 Anny Camargo1,2
Anny Camargo1,2 Enzo Guerrero-Araya3
Enzo Guerrero-Araya3 Sergio Castañeda1
Sergio Castañeda1 Laura Vega1
Laura Vega1 María X. Cardenas-Alvarez1,4
María X. Cardenas-Alvarez1,4 César Rodríguez5
César Rodríguez5 Daniel Paredes-Sabja3,6
Daniel Paredes-Sabja3,6 Juan David Ramírez1,7
Juan David Ramírez1,7 Marina Muñoz1,3*
Marina Muñoz1,3*- 1Centro de Investigaciones en Microbiología y Biotecnología-UR (CIMBIUR), Facultad de Ciencias Naturales, Universidad del Rosario, Bogotá, Colombia
- 2Faculty of Health Sciences, Universidad de Boyacá, Tunja, Colombia
- 3ANID, Millennium Science Initiative Program, Millennium Nucleus in the Biology of the Intestinal Microbiota, Santiago, Chile
- 4Department of Pharmacology, University of North Carolina, Chapel Hill, NC, United States
- 5Laboratorio de Investigación en Bacteriología Anaerobia, Facultad de Microbiología, Centro de Investigación en Enfermedades Tropicales, Universidad de Costa Rica, San José, Costa Rica
- 6Department of Biology, Texas A&M University, College Station, TX, United States
- 7Molecular Microbiology Laboratory, Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Clostridium perfringens is the causative agent of many enterotoxic diseases in humans and animals, and it is present in diverse environments (soil, food, sewage, and water). Multilocus Sequence Typing (MLST) and Whole Genome Sequencing (WGS) have provided a general approach about genetic diversity of C. perfringens; however, those studies are limited to specific locations and often include a reduced number of genomes. In this study, 372 C. perfringens genomes from multiple locations and sources were used to assess the genetic diversity and phylogenetic relatedness of this pathogen. In silico MLST was used for typing the isolates, and the resulting sequence types (ST) were assigned to clonal complexes (CC) based on allelic profiles that differ from its founder by up to double-locus variants. A pangenome analysis was conducted, and a core genome-based phylogenetic tree was created to define phylogenetic groups. Additionally, key virulence factors, toxinotypes, and antibiotic resistance genes were identified using ABRicate against Virulence Factor Database (VFDB), TOXiper, and Resfinder, respectively. The majority of the C. perfringens genomes found in publicly available databases were derived from food (n = 85) and bird (n = 85) isolates. A total of 195 STs, some of them shared between sources such as food and human, horses and dogs, and environment and birds, were grouped in 25 CC and distributed along five phylogenetic groups. Fifty-three percent of the genomes were allocated to toxinotype A, followed by F (32%) and G (7%). The most frequently found virulence factors based on > 70% coverage and 99.95% identity were plc (100%), nanH (99%), ccp (99%), and colA (98%), which encode an alpha-toxin, a sialidase, an alpha-clostripain, and a collagenase, respectively, while tetA (39.5%) and tetB (36.2%), which mediate tetracycline resistance determinants, were the most common antibiotic resistance genes detected. The analyses conducted here showed a better view of the presence of this pathogen across several host species. They also confirm that the genetic diversity of C. perfringens is based on a large number of virulence factors that vary among phylogroups, and antibiotic resistance markers, especially to tetracyclines, aminoglycosides, and macrolides. Those characteristics highlight the importance of C. perfringens as a one of the most common causes of foodborne illness.
Introduction
Clostridium perfringens is an anaerobic, Gram-positive, spore-forming bacillus capable of surviving extreme conditions such as high temperature (> 60°C) and low nutrient levels (Hassan et al., 2015). In 2002, the first C. perfringens genome was sequenced showing 2,660 protein-codifying regions, ten rRNA genes, and a low G + C content (28.6%; Shimizu et al., 2002). Since then, many studies have revealed the presence of genes that codify to multiple virulence factors involved in the pathogenicity of this species (Li and BA, 2014; Revitt-Mills et al., 2015; Kiu et al., 2017; Abdel-Glil et al., 2021), such as the alpha (CPA), beta (CPB), epsilon (ETX), iota (ITX), enterotoxin (CPE), and necrotic B-like (NetB) toxins (Awad et al., 1995; Sarker et al., 1999; Keyburn et al., 2008; Garcia et al., 2013; Rood et al., 2018), that contribute to neurologic, histotoxic, and intestinal infections in animals and humans and are used to classify strains in seven different toxin types (A to G; Kiu and Lindsay, 2018).
Despite the usefulness of toxin typing in epidemiology and diagnosis, Multilocus Sequence Typing (MLST) has been implemented as an alternative approach for C. perfringens typing (Chalmers et al., 2008; Deguchi et al., 2009; Verma et al., 2020). This method is based on the presence and combination of internal fragments of eight housekeeping genes, which in C. perfringens includes colA, groEL, sodA, plc, gyrB, sigK, pgk, and nadA. The sequences are assigned as distinct alleles creating a unique allelic profile or sequence type (ST). Using the goeBURST algorithm, the STs are classified into groups of genetically related organisms called clonal complexes (CC), which provide insights about evolution and diversification processes and allow intra-species analyses (Larsen et al., 2012; Uzal et al., 2014; Page et al., 2017) such as epidemiological and phylogenetical associations (Maiden, 2006). In addition, the identification of shared STs allows the assessment of possible transmission routes between hosts, a third shared source, and genetic stability in the lineage (Xiao et al., 2012).
