 Ana Durán-Viseras
Ana Durán-Viseras Adrian-Stefan Andrei
Adrian-Stefan Andrei Rohit Ghai
Rohit Ghai Cristina Sánchez-Porro
Cristina Sánchez-Porro Antonio Ventosa
Antonio Ventosa- 1Department of Microbiology and Parasitology, Faculty of Pharmacy, University of Seville, Seville, Spain
- 2Department of Aquatic Microbial Ecology, Institute of Hydrobiology, Biology Centre of the Academy of Sciences of the Czech Republic, České Budějovice, Czechia
Hypersaline aquatic and terrestrial ecosystems display a cosmopolitan distribution. These environments teem with microbes and harbor a plethora of prokaryotic lineages that evaded ecological characterization due to the prior inability to cultivate them or to access their genomic information. In order to close the current knowledge gap, we performed two sampling and isolation campaigns in the saline soils of the Odiel Saltmarshes and the salterns of Isla Cristina (Huelva, Spain). From the isolated haloarchaeal strains subjected to high-throughput phylogenetic screening, two were chosen (F15BT and F9-27T) for physiological and genomic characterization due of their relatedness to the genus Halonotius. Comparative genomic analyses were carried out between the isolated strains and the genomes of previously described species Halonotius pteroides CECT 7525T, Halonotius aquaticus F13-13T and environmentaly recovered metagenome-assembled representatives of the genus Halonotius. The topology of the phylogenomic tree showed agreement with the phylogenetic ones based on 16S rRNA and rpoB′ genes, and together with average amino acid and nucleotide identities suggested the two strains as novel species within the genus. We propose the names Halonotius terrestris sp. nov. (type strain F15BT = CECT 9688T = CCM 8954T) and Halonotius roseus sp. nov. (type strain F9-27T = CECT 9745T = CCM 8956T) for these strains. Comparative genomic analyses within the genus highlighted a typical salt-in signature, characterized by acidic proteomes with low isoelectric points, and indicated heterotrophic aerobic lifestyles. Genome-scale metabolic reconstructions revealed that the newly proposed species encode all the necessary enzymatic reactions involved in cobalamin (vitamin B12) biosynthesis. Based on the worldwide distribution of the genus and its abundance in hypersaline habitats we postulate that its members perform a critical function by being able to provide “expensive” commodities (i.e., vitamin B12) to the halophilic microbial communities at large.
Introduction
Haloarchaea are extremely halophilic archaea belonging to the phylum Euryarchaeota. They constitute a diverse group of archaea that are widely distributed in different extreme habitats with high salt concentrations, from aquatic systems (saline lakes, salterns) to saline soils, salt mines and salted foods (Ventosa, 2006). Haloarchaea are placed in the class Halobacteria, which comprises three orders: Halobacteriales, Haloferacales and Natrialbales, encompassing five families and more than 50 genera (Amoozegar et al., 2017). Despite their ecological role (in hypersaline habitats they constitute a dominant population) (Ghai et al., 2011; Fernández et al., 2014a, b; Ventosa et al., 2015; Vera-Gargallo et al., 2019) and their biotechnological potential (Amoozegar et al., 2017), little information is available on the genomic architecture and metabolic potential of cultured haloarchaea representatives (Bolhuis et al., 2006; Pfeiffer et al., 2008). The genus Halonotius belongs to the class Halobacteria, order Haloferacales, family Halorubraceae (Amoozegar et al., 2017) and currently it includes two species: the type species, Halonotius pteroides, isolated from a crystallizer of an Australian saltern, which represented up to 16% of the prokaryotic population of this habitat (Burns et al., 2004, 2010), and Halonotius aquaticus, recently isolated from a marine saltern in Spain (Durán-Viseras et al., 2019). Although the genus comprises only two described species, it has been previously reported in several other habitats, such as saline lakes in Australia and China (Podell et al., 2014; Han et al., 2017), solar salterns in Turkey and Spain (Çınar and Mutlu, 2016; Durán-Viseras et al., 2019) and food-grade salt samples (Henriet et al., 2014), indicative of a world-wide environmental distribution. Moreover, three metagenome-assembled genomes previously recovered from Lake Tyrell (Australia), appeared to be phylogenetically close to Halonotius (Podell et al., 2013).
In this work, we have carried out a comparative genomic analysis of the genus Halonotius, for which we used the genomes of the two species belonging to the genus (i.e., Halonotius pteroides and Halonotius aquaticus), the ones from the two new strains that we isolated, and two metagenome-assembled genomes mentioned above. Our data indicate that Halonotius spp., like many other haloarchaea [e.g., Haloquadratum (Bolhuis et al., 2006), Halobacterium (Pfeiffer et al., 2008)], are photoheterotrophs with a typical aerobic electron transport chain and harbor a rhodopsin-related gene pool, specifically haloarchaeal proton-pumps and sensory rhodopsins. Remarkably, all analyzed Halonotius genomes were found to encode a complete cobalamin biosynthesis pathway. Moreover, our results show that this genus is abundant and diverse in hypersaline environments and support the description of two new species, for which we propose the designations Halonotius roseus sp. nov., and Halonotius terrestris sp. nov., respectively. Additionally, the detailed study of the polar lipids profiles of these strains justify an emendation of the genus Halonotius.
Materials and Methods
Strains Isolation
Two Halonotius strains were isolated during two sampling campaigns, conducted in September 2014 and June 2016, as previously described (Vera-Gargallo and Ventosa, 2018; Durán-Viseras et al., 2019). Strain F15BT was isolated from a hypersaline soil (36.1 g/L salinity) of the Odiel Saltmarshes (Huelva, Spain, 37°22′N 6°98′W), while strain F9-27T was isolated from a pond water sample (310 g/L salinity and pH 7.1) collected from the saltern of Isla Cristina (Huelva, Spain, 37°21′N 7°33′W). Samples were diluted, plated and incubated at 37°C for 8 weeks. Strains were isolated in R2A medium (Difco) supplemented with 25% (w/v) seawater salt solution prepared by dilution of SW 30% stock solution (Subow, 1931) which contained (g/L): NaCl, 195; MgCl2.6H2O, 32.5; MgSO4.7H2O, 50.8; CaCl2, 0.83; KCl, 5.0; NaHCO3, 0.17; NaBr, 0.58 and solidified with 2% (w/v) purified agar when necessary. The cultures were routinely grown in R2A 25% (w/v) medium adjusted to pH 7.5 and incubated at 37°C. They were maintained at −80°C in this medium containing 50% (v/v) glycerol.
Halonotius pteroides CECT 7525T, type species of the genus Halonotius and Halonotius aquaticus F13-13T were also used in this study. These strains were also grown in R2A 25% (w/v) medium as described above.
DNA Extraction, Purification and Sequencing
Genomic DNA was extracted and purified following the methodology previously described by Durán-Viseras et al. (2019). DNA quantification was determined using spectrophotometric (DeNovix DS-11 FX, DeNovix Technologies, Wilmington, DE, United States) and fluorometric (Qubit 3.0 Fluorometer, Thermofisher Scientific, United States) assays. DNA quality was checked by agarose gel (1%) electrophoresis.
The 16S rRNA and the rpoB′ genes were PCR-amplified, using the primer pairs ArchF/ArchR (DeLong, 1992; Arahal et al., 1996) and rpobF/rpobR (Fullmer et al., 2014), respectively, and sequenced by Sanger technology at a commercial company (Stabvida, Oeiras, Portugal). The shotgun sequencing of strains F15BT and F9-27T was also performed by the same commercial company (Stabvida, Oeiras, Portugal) on the Hiseq 4000 platform, using 150 bp paired-end sequencing. The genomes of the strains Halonotius aquaticus F13-13T and Halonotius pteroides CECT 7525T were recovered from a previous study (Durán-Viseras et al., 2019).
Genome Assembly and Annotation
Preprocessing of raw Illumina reads of strains F15BT, F9-27T, Halonotius aquaticus F13-13T and Halonotius pteroides CECT 7525T was carried out by using a combination of software tools implemented in the BBMap project (Bushnell, 2016). Briefly, bbduk.sh was used to: (i) remove poor quality sequences from the obtained interleaved files (qtrim = rl trimq = 18), (ii) rename the reads, (iii) identify and remove phiX and p-Fosil2 controls (k = 21 ref = vectorfile ordered cardinality) and (iv) remove Illumina adapters (k = 21 ref = adapterfile ordered cardinality). bbmerge.sh was used for de novo identification of any other potential adapters. A total of, 5,402,062, 14,332,336, 13,117,040, and 14,471,142 reads were generated for the strains F15BT, F9-27T, Halonotius aquaticus F13-13T and Halonotius pteroides CECT 7525T, respectively.
The quality-checked paired-end Illumina reads for the strains F15BT, F9-27T, Halonotius aquaticus F13-13T and Halonotius pteroides CECT 7525T were assembled using Spades (v3.12.0) (Bankevich et al., 2012) in 26, 4, 10, and 42 contigs, respectively. Assembly quality and genomic G + C content was checked using QUAST v2.3 (Gurevich et al., 2013). Genome completeness, contamination and strain heterogeneity were estimated using CheckM v1.0.5 (Parks et al., 2015). Genome sequences were annotated with Prokka (Seemann, 2014). BlastKOALA (Aziz et al., 2008) was used to assign KO identifiers (K numbers) to orthologous genes present in the genomes. Inferences of metabolic pathways were conducted by mapping to the KEGG pathways and KEGG modules the assigned KO numbers of each individual genome. Additionally, selected proteins were analyzed using jackhmmer (Finn et al., 2015), CDD (Marchler-Bauer et al., 2015), and Phobius (Kall et al., 2007).
Phylogenetic Analyses
The 16S rRNA gene-based phylogenetic analyses were performed using the maximum-likelihood and maximum-parsimony algorithms integrated in the ARB software package (Ludwig et al., 2004). The 16S rRNA gene sequences alignment was confirmed and checked against both the primary and secondary structures of the 16S rRNA sequence using the alignment tool of the ARB software package. In order to evaluate the robustness of the obtained phylogenetic tree, a bootstrap analysis (1000 replications) was performed.
The rpoB′ gene-based phylogenetic analyses were performed by MEGA 6.0 software (Tamura et al., 2013) using the maximum-likelihood. In order to evaluate the robustness of the obtained phylogenetic tree, a bootstrap analysis (1000 replications) was performed.
For phylogenomic analyses a set of 257 conserved proteins, identified based on COG annotations (Tatusov et al., 2001), were individually aligned using PRANK (Löytynoja, 2014) (−protein + F), trimmed with BMGE (Criscuolo and Gribaldo, 2010) (−m BLOSUM30 –t AA –g 0.5) and concatenated. The obtained alignment (containing 40,476 positions) was used for maximum-likelihood phylogeny reconstruction using IQ-TREE (version 1.6.7) (Nguyen et al., 2015): amino-acid exchange rate matrix LG (Le and Gascuel, 2008), empirical base frequencies and discrete Gamma rate heterogeneity (as selected by ModelFinder as the best-fit substitution model; Kalyaanamoorthy et al., 2017). The tree topology was tested using SH-Alrt and ultrafast bootstrap approximation (1000 replications).
Additional 16S rRNA gene sequences, rpoB′ sequences and genomes used for phylogenetic analyses were obtained from the GenBank database, and their strain designations and accession numbers are shown in Figures 1, 2 and Table 1. The trees were rooted using Candidatus Nanosalinarum J07AB56 and Candidatus Nanosalina J07AB43 as outgroups.
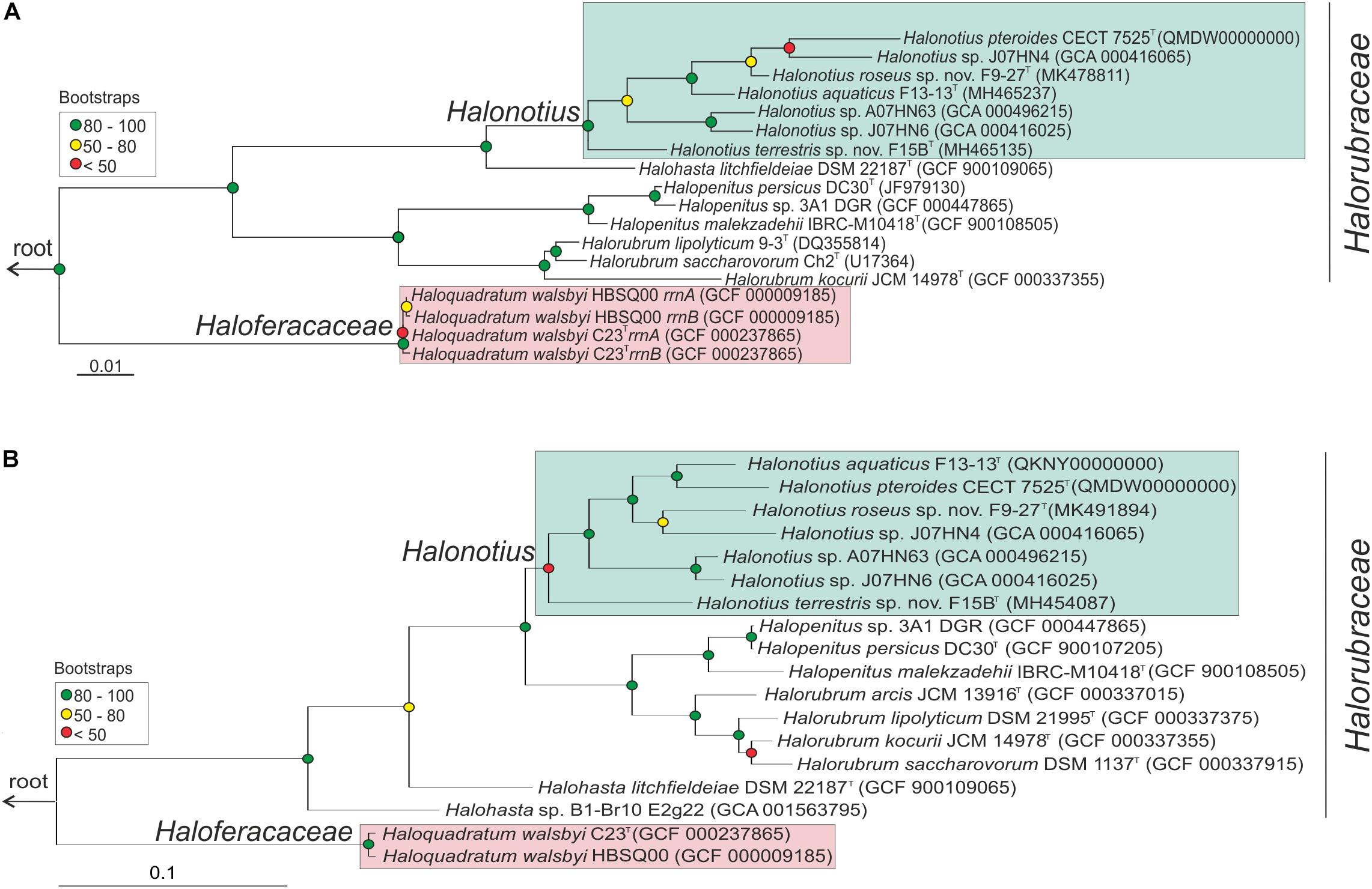
Figure 1. Halonotius phylogenetic analysis. (A) Maximum-likelihood phylogenetic tree based on the 16S rRNA gene sequences. Bar, 0.01 substitutions per nucleotide position. (B) Maximum-likelihood phylogenetic tree based on amino acid sequences of the rpoB′ genes. Bar, 0.1 substitution per nucleotide position. Sequence accession numbers are shown in parentheses. Red circles highlight Bootstrap/UFBootstrap values lower than 50, the yellow ones between 50 and 80 and green color is used to depict values higher than 80. The trees were rooted using the DPANN representatives Candidatus Nanosalinarum J07AB56 and Candidatus Nanosalina J07AB43.
