 Kamyar Mogodiniyai Kasmaei
Kamyar Mogodiniyai Kasmaei John Sundh
John Sundh- 1Department of Animal Nutrition and Management, Swedish University of Agricultural Sciences, Uppsala, Sweden
- 2Science for Life Laboratory, Department of Biochemistry and Biophysics, National Bioinformatics Infrastructure Sweden, Stockholm University, Solna, Sweden
Feruloyl esterases (FAEs) can reduce the recalcitrance of lignocellulosic biomass to enzymatic hydrolysis, thereby enhancing biorefinery potentials or animal feeding values of the biomass. In addition, ferulic acid, a product of FAE activity, has applications in pharmaceutical and food/beverage industries. It is therefore of great interest to identify new FAEs to enhance understanding about this enzyme family. For this purpose, we used whole-genome shotgun metagenomics and genome binning to explore rumens of dairy cows, large intestines of horses, sediments of freshwater and forest topsoils to identify novel prokaryotic FAEs and trace the responsible microorganisms. A number of prokaryotic genomes were recovered of which, genomes of Clostridiales order and Candidatus Rhabdochlamydia genus showed FAE coding capacities. In total, five sequences were deemed as putative FAE. The BLASTP search against non-redundant protein database of NCBI indicated that these putative FAEs represented novel sequences within this enzyme family. The phylogenetic analysis showed that at least three putative sequences shared evolutionary lineage with FAEs of type A and thus could possess specific activities similar to this type of FAEs, something that is not previously found outside fungal kingdom. We nominate Candidatus Rhabdochlamydia genus as a novel FAE producing taxonomic unit.
Introduction
Production of biofuels and biochemicals from lignocellulosic biomass, a non-food renewable carbon resource, has increasingly become of great importance due to increasing global demands for energy and chemicals, increasing prices of fossil fuels and environmental concerns associated with fossil fuels. Lignocellulosic biomass mainly comprises three structural polymers namely cellulose, hemicellulose and lignin. In the cell walls of monocots (e.g., grass, cereals), lignin and hemicellulose interconnect, forming a matrix that encrusts the cellulose (Wong, 2006; Rubin, 2008). This configuration creates a complex structure, believed to be the main cause of recalcitrance of lignocellulosic biomass to enzymatic hydrolysis (Rubin, 2008; Pu et al., 2013).
The linkage between lignin and hemicellulose is mainly mediated by ferulic acid (FA), forming ester bonds with hemicellulose from the carboxylic side and ether bonds with lignin from the phenolic side of the molecule. These ester bonds in the cell walls of plants can be cleaved with feruloyl esterases (FAEs) (EC 3.1.1.73), member of carboxylic ester hydrolases (EC 3.1.1.-) (Jeske et al., 2019), to reduce complexity of cell wall configuration, thereby enhancing utilization of lignocellulosic biomass (Wong, 2006). Further importance of FAEs is in pharmaceutical and food/beverage industries as FA, a product of FAE activity, has evidently antioxidant properties (Pohl and Lin, 2018) and can also be used to produce vanillin (Chen et al., 2016). In addition, several attempts have already been made to improve digestibility of forages in dairy cattle rations by use of FAE producing lactic acid bacteria (Muck et al., 2018).
Feruloyl esterases are classified into four types (A, B, C, and D) based on substrate specificity against model methyl esters and release of diferulic acid (5–5’) from plant cell walls (Crepin et al., 2004). The efficiency of FAEs in breaking lignin-hemicellulose interconnections seems to differ among different FAEs. FAEs-A break these interconnections in the cell walls of cereals at higher rates than FAEs-B (Crepin et al., 2004). Based on phylogenetic analysis, fungal FAEs were classified into seven subfamilies (Benoit et al., 2008) but the phylogeny was further improved in a later attempt, with recognition of 13 subfamilies of fungal FAEs (Dilokpimol et al., 2016). These attempts showed that FAEs did not evolve from a common ancestor (Benoit et al., 2008; Dilokpimol et al., 2016). In a novel approach, protein descriptors, derived from amino acid sequences, were used in conjunction with a machine learning method to classify fungal, bacterial and plant FAEs, which resulted in formation of 12 families of FAEs (Udatha et al., 2011). There is to some extent agreement between the A–D classification and 1–13 subfamily classification as for instance subfamilies 6 and 7 solely include FAEs-B and FAEs-A, respectively. However, the subfamily 1 includes both FAEs-B and FAEs-C and subfamily 5 contains FAEs-A and FAEs-D. It appears that the classification of FAEs can further be improved in the near future when more data is available.
Several fungal and bacterial species are known to produce FAEs, including Aspergillus spp., a number of anaerobic fungal species, Bacillus spp., Lactobacillus spp., etc. (Donaghy et al., 1998; Dilokpimol et al., 2016). Due to industrial significance of FAEs, there is an ever-growing interest to identify new FAEs and new microorganisms with this ability. Potential habitats of FAE producing microorganisms are ecosystems in which, plants are degraded, such as digestive tract of herbivores, soil or aquatic ecosystems. The rapid development of sequencing platforms and metagenomic methodologies has enabled to effectively explore these ecosystems for such purpose. In this work, we explored rumens of dairy cows, large intestines of horses, sediments of freshwater and topsoils of forests by means of whole-genome shotgun metagenomics and genome binning to study prokaryotic capacities for FAE production and potential novelty of the predicted FAEs.
Materials and Methods
Sampling
Approximately 50 mL rumen content was sampled from four adult Swedish Red and White breed dairy cows through permanent rumen fistula. Cows had been fed standard diets, containing forage and concentrate, based on their production levels. These cows had been fitted with fistula previously, approved by the Uppsala Ethics Committee (C 93/12 and C 142/14) and were maintained at the Livestock Research Centre of the Swedish University of Agricultural Sciences (SLU) for research/education purposes. Horse fecal samples (ca. 75 g) were directly taken from rectum of four adult horses, fed conventional forage-based diet. These horses were maintained at SLU for research/education purposes approved by the Uppsala Ethics Committee (C 148/13). All the animals used were maintained under SLU policy for use of animals in research and education (SLU.ua 2015.1.1.1-4840). Sediment samples were collected from one stream, one river, one lake and one pond from shallow locations in where, water was still and sediment contained dead plant biomass and thus, sampling locations were considered ecologically similar. Four topsoil samples were obtained from four pine-deciduous forests from locations with decaying plant biomass. All samples were collected in the region of Uppsala, Sweden during spring 2017.