Other methods, like Whole Genome Sequencing (WGS) have enabled a better understanding of bacterial pathogens through a high-resolution characterization of their genetic variation and evolution (Raskin et al., 2006). Recently, a genomic analysis allocated 206 publicly available C. perfringens genomes into five phylogroups (I–V) linked to different disease outcomes and hosts. Phylogroup I included human food poisoning strains and phylogroup II mostly grouped isolates from enteric lesions in horses and dogs. Phylogroup III was the most abundant and heterogeneous group, containing a variety of strains from different hosts causing multiple diseases, while phylogroups IV and V were less abundant (Abdel-Glil et al., 2021). Additionally, the use of WGS has allowed the identification of virulence factors such as sialidases and hyaluronidases along with other toxins associated with clinical outcomes (Kiu et al., 2017; Abdel-Glil et al., 2021). Furthermore, over the last few years molecular markers linked to antibiotic resistance to tetracycline, rifamycin, and aminoglycoside among others, have been recognized as a potential risk for treatment of the infections caused by C. perfringens (Kiu and Lindsay, 2018).
So far, C. perfringens genomic studies have been limited to a few geographic locations or to a small number of genomes. Hence, to better understand the global diversity of C. perfringens, we conducted a comparative genome analysis of 372 genomes from multiple locations and sources. Our goal was to determine the intra-species diversity and phylogenetic relationships of C. perfringens, as well as to identify and characterize key molecular markers associated with its pathogenicity, virulence, and antibiotic resistance from whole genome analyses.
Materials and methods
Strain and genome collection
A total of 372 C. perfringens genomes were included in our analysis. Two strains sequenced specifically for this study were recovered from human feces and water, in 2013 and 2019, respectively. Briefly, samples were collected in sterile screw cap containers to avoid direct oxygen exposure (Siah et al., 2014) and were grown on tryptose sulfite cycloserine (TSC) agar (Merck) under anaerobic conditions by using anaerobic jars with anaerobic atmosphere generation pouches (AnaeroGen, Thermo Scientific, Oxoid) at 37°C for 24 h. Genomic DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega). The quantity of the extracted DNA was assessed using a Qubit 2.0 Fluorometer. DNA library preparation and paired-end whole genome sequencing (2 × 150 bp) were conducted on the Illumina NextSeq platform 2000 at the Microbial Genome Sequencing Center (MiGS). The assembled genomes were deposited at DDBJ/ENA/GenBank as part of a Bioproject under the accession number PRJNA836622.
Publicly available sequence data of 370 C. perfringens genomes was also included in our study for comparative analyses. These data comprised 197 genome assemblies from the Pathosystems Resource Integration Center (PATRIC; Wattam et al., 2014; accessed on May 20th, 2021), 40 assemblies from the Public databases for molecular typing and microbial genome diversity (PubMLST; Jolley et al., 2018; accessed on May 14th, 2021), and 133 reads that were retrieved from the European Nucleotide Archive (ENA) database1 using the keywords “C. perfringens” (accessed on Jun 20th, 2021). Metadata and access numbers were verified to avoid duplicates (Supplementary Table S1).
Taxonomic classification, genome assembly and annotation
Quality stats of reads were collected using FastQC.2 The 133 raw read pairs from ENA were processed with Trimmomatic v. 0.38 to remove low-quality bases below Phred 30 and adapter sequences (Bolger et al., 2014). De novo assembly was conducted using SPAdes v. 3.14.1(Bankevich et al., 2012) with the default settings. The quality of the resulting assemblies was assessed with Quast v5.0.2 (Gurevich et al., 2013).
The taxonomic classification of the assemblies was verified using Kraken v1.1.1 (Wood and Salzberg, 2014) with default parameters and using standard databases. Additionally, average nucleotide identity (ANI; Richter and Rossello-Mora, 2009) was calculated with pyANI.3 An average identity percentage above 95% denotes strains that belong to the same species. C. perfringens ATCC 13124 was used as a reference strain for this analysis. The assemblies generated by SPAdes and the ones obtained from public databases were annotated with PROKKA v. 1.11 (Seemann, 2014) with the default parameters and using the UniprotKB (SwissProt) database (Research UCJNa, 2017), considering kingdom specific databases for Bacteria. Annotated assemblies were taken to calculate the pangenome using Roary (Page et al., 2015). Genes present in 95% of the genomes with at least 95% of identity were designated as core genes.
In silico MLST assignment
To assign isolates to sequence type (ST), in silico MLST was performed using FastMLST v0.0.144 (Guerrero-Araya et al., 2020) with the default parameters. Concatenated sequences of the eight housekeeping genes of the scheme were extracted (colA, groEL, sodA, plc, gyrB, sigK, pgk, and nadA) and novel STs were submitted to the PubMLST database for identification (accessed on May 14th, 2021). Global optimal eBURST (goeBURST)5 was used to visualize CC, defined in this study as closely related STs that differ from a common founder at up to two of the eight loci used in the MLST scheme (DLV, double locus variant; Francisco et al., 2012).