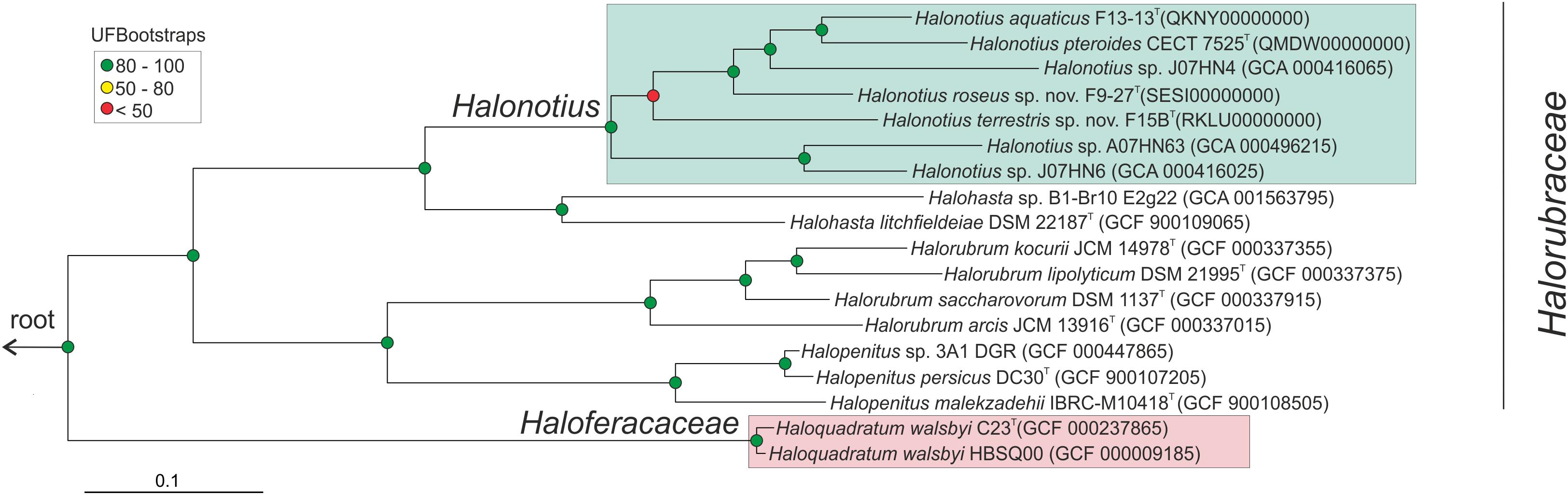
Figure 2. Halonotius phylogenomic tree. Maximum-likelihood phylogenomic tree based on the alignment of 257 shared orthologous genes. Bar, 0.1 substitution per nucleotide position. Sequence accession numbers are shown in parentheses. Red circles highlight Bootstrap/UFBootstrap values lower than 50, the yellow ones between 50 and 80 and green color is used to depict values higher than 80. The trees were rooted using the DPANN representatives Candidatus Nanosalinarum J07AB56 and Candidatus Nanosalina J07AB43.
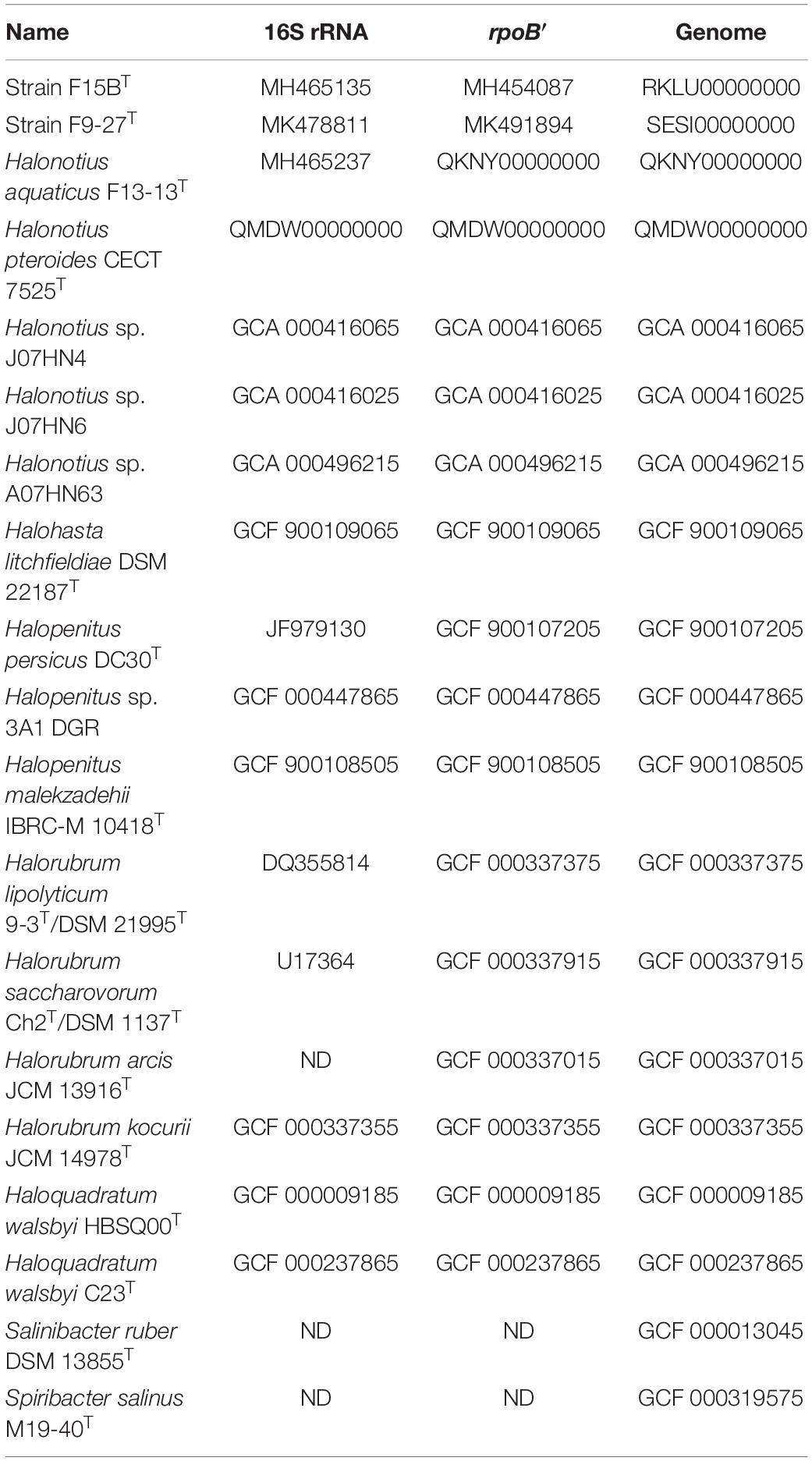
Table 1. Accession numbers of reference sequences used in this study.
Rhodopsins Tree
The genomes were scanned for the presence of rhodopsin sequences by using hmmsearch (Eddy, 2011) and a profile hidden Markov model (HMM) of the bacteriorhodopsin-like protein family (Pfam accession: PF01036). To align the highly similar identified sequences and a curated database composed of a collection of type-1 rhodopsins, MAFFT with the L-INS-i accuracy model was used (Katoh and Standley, 2013). A maximum likelihood tree with 100 bootstrap replicates was constructed based on this protein alignment, by using FastTree2 (Price et al., 2010).
Abundance Estimation for Euryarchaeota and Halonotius
Preprocessed Illumina reads from public metagenomes were queried for putative RNA sequences by using UBLAST (Edgar, 2010) against the non-redundant SILVA (Pruesse et al., 2007) SSURef_NR99_132 database, that was clustered at 85% sequence identity by UCLUST (Edgar, 2010). Identified putative 16S rRNA sequences (e-value < 1e-5) were screened using SSU-ALIGN. Resulting bona fide 16S rRNA sequences were compared by blastn (Altschul et al., 1990) (e-value < 1e-5) against the curated SILVA (Pruesse et al., 2007) SSURef_NR99_132 database. Matches with identity ≥80% and alignment length ≥90 bp were considered for downstream analyses. Sequences assigned to the phylum Euryarchaeota and genus Halonotius were used to calculate the environmental abundances for these taxonomic categories.
Fragment Recruitment
The presence of haloarchaeal strains related (at species level) to the obtained Halonotius strains (F15BT, F9-27T and Halonotius aquaticus F13-13T) was assessed by performing fragment recruitments with environmental metagenomic datasets (Table 2). Briefly, in order to avoid analysis bias, we concatenated the contigs belonging to each genome and masked all the rRNA gene sequences present. Subsequently, blastn (with the cut-offs: alignment length ≥ 30 nt, identity > 95%, E value < = 1e-5) was used in order to align the metagenomic quality-filtered shotgun reads (Table 2) against the Halonotius genomes. Figures were constructed by using the best-hits results obtained after blastn analyses.
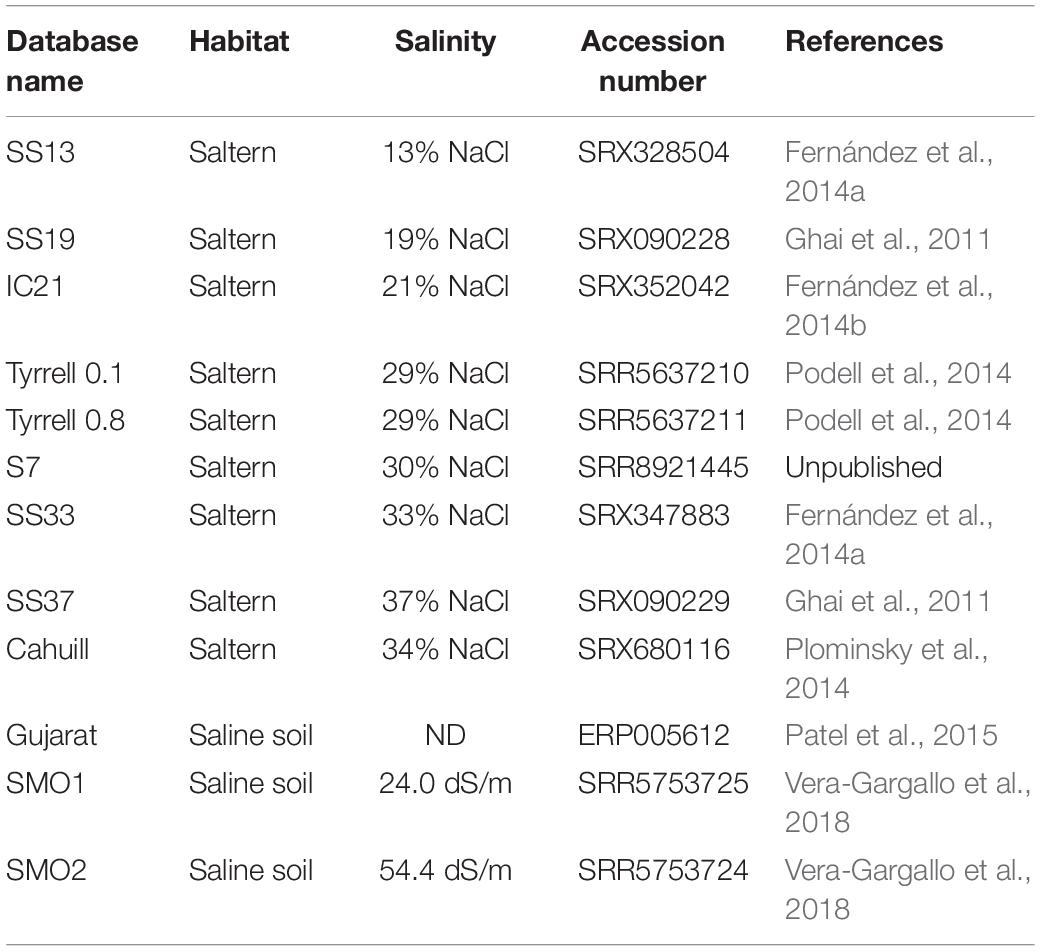
Table 2. Features of the different databases from hypersaline habitats analyzed in this study.