Library Preparation and Sequencing
DNA extraction was done with NucleoSpin® soil (MACHEREY-NAGEL, Düren, Germany). DNA quality and quantity were checked with Agilent 2200 TapeStation System (Agilent, Santa Clara, CA, United States) and Qubit® 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, United States), respectively by the Science for Life Laboratory (SciLifeLab), Uppsala, Sweden. Library preparation was done with TruSeq DNA PCR-Free kit (Illumina, Inc., San Diego, CA, United States) and paired-end sequencing (2 × 125) was performed using Illumina HiSeq2500 system and v4 sequencing chemistry (Illumina, Inc., San Diego, CA, United States) in one lane by the SciLifeLab.
Bioinformatic Analysis
Assembly and Binning
Reads were quality checked with Trimmomatic (Bolger et al., 2014) (vs. 0.36, LEADING:3, TRAILING:3, SLIDINGWINDOW: 4:15, MINLEN: 36) before pooling into four datasets, referred to as Cow, Horse, Sediment, and Soil. The pooled datasets were de novo assembled with Megahit (Li et al., 2015) (vs. v1.1.2, default settings) after which, reads were aligned to contigs ≥1500 bp with bbmap (Bushnell, 2014) (vs. 37.53, default settings). Binning was done with Metabat (Kang et al., 2015) (vs. v2.12.1, minContig: 1500) and bin redundancy was checked by calculating Average Nucleotide Identity (ANI) with FastANI1 (vs. v1.1, default setting). A threshold of 95% ANI was used to merge binned genomes. Completeness and contamination of binned genomes were estimated with CheckM (Parks et al., 2015) (vs. v1.0.11) using lineage specific marker genes. Binned genomes with a contamination <10% and a completeness >30% were selected for downstream analyses.
Annotation
Annotation of recovered genomes was done with Prokka (Seemann, 2014) (vs. 1.12, default settings) and predicted proteins were further annotated with InterProScan (Jones et al., 2014) (vs. 5.30–69.0, default settings). The InterPro database classifies proteins with similar domains/sites into single entries. The IPR011118 family includes FAE of faeB gene (accession ID: AJ309807), tannase and some other proteins. The IPR010126 family contains some lipases, FAE of faeC gene (accession ID: AJ505939) and acetyl xylan esterase. The IPR034429 family comprises FAEs-C and IPR002921 domain corresponds to a domain in FAEs-A, similar to a domain in fungal lipases.
Proteins classified as members of IPR011118, IPR034429, IPR010126 or IPR002921 entries were scanned with ScanProsite (de Castro et al., 2006) to identify sequence motifs. They were further queried with BLASTP (vs. 2.7.1, default settings) against a set of reviewed FAEs of the UniProt database (2019-10-03) (The UniProt Consortium, 2019). The protocol used to select these reference FAEs was: searching for “ec:3.1.1.73” at the UniProt database and filtering by “Reviewed.” This resulted in 44 sequences among which, one sequence was incomplete (UniProt ID: P0CT85) and was thus excluded, resulting in total of 41 fungal sequences and 2 bacterial sequences. In addition, the predicted proteins belonging to the FAE-containing entries of InterPro database were subjected to BLASTP search (vs. 2.7.1, default settings) against non-redundant protein database of NCBI (2019-04-03) to assess their novelty.