Phylogenetic reconstruction based on MLST and core genome gene sequences
Concatenated sequences of the eight housekeeping genes belonging to the MLST scheme were used as input for a multiple alignment. Likewise, core genes were concatenated and aligned using MAFFT v7.407. The best-fit model for base substitution in IQ-TREE v2.0.3 was selected to infer phylogenetic relationships by maximum likelihood (ML) and ultrafast bootstrap (1,000 repetitions; Nguyen et al., 2015). A clade was considered well supported when the bootstrap was ≥ 80%, as previously indicated (Wróbel, 2008; Minh et al., 2013). The resulting trees were visualized and edited using iTOL v.5 (Letunic and Bork, 2021). To confirm the phylogenetic clades generated by IQ-TREE, a NeighbourNet network was reconstructed in SplitsTree4 software v4.17.0 (Huson and Bryant, 2006) from a core genome alignment and producing a splits graph representing sequence distances (Huson, 1998).
Reference genomes were included to classify isolates into phylogenetic groups. For Phylogroup I, C. perfringens Darmbrand NCTC8081 (ERR1656460) was selected, which is responsible for human enteritis Necroticans cases in Germany in the 1940s and is genetically related to strains carrying chromosomal cpe (Lindström et al., 2011; Ma et al., 2012). For Phylogroup II, strains involved in necrotizing enteritis in foals and hemorrhagic gastroenteritis in dogs, and denoted with the prefix JFP were chosen (GCA_001949795.1, GCA_001949805.1, GCA_001949775.1; Abdel-Glil et al., 2021). For Phylogroup III, C. perfringens ATCC3626 (GCA_000171155.1) and C. perfringens ATCC13124 (GCA_000013285.1) were used, and for Phylogroup IV, C. perfringens type D (GCA_006385425.1) isolated from a sheep. Phylogroup V was established based on the strain C. perfringens Tumat (GCA_003990265.1), Which was isolated from the mummified remains of an ancient puppy found in Siberian Permafrost (Abdel-Glil et al., 2021).
In silico identification of virulence and antimicrobial resistance genes
Toxin detection (CPA, CPB, ETX, IAP, CPE, and netB) and typing of the 372 isolates were conducted with ABRicate v. 0.5 using the TOXIper database6 (available until October 20th, 2020). Additional virulence factors such as sialidases, collagenases, and secondary toxins like pfo, iam, and cpb2 were screened using the Virulence Factor Database (VFDB; March 17th, 2017; Chen et al., 2016). The Resfinder database (available until July 8th, 2017; Zankari et al., 2012) was used for the Antimicrobial Resistance (AMR) evaluation since it was the most complete database available with 2,228 AMR sequences at the moment of the analysis.
Results
Long-range dispersion of Clostridium perfringens
A total of 372 genomes from different sources were collected for our study: two de novo sequenced genomes from Chile, 237 previously assembled sequences, and 133 raw reads (Supplementary Table S1). The number of contigs among the assemblies was variable, with an average of 82 contigs per genome. On overage, the genome size was 3.3 Mb, with a low GC content of around 28% (Supplementary Table S2). In addition, ANI values were invariably > 95% for the 372 genomes when compared to the reference strain C. perfringens ATCC 13124, confirming their classification as C. perfringens (Supplementary Figure S1). Most of the publicly available genomes were obtained from the US (123/372, 33.1%), France (48/372, 12.9%), and China (44/372, 11.8%; Figure 1A), and were recovered mainly from food (85/372, 22.8%), birds (85/372, 22.8%), and humans (74/372, 19.8%; Figure 1B).

Figure 1. Origin and toxinotype profile of 372 Clostridium perfringens genomes. (A) Distribution of isolates by country of origin. (B) Distribution of isolates by source. (C) Distribution of isolates by toxin type. (D) Toxinotype diversity among hosts. All figures were created with R software.
Wide toxinotype diversity among multiple hosts
The seven different toxinotypes defined until now were seen among our 372 isolates. Toxinotype A was the most frequently found (198/372, 53%), followed by toxinotype F (120/372, 32%), and G (24/372, 7%; Figure 1C). Interestingly, toxinotype distribution varied among hosts. In this regard, 60% (45/74) and 32% (24/74) of the human isolates were classified as toxinotype A and F, respectively. Two human isolates (2.7%) belonged to toxinotype E, and toxinotype C, D, and G had one (1.3%) isolate each. Food isolates were also distributed in a similar way, as 32% (27/85) were toxinotype A, 67% (57/85) F, and 1% (1/85) G.
As for animals, 72% (61/85) of the bird isolates were classified as toxinotype A, 26% (22/85) as G, and 1% as C (1/85) and F (1/85). Fifty percent of the swine isolates (8/16) corresponded to toxinotype A, 43.7% (7/16) to C, and 6.3% (1/16) to F. Likewise, the ovine isolates were classified as toxinotype A (2/15, 14%), B (3/15, 20%), C (5/15, 33%), and D (5/15, 33%). Isolates from canines (16/22, 73%) and equines (15/16, 94%) belonged to toxinotype F and only a few to toxinotype A (6/22, 27% and 1/16, 6%, respectively). The high diversity of toxinotypes in the multiple evaluated hosts is an indicator of heterogeneous toxins that can support the differential pathogenic effect of C. perfringens populations circulating in each of these species.
Clostridium perfringens clones of heterogeneous origins
Multilocus sequence typing (MLST) was used to classify C. perfringens genomes. A total of 195 STs were identified among 322 genomes. One or more genes from the MLST scheme could not be recovered from 50 genomes, thus they were not assigned an ST. cpa, colA, and nadA were the genes with the most alleles (84, 84, and 73 respectively) compared to other MLST genes, which had less than 50 alleles (Supplementary Table S3).