Identification of B12-Dependent Enzymes in Metagenomic Datasets
Three metagenomic datasets (i.e., IC21, SMO2 and S7; Table 2) were selected for B12-dependent enzyme screening, based on the results of the fragment recruitment analyses (Figure 3). Preprocessing of raw sequencing reads was performed as described above. MEGAHIT software (v1.1.5) (Li et al., 2015) with the k-mer sizes: 396 49,69,89,109,129,149, and default settings was used for metagenomic assembly. Protein-coding genes were predicted by MetaProdigal (Hyatt et al., 2012). hmmsearch (-E -evalue 1e-5 -prcov 50 -hmcov 50) (Eddy, 2011) was used for identification of B12-dependent enzymes in the assembled metagenomic datasets. The identified protein sequences and the used HMMs are deposited in figshare1.
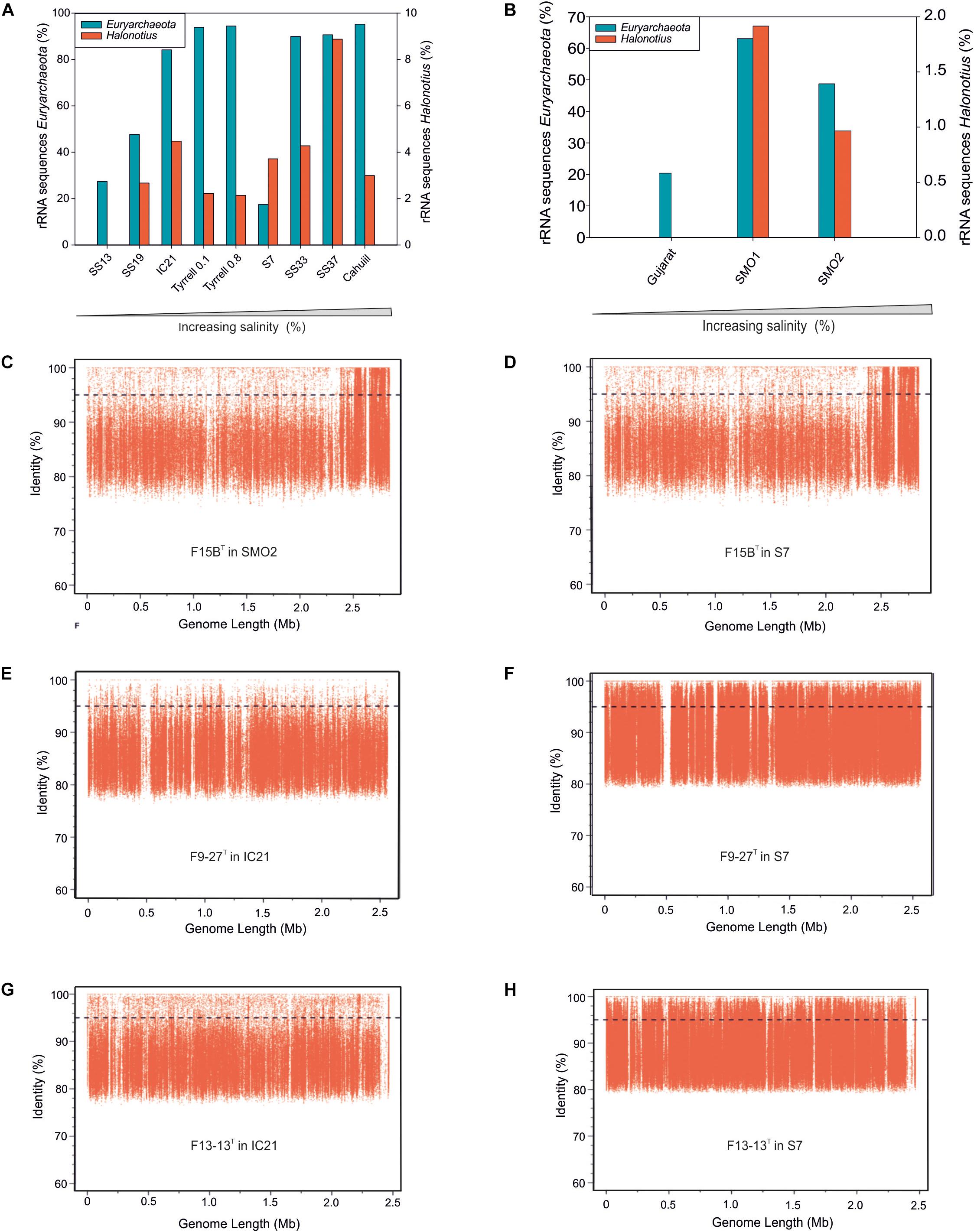
Figure 3. Halonotius sequences abundance. (A,B) Percentage of rRNA sequences related to Euryarchaeota and Halonotius recovered from 12 shotgun metagenomes. The metagenomic datasets are ordered by salinity gradient. (C–H) Recruitment plots of Halonotius strains F15BT, F9-27T and Halonotius aquaticus F13-13T against different hypersaline habitats. In each panel the Y axis represents the identity percentage and X axis represents the genome length. A restrictive cut-off 95% of nucleotide identity in at least 30 bp of the metagenomic read was used. The black dashed line shows the threshold for presence of same species (95% identity). SS13, Metagenome from Santa Pola solar saltern (Alicante, Spain), 13% salinity (SRX328504); SS19, Metagenome from Santa Pola solar saltern (Alicante, Spain), 19% salinity (SRX090228); IC21, Metagenome from Isla Cristina solar saltern (Huelva, Spain), 21% salinity (SRX352042); Tyrrell 0.1, Metagenome from Lake Tyrrell (Victoria, Australia), 29% salinity (SRR5637210); Tyrrell 0.8, Metagenome from Lake Tyrrell (Victoria, Australia), 29% salinity (SRR5637211); S7, Metagenome from Fara Fund hypersaline meromictic lake, 30% salinity (SRR8921445); SS33, Metagenome from Santa Pola solar saltern (Alicante, Spain), 33% salinity (SRX347883); SS37, Metagenome from Santa Pola solar saltern (Alicante, Spain), 37% salinity (SRX090229); Cahuill, Metagenome from Cahuil lagoon (Chile), 34% salinity (SRX680116); Gujarat, Metagenome from Little Rann of Kutch hypersaline soil (Gujarat, India), (ERP005612); SMO1, Metagenome from Marismas del Odiel Salt Marshes hypersaline soil (Huelva, Spain), 24 mS/cm salinity (SRR5753725); SMO2, Metagenome from Marismas del Odiel Salt Marshes hypersaline soil (Huelva, Spain), 54 mS/cm salinity (SRR5753724).
ANI, AAI, DDH and Protein Isoelectric Points
Average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated as previously described by Konstantinidis and Tiedje (2005a, b), respectively. Digital DDH values were determined using the Genome-to-Genome Distance Calculator (GGDC) website using formula 2 as described by Auch et al. (2010) and Meier-Kolthoff et al. (2013). The isoelectric points of proteins were calculated using the iep program implemented in the EMBOSS package (Rice et al., 2000).
Chemotaxonomic Analysis
Cultures of the strains F15BT, F9-27T, Halonotius aquaticus F13-13T and Halonotius pteroides CECT 7525T [obtained after 14 days of aerobic incubation in R2A with 25% (w/v) salts liquid medium under optimal conditions] were used for chemotaxonomic analyses. For lipid identification Halobacterium salinarum DSM 3754T and Halorubrum saccharovorum DSM 1137T were used as reference. The lipids of these two species have been previously experimentally identified (Kushwaha et al., 1982; Torreblanca et al., 1986). The strains used for comparisons were cultured according to the original descriptions for each species and standardized to the same incubation conditions. Lipid extraction was carried out following the method described by Corcelli and Lobasso (2006). Total lipid extracts were analyzed by high-performance TLC using HPTLC silica gel 60 plates crystal back (10 × 20 cm; Merck art. 5626); the plates were eluted in the solvent system chloroform/methanol/90% acetic acid (65: 4: 35, by vol.). For detection of all lipids, phospholipids and polar lipids, the plate was sprayed with sulfuric acid 5% in water, molybdenum blue spray reagent and phosphomolybdic acid, respectively and compared with the ones from Halobacterium salinarum DSM 3754T and Halorubrum saccharovorum DSM 1137T.
Phenotypic Characterization
Phenotypic tests for strains F15BT and F9-27T and Halonotius aquaticus F13-13T and Halonotius pteroides CECT 7525T, were performed according to the minimal standards delineated for the taxonomic description of novel taxa of the Halobacteria (Oren et al., 1997), following the same methodology previously described (Durán-Viseras et al., 2019).
Results and Discussion
Phylogenetic Analyses
After extensive haloarchaeal strains isolations from two hypersaline environments located in South west Spain (Odiel Saltmarshes and Isla Cristina), the strains F15BT and F9-27T were selected for further analyses. Both strains proved to be closely related to members of the genus Halonotius, as shown by 16S rRNA gene sequence comparison with available sequences in the public databases and by phylogenetic reconstruction (Figure 1A). While the 16S rRNA gene sequence of strain F15BT (1400 bp) was most closely related to Halonotius aquaticus F13-13T (96.4% sequence similarity), strain F9-27T (1394 bp) was more similar to Halonotius pteroides CECT 7525T (96.9% sequence similarity). The 16S rRNA gene sequence similarity between strains F15BT and F9-27T was 95.8%. The 16S rRNA sequence obtained by PCR and the one extracted from the genome for strains F15BT and F9-27T were compared, and proved to be identical. The 16S rRNA gene sequence similarity to other genera such as Halohasta or Halorubrum, was lower than 94 and 87%, respectively. The maximum-likelihood 16S rRNA phylogenetic tree showed that strains F15BT and F9-27T grouped within the Halonotius cluster, but in independent branches from Halonotius pteroides CECT 7525T and Halonotius aquaticus F13-13T, respectively, suggesting that they could constitute different species within the genus Halonotius (Figure 1A).
Limitations of the 16S rRNA gene sequence analysis to discriminate species of haloarchaea have been reported (Boucher et al., 2004; Sun et al., 2013), and thus a phylogenetic tree based on the comparison of the rpoB’ gene sequences was reconstructed (Figure 1B). In concordance to the 16S rRNA tree, both strains clustered with species of the genus Halonotius, but were located in different branches (Figure 1B). Strain F15BT (1827 bp) showed a nucleotide similarity of 89.1% with Halonotius aquaticus F13-13T and strain F9-27T (1827 bp) shows a nucleotide similarity of 92.0% with Halonotius sp. J07HN4 and 91.8% with Halonotius aquaticus F13-13T. The rpoB′ gene sequence similarity between strains F15BT and F9-27T was 89.3%.
Genome Sequence Characteristics
The genomes of the isolated strains, F15BT and F9-27T were successfully sequenced, and in conjunction with those of the two currently described species of the genus, Halonotius pteroides CECT 7525T and Halonotius aquaticus F13-13T, were assembled de novo. The characteristics of all these genomes are shown in Table 3. As most members of the class Halobacteria (phylum Euryarchaeota), all genomes had a high G + C content, between 59.5–62.7 mol%, with F9-27T having the highest. All four Halonotius strains studied have similar genome size, ranging from 2.5–3.0 Mb, making them the smallest ones reported for members of the family Halorubraceae. Additional genomic features are shown in Supplementary Figure 1.
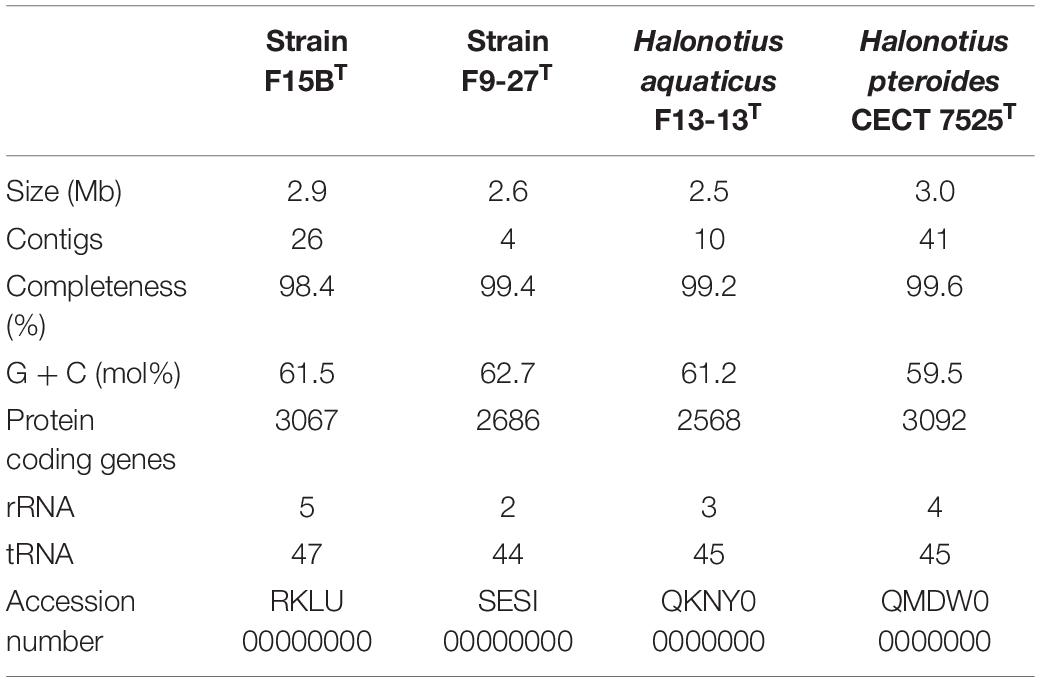
Table 3. General features of sequenced genomes.