Two conditions were opted for annotation as a putative FAE: more than 90% primary sequence similarity to reference FAEs or possession of the serine active site motif2. The consensus pattern of this motif is [LIV]-{KG}-[LIVFY]-[LIVMST]-G-[HYWV]-S-{YAG}-G-[GSTAC], with square and curly brackets indicating acceptable and unacceptable amino acids in the respective positions, respectively. An overview of the annotation protocol is in Figure 1.
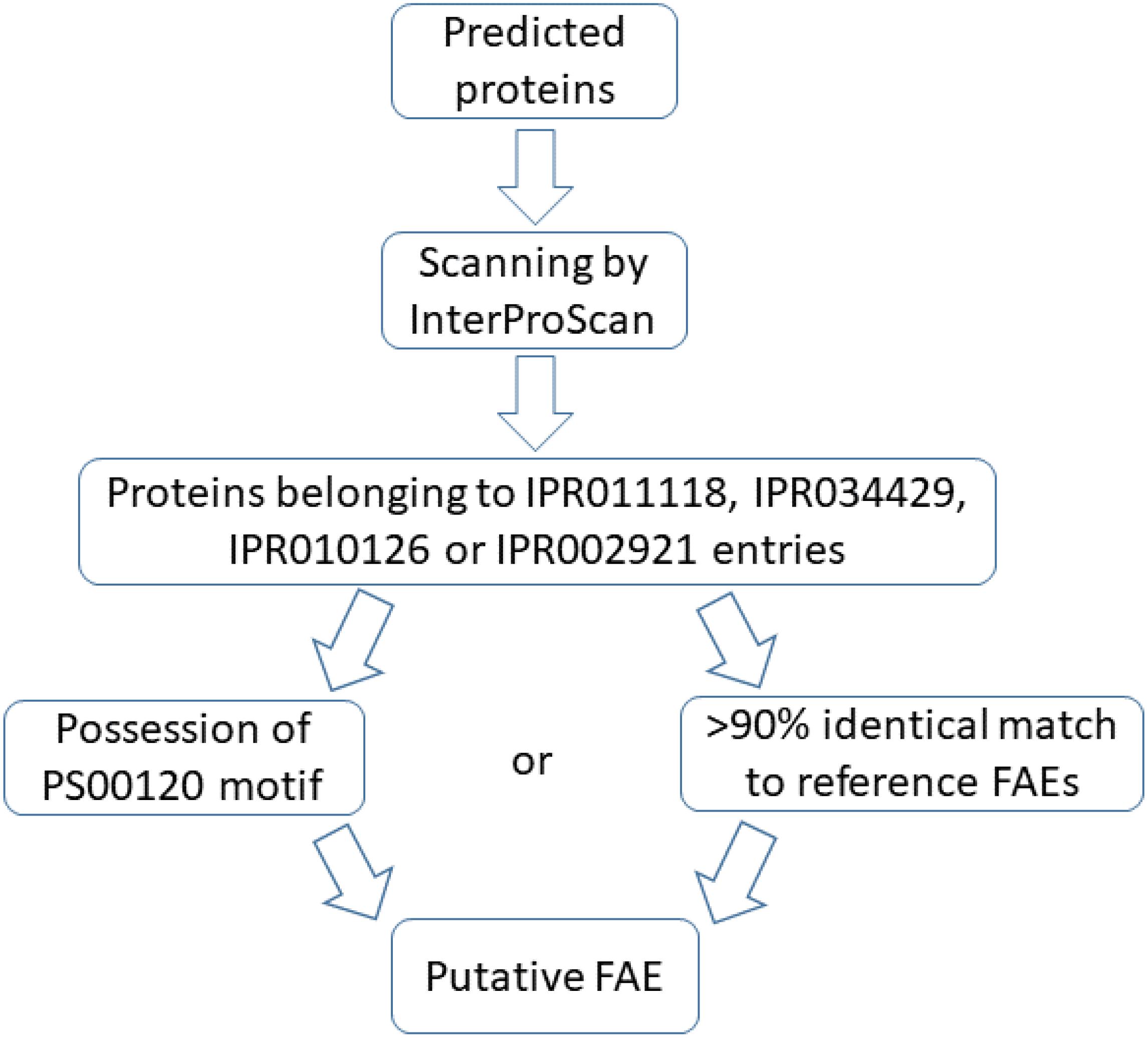
Figure 1. Protocol used to annotate predicted proteins as putative feruloyl esterases (FAEs).
Community Analysis and Taxonomic Classification
Prokaryotic community composition in pooled datasets was estimated by means of taxonomic classification of reads using Kaiju (Menzel et al., 2016) (vs. 1.7.2, default settings) and non-redundant protein database of NCBI (2019-06-25). Recovered genomes were assigned taxon with phylophlan (Segata et al., 2013) (vs. 0.99, default settings), using predicted proteins from Prokka annotation as input. The taxonomic assignments were further evaluated with CheckM and METAXA2 (Bengtsson-Palme et al., 2015) (vs. 2.2 beta 9, default settings). In case of agreement among predictions, the lowest taxonomic rank given by any of the software was reported and in case of disagreement, the lowest common taxonomic rank was assigned.
Phylogenetic Analysis
Signal peptides of putative FAEs were predicted at the SignalP-5.0 server (Almagro Armenteros et al., 2019) and trimmed. The query, reference FAEs (without signal peptides) and three Glucuronoyl esterases (without signal peptides), as an outgroup (Dilokpimol et al., 2016), were aligned with Clustal Omega (Sievers et al., 2011) (vs. 1.2.4) on The European Bioinformatics Institute (EMBL-EBI) Web server (Madeira et al., 2019) using the default settings. Phylogenetic analysis was made with two different methods: maximum likelihood (ML) and neighbor joining (NJ). For the former method, FastTree (Price et al., 2009) (vs. 2.1.10, default settings) was used with 1,000-time resampling and the Shimodaira–Hasegawa test. For the latter, the alignment was first converted to Phylip format on the NGPhylogeny.fr Web server (Lemoine et al., 2019) before using FastME (Lefort et al., 2015) (vs. 2.0) with 1,000-time bootstrapping at the ATGC bioinformatics platform3. Both trees were visualized with ETE toolkit (Huerta-Cepas et al., 2016) (vs. 3.1.1).
Results
Assembly and Binning
The Cow, Horse, Sediment, and Soil datasets had 2 × 85,285,247, 2 × 46,961,631, 2 × 79,657,128, and 2 × 72,876,571 reads, respectively. Assembly statistics is shown in Table 1. The longest contig was assembled in the Soil dataset with a length of 514,904 bp. More contigs were assembled in the Cow and Horse datasets than in the Sediment and Soil datasets and the Sediment dataset had the poorest assembly statistics. Binning resulted in formation of 87, 83, 15, and 10 binned genomes in the Cow, Horse, Sediment, and Soil datasets, respectively. There was no genome redundancy based on ANI. For downstream analyses, 31, 44, 7, and 6 genomes were selected from the Cow, Horse, Sediment, and Soil datasets, respectively (Supplementary Table S1).
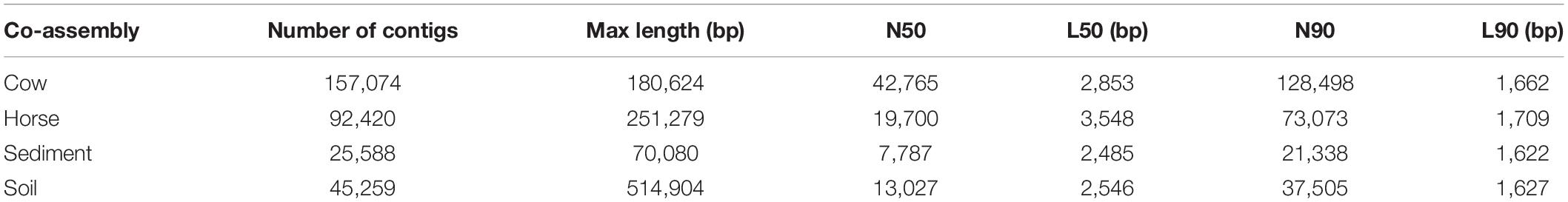
Table 1. Assembly statistics of Cow, Horse, Sediment, and Soil datasets.