MLST loci from the 322 genomes, were concatenated and aligned to generate a phylogenetic tree (Figure 2A), where five clusters were identified using the phylogenetic relationships and biological features as criteria, as reported before for C. perfringens (Abdel-Glil et al., 2021) Overall, Cluster III isolates were more frequent in our dataset (226/322, 70%). This cluster grouped isolates from diverse sources, including animals (120/226, 53%), humans (44/226, 20%), food (34/226, 15%), unknown sources (21/226, 9%), and environment (21/226, 3%). Cluster I (67/322, 20%) grouped mostly isolates from food (42/67, 63%) along with isolates from human (19/67, 28%), animal (3/67, 4.5%), and unknown sources (3/67, 4.5%). Interestingly, Cluster II (23/322, 7%) only grouped isolates from animals (19/23, 83%) and humans (4/23, 17%), whereas Cluster IV was formed by 3 isolates (3/322, 1%) from animal origin, and Cluster V by 3 isolates (3/322, 1%) from animal (2/3, 66%) and food (1/3, 34%) sources (Figure 2A).
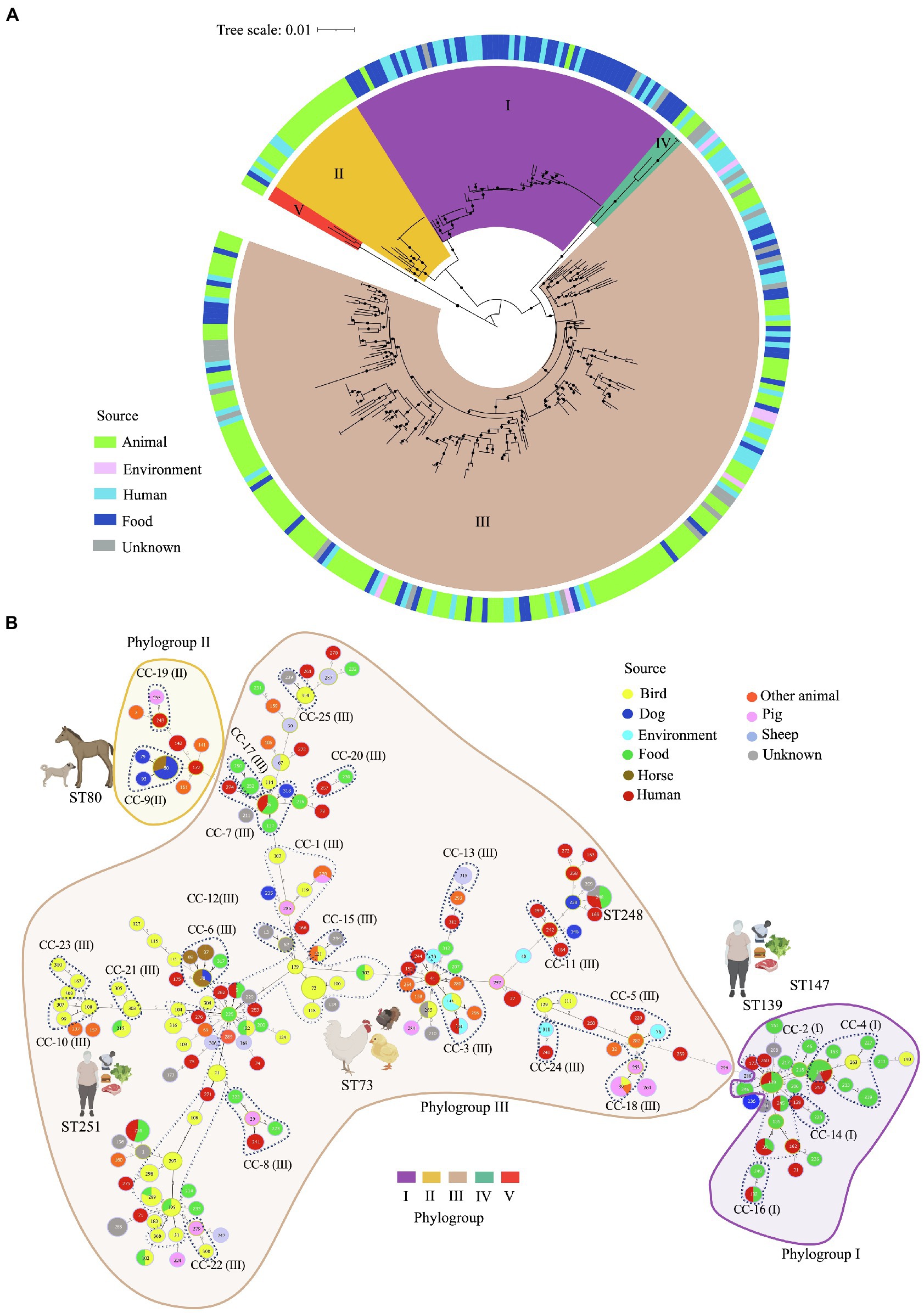
Figure 2. Multilocus sequence typing (MLST)-based phylogeny and goeBURST full minimum spanning tree of 195 C. perfringens MLST profiles among 322 genomes. (A) MLST-based phylogeny tree obtained with fastMLST. The outer ring shows the origin of the isolates as indicated in the legend. Clusters are represented by different colors on the inside of the ring. (B) MLST-based minimum spanning tree obtained with goeBURST. Sequence types (STs) are displayed as circles. Founder STs were defined as the STs with the greatest number of single-locus variants. Circle size indicates the number of isolates in every particular ST, with each color representing a different source type. Lines represent closely related isolates and line length illustrates STs that vary by one, two, or more alleles in their MLST profile. Clonal complexes (CC), defined as closely related STs that differ up to two of the eight loci used in the MLST scheme from a common founder (DLV, double locus variant), are designated by dashing lines.