Phylogenomic Analysis
To confirm the phylogenomic relationships previously obtained by 16S rRNA and rpoB′ genes sequence comparison between Halonotius strains, Halonotius metagenome-assembled genomes (MAGs) and other related haloarchaeal species, a phylogenomic tree based on core orthologous genes was constructed. Candidatus Nanosalina J07AB43 (GCA_000220375) and Candidatus Nanosalinarum J07AB56 (GCA_000220355) MAGs were used as outgroups. A total of 257 core orthologous genes were shared between all genomes. The phylogenomic tree based on the concatenated alignment, showed that all Halonotius genomes clustered together in a single branch, well separated from other haloarchaeal groups and supported by high bootstrap values (Figure 2). Additionally, also supported by high bootstrap values, within the Halonotius cluster, all Halonotius strains appeared monophyletically separated, suggesting they represent different species within this genus. The results are in concordance with the initial phylogeny described by 16S rRNA and rpoB′ genes sequence comparison, which suggest that for this genus both genes could be used as reliable phylogenetical markers.
Ecological Distribution and Abundance of the Genus Halonotius
Metagenomic studies carried out in hypersaline environments have enabled the determination of the microbial diversity of these habitats, suggesting that a large proportion of haloarchaeal members, abundant in these systems, had not been isolated or characterized before (Ventosa et al., 2015). Based on 16S rRNA gene sequence reads abundances from different aquatic and terrestrial hypersaline systems with different salinities (see Materials and Methods), representatives of the genus Halonotius were found to be present in almost all metagenomes from habitats with intermediate to high salinity concentrations. The genus Halonotius comprised up to 9% of the prokaryotic community in aquatic saline environments (Figure 3A) and up to 1.8% of the prokaryotic community in saline soils (Figure 3B). The data confirm that members of the genus Halonotius are abundant microorganisms in hypersaline environments, especially at high salinities (from 19 to 37% total salts) but are completely absent in habitats without or with lower salt concentrations. In this sense, we can consider them as obligately extreme halophiles that prefer habitats with high salinities.
To assess the distribution of the cultured Halonotius strains, fragment recruitment was carried out against all those metagenomic datasets mentioned above. The metagenomes which provided significant recruitment values are shown in Figures 3C–H. All Halonotius strains used in this study showed high recruitment values in their environment of origin: the metagenomic dataset SMO2 reported from the Odiel Saltmarshes saline soil in Huelva, Spain for strain F15BT, and dataset IC21 obtained from Isla Cristina salterns in Huelva, Spain, for strains F13-13T and F9-27T, where strain F13-13T seemed to be more abundant. Remarkably, highest recruitment values were obtained against the metagenomic dataset S7, which corresponds to a hypersaline meromictic pit lake located in Transylvanian Basin, Romania; suggesting a worldwide distribution for this genus. Even the highest abundance of 16S rRNA Halonotius reads was obtained in SS37 metagenome, none of the Halonotius isolates recruited significantly in that metagenome, only Halonotius pteroides CECT 7525T seemed to be present in that metagenome dataset (Supplementary Figure 2). The abundance of reads above 95% similarity for Halonotius pteroides CECT 7525T against metagenomic database SS37 and the above mentioned 16S rRNA reads abundance in this metagenome, indicates that there might be other Halonotius species present in significant amounts in this specific habitat.
Furthermore, to evaluate the presence of Halonotius worldwide, we have searched the SILVA database for 16S rRNA sequences belonging to this genus. Results indicate that members of the genus Halonotius are found in a variety of hypersaline environments from different countries, such as: Canada, United States, Bolivia, Tunisia, Spain, Romania, Turkey, Russia, China, and Australia (Supplementary Figure 3). All these data, in addition to the 16S rRNA reads abundance along the different metagenomes, support a worldwide distribution of this genus.
Metabolism
Halonotius strains were isolated on complex media for heterotrophic aerobic microbes (see Materials and Methods). Genome annotation of Halonotius members confirm their heterotrophic metabolism. No evidences for photosynthetic or chemolithotrophic capabilities were found in any of these genomes. In line with their heterotrophic capabilities, several ABC and other transporters for uptake of carbohydrates were identified, forming a large fraction of the genes in all Halonotius genomes (Figure 4).
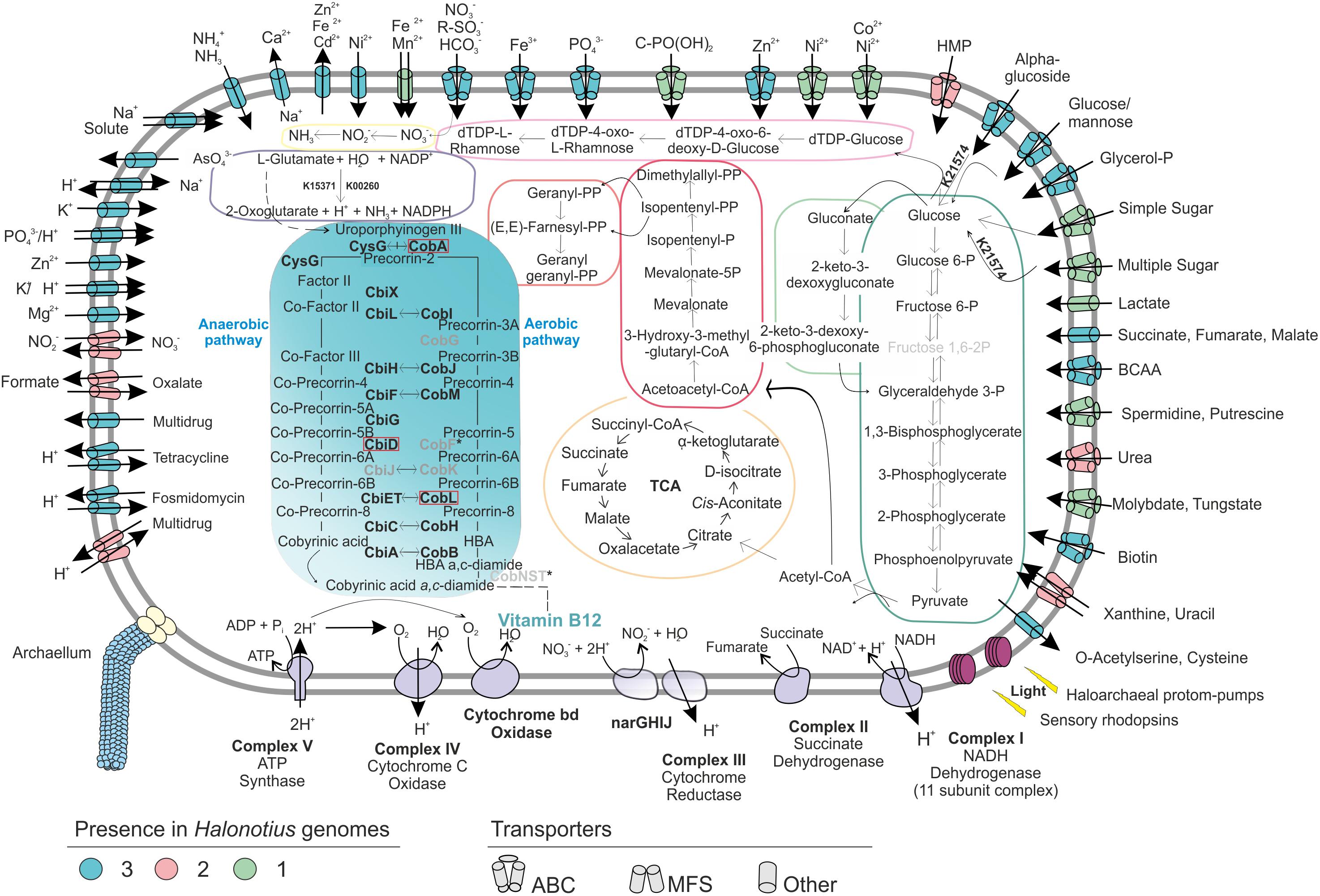
Figure 4. Metabolic reconstruction of the isolated strains Halonotius terrestris sp. nov. F15BT, Halonotius roseus sp. nov. F9-27T and Halonotius aquaticus F13-13T. The turquoise panel depicts the simplified metabolic pathway of vitamin B12 biosynthesis; the gene names highlighted in boldface were present in the genomes, while the ones in gray are inferred to be absent in archaea. Horizontal arrows indicate homology between aerobic and anaerobic pathway enzymes. The discontinuous lines correspond to complete pathways not represented in the figure. The dark green box highlights glycolysis (Embden–Meyerhof–Parnas), while the light green one shows the Entner–Doudoroff pathway modified for haloarchaea. The orange depicts the tricarboxylic acid cycle (TCA); pink color corresponds to isoprenoid biosynthesis: C5 isoprenoid (dark) and C10–C20 isoprenoid (light). The light violet box corresponds to dTDP-L-rhamnose biosynthesis; the yellow color shows the assimilatory nitrate reduction pathway; dark purple is used for the L-glutamate degradation by glutamate dehydrogenase (GdhA). Alpha-amylase enzyme for glucose degradation is also represented by its KO identifier. Transparency was used to show the inferred components of the complex III of electron transport chain. Red boxes are used for genes not present in the studied genomes. BCAA, branched-chain amino acid; HMP, hydroxymethylpyrimidine; TCA, tricarboxylic acid cycle.
Representative genes for central carbohydrate metabolism like the tricarboxylic acid cycle, pentose phosphate, gluconeogenesis and Entner–Doudoroff pathway were present (Figure 4). For the oxidation of pyruvate to acetyl CoA (Figure 4), only the anaerobic route via pyruvate ferredoxin oxidoreductase (porA and porB) seemed to be present, while the aerobic route via pyruvate dehydrogenase (PDH) was absent. Additionally, genes encoding an α-amylase (K21574), which can degrade complex carbohydrates to glucose were detected (Figure 4). Remarkably, in agreement with previous studies on haloarchaea (Falb et al., 2008; Anderson et al., 2011), the standard Embden-Meyerhof pathway of glycolysis appears to be incomplete, with 6-phosphofructokinase (the key enzyme of the classical pathway), found absent in all studied genomes (Figure 4). This suggest that haloarchaea could utilize the Entner-Doudoroff pathway for glucose degradation or that alternative enzymes could be used for this step.
Regarding nitrogen metabolism, Halonotius genomes encode the essential genes involved in the assimilation of ammonia and amino acid metabolism, glutamine synthetase and glutamate synthase. Accordingly, all genomes also presented the gene encoding the high-affinity ammonium transporter Amt, indicating that nitrogen uptake occurs in its most reduced form, ammonia (Figure 4). Along similar lines, strains F15BT and F9-27T showed a major facilitator superfamily (MFS) antiporter for nitrate/nitrite uptake (Figure 4). Genes encoding the enzymes for nitrate reduction assimilation to ammonia, nitrate reductase and nitrite reductase, were also present. Besides, several ABC transporters for nitrogen sources, such as branched-chain amino acids, urea, putrescine/spermidine and nitrate-nitrite/taurine were also found (Figure 4). In addition, Halonotius genomes showed other genes which could provide nitrogen rich compounds, by degradation of amino acids, such as L-glutamate, to ammonia generating NADPH (glutamate dehydrogenase) (Figure 4) or by urea degradation, which was only absent in strain F15BT. Most pathways for amino acid biosynthesis were also found in the studied genomes (Supplementary Table 1).
The presence of the high-affinity phosphate transport system (PstSCAB), suggests that this is the main route of uptake of inorganic phosphate (Figure 4). All studied genomes of Halonotius also contained the ABC transporter, PhnCDE, a high affinity uptake system for phosphonates (Figure 4), which are organophosphorous compounds with a carbon–phosphorous bond that under phosphate starvation conditions could be used also as a nutritional source of phosphorous (Villarreal-Chiu et al., 2012).
In addition, several antimicrobial compounds and multidrugs transport systems were found and genes encoding archaella were also discovered in all Halonotius genomes.
Cobalamin (Vitamin B12) Biosynthesis
Genome-scale metabolic reconstructions performed for the Halonotius members brought to light the complete cobalamin biosynthesis pathway (Figures 4, 5), suggesting the genetic capability of the strains of this genus for its de novo synthesis. Cobalamin (vitamin B12) is a complex metabolite, produced only by a subset of bacteria and archaea, and essential cofactor required by many organisms from all the domains of life. Cobalamin can be synthesized by either an aerobic or anaerobic pathway from uroporphyrinogen III as precursor, involving 20 enzymatic steps. Several enzymes are homologous and shared by both pathways, while only few are specifically oxygen-requiring or oxygen-sensitive (Moore and Warren, 2012). The complex structure of cobalamin and the high metabolic toll of its biosynthesis pathway (Morris, 2012; Doxey et al., 2015) fueled a ‘race’ toward a genomic removal, thus designating the few taxa capable of its biosynthesis as ‘losers’ that perform a Black Queen function (Morris, 2015). This term alludes to the game of Hearts, where players try to avoid being stuck with the queen of spades, and has been used to describe costly genes or functions that members of the community benefit from losing. Thus, generating individual “beneficiaries” of reduced genomic content depending on luckless “helpers” that provide goods to the whole community (Morris et al., 2012). The role of representatives of the Archaea in cobalamin production has been poorly studied and limited to Thaumarchaeota, halophilic Crenarchaeaota and marine methanogenic Euryarchaeota (Rodionov et al., 2003; Doxey et al., 2015).

Figure 5. Halonotius cobalamin synthesis gene clusters involved in cobalamin synthesis. The figure highlights the co-occurrence of cobalamin synthesis genes and their conserved order in the analyzed Halonotius genomes (n = 4). (A) Represents the genes from the first stage of the pathway, from uroporphyrinogen III to a,c-diamine. (B) Represents the genes from the second stage of the pathway, from a,c-diamine to vitamin B12. The red star marks genes that are present in two copies. The genes depicted in white color were found not to be conserved in the gene cluster. The scale bar indicates gene lengths (in bp).