Community Composition and Recovered Taxa
The prokaryotic community composition was notably similar between the Cow and Horse samples, with the dominance of Prevotellaceae, Lachnospiraceae, Ruminococcaceae, and Clostridiaceae (Figure 2). There were also similarities between the Sediment and Soil samples, with the community comprising a wide range of taxa from Acidobacteria and Actinobacteria to Alphaproteobacteria and Betaproteobacteria (Figure 2). Viruses and archaea accounted for a combined total of at most 1.2% in each sample (data not shown).
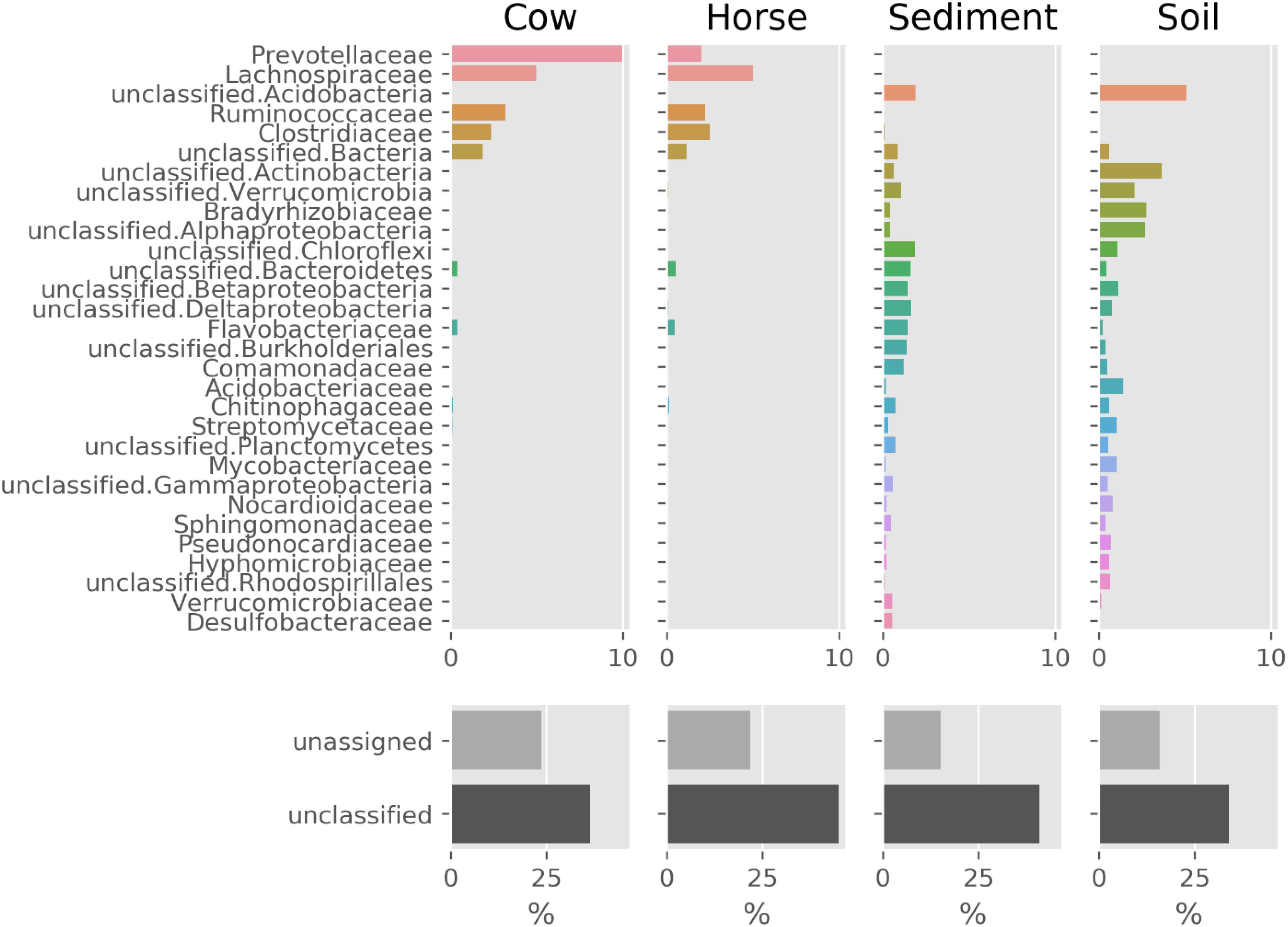
Figure 2. Prokaryotic composition (family level) of Cow, Horse, Sediment, and Soil ecosystems based on classification of reads by Kaiju. Families shown make up at least 75% of the community (together with the unclassified and unassigned sequences). “unclassified” sequences have no classification by Kaiju and “unassigned” sequences are those not assigned to a non-viral species.
In the Cow and Horse datasets, high (≥90% completeness) and/or low (<90% completeness) quality genomes of Lachnospiraceae family (e.g., Cow.1, Cow.7, Horse.8, and Horse.31), within the Clostridiales order, and Prevotellaceae family (e.g., Cow.2, Cow.19, Horse.5, and Horse.40), within the Bacteroidales order, were frequently recovered (Supplementary Table S1). High and/or low quality genomes of Ruminococcaceae family, within the Clostridiales order, were also frequent in the Horse dataset (Horse.7, Horse.14, and Horse.24). In the Sediment and Soil datasets, high and/or low quality genomes of bacteria typically inhabiting fresh water (e.g., Sediment.1) and soil (e.g., Soil.1 and Soil.4) were reconstructed. Archaeal genomes of Euryarchaeota phylum (Cow.14) and Methanomicrobiales order (Horse.32) were also partially reconstructed (Supplementary Table S1). An overview of taxa recovered from different datasets is in Figure 3.