Of the 195 STs detected, five were the most common: ST-147, -248, -80, -251, and ST-73 (Supplementary Figure S2). Isolates within these STs tended to have the same toxinotype, however, they were located across different clusters. For example, 14 toxinotype F isolates from food and human sources from the US and Italy isolated during 2017 and 2019 were classified as ST-147 and grouped in Cluster I. In the same way, 13 toxinotype F isolates from dogs and horses from Canada and the US, were classified as ST-80 located in Cluster II. Furthermore, ST-251 included 11 isolates from toxinotype F in cluster I recovered in the US. In contrast, ST-73 grouped 11 isolates from toxinotypes G and A from birds, all of them grouped in Cluster III.
The MLST-based minimum spanning tree built using the goeBURST algorithm led to evolutionary inferences. By identifying founder STs in the 25 CC and 95 singletons (Figure 2B; Supplementary Figure S3). Overall, CC1 was the most commonly represented CC. It was found within Cluster III, grouping 23 STs from 50 genomes mostly from birds (ST-73, -106, -118), human stool and blood samples (ST-5, -262 y ST-271), and food (ST-5, -195, -225, -299 y ST-302). The founder ST for this group was ST-225, which is associated with food. Usually, the predicted founder corresponds to the most predominant ST in a CC (Feil et al., 2004), however this ST contained only one isolate whereas DLV descendant ST-73 contained 11 isolates (Supplementary Figure S3). This interesting finding might be caused by an origin bias due to either the relatively low number of samples used or by the natural selection pressure within the population leading to the emergence of strains with a strong adaptive advantage (Feil et al., 2004).
Most of the isolates from human stools and food were grouped in Cluster I as CC-2 along with ST-139 as founder ST. ST-39 and ST-253 from swine farms in China were classified as CC-18 in Cluster III, and founder ST-80, -93, and -79 from canine and equine isolates were in cluster II as CC-9. Some of the STs varied in terms of hosts, for instance ST-80 and ST-78 included isolates from dogs and horses, ST-5, -33, -132, -139, -147, -248, and -251 included human and food isolates, and ST-39 grouped isolates from swine and birds, while ST-143 grouped birds and environment, and ST-215 birds and food (Figure 2B).
Five Clostridium perfringens phylogroups identified by core genome-based phylogenetic analysis