Halonotius genomes contained almost all genes involved in aerobic and anaerobic pathways, with only a few gaps (i.e., CobG and CobK were missing from the aerobic pathway; CbiD and CbiJ were missing from the anaerobic pathway) (Figure 4). Additionally, all genes encoding the conversion of cobyrinic acid a,c-diamide to vitamin B12 were also found and are represented by a discontinuous line on the metabolic map (Figures 4, 5B). None of the absent genes were either detected in any known archaeal cobalamin producers and therefore should not be considered as essential genes for this pathway (Doxey et al., 2015). Furthermore, previous studies considered the presence of cbiA/cobB, cbiC/cobH or cobT genes in a microorganism, as reliable indicators for the complete pathway (Bertrand et al., 2011). As far as the authors are aware, despite a previous report on Halobacterium salinarum NRC-1 cobamide production (Woodson et al., 2003), little is known about the cobalamin biosynthesis pathway in hypersaline environments, or halophilic archaea at large. Due to the detection of the cobalamin biosynthesis genes clusters in all the analyzed Halonotius genomes (Figure 5), and the widespread distribution and abundance (Figure 3 and Supplementary Figure 3) of this genus in hypersaline environments, we hypothesize that these organisms perform a critical role in the microbial community, where they contribute to the maintenance of the environmental cobalamin supply and modulate trophic interactions. Moreover, the detection of B12-dependent enzymes (e.g., methylmalonyl-CoA mutases; metH, MTR 5-methyltetrahydrofolate-homocysteine methyltransferases; nrdA/nrdE ribonucleoside-diphosphate reductases; pduC propanediol dehydratases) (Supplementary Table 2) in metagenomic datasets where Halonotius genomes were inferred to be present, pinpoints toward metabolic dependencies between the cobalamin suppliers and consumers.
Osmoregulation
The mechanisms used by microorganisms to deal with their cellular adjustment to high salinity environments are diverse and can be classified in salt-in and salt-out strategies (Galinski and Trüper, 1994; Wood et al., 2001), physiological osmostress adaptations which are not necessarily mutually exclusive (Deole et al., 2013; Oren, 2013; Youssef et al., 2014). The osmotic balance in microorganisms that use the salt-in strategy is achieved by accumulation of a large fraction of inorganic ions in the medium, as K+ and Cl– via transport (Oren, 2011), while Na+ ions are excluded as much as possible from cells (Gunde-Cimerman et al., 2018). However, in order to keep the proteins solubility and to ensure the functionality of key cellular activities, the entire proteome of the microorganism required compensatory changes for the cytoplasm′s adaptation to the high ionic strength (Oren, 2013; Saum et al., 2013; Talon et al., 2014; Warden et al., 2015). This has left an acidic signature on the proteome of those salt-in microorganisms, reducing its surface hydrophobicity and limiting these microorganisms to hypersaline habitats where the external salinity does not frequently fluctuate.
We have characterized the proteome of all available Halonotius genomes (Figure 6), and to analyze the differences in protein acidity we have compared them to proteomes from salt-in halophilic archaea [Haloquadratum walsbyi HBSQ00 (Bolhuis et al., 2006) and Halorubrum saccharovorum DSM 1137T (Becker et al., 2014)], a salt-in halophilic bacterium [Salinibacter ruber DSM 13855T (Mongodin et al., 2005)] and to that of a salt-out bacterium [Spiribacter salinus M19-40T (López-Pérez et al., 2013)] (Figure 6). The proteome pI plot showed a single peak around 4.0 for all haloarchaeal genomes and for Salinibacter ruber DSM 13855T, which follow the trend of amino acid use in the hypersaline system (Fernández et al., 2014b) being acidic residues more frequently employed and thus suggesting a salt-in strategy for Halonotius strains. Only Spiribacter salinus M19-40T, microorganism employing a salt-out strategy, resulted in a different single peak around 4.5.
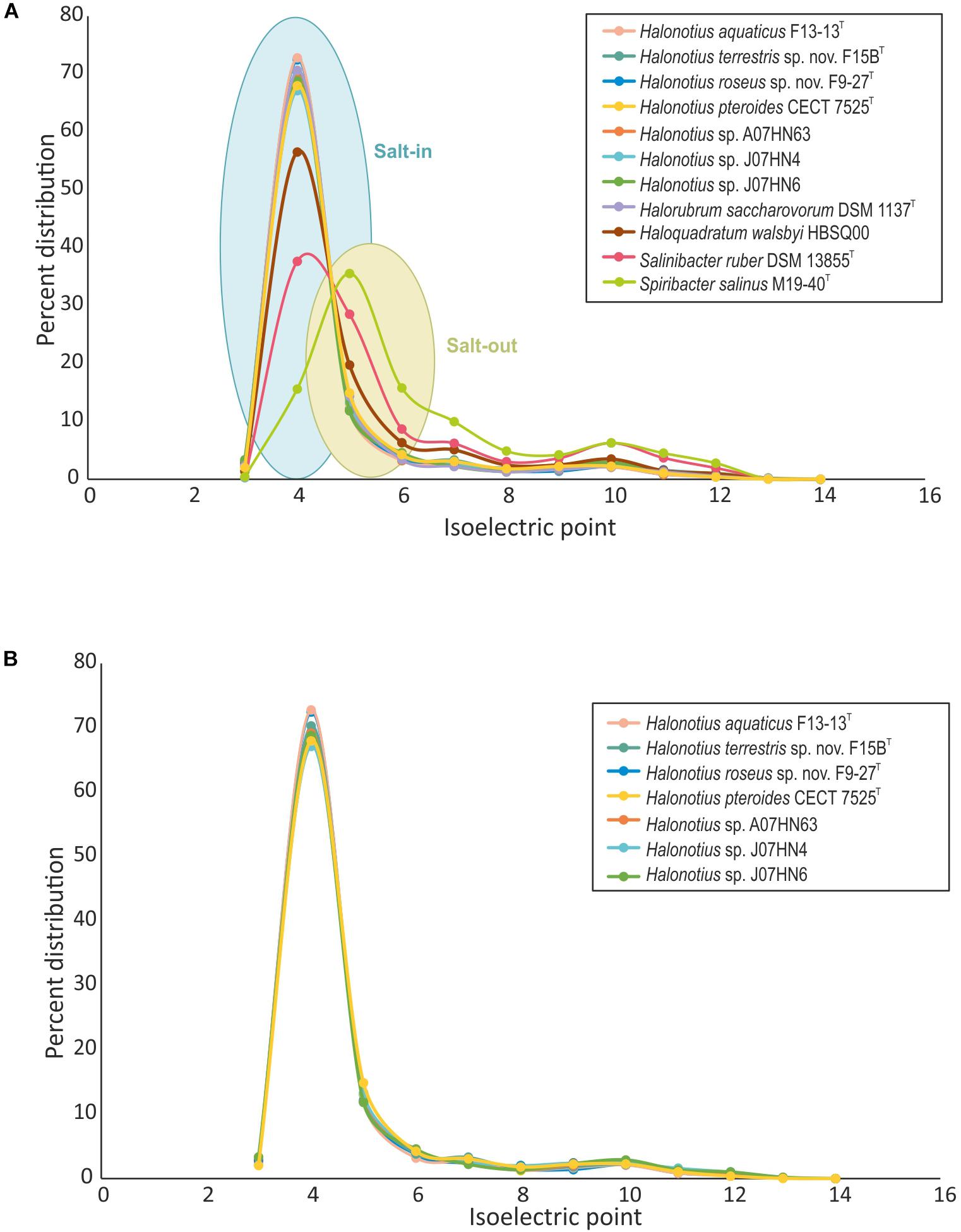
Figure 6. Comparison of isoelectric profiles of Halonotius proteomes with those of other prokaryotic species. (A) Comparison of isoelectric point of predicted proteins for Halonotius and other prokaryotic species, computed for each translated genome and shown as a percentage of distribution. (B) Comparison of isoelectric point of predicted proteins between proteomes of cultured Halonotius genomes and environmental Halonotius MAGs, shown as a percentage of distribution.
Another method to determine the salt adaptation of a microorganism is the reliance on the Na+ gradient to transport actively nutrients into the cell (Oren, 2011; Gunde-Cimerman et al., 2018). All Halonotius strains have several secondary transporters that catalyze the translocation of solutes across the cytoplasmic membrane using electrochemical ion gradients. Na+ is extruded from the cells by Na+/H+ and Na+/Ca2+ antiporters systems while K+ can enter the cells passively through K+ channels in the membrane and by K+/H+ symporters systems (Figure 4), consistent with salt-in adaptations to a hypersaline environment. The necessary energy is derived from the proton gradient over the membrane, generated by respiratory electron transport and from the light-dependent proton pumping rhodopsin (Figure 4).
In contrast, the salt-out strategy is based in keeping most of inorganic ions out and using organic compatible solutes to balance the high salinity of the external environment (Empadinhas and Da Costa, 2008; Gunde-Cimerman et al., 2018). In the case of Halonotius, ABC-type transport systems or other transporters for compatible solutes uptaking were not observed in their genomes. In the same way, none of the genes encoding key enzymes for the synthesis of organic compatible solutes, such as glycine betaine, ectoine, hydroxyectoine, proline, biphosphoglycerate or trehalose were found in any of the studied Halonotius genomes. This suggests that members of the genus Halonotius do not use this mechanism to balance the high salinity of the environment.
Rhodopsin Analysis
Microbial rhodopsins are a family of photoactive retinylidene proteins widely distributed within the microbial world. They were first discovered in haloarchaea in the early’s 1970 (Oesterhelt and Stoecknius, 1971; Oesterhelt and Stoeckenius, 1973), however, later genomic and metagenomic sequencing revealed homologs in many disparate eukaryotes and bacteria, and later in the marine group II euryarchaeota (Frigaard et al., 2006). These kind of proteins are characterized by their diversity of function, using variations of a shared seven-transmembrane helix structure and similar photochemical reactions to carry out distinctly different light-driven energy and sensory traduction processes (Govorunova et al., 2017). Their biological functions fall into two categories: (a) photoenergy transducers that convert light into electrochemical potential to energize cells (light-driven ion pumps), which catalyze outward active transport of protons, and (b) photosensory receptors, that use light to gain information about the environment to regulate cell processes (Govorunova et al., 2017).
Rhodopsin-like sequences were found in all members of the genus Halonotius (Figure 7B). Specifically, haloarchaeal proton-pumps were found in all sequenced Halonotius genomes, which indicates that ATP synthesis light-mediated is a typical feature of species of this genus. Additionally, strains F13-13T and F15BT also encoded sensory rhodopsins, suggesting additional metabolic flexibility in illuminated conditions for these two strains.
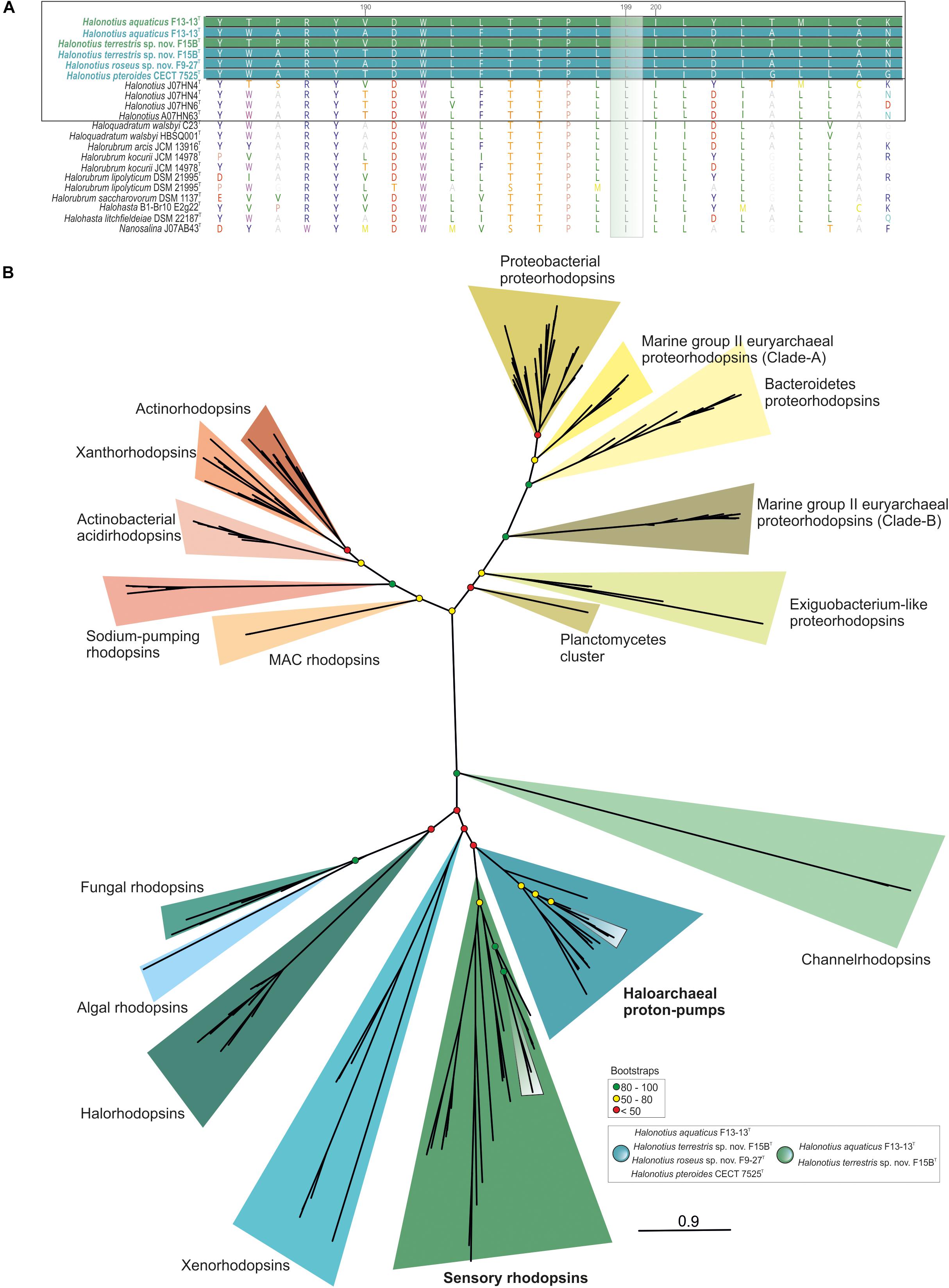
Figure 7. Halonotius rhodopsin tree. (A) Alignment comparison of rhodopsin protein sequence from Halonotius and other haloarchaea. Sequence accession numbers are shown in parentheses. Cultured Halonotius sequences are highlighted in boldface; blue corresponds to haloarchaeal proton-pump rhodopsins, while green highlights the sensory ones. The green box shows the position 199 of the rhodopsin alignment. The leucine (L) variant absorbs maximally in the green spectrum. (B) Maximum-likelihood phylogenetic tree constructed using 220 rhodopsins sequences. Boxes were used to highlight rhodopsin sequences belonging to Halonotius (see box at bottom right side of the figure). Bootstrap values on nodes are indicated by colored circles. Red circles show values lower than 50, the yellow ones between 50 and 80; green color is used to depict values higher than 80.