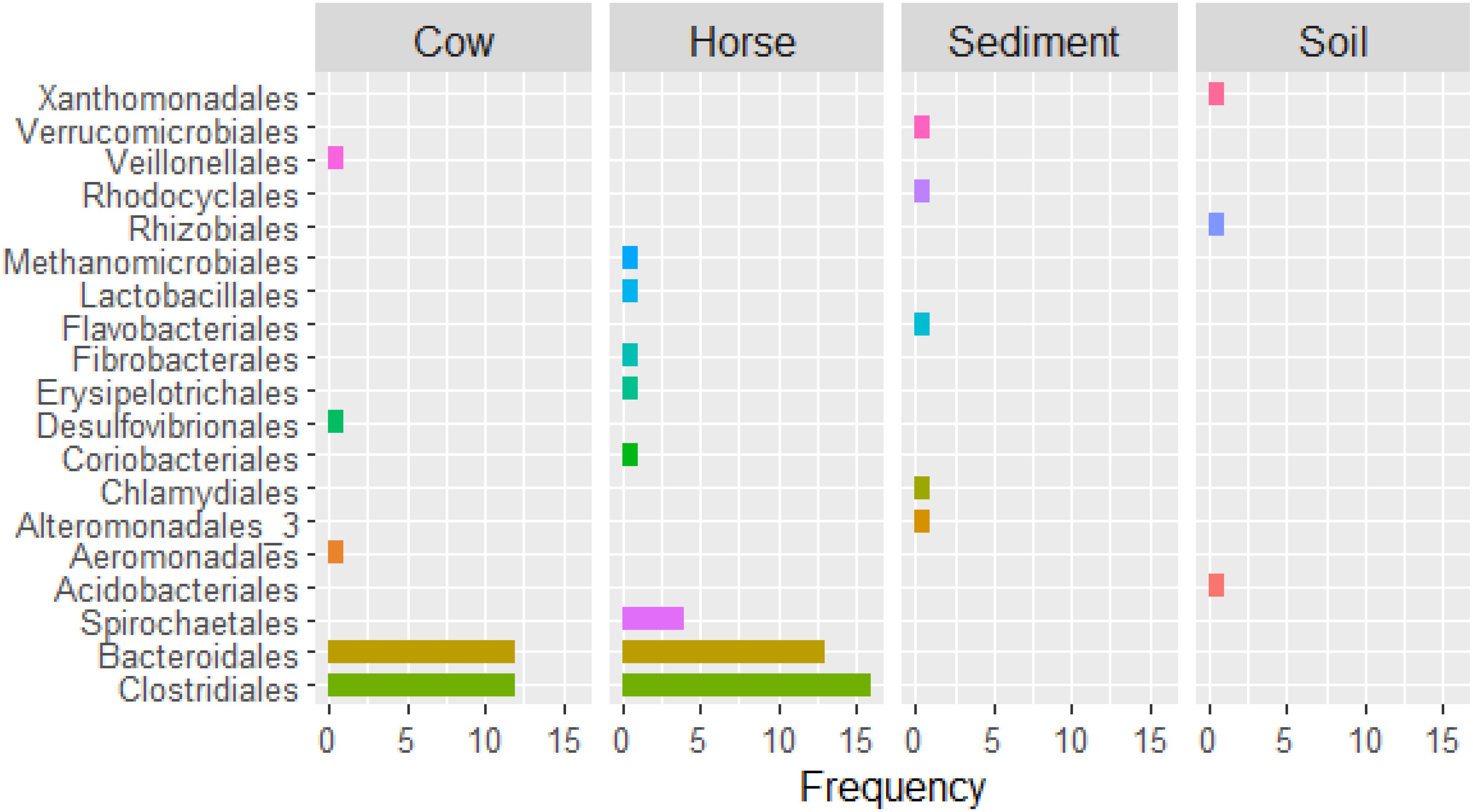
Figure 3. Number of binned genomes per taxon (order level) recovered from Cow, Horse, Sediment, and Soil datasets.
Annotation and Phylogenetic Relationship
In total, 35 hypothetical proteins were classified as members of IPR011118, IPR010126, IPR002921 entries (Table 2). None of the predicted proteins belonged to the IPR034429 entry. The BLASTP bitscores against reference FAEs were generally low, as were the sequence identities which ranged from 21 to 46% (Table 2). In the BLASTP search against the full non-redundant protein database, the scores were higher, with the sequence identities ranging from 27 to 100%. One of the predicted proteins (Horse.16: FOA763) identically matched to a bacterial FAE and two others (Horse.14: NAH160; Soil.2: DAH257) had slight similarities (32 and 50%, respectively) to bacterial FAEs. Five proteins of IPR002921 entry contained the serine active site motif and were thus considered as putative FAEs (Table 3). Taxonomic classifications of binned genomes with FAE coding capacities are in Table 4. Genomes of the Clostridiales order in the Cow and Horse datasets coded for FAEs, as did a genome of Candidatus Rhabdochlamydia genus in the Sediment dataset.