A total of 35,876 genes are included in the pangenome of this dataset. Of them, 34,917 (97.3%) genes are accessory and only 959 (2.7%) are considered part of the core genome. In the resulting tree, five main branches were observed matching the clusters that were established by using the reference genomes and a bootstrap ≥ 80% (Figure 3A). The core genome-based phylogenetic tree matches with the MLST-based topology, as well as with the phylogenetic network generated using neighbor-joining, confirming the classification of C. perfringens population in five distinct clusters as reported previously (Figure 3B).
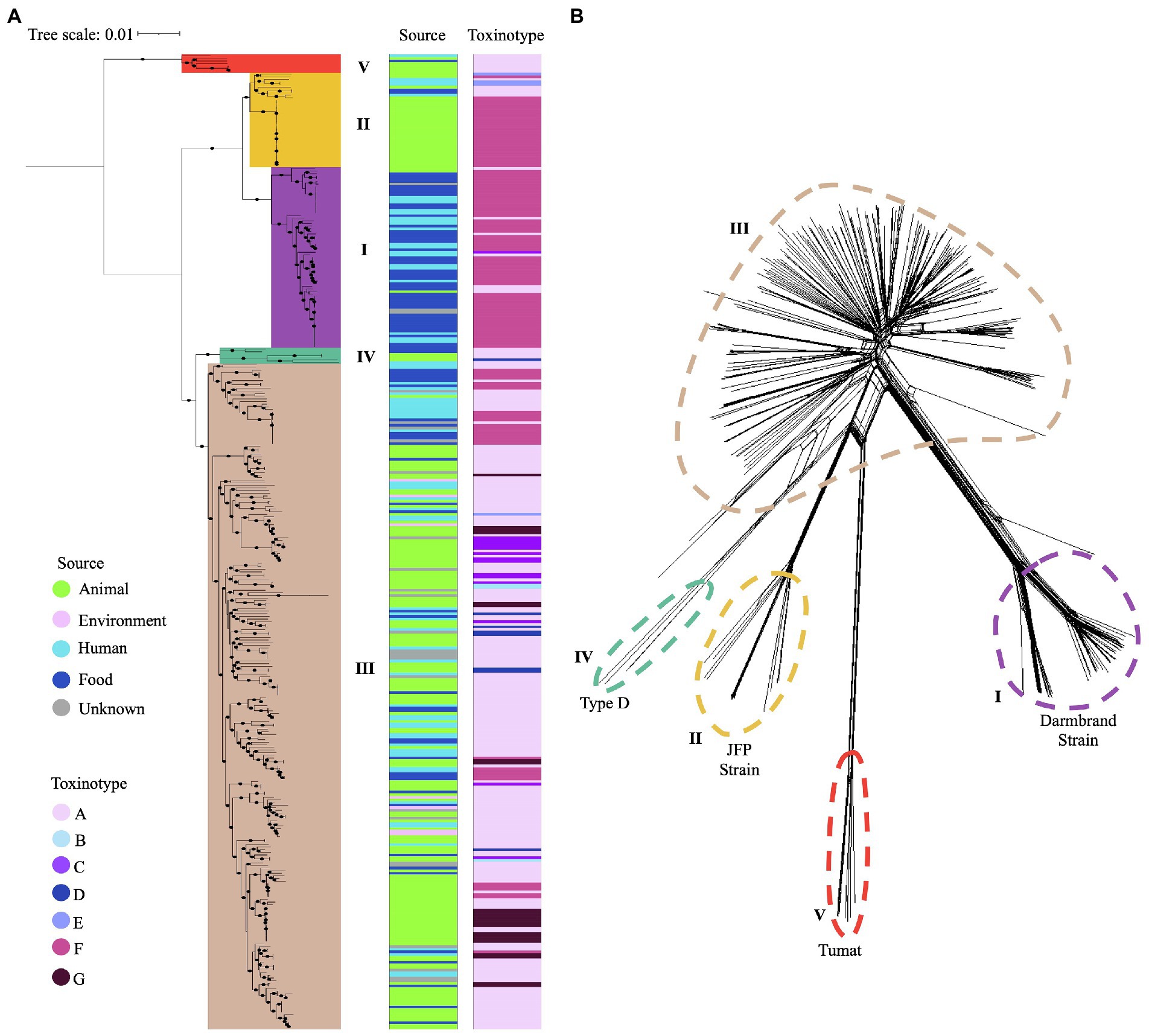
Figure 3. Phylogenetic grouping of C. perfringens using two different approaches. (A) Phylogenetic tree based on the core genome of 372 genomes. Five main phylogroups are highlighted in different colors. Source type and toxinotype are shown on the right panels. (B) Five phylogenetic networks based on the Neighbor-Joining (NJ) algorithm are shown with different colors.
Phylogroup I (n = 69) clustered most of the isolates from humans and food from toxinotype F (n = 57) that were cpa and cpe positive, along with some isolates from toxinotype A (n = 7) from humans and animals. Phylogroup II (n = 36) grouped canine and equine isolates from toxinotype F (n = 26), as well as one cattle and two human isolates from toxinotype E. Phylogroup III (n = 257) was the most diverse group, where the novel isolates recovered from water and a human in Chile were located. In this phylogroup isolates from birds, food, environment, and humans classified as toxinotype A (n = 177), toxinotype F (n = 31), toxinotype G (n = 24), and in less proportion toxinotype C (n = 14), D (n = 7), and B (n = 3) were also found. Phylogroup IV (n = 3) and V (n = 7) included a majority of toxinotype A isolates (Figure 3A).
Clostridium perfringens is a highly versatile pathogen with a large number of virulence factors
A total of 372 WGS were analyzed to evaluate the distribution of virulence genes. The alpha-toxin gene plc associated with gangrene in humans and several animals, and possibly involved in enterotoxemia and gastrointestinal disease in ruminants, horses, and swine was present in most of the isolates, along with genes encoding extracellular enzymes such as alpha-clostripain (ccp), hyaluronidases (nagH, nagI and nagJ), and alpha-clostripain collagenase (colA), as expected (Canard et al., 1994; Sakurai et al., 2004; Goossens et al., 2017; Geier et al., 2021). Other genes present in the majority of the isolates were pfoA (perfringolysin O), tpeL (toxin perfringens large), and cpb2 (beta2 toxin), which are protein coding genes involved in gastrointestinal outcomes and gangrene (Coursodon et al., 2012; Bueschel et al., 2013; Chen and McClane, 2015), and the sialidases nanH, nanI y nanJ, which play an important role in colonization and immunomodulation (Figure 4A).