According to major absorption light wavelength, rhodopsins could be classified as “green-absorbing” and “blue-absorbing.” A single amino acid residue determines this preference. The leucine (L) and methionine (M) variants absorb maximally in the green spectrum while the glutamine (Q) variant absorbs maximally in the blue spectrum (Man et al., 2003). Alignment of rhodopsins sequences found in Halonotius genomes showed a leucine amino acid in this position, indicating that they show a green light absorption (Figure 7A).
Amino Acid Identity (AAI), Average Nucleotide Identity (ANI) and Digital DNA-DNA Hybridization (DDH)
The AAI, the ANI, and the digital DNA–DNA hybridization (DDH) parameters between strains F15BT, F9-27T, Halonotius pteroides CECT 7525T, Halonotius aquaticus F13-13T and other environmental Halonotius MAGs, were calculated as described in the “Materials and Methods” and the results are shown in Figure 8 and in Table 4. A threshold AAI value of 65% has been established for genus delineation (Klappenbach et al., 2007; Konstantinidis et al., 2017). AAI values for all Halonotius genomes were in all cases higher than 75% (Figure 8), indicating that all of them belongs to the genus Halonotius.
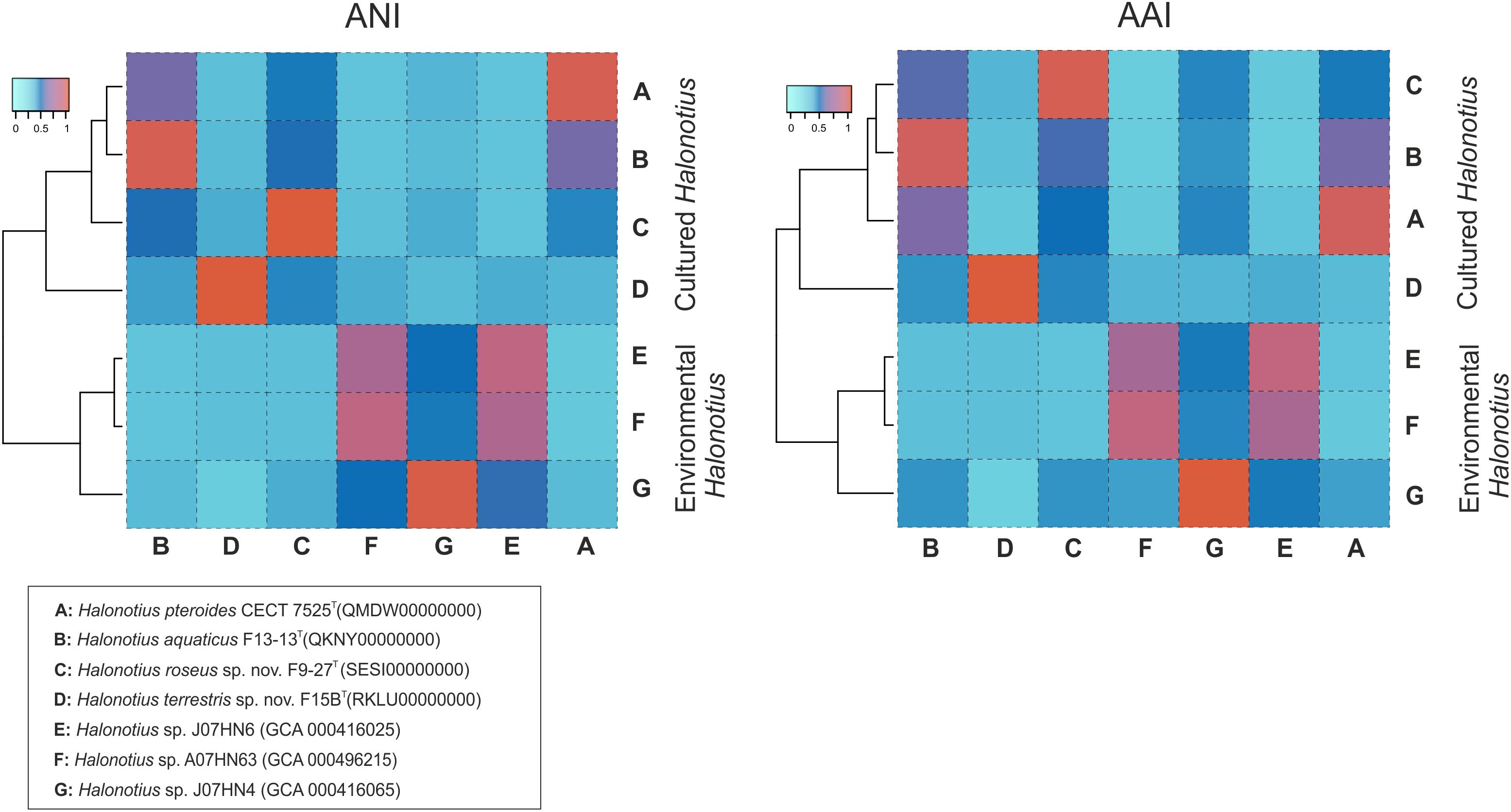
Figure 8. Amino acid identities and average nucleotide identities between Halonotius genomes. Hierarchical clustering relationships between cultured Halonotius genomes and environmental Halonotius MAGs. Amino acid identities (AAI) and average nucleotide identities (ANI) are represented by heat maps, where similarity values are represented by the color key histograms on the upper panels. Strains and sequence accession numbers are shown in the box at the bottom of the figure.
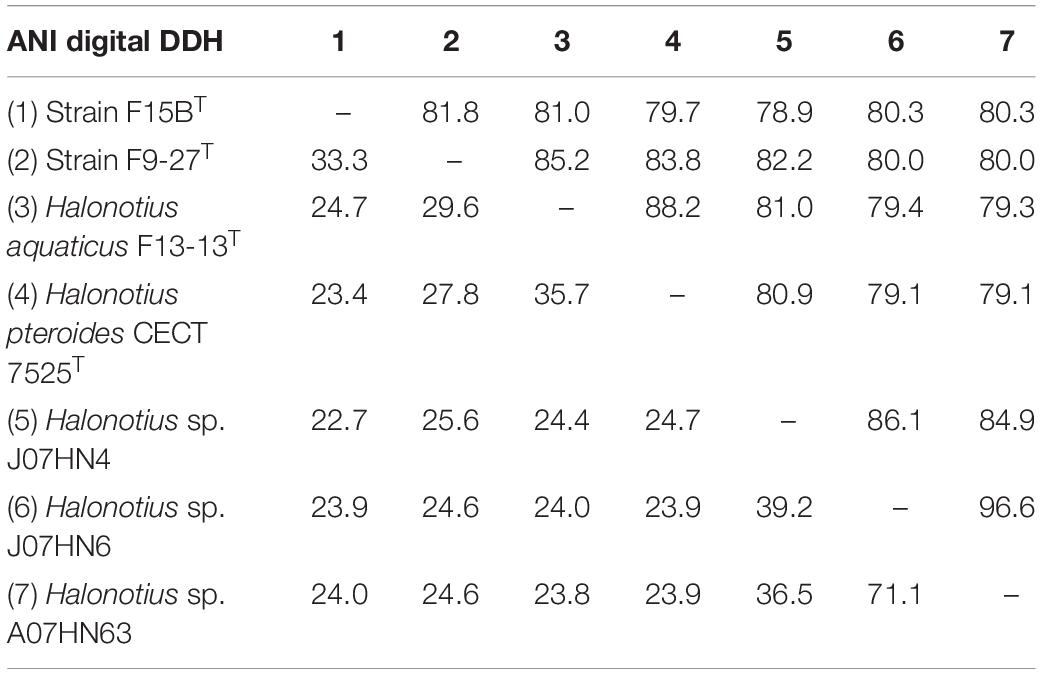
Table 4. ANI and DDH values between genomes of Halonotius.
For species delineation, a threshold ANI value of 95–96% (Konstantinidis and Tiedje, 2004; Richter and Rosselló-Móra, 2009) and a DDH value of 70% (Stackebrandt and Goebel, 1994; Auch et al., 2010) has been established. ANI values for all four strains of Halonotius studied were in all cases lower than 88% (Figure 8 and Table 4) and DDH values lower than 35.7% (Table 4), suggesting that they constitute four separate taxa at the species level. We can conclude that strains F9-27T and F15BT represent two different species of the genus Halonotius, for which we propose the new designations Halonotius roseus sp. nov. and Halonotius terrestris sp. nov., respectively.
Phenotypic Characterization
The different phenotypic features between strains F9-27T and F15BT and the other members of the genus Halonotius are shown in Table 5. More phenotypic features of strains F9-27T and F15BT are detailed in their species descriptions given below.
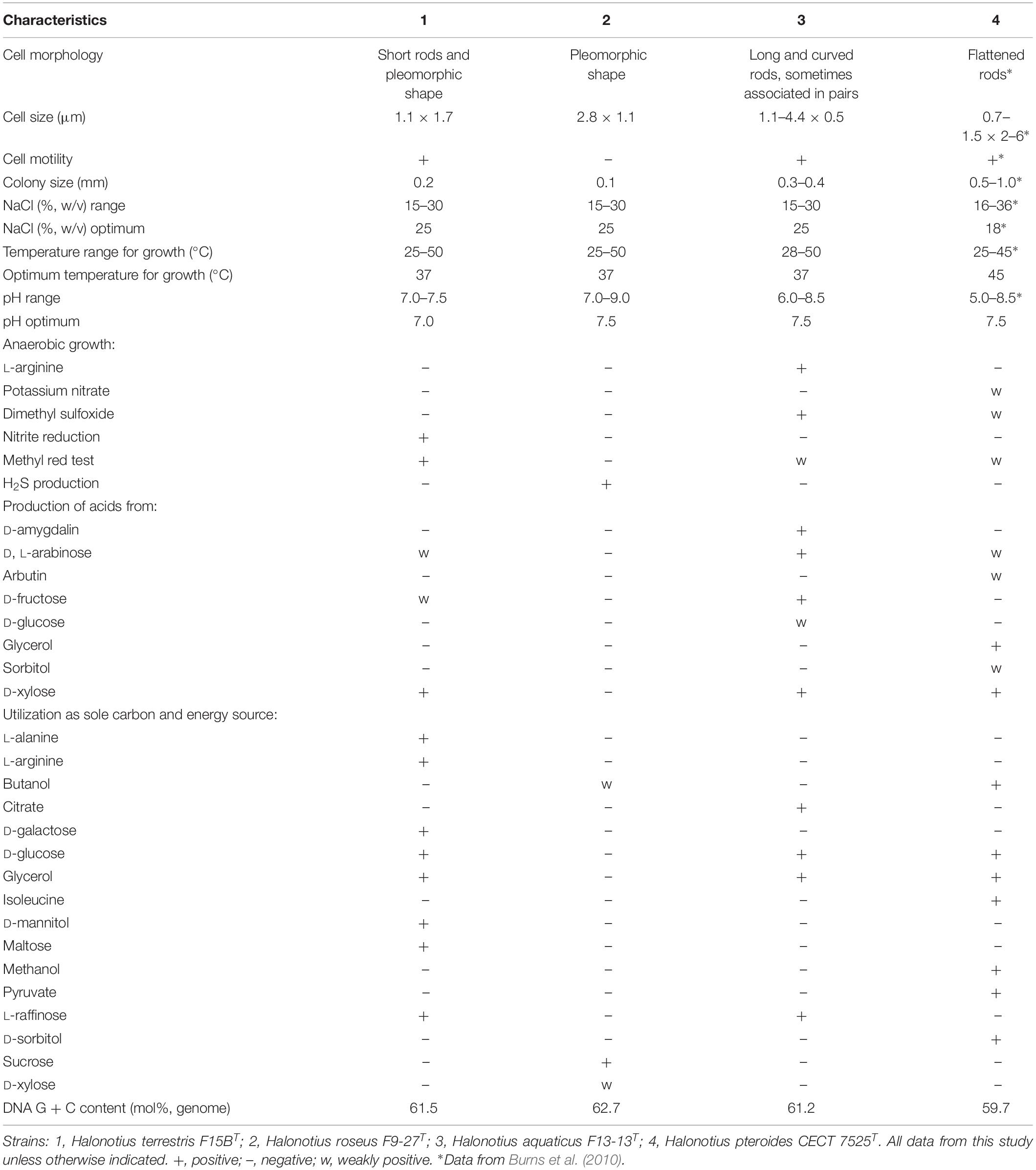
Table 5. Characteristics that differentiates Halonotius terrestris F15BT and Halonotius roseus F9-27T from related species of the genus Halonotius.
Chemotaxonomic Characterization
An exhaustive comparative analysis of the total lipids, phospholipids and polar lipids, between strains F9-27T and F15BT and the previously described Halonotius species, was carried out (Supplementary Figure 4). Results revealed that all of them possess phosphatidylglycerol (PG), phosphatidylglycerol phosphate methyl ester (PGP-Me), phosphatidylglycerol sulfate (PGS) and one glycolipid chromatographically identical to sulfated diglycosil diether (S-DGD-1) as major polar lipids. As the presence of PGS was not reported at the description of the genus Halonotius and Halonotius pteroides (Burns et al., 2010), here we also propose the emended description of this genus in order to include this and other features of species of the genus.