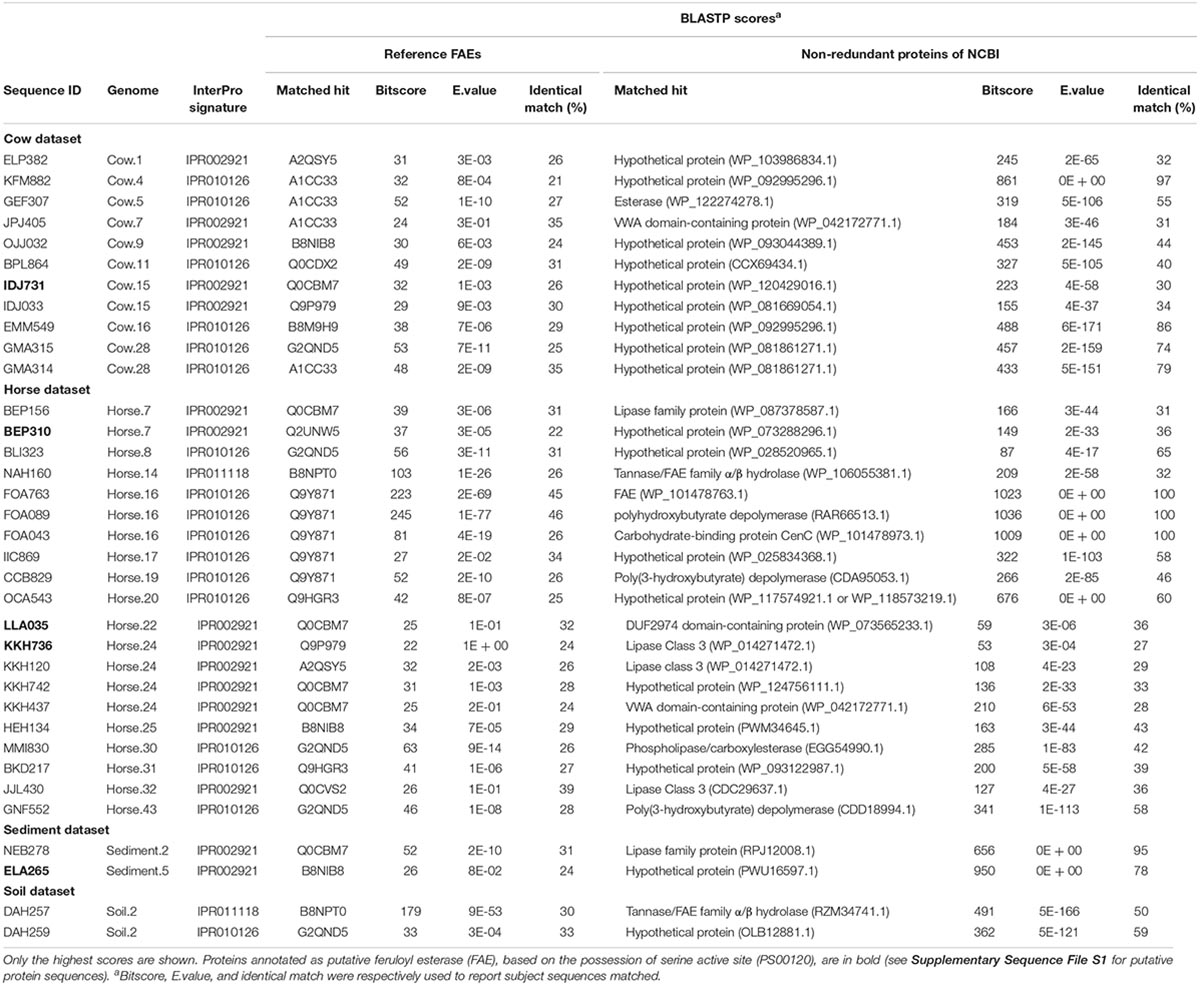
Table 2. BLASTP scores of putative proteins annotated as members of IPR011118, IPR010126, and IPR002921 entries of InterPro database.
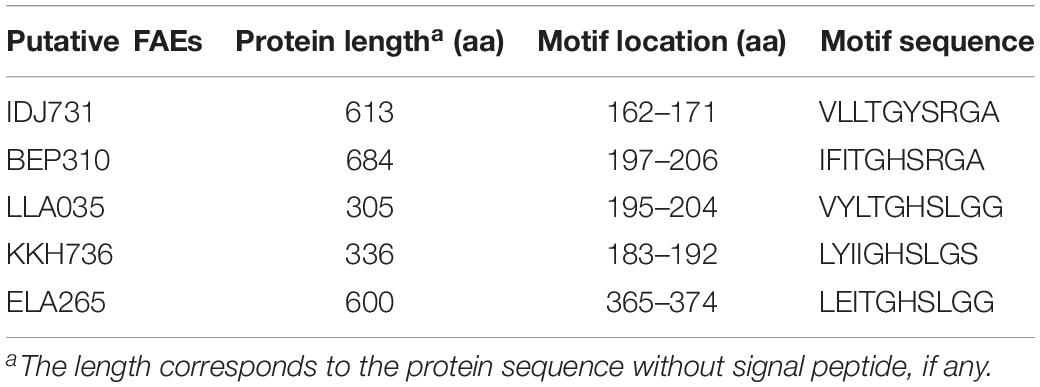
Table 3. Location of the serine active site motif (PS00120) in putative feruloyl esterases (FAEs) (see Supplementary Sequence File S1 for complete sequences).
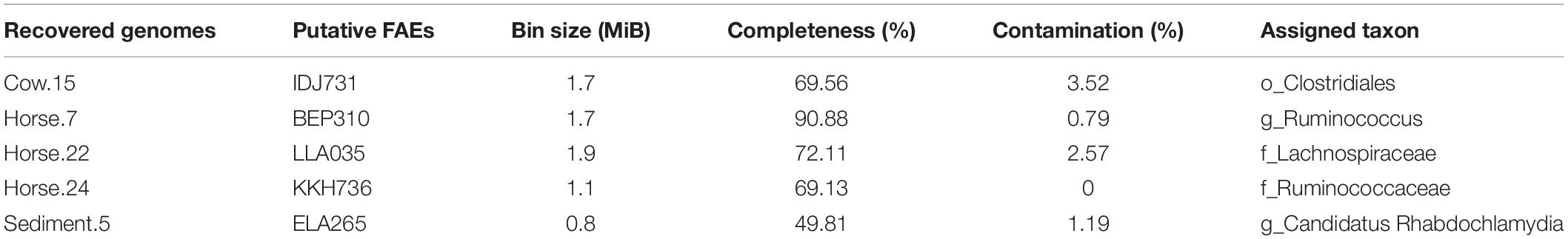
Table 4. Taxonomic classification of binned genomes with feruloyl esterase (FAE) coding capacities.
Overall, the topologies of ML (Figure 4A) and NJ (Figure 4B) trees were similar with formation of three main clades. In the ML tree, the putative FAEs and FAEs-A formed a clade that had a moderate support (0.443). The other two clades had high supports and collectively included FAEs-B, FAEs-C and a FAE-D (Q7RWX8) that was included in our reference dataset. The main difference in the NJ tree was that the putative FAEs of Horse.7 and Horse.24 were not placed with the other putative sequences and FAEs-A in one clade but were placed basal to the other two main clades.