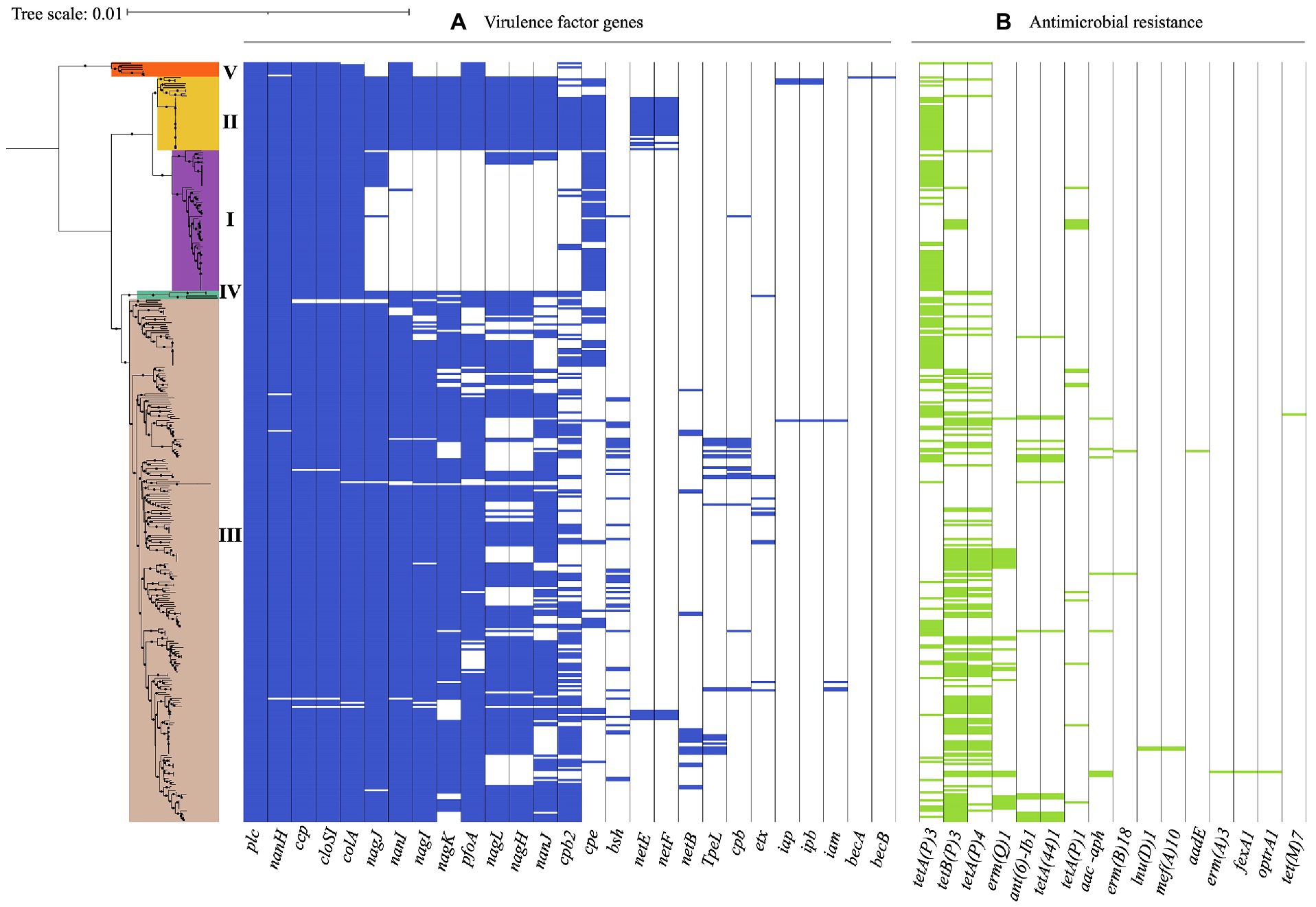
Figure 4. Virulence-associated and antimicrobial resistance (AMR) genes in 372 C. perfringens genomes. The core genome-based phylogenetic tree showed phylogroups highlighted in different colors. Matrix of absence and presence of genes shows (A) virulence genes (B) antimicrobial resistance (AMR) genes. The minimum coverage threshold needed for detection of these genes was 70% and the percentage of identity was 99.95%. The resulting trees were visualized and edited using iTOL v.5 (Letunic and Bork, 2021).
The presence of virulence genes varied among the phylogroups. For example, all of the isolates in phylogroup I carried nanH, however nanI, nanJ, nagH, pfoA, and cpb2 were absent in this group, in contrast to phylogroup II, where these genes were found in all of the isolates along with nagI and nagK. Additionally, 64% (23/36) of the isolates in this phylogroup also carried netE and 58% (21/36) netF. While the presence of these virulence genes in phylogroup III was variable, this was the only group where TpeL, a member of the large clostridial toxin (LCT) family involved in cell cytotoxicity was present, especially in isolates of toxinotype A, B, C, and G. However, this gene was not detected in type D or F isolates carrying the cpe and itx toxin genes. This difference in toxin profiles can be attributed to a potential incompatibility between plasmids carrying these genes (Chen and McClane, 2015).
Isolates in phylogroup IV carried the coding genes for the hyaluronidases nagH, nagI, and nagJ, as well as nanI and pfoA. Phylogroup V, which cluster toxinotype A isolates carried nanI and pfoA, unlike the toxinotype A isolates in phylogroup I (Figure 4A). These findings suggest that the differential presence of virulence factors in phylogroups may be due to selective advantage conferred by determinants in different niches (Sawires and Songer, 2006), routine treatment with clostridial toxoids especially in ruminants, or even environmental differences in the geographical regions (Simpson et al., 2018).
Prevalence of antibiotic resistance genes in Clostridium perfringens
AMR gene were found in 72.8% (271/372) of the genomes, with tetA (107/271, 39.5%) and tetB (98/271, 36.2%) involved in tetracycline resistance, being the most frequent and commonly found in phylogroup III isolates (Figure 4B). Interestingly, the water isolate from Chile harbors genes for tetracycline resistance (tetA), while the one from human stool possesses genes that encoded tetracycline (tetA, tet44) and aminoglycoside resistance (ant(6)-Ib1). Likewise, this approach identified genomes carrying ermQ, (28/372, 7.5%) an erythromycin resistance methylase gene mainly found in birds and environment isolates grouped in phylogroup III and classified as toxinotype A. ant(6)-Ib1 genes encoding aminoglycoside resistance were found in some toxinotype A and C isolates from swine and birds from phylogroup III (Figure 4B).