Conclusion
This study provides a complete comparative genomic analysis of the genus Halonotius, showing evidence of the ubiquity and abundance of species of this genus in different hypersaline environments and significantly expanding our knowledge of this almost unknown haloarchaeal genus. Results from Halonotius 16S rRNA read abundances from different aquatic and terrestrial hypersaline systems and Halonotius genomes recruitments against these systems, confirm Halonotius abundance in these environments. Remarkably, results from the complete metabolic analysis of this genus brought to light that Halonotius members present the complete cobalamin biosynthesis cluster, suggesting their capability to produce cobalamin. Thus, we suggest that species of this genus could play a relevant environmental role for the community in de novo cobalamin synthesis on these saline environments, and thus, the members of Halonotius could perform a Black Queen function among members of the haloarchaea in hypersaline habitats (Morris et al., 2012; Mas et al., 2016).
Additionally, this study indicates that members of the genus Halonotius might have a osmoregulatory salt-in strategy, based on their acidic proteomes, the presence of typically salt-in transporters and by the absence of compatible solutes transporters or pathways for their biosynthesis which could suggest a salt-out strategy. We could find proton-pumping rhodopsins in all Halonotius genomes and sensory rhodopsins in some, all absorbing in the green spectrum.
Finally, this study has also permitted the emended description of the genus Halonotius and the identification of the isolates as two novel species of this genus, for which we propose the new names Halonotius terrestris sp. nov. and Halonotius roseus sp. nov., whose descriptions are given below.
Description of Halonotius terrestris sp. nov.
Halonotius terrestris (ter.res’ tris. L. fem. adj. terrestris of or belonging to the earth, terrestrial).
Cells are Gram-stain-negative, motile, short rods, sometimes pleomorphic with 1.1 × 1.7 μm (Supplementary Figure 5). Colonies are circular, entire, red-pigmented with 0.2 mm in diameter on R2A25% medium after 10 days of incubation at 37°C. Extremely halophilic, able to grow in media with 15–30% (w/v) salts, with optimal growth at 25% (w/v) salts. No growth occurs in the absence of NaCl. Able to grow in the pH range of 7.0–7.5 and from 25 to 50°C, with optimal growth at pH 7.0 and at 37°C. Catalase and oxidase positive. Gelatin, starch, Tween 80 and esculin are not hydrolyzed. Nitrate and nitrite are reduced without gas production. H2S production is negative. Simmons’ citrate and Voges-Proskauer tests are negative. Methyl red test is positive. Indole is not produced. L-arginine, L-lysine and L-ornithine decarboxylase tests are negative. Acid is produced from D, L-arabinose, D-fructose, D-ribose and D-xylose but not from D-amygdalin, arbutin, D-cellobiose, L-citruline, dulcitol, D, L-ethionine, D-galactose, glycerol, D-glucose, inulin, lactose, D-maltose, D-mannitol, D-mannose, D-melezitose, D-melibiose, D-raffinose, sorbitol, D-sucrose, D-trehalose and L-xylitol. D-galactose, D-glucose, gycerol, maltose, D-mannitol, L-raffinose and tartrate are used as carbon and energy source but not D-arabinose, butanol, D-cellobiose, D-dulcitol, D-ethanol, fructose, lactose, D-mannose, D-melibiose, methanol, D-melezitose, D-ribose, salicin, sorbitol, sucrose, D-trehalose, xylitol and D-xylose. Not able to use L-alanine, L-cysteine, glutamine, L-methionine, L-glycine, L-lysine, isoleucine and valine as sole carbon, nitrogen and energy source but it uses L-arginine.
The major polar lipids are PG, PGP-Me, PGS and one glycolipid chromatographically identical to sulfated diglycosil diether (S-DGD-1). The DNA G + C content is 61.9 mol% (genome).
The type strain is F15BT (= CECT 9688T = CCM 8954T), isolated from a hypersaline soil of the Odiel Saltmarshes (Huelva, Spain).
The GenBank/EMBL/DDBJ accession number for the 16S rRNA and rpoB′ gene sequences of Halonotius terrestris F15BT are MH465135 and MH454087, respectively, and that of the complete genome is RKLU00000000.
Description of Halonotius roseus sp. nov.
Halonotius roseus (ro’se.us. L. masc. adj. roseus rose colored, pink).
Cells are Gram-stain-negative, non-motile, pleomorphic with 2.8 × 1.1 μm (Supplementary Figure 6). Colonies are circular, entire, red pigmented with 0.1 mm in diameter on R2A25% medium after 10 days of incubation at 37°C. Extremely halophilic, able to grow in media with 15–30% (w/v) salts, with optimal growth at 25% (w/v) salts. No growth occurs in the absence of NaCl. Able to grow in the pH range of 7.0–9.0 and from 25 to 50°C, with optimal growth at pH 7.5 and at 37°C. Catalase and oxidase positive. Gelatin, starch, Tween 80 and esculin are not hydrolyzed. Nitrate is reduced to nitrite, but nitrite is not reduced. Able to produce H2S. Simmons’ citrate, Voges-Proskauer and methyl red tests are negative. Indole is not produced. L-Arginine, L-lysine, and L-ornithine decarboxylase tests are negative. Acid is produced from D-ribose but not from D-amygdalin, D, L-arabinose, arbutin, D-cellobiose, L-citruline, dulcitol, D, L-ethionine D-fructose, D-galactose, glycerol, D-glucose, inulin, lactose, D-maltose, D-mannitol, D-mannose, D-melezitose, D-melibiose, D-raffinose, sorbitol, D-sucrose, D-trehalose, L-xylitol and D-xylose. Sucrose, butanol, tartrate, and D-xylose are used as carbon and energy source but not D-arabinose, D-cellobiose, D-dulcitol, D-ethanol, fructose, galactose, D-glucose, glycerol, lactose, maltose, D-mannitol, D-mannose, D-melezitose, D-melibiose, methanol, L-raffinose, D-ribose, salicin, sorbitol, D-trehalose and xylitol. Not able to use L-arginine, L-cysteine, glutamine, L-methionine, L-glycine, L-lysine, isoleucine and valine as sole carbon, nitrogen and energy source but it uses L-alanine.
The major polar lipids are PG, PGP-Me, PGS and one glycolipid chromatographically identical to sulfated diglycosil diether (S-DGD-1). The DNA G + C content is 62.7 mol% (genome).
The type strain is F9-27T (= CECT 9745T = CCM 8956T), isolated from the water of a pond from Isla Cristina saltern (Huelva, Spain).
The GenBank/EMBL/DDBJ accession number for the 16S rRNA and rpoB’ gene sequences of Halonotius roseus F9-27T are MK478811 and MK491894, respectively, and that of the complete genome is SESI00000000.
Emended Description of the Genus Halonotius Burns et al. (2010)
Characteristics are as given in the description of the genus by Burns et al. (2010). The genus belongs phylogenetically to the family Halorubraceae, within the order Haloferacales, class Halobacteria. The major polar lipids are PG, PGP-Me, PGS and one glycolipid chromatographically identical to sulfated diglycosil diether (S-DGD-1). The DNA G + C content ranges from 59.5 to 62.7 mol% (genome).
Data Availability
The datasets generated for this study can be found in GenBank, RKLU00000000 and SESI00000000.
Author Contributions
AV, CS-P, and RG conceived and designed the study. AD-V, CS-P, and AV designed and performed the acquisition of environmental isolates. AD-V performed the microbial experiments and obtained the genomes. AD-V performed the bioinformatic analyses under close guidance of A-SA and RG. AD-V, A-SA, RG, CS-P, and AV analyzed and interpreted the data. AD-V, CS-P, and AV drafted the manuscript. AD-V, A-SA, RG, CS-P, and AV critically revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by projects from the Spanish Ministry of Economy and Competitiveness (MINECO) (Project CGL2017-83385-P) and from the Junta de Andalucía (BIO-213), both with FEDER funds. AD-V was recipient of a predoctoral fellowship (FPU14/05128) from the Spanish Ministry of Education, Culture and Sports and of a short-stay FEMS research and training grant (FEMS-GO-2018-139). RG was supported by the research grant 17-04828S (Grant Agency of the Czech Republic). A-SA was supported by the research grants 17-04828 S (Grant Agency of the Czech Republic) and MSM200961801 (Academy of Sciences of the Czech Republic).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01928/full#supplementary-material
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Amoozegar, M. A., Siroosi, M., Atashgahi, S., Smidt, H., and Ventosa, A. (2017). Systematics of haloarchaea and biotechnological potential of their hydrolytic enzymes. Microbiology 163, 623–645. doi: 10.1099/mic.0.000463
Anderson, I., Scheuner, C., Göker, M., Mavromatis, K., Hooper, S. D., Porat, I., et al. (2011). Novel insights into the diversity of catabolic metabolism from ten haloarchaeal genomes. PLoS One 6:e20237. doi: 10.1371/journal.pone.0020237
Arahal, D. R., Dewhirst, F. E., Paster, B. J., Volcani, B. E., and Ventosa, A. (1996). Phylogenetic analyses of some extremely halophilic archaea isolated from Dead Sea water, determined on the basis of their 16S rRNA sequences. Appl. Environ. Microbiol. 62, 3779–3786.
Auch, A. F., Klenk, H.-P., and Göker, M. (2010). Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2, 142–148. doi: 10.4056/sigs.541628
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Becker, E. A., Seitzer, P. M., Tritt, A., Larsen, D., Krusor, M., Yao, A. I., et al. (2014). Phylogenetically driven sequencing of extremely halophilic archaea reveals strategies for static and dynamic osmo-response. PLoS Genet. 10:e1004784. doi: 10.1371/journal.pgen.1004784
Bertrand, E. M., Saito, M. A., Jeon, Y. J., and Neilan, B. A. (2011). Vitamin B12 biosynthesis gene diversity in the Ross Sea: the identification of a new group of putative polar B12 biosynthesizers. Environ. Microbiol. 13, 1285–1298. doi: 10.1111/j.1462-2920.2011.02428.x
Bolhuis, H., Palm, P., Wende, A., Falb, M., Rampp, M., Rodriguez-Valera, F., et al. (2006). The genome of the square archaeon Haloquadratum walsbyi: life at the limits of water activity. BMC Genomics 7:169. doi: 10.1186/1471-2164-7-169
Boucher, Y., Douady, C. J., Sharma, A. K., Kamekura, M., and Doolittle, W. F. (2004). Intragenomic heterogeneity and intergenomic recombination among haloarchaeal rRNA genes. J. Bacteriol. 186, 3980–3990. doi: 10.1128/JB.186.12.3980-3990.2004
Burns, D. G., Camakaris, H. M., Janssen, P. H., and Dyall-Smith, M. L. (2004). Combined use of cultivation-dependent and cultivation-independent methods indicates that members of most haloarchaeal groups in an Australian crystallizer pond are cultivable. Appl. Environ. Microbiol. 70, 5258–5265. doi: 10.1128/AEM.70.9.5258-5265.2004
Burns, D. G., Janssen, P. H., Itoh, T., Kamekura, M., Echigo, A., and Dyall-Smith, M. L. (2010). Halonotius pteroides gen. nov., sp. nov., an extremely halophilic archaeon recovered from a saltern crystallizer. Int. J. Syst. Evol. Microbiol. 60, 1196–1199. doi: 10.1099/ijs.0.010017-0
Bushnell, B. (2016). BBMap Project. Available at: http://sourceforge.net/projects/bbmap (accessed October, 2018).