Figure 4. Phylogenetic relationships of feruloyl esterases (FAEs) predicted in this study (in bold) with reference FAEs, using maximum likelihood (A) and neighbor joining (B) methods. Confidence values in panels (A) and (B) represent Shimodaira–Hasegawa support values (1000 resampling) and bootstrap values (1000 bootstraps), respectively. Leaf nodes in green are the outgroup. Leaf naming is “<species> (<UniProt ID>)” for the reference/outgroup sequences and “<bin id><predicted taxon> (<protein id>)” for the putative sequences.
Discussion
In this study, we used whole-genome shotgun metagenomics combined with de novo assembly and genome binning to study prokaryotic FAEs of anaerobic (cow rumen, large intestine of horse and sediment of fresh water) and microaerobic (topsoil) ecosystems. The lower assembly quality in the Sediment and Soil datasets (Table 1) suggests that there was insufficient coverage of microbial genomes in these samples, likely due to a high microbial diversity in these two ecosystems, something evident from Figure 2.
Although several proteins from the binned genomes matched the FAE-containing entries of the InterPro database, these proteins showed very low primary sequence similarities to our reference FAEs. We therefore, explored the reference FAEs to identify sequence motifs of this enzyme family to enable a functional annotation of our predicted proteins. Surprisingly, only FAEs-A consistently contained a motif, i.e., the serine active site (PS00120), a signature of some lipases4. It was previously reported that FAEs-A have sequence similarities to lipases (Crepin et al., 2004) and therefore, this finding may not be entirely unexpected. The PS00120 motif is also detected in FAEs of Lactobacillus spp. (Xu et al., 2017). Our finding here indicates that attempts should be made to identify sequence features unique to FAEs to facilitate functional annotation of novel FAEs.
The PS00120 motif was only found in the protein sequences classified as members of IPR002921 domain but not in all of them (only 5 out of 16 sequences). As the IPR002921 entry describes a domain in FAEs-A, we considered these five sequences as putative FAE. The results from BLASTP search against non-redundant proteins (Table 2) suggest that these putative FAEs represent novel sequences in this enzyme family.
Species belonging to the Clostridiales order were previously reported to produce FAEs, including Butyrivibrio fibrisolvens (Dalrymple et al., 1996), B. proteoclasticus (Goldstone et al., 2010), Ruminococcus albus, and R. flavefaciens (McSweeney et al., 1998). To the best of our knowledge, this is the first report about FAE coding capacity within the Candidatus Rhabdochlamydia genus. It should be pointed out that although the Sediment dataset was constructed from a mix of different sources, the genome quality analysis showed that the considered genome had a very low contamination (∼1%, Table 4), indicating that our sampling strategy was adequate.
The association between specific activities of FAEs, summarized as the A–D classification scheme (Crepin et al., 2004), and phylogenetic relationships of these enzymes is not straightforward (Dilokpimol et al., 2016), something also evident from our phylogenetic analysis. In both ML and NJ trees, two clusters were formed with each comprising FAEs of mixed specific activities. It is possible that ecological niches and specific needs of individual species largely determine the specific activity of FAEs, also pointed out by Benoit et al. (2008). This was however not the case for the FAEs-A, as in both trees the FAEs-A formed a distinct cluster, not showing close evolutionary relationships with other types of FAEs. The different evolutionary lineage of FAEs-A is further evidenced from that the PS00120 motif was only found in this type of FAEs. Interestingly, three putative sequences were consistently clustered with FAEs-A in both phylogenetic trees, suggesting that these putative FAEs may have specific activities similar to this type of FAEs, something that should be verified experimentally. Production of FAEs-A is until now only found in fungi and in particular in Aspergillus spp.
Conclusion
In total, 31, 44, 7, and 6 prokaryotic genomes were reconstructed from the Cow, Horse, Sediment, and Soil datasets, respectively, and were explored for FAE coding capacities. Four genomes of Clostridiales order in the Cow and Horse datasets and one genome of Candidatus Rhabdochlamydia genus in the Sediment dataset were found to have such capacity. In total, five FAEs were predicted. The results from BLASTP against non-redundant protein database of NCBI suggested that these putative FAEs are novel. Phylogenetic analysis suggested that at least three putative sequences might have specific activities similar to FAEs-A.
Data Availability Statement
Raw data is deposited at the Sequence Read Archive database under PRJNA543979 accession number.
Author Contributions
KM designed the study and did the sampling, DNA extraction, bioinformatic analyses and preparation of the first draft of the manuscript. JS provided bioinformatic expertise and contributed to the manuscript preparation.
Funding
This study was partially supported by the Swedish Research Council Formas under project registration number: 2016-01449. JS was supported by the Knut and Alice Wallenberg Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Prof. Anna Schnürer from the SLU, Sweden, and Dr. Sabine Kleinsteuber from the Helmholtz Centre for Environmental Research–UFZ, Germany, for their valuable inputs on experimental setup. Sequencing was performed by the SNP and SEQ Technology Platform in Uppsala. The facility is part of the National Genomics Infrastructure (NGI) Sweden and SciLifeLab. The SNP and SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation. The computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under Project SNIC 2017/7-306.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02673/full#supplementary-material
TABLE S1 | Genome quality and taxonomic classifications of recovered genomes searched for feruloyl esterase coding capacities.
SEQUENCE FILE S1 | Putative protein sequences annotated as members of IPR011118, IPR010126, and IPR002921 entries of InterPro database. Sequences annotated as putative feruloyl esterases are IDJ731 (1–30), BEP310 (1–25), LLA035, KKH736 (1–22), and ELA265, with location of predicted signal peptides, if any, in parentheses.
Footnotes
- ^ https://github.com/ParBLiSS/FastANI
- ^ https://prosite.expasy.org/PS00120
- ^ http://www.atgc-montpellier.fr/
- ^ https://prosite.expasy.org/PDOC00110
References
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Bengtsson-Palme, J., Hartmann, M., Eriksson, K. M., Pal, C., Thorell, K., Larsson, D. G. J., et al. (2015). METAXA2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Res. 15, 1403–1414. doi: 10.1111/1755-0998.12399
Benoit, I., Danchin, E. G. J., Bleichrodt, R. J., and de Vries, R. P. (2008). Biotechnological applications and potential of fungal feruloyl esterases based on prevalence, classification and biochemical diversity. Biotechnol. Lett. 30, 387–396. doi: 10.1007/s10529-007-9564-6
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bushnell, B. (2014). “BBMap: a fast, accurate, splice-aware aligner,” in Proceedings of the 9th Annual Genomics of Energy & Environment Meeting, (Walnut Creek, CA).
