution. Ann. Rev. Phytopathol. 48, 205–223. doi: 10.1146/annurev-phyto-073009-114404
Gibbs, A. J., Ohshima, K., Phillips, M. J., and Gibbs, M. J. (2008). The prehistory of Potyviruses: their initial radiation was during the dawn of agriculture. PLoS One 3:e2523. doi: 10.1371/journal.pone.0002523
Gibbs, A. J., Ohshima, K., Yasaka, R., Mohammadi, M., Gibbs, M. J., and Jones, R. A. C. (2017). The phylogenetics of the global population of Potato virus Y and its necrogenic recombinants. Virus Evol. 3:vex002. doi: 10.1093/ve/vex002
Green, K. J., Brown, C. J., Gray, S. M., and Karasev, A. V. (2017). Phylogenetic study of recombinant strains of Potato virus Y. Virology 507, 40–52. doi: 10.1016/j.virol.2017.03.018
Grzela, R., Szolajska, E., Ebel, C., Madern, D., Favier, A., Wojtal, I., et al. (2008). Virulence factor of Potato virus Y, genome-attached terminal protein VPg, is a highly disordered protein. J. Biol. Chem. 283, 213–221. doi: 10.1074/jbc.M705666200
Han, S., Gao, Y., Fan, G., Zhang, W., Qiu, C., Zhang, S., et al. (2017). A novel recombined Potato virus Y isolate in China. Plant Pathol. J. 33, 382–392. doi: 10.5423/PPJ.OA.09.2016.0189
Hunt, B. G., Ometto, L., Wurm, Y., Shoemaker, D., Yi, S. V., Keller, L., et al. (2011). Relaxed selection is a precursor to the evolution of phenotypic plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 15936–15941. doi: 10.1073/pnas.1104825108
Huson, D. H. (1998). SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14, 68–73. doi: 10.1093/bioinformatics/14.1.68
Ivanov, K. I., Eskelin, K., Lohmus, A., and Makinen, K. (2014). Molecular and cellular mechanisms underlying potyvirus infection. J. Gen. Virol. 95, 1415–1429. doi: 10.1099/vir.0.064220-64220
Jombart, T., Devillard, S., and Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics 11:94. doi: 10.1186/1471-2156-11-94
Karasev, A. V., and Gray, S. M. (2013). Continuous and emerging challenges of Potato virus Y in potato. Ann. Rev. Phytopathol. 51, 571–586. doi: 10.1146/annurev-phyto-082712-102332
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, J., Cha, D. J., Kwon, M., and Maharjan, R. (2016). Potato virus Y (PVY) detection in a single aphid by one-step RT-PCR with boiling technique. Entomol. Res. 46, 278–285. doi: 10.1111/1748-5967.12170
Lacomme, C., Glais, L., Bellstedt, D. U., Dupuis, B., Karasev, A. V., and Jacquot, E. (2017). Potato Virus Y: Biodiversity, Pathogenicity, Epidemiology and Management. Basel: Springer International Publishing.
Lefkowitz, E. J., Dempsey, D. M., Hendrickson, R. C., Orton, R. J., Siddell, S. G., and Smith, D. B. (2018). Virus taxonomy: the database of the international committee on taxonomy of viruses (ICTV). Nucleic Acids Res. 46, D708–D717. doi: 10.1093/nar/gkx932
Lorenzen, J. H., Piche, L. M., Gudmestad, N. C., Meacham, T., and Shiel, P. (2006). A multiplex PCR assay to characterize Potato virus Y isolates and identify strain mixtures. Plant Dis. 90, 935–940. doi: 10.1094/pd-90-0935
MacKenzie, T. D. B., Arju, I., Gallagher, A., Nie, X., and Singh, M. (2018). Evidence of Potato virus Y spread through post-emergence management practices in commercial potato fields. Am. J. Potato Res. 95, 720–728. doi: 10.1007/s12230-018-9632-9636
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003. doi: 10.1093/ve/vev003
Moury, B., Charron, C., Janzac, B., Simon, V., Gallois, J. L., Palloix, A., et al. (2014). Evolution of plant eukaryotic initiation factor 4E (eIF4E) and potyvirus genome-linked protein (VPg): a game of mirrors impacting resistance spectrum and durability. Infect. Genet. Evol. 27, 472–480. doi: 10.1016/j.meegid.2013.11.024
Moury, B., Morel, C., Johansen, E., Guilbaud, L., Souche, S., Ayme, V., et al. (2004). Mutations in Potato virus Y genome-linked protein determine virulence toward recessive resistances in Capsicum annuum and Lycopersicon hirsutum. Mol. Plant Microbe Interact. 17, 322–329. doi: 10.1094/mpmi.2004.17.3.322
Murray, G. G. R., Wang, F., Harrison, E. M., Paterson, G. K., Mather, A. E., Harris, S. R., et al. (2016). The effect of genetic structure on molecular dating and tests for temporal signal. Methods Ecol. Evol. 7, 80–89. doi: 10.1111/2041-210X.12466