Çınar, S., and Mutlu, M. B. (2016). Comparative analysis of prokaryotic diversity in solar salterns in eastern Anatolia (Turkey). Extremophiles 20, 589–601. doi: 10.1007/s00792-016-0845-7
Corcelli, A., and Lobasso, S. (2006). 25 characterization of lipids of Halophilic Archaea. Methods Microbiol. 35, 585–613. doi: 10.1016/S0580-9517(08)70028-X
Criscuolo, A., and Gribaldo, S. (2010). BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 10:210. doi: 10.1186/1471-2148-10-210
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 5685–5689. doi: 10.1073/PNAS.89.12.5685
Deole, R., Challacombe, J., Raiford, D. W., and Hoff, W. D. (2013). An extremely halophilic proteobacterium combines a highly acidic proteome with a low cytoplasmic potassium content. J. Biol. Chem. 288, 581–588. doi: 10.1074/jbc.M112.420505
Doxey, A. C., Kurtz, D. A., Lynch, M. D., Sauder, L. A., and Neufeld, J. D. (2015). Aquatic metagenomes implicate Thaumarchaeota in global cobalamin production. ISME J. 9, 461–471. doi: 10.1038/ismej.2014.142
Durán-Viseras, A., Ventosa, A., and Sánchez-Porro, C. (2019). Halonotius aquaticus sp. nov., a new haloarchaeon isolated from a marine saltern. Int. J. Syst. Evol. Microbiol. 69, 1306–1312. doi: 10.1099/ijsem.0.003309
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Empadinhas, N., and Da Costa, M. S. (2008). Osmoadaptation mechanisms in prokaryotes: distribution of compatible solutes. Int. Microbiol. 11, 151–161. doi: 10.2436/20.1501.01.55
Falb, M., Müller, K., Königsmaier, L., Oberwinkler, T., Horn, P., von Gronau, S., et al. (2008). Metabolism of halophilic archaea. Extremophiles 12, 177–196. doi: 10.1007/s00792-008-0138-x
Fernández, A. B., Ghai, R., Martín-Cuadrado, A.-B., Sánchez-Porro, C., Rodríguez-Valera, F., and Ventosa, A. (2014a). Prokaryotic taxonomic and metabolic diversity of an intermediate salinity hypersaline habitat assessed by metagenomics. FEMS Microbiol. Ecol. 88, 623–635. doi: 10.1111/1574-6941.12329
Fernández, A. B., Vera-Gargallo, B., Sánchez-Porro, C., Ghai, R., Papke, R. T., Rodríguez-Valera, F., et al. (2014b). Comparison of prokaryotic community structure from Mediterranean and Atlantic saltern concentrator ponds by a metagenomic approach. Front. Microbiol. 5:196. doi: 10.3389/fmicb.2014.00196
Finn, R. D., Clements, J., Arndt, W., Miller, B. L., Wheeler, T. J., Schreiber, F., et al. (2015). HMMER web server: 2015 update. Nucleic Acids Res. 43, W30–W38. doi: 10.1093/nar/gkv397
Frigaard, N.-U., Martinez, A., Mincer, T. J., and DeLong, E. F. (2006). Proteorhodopsin lateral gene transfer between marine planktonic bacteria and Archaea. Nature 439, 847–850. doi: 10.1038/nature04435
Fullmer, M. S., Soucy, S. M., Swithers, K. S., Makkay, A. M., Wheeler, R., Ventosa, A., et al. (2014). Population and genomic analysis of the genus Halorubrum. Front. Microbiol. 5:140. doi: 10.3389/fmicb.2014.00140
Galinski, E. A., and Trüper, H. G. (1994). Microbial behaviour in salt-stressed ecosystems. FEMS Microbiol. Rev. 15, 95–108. doi: 10.1111/j.1574-6976.1994.tb00128.x
Ghai, R., Pašić, L., Fernández, A. B., Martin-Cuadrado, A.-B., Mizuno, C. M., McMahon, K. D., et al. (2011). New abundant microbial groups in aquatic hypersaline environments. Sci. Rep. 1:135. doi: 10.1038/srep00135
Govorunova, E. G., Sineshchekov, O. A., Li, H., and Spudich, J. L. (2017). Microbial rhodopsins: diversity, mechanisms, and optogenetic applications. Annu. Rev. Biochem. 86, 845–872. doi: 10.1146/annurev-biochem-101910-144233
Gunde-Cimerman, N., Plemenitaš, A., and Oren, A. (2018). Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 42, 353–375. doi: 10.1093/femsre/fuy009.Review
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Han, R., Zhang, X., Liu, J., Long, Q., Chen, L., Liu, D., et al. (2017). Microbial community structure and diversity within hypersaline Keke Salt Lake environments. Can. J. Microbiol. 63, 895–908. doi: 10.1139/cjm-2016-0773
Henriet, O., Fourmentin, J., Delincé, B., and Mahillon, J. (2014). Exploring the diversity of extremely halophilic archaea in food-grade salts. Int. J. Food Microbiol. 191, 36–44. doi: 10.1016/J.IJFOODMICRO.2014.08.019
Hyatt, D., LoCascio, P. F., Hauser, L. J., and Uberbacher, E. C. (2012). Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28, 2223–2230. doi: 10.1093/bioinformatics/bts429
Kall, L., Krogh, A., and Sonnhammer, E. L. L. (2007). Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 35, W429–W432. doi: 10.1093/nar/gkm256
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Klappenbach, J. A., Goris, J., Vandamme, P., Coenye, T., Konstantinidis, K. T., and Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Konstantinidis, K. T., Rosselló-Móra, R., and Amann, R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. doi: 10.1038/ismej.2017.113
Konstantinidis, K. T., and Tiedje, J. M. (2004). Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. U.S.A. 101, 3160–3165. doi: 10.1073/pnas.0308653100
Konstantinidis, K. T., and Tiedje, J. M. (2005a). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Konstantinidis, K. T., and Tiedje, J. M. (2005b). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Kushwaha, S. C., Juez-Perez, G., Rodriguez-Valera, F., Kates, M., and Kushner, D. J. (1982). Survey of lipids of a new group of extremely of halophilic bacteria from salt ponds in Spain. Can. J. Microbiol. 28, 1365–1372. doi: 10.1139/m82-203
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
López-Pérez, M., Ghai, R., Leon, M. J., Rodríguez-Olmos, Á, Copa-Patiño, J. L., Soliveri, J., et al. (2013). Genomes of “Spiribacter”, a streamlined, successful halophilic bacterium. BMC Genomics 14:787. doi: 10.1186/1471-2164-14-787
Löytynoja, A. (2014). Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 1079, 155–170. doi: 10.1007/978-1-62703-646-7_10
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Man, D., Wang, W., Sabehi, G., Aravind, L., Post, A. F., Massana, R., et al. (2003). Diversification and spectral tuning in marine proteorhodopsins. EMBO J. 22, 1725–1731. doi: 10.1093/emboj/cdg183
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Mas, A., Jamshidi, S., Lagadeuc, Y., Eveillard, D., and Vandenkoornhuyse, P. (2016). Beyond the Black Queen hypothesis. ISME J. 10, 2085–2091. doi: 10.1038/ismej.2016.22
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14:60. doi: 10.1186/1471-2105-14-60
Mongodin, E. F., Nelson, K. E., Daugherty, S., Deboy, R. T., Wister, J., Khouri, H., et al. (2005). The genome of Salinibacter ruber: converfence and gene exchange among hyperhalophilic bacteria and archaea. Proc. Natl. Acad. Sci. U.S.A. 102, 18147–18152. doi: 10.1073/pnas.0509073102
Moore, S. J., and Warren, M. J. (2012). The anaerobic biosynthesis of vitamin B12. Biochem. Soc. Trans. 40, 581–586. doi: 10.1042/BST20120066
Morris, J. J. (2015). Black Queen evolution: the role of leakiness in structuring microbial communities. Trends Genet. 31, 475–482. doi: 10.1016/j.tig.2015.05.004
Morris, J. J., Lenski, R. E., and Zinser, E. R. (2012). The black queen hypothesis: evolution of dependencies through adaptive gene loss. mBio 3:e00036-12. doi: 10.1128/mBio.00036-12
Morris, M. S. (2012). The role of B vitamins in preventing and treating cognitive impairment and decline. Adv. Nutr. 3, 801–812. doi: 10.3945/an.112.002535
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Oesterhelt, D., and Stoeckenius, W. (1973). Functions of a new photoreceptor membrane. Proc. Natl. Acad. Sci. U.S.A. 70, 2853–2857. doi: 10.1073/PNAS.70.10.2853
Oesterhelt, D., and Stoecknius, W. (1971). Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat. New Biol. 233, 149–152. doi: 10.1038/newbio233149a0
Oren, A. (2011). Thermodynamic limits to microbial life at high salt concentrations. Environ. Microbiol. 13, 1908–1923. doi: 10.1111/j.1462-2920.2010.02365.x
Oren, A. (2013). Life at high salt concentrations, intracellular KCl concentrations, and acidic proteomes. Front. Microbiol. 4:315. doi: 10.3389/fmicb.2013.00315
Oren, A., Ventosa, A., and Grant, W. D. (1997). Proposed minimal standards for description of new taxa in the order Halobacteriales. Int. J. Syst. Bacteriol. 47, 233–238. doi: 10.1099/00207713-47-1-233
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patel, R., Mevada, V., Prajapati, D., Dudhagara, P., Koringa, P., and Joshi, C. G. (2015). Metagenomic sequence of saline desert microbiota from wild ass sanctuary, Little Rann of Kutch, Gujarat, India. Genome Data 3, 137–139. doi: 10.1016/j.gdata.2015.01.003
Pfeiffer, F., Schuster, S. C., Broicher, A., Falb, M., Palm, P., Rodewald, K., et al. (2008). Evolution in the laboratory: the genome of Halobacterium salinarum strain R1 compared to that of strain NRC-1. Genomics 91, 335–346. doi: 10.1016/j.ygeno.2008.01.001
Plominsky, A. M., Delherbe, N., Ugalde, J. A., Allen, E. E., Blanchet, M., Ikeda, P., et al. (2014). Metagenome sequencing of the microbial community of a solar saltern crystallizer pond at Cáhuil lagoon, Chile. Genome Announc. 2:e01172-14. doi: 10.1128/genomeA.01172-14
Podell, S., Emerson, J. B., Jones, C. M., Ugalde, J. A., Welch, S., Heidelberg, K. B., et al. (2014). Seasonal fluctuations in ionic concentrations drive microbial succession in a hypersaline lake community. ISME J. 8, 979–990. doi: 10.1038/ismej.2013.221
Podell, S., Ugalde, J. A., Narasingarao, P., Banfield, J. F., Heidelberg, K. B., and Allen, E. E. (2013). Assembly-driven community genomics of a hypersaline microbial ecosystem. PLoS One 8:e61692. doi: 10.1371/journal.pone.0061692
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Rice, P., Longden, L., and Bleasby, A. (2000). EMBOSS: the european molecular biology open software suite. Trends Genet. 16, 276–277. doi: 10.1016/S0168-9525(00)02024-2
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rodionov, D. A., Vitreschak, A. G., Mironov, A. A., and Gelfand, M. S. (2003). Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J. Biol. Chem. 278, 41148–41159. doi: 10.1074/jbc.M305837200
Saum, S. H., Pfeiffer, F., Palm, P., Rampp, M., Schuster, S. C., Müller, V., et al. (2013). Chloride and organic osmolytes: a hybrid strategy to cope with elevated salinities by the moderately halophilic, chloride-dependent bacterium Halobacillus halophilus. Environ. Microbiol. 15, 1619–1633. doi: 10.1111/j.1462-2920.2012.02770.x
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Stackebrandt, E., and Goebel, B. M. (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Subow, N. N. (1931). Oceanographical Tables. Comissariat of agriculture of USSR. Hydro-Meteorogical Committee of USSR. Moscow: Oceanographical Institute of USSR.
Sun, D.-L., Jiang, X., Wu, Q. L., and Zhou, N.-Y. (2013). Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol. 79, 5962–5969. doi: 10.1128/AEM.01282-13
Talon, R., Coquelle, N., Madern, D., and Girard, E. (2014). An experimental point of view on hydration/solvation in halophilic proteins. Front. Microbiol. 5:66. doi: 10.3389/fmicb.2014.00066
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., et al. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22–28. doi: 10.1093/nar/29.1.22
Torreblanca, M., Rodríguez-Valera, F., Juez, G., Ventosa, A., Kamekura, M., and Kates, M. (1986). Classification of non-alkaliphilic halobacteria based on numerical taxonomy and polar lipid composition and description of Haloarcula gen. nov. and Haloferax gen. nov. Syst. Appl. Microbiol. 8, 89–99. doi: 10.1016/s0723-2020(86)80155-2
Ventosa, A. (2006). “Unusual micro-organisms from unusual habitats: hypersaline environments,” in Prokaryotic Diversity: Mechanisms and Significance: Published for the Society for General Microbiology, eds N. A. Logan, H. M. Lappin-Scott, and P. C. F. Oyston (Cambridge: Cambridge University Press), 223–254. doi: 10.1017/CBO9780511754913.015
Ventosa, A., de la Haba, R. R., Sánchez-Porro, C., and Papke, R. T. (2015). Microbial diversity of hypersaline environments: a metagenomic approach. Curr. Opin. Microbiol. 25, 80–87. doi: 10.1016/J.MIB.2015.05.002
Vera-Gargallo, B., Chowdhury, T. R., Brown, J., Fansler, S. J., Durán-Viseras, A., Sánchez-Porro, et al. (2019). Spatial distribution of prokaryotic communities in hypersaline soils. Sci. Rep. 9:1769. doi: 10.1038/s41598-018-38339-z
Vera-Gargallo, B., Navarro-Sampedro, L., Carballo, M., and Ventosa, A. (2018). Metagenome sequencing of prokaryotic microbiota from two hypersaline soils of the odiel salt marshes in Huelva, Southwester Spain. Genome Announc. 6:e00140-18. doi: 10.1128/genomeA.00140.18
Vera-Gargallo, B., and Ventosa, A. (2018). Metagenomic insights into the phylogenetic and metabolic diversity of the prokaryotic community dwelling in hypersaline soils from the Odiel Saltmarshes (SW Spain). Genes 9:E152. doi: 10.3390/genes9030152
Villarreal-Chiu, J. F., Quinn, J. P., and McGrath, J. W. (2012). The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front. Microbiol. 3:19. doi: 10.3389/fmicb.2012.00019
Warden, A. C., Williams, M., Peat, T. S., Seabrook, S. A., Newman, J., Dojchinov, G., et al. (2015). Rational engineering of a mesohalophilic carbonic anhydrase to an extreme halotolerant biocatalyst. Nat. Commun. 6:10278. doi: 10.1038/ncomms10278
Wood, J. M., Bremer, E., Csonka, L. N., Kraemer, R., Poolman, B., van der Heide, T., et al. (2001). Osmosensing and osmoregulatory compatible solute accumulation by bacteria. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 130, 437–460. doi: 10.1016/S1095-6433(01)00442-1
Woodson, J. D., Peck, R. F., Krebs, M. P., and Escalante-Semerena, J. C. (2003). The cobY gene of the archaeon Halobacterium sp. strain NRC-1 is required for de novo Cobamide synthesis. J. Bacteriol. 185, 311–316. doi: 10.1128/JB.185.1.311-316.2003
Keywords: Halonotius, haloarchaea, comparative genomic analysis, hypersaline environment, Halonotius terrestris sp. nov., Halonotius roseus sp. nov.
Citation: Durán-Viseras A, Andrei A-S, Ghai R, Sánchez-Porro C and Ventosa A (2019) New Halonotius Species Provide Genomics-Based Insights Into Cobalamin Synthesis in Haloarchaea. Front. Microbiol. 10:1928. doi: 10.3389/fmicb.2019.01928
Received: 30 April 2019; Accepted: 05 August 2019;
Published: 27 August 2019.
Edited by:
André Antunes, Edge Hill University, United KingdomReviewed by:
Ramprasad E. V. V., University of Hyderabad, IndiaUri Gophna, Tel Aviv University, Israel
Copyright © 2019 Durán-Viseras, Andrei, Ghai, Sánchez-Porro and Ventosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Sánchez-Porro, sanpor@us.es; Antonio Ventosa, ventosa@us.es