Chen, P., Yan, L., Wu, Z., Li, S., Bai, Z., Yan, X., et al. (2016). A microbial transformation using Bacillus subtilis B7-S to produce natural vanillin from ferulic acid. Sci. Rep. 6:20400. doi: 10.1038/srep20400
Crepin, V. F., Faulds, C. B., and Connerton, I. F. (2004). Functional classification of the microbial feruloyl esterases. Appl. Microbiol. Biotechnol. 63, 647–652. doi: 10.1007/s00253-003-1476-3
Dalrymple, B. P., Swadling, Y., Cybinski, D. H., and Xue, G. P. (1996). Cloning of a gene encoding cinnamoyl ester hydrolase from the ruminal bacterium Butyrivibrio fibrisolvens E14 by a novel method. FEMS Microbiol. Lett. 143, 115–120. doi: 10.1016/0378-1097(96)00306-0
de Castro, E., Sigrist, C. J. A., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365.
Dilokpimol, A., Mäkelä, M. R., Aguilar-Pontes, M. V., Benoit-Gelber, I., Hildén, K. S., and de Vries, R. P. (2016). Diversity of fungal feruloyl esterases: updated phylogenetic classification, properties, and industrial applications. Biotechnol. Biofuels 9:231.
Donaghy, J., Kelly, P. F., and Mckay, A. M. (1998). Detection of ferulic acid esterase production by Bacillus spp. and Lactobacilli. Appl. Microbiol. Biotechnol. 50, 257–260. doi: 10.1007/s002530051286
Goldstone, D. C., Villas-Bôas, S. G., Till, M., Kelly, W. J., Attwood, G. T., and Arcus, V. L. (2010). Structural and functional characterization of a promiscuous feruloyl esterase (Est1E) from the rumen bacterium Butyrivibrio proteoclasticus. Proteins 78, 1457–1469. doi: 10.1002/prot.22662
Huerta-Cepas, J., Serra, F., and Bork, P. (2016). ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638. doi: 10.1093/molbev/msw046
Jeske, L., Placzek, S., Schomburg, I., Chang, A., and Schomburg, D. (2019). BRENDA in 2019: a European ELIXIR core data resource. Nucleic Acids Res. 47, D542–D549. doi: 10.1093/nar/gky1048
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Lefort, V., Desper, R., and Gascuel, O. (2015). FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. doi: 10.1093/molbev/msv150
Lemoine, F., Correia, D., Lefort, V., Doppelt-Azeroual, O., Mareuil, F., Cohen-Boulakia, S., et al. (2019). NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res. 47, W260–W265. doi: 10.1093/nar/gkz303
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, w636–w641. doi: 10.1093/nar/gkz268
McSweeney, C. S., Dulieu, A., and Bunch, R. (1998). Butyrivibrio spp. and other xylanolytic microorganisms from the rumen have cinnamoyl esterase activity. Anaerobe 4, 57–65. doi: 10.1006/anae.1997.0130
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7:11257. doi: 10.1038/ncomms11257
Muck, R. E., Nadeau, E. M. G., McAllister, T. A., Contreras-Govea, F. E., Santos, M. C., and Kung, L. Jr. (2018). Silage review: recent advances and future uses of silage additives. J. Dairy Sci. 101, 3980–4000. doi: 10.3168/jds.2017-13839
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pohl, F., and Lin, P. K. T. (2018). The potential use of plant natural products and plant extracts with antioxidant properties for the prevention/treatment of neurodegenerative diseases: In Vitro, In Vivo and clinical trials. Molecules 23:3283. doi: 10.3390/molecules23123283
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Pu, Y., Hu, F., Huang, F., Davison, B. H., and Ragauskas, A. J. (2013). Assessing the molecular structure basis for biomass recalcitrance during dilute acid and hydrothermal pretreatments. Biotechnol. Biofuels 6:15. doi: 10.1186/1754-6834-6-15
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Segata, N., Börnigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4:2304. doi: 10.1038/ncomms3304
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
The UniProt Consortium (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. doi: 10.1093/nar/gky1049
Udatha, D. B., Kouskoumvekaki, I., Olsson, L., and Panagiotou, G. (2011). The interplay of descriptor-based computational analysis with pharmacophore modeling builds the basis for a novel classification scheme for feruloyl esterases. Biotechnol. Adv. 29, 94–110. doi: 10.1016/j.biotechadv.2010.09.003
Wong, D. W. S. (2006). Feruloyl Esterase - a key enzyme in biomass degradation. Appl. Biochem. Biotechnol. 133, 87–112. doi: 10.1385/abab:133:2:87
Keywords: biorefinery, de novo assembly, lignocellulosic biomass, phylogenetic analysis, sequence motif, taxonomic classification
Citation: Mogodiniyai Kasmaei K and Sundh J (2019) Identification of Novel Putative Bacterial Feruloyl Esterases From Anaerobic Ecosystems by Use of Whole-Genome Shotgun Metagenomics and Genome Binning. Front. Microbiol. 10:2673. doi: 10.3389/fmicb.2019.02673
Received: 24 June 2019; Accepted: 04 November 2019;
Published: 20 November 2019.
Edited by:
Daniel Yero, Autonomous University of Barcelona, SpainReviewed by:
Zhihong Xin, Nanjing Agricultural University, ChinaAdiphol Dilokpimol, Westerdijk Fungal Biodiversity Institute, Netherlands
Copyright © 2019 Mogodiniyai Kasmaei and Sundh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kamyar Mogodiniyai Kasmaei, kamyar.mogodiniyai.kasmaei@slu.se