Nagy, P. D. (2008). “Recombination in plant RNA viruses,” in Plant Virus Evolution, ed. M. J. Roossinck (Heidelberg: Springer), 133–156. doi: 10.1007/978-3-540-75763-4_8
Navascues, M., and Emerson, B. C. (2009). Elevated substitution rate estimates from ancient DNA: model violation and bias of Bayesian methods. Mol. Ecol. 18, 4390–4397. doi: 10.1111/j.1365-294X.2009.04333.x
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2014). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nie, B., Singh, M., Sullivan, A., Murphy, A., Xie, C., and Nie, X. (2012). Response of potato cultivars to five isolates belonging to four strains of Potato virus Y. Plant Dis. 96, 1422–1429. doi: 10.1094/PDIS-01-12-0018-RE
Nie, X., Singh, M., Pelletier, Y., and McLaren, D. (2013). Recent advances on Potato virus Y research in Canada. Am. J. Potato Res. 90, 14–20. doi: 10.1007/s12230-012-9288-9286
Nie, X., Singh, R. P., and Singh, M. (2004). Molecular and pathological characterization of N: O isolates of the Potato virus Y from Manitoba. Can. J. Plant Pathol. 26, 573–583. doi: 10.1080/07060660409507178
Parker, J., Rambaut, A., and Pybus, O. G. (2008). Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect. Genet. Evol. 8, 239–246. doi: 10.1016/j.meegid.2007.08.001
Quenouille, J., Vassilakos, N., and Moury, B. (2013). Potato virus Y: a major crop pathogen that has provided major insights into the evolution of viral pathogenicity. Mol. Plant Pathol. 14, 439–452. doi: 10.1111/mpp.12024
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarisation in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Sagulenko, P., Puller, V., and Neher, R. A. (2018). TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. 4:vex042. doi: 10.1093/ve/vex042
Scholthof, K. B., Adkins, S., Czosnek, H., Palukaitis, P., Jacquot, E., Hohn, T., et al. (2011). Top 10 plant viruses in molecular plant pathology. Mol. Plant Pathol. 12, 938–954. doi: 10.1111/j.1364-3703.2011.00752.x
Singh, R. P., Valkonen, J. P., Gray, S. M., Boonham, N., Jones, R. A., Kerlan, C., et al. (2008). Discussion paper: the naming of Potato virus Y strains infecting potato. Arch. Virol. 153, 1–13. doi: 10.1007/s00705-007-1059-1051
Sironi, M., Cagliani, R., Forni, D., and Clerici, M. (2015). Evolutionary insights into host-pathogen interactions from mammalian sequence data. Nat. Rev. Genet. 16, 224–236. doi: 10.1038/nrg3905
Storey, J. D., and Tibshirani, R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100:9440. doi: 10.1073/pnas.1530509100
Visser, J. C., Bellstedt, D. U., and Pirie, M. D. (2012). The recent recombinant evolution of a major crop pathogen, Potato virus Y. PLoS One 7:e50631. doi: 10.1371/journal.pone.0050631
Xie, L., Zhang, X., Zheng, S., Zhang, L., and Li, T. (2017). Molecular identification and specific detection of Telosma mosaic virus infecting passion fruit. Scientia Agricultura Sinica 50, 4725–4734.
Yang, Z. (1998). Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573. doi: 10.1093/oxfordjournals.molbev.a025957
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., and Nielsen, R. (1998). Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 46, 409–418. doi: 10.1007/PL00006320
Yang, Z., and Nielsen, R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Keywords: potato virus Y, viral genome linked protein, Bayesian phylogenetic analysis, selective constraint, Bayesian tip-association significance
Citation: Mao Y, Sun X, Shen J, Gao F, Qiu G, Wang T, Nie X, Zhang W, Gao Y and Bai Y (2019) Molecular Evolutionary Analysis of Potato Virus Y Infecting Potato Based on the VPg Gene. Front. Microbiol. 10:1708. doi: 10.3389/fmicb.2019.01708
Received: 08 March 2019; Accepted: 10 July 2019;
Published: 26 July 2019.
Edited by:
Ludmila Chistoserdova, University of Washington, United StatesReviewed by:
Diego Santos-Garcia, UMR5558 Biométrie et Biologie Evolutive (LBBE), FranceC. Martin Lawrence, Montana State University, United States
Copyright © 2019 Mao, Sun, Shen, Gao, Qiu, Wang, Nie, Zhang, Gao and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fangluan Gao, raindy@fafu.edu.cn; Yanju Bai, yanjubai@163.com
†These authors have contributed equally to this work