Discussion
Clostridium perfringens is a clinically relevant pathogen due to its presence across several host species and its capacity to cause numerous medically important intestinal diseases in humans and animals (Mehdizadeh Gohari et al., 2021). To assess the genetic variation within the species, as well as to establish the phylogenetic relatedness, we collected the publicly available genomes of 370 isolates collected between 2010 and 2020 from different countries, with a majority of the isolates obtained in developed countries such as the US, France, and China (Figure 1A). There is a poor representation of isolates from developing countries in our dataset, possibly due to limited epidemiologic surveillance and genomic data collection, especially in South America, from where only one genome from Argentina was found and two more from Chile were added in this study.
The dataset also has a high percentage of strains isolated from stool samples of animals used for human consumption as well as isolates from food origin (Figures 1B, 2B), which along with the evidence of food products such as milk, meat, poultry, and pork among others as a source of infection, confirm the key role that C. perfringens plays as one of the most common causes of foodborne illness (Brynestad et al., 1997; Grass et al., 2013; Bintsis, 2017; Xu et al., 2021). However, despite the high percentage of isolates of human and food origin, it should be noted that environmental or commensal isolates are scarce (Figure 1B). In addition, some metadata variables are unknown, such as the possible association of genomes with outbreaks, which could underestimate the diversity of C. perfringens and affect the results of AMR prevalence, emphasizing the need for further studies with a greater number of genomes.
Clostridium perfringens toxinotypes are associated with heterogeneous diseases such as clostridial myonecrosis (gas gangrene) or gastrointestinal infections in humans and animals (Rood et al., 2018). Toxinotype G, associated with necrotic enteritis and gangrenous dermatitis in birds (Yang et al., 2019; Kiu et al., 2019a), and toxinotypes D and E that cause illness in sheep and cattle (Layana et al., 2006; Uzal and Songer, 2008; Nazki et al., 2017; Rood et al., 2018), have not been described in humans before, however, we identified one isolate from a raw chicken patty and another from human blood as toxinotype G (Figure 1D). Likewise, we found toxinotype D and E strains isolated from human stools. These findings support previous studies that have reported C. perfringens type D and E strains harboring etx and iap in humans (Al-Shukri et al., 2021), which can explain potential routes of transmission in subjects that have been in contact with infected animals or have consumed contaminated food (Songer, 2010; Kiu et al., 2019b). Furthermore, these findings might be related to C. perfringens strains considered as normal intestinal microbiota that are in contact with acquired strains carrying conjugative plasmids that are often associated with insertion sequences that can mobilize toxin genes between different strains. This could lead to the conversion into virulent toxin-producing strains and the emergence of specific toxinotypes in new hosts (Freedman et al., 2015). Future studies should include genomes assembled with a standard pipeline that includes an approach to recover extrachromosomal elements in order to describe the plasmidome of C. perfringens and thus contribute to the biological knowledge of this species.
MLST and geoBURST analyses have not been widely used for C. perfringens: However, the identification of the founder STs, that differs from other STs at only one locus, provide a tool for epidemiological and evolutionary investigations of emergent pathogens (Feil and Enright, 2004). Furthermore, the identification of 25 CC in this work allowed us to compare the distribution of C. perfringens isolates from animals, humans, food and environment (Figure 2B; Supplementary Figure S2), evidencing a close relationship between isolates from different hosts. These findings support the strong association with foodborne diseases and suggesting their zoonotic potential and high diversification of this specie, as previously described (Verma et al., 2020; Hassani et al., 2022; Xiu et al., 2022).
The CC including toxinotype G isolates from human and birds, and the evidence of the distribution of human, food, and animal isolates in the same ST or CC matches the zoonotic potential of C. perfringens as demonstrated by other published studies (Immerseel et al., 2004; Mwangi et al., 2019; Verma et al., 2020). Population structure analyses based on MLST (Figure 2A) revealed five clusters in line with those generated here using the core genome (Figures 3A,B), and with results from previous studies (Abdel-Glil et al., 2021). Although MLST has a limited ability to establish phylogenetic relations since it only uses fragments from eight housekeeping genes, it is still a useful tool for interspecies C. perfringens typing due to its reproducibility, high discriminatory power, and easy accessibility (Pérez-Losada et al., 2013; Jolley et al., 2018; Guerrero-Araya et al., 2020). Despite these advantages, WGS has emerged as a more robust and complete tool contributing to the investigation of phylogenetic relatedness among isolates and allowing a deeper understanding of transmission dynamics, emerging clones, key virulence loci, and the presence of AMR genes (Salipante et al., 2015; Quainoo et al., 2017; Pightling et al., 2018).
The pangenome analysis of C. perfringens conducted in this study showed an accessory genome of 97.32%, an extremely high percentage in comparison with other closely related bacteria such as C. paraputrificum, species with an accessory genome of 67%, C. tertium with 37.6% of accessory genes (Muñoz et al., 2019), or C. baratii with an accessory genome of 24.43% (Silva-Andrade et al., 2022). This high level of genome plasticity is similar to the one found in Clostridioides difficile, which has an accessory genome of 87.2% (Knight et al., 2021). C. difficile and C. perfringens are part of the normal intestinal flora but can become gastrointestinal pathogens, which can be explained by the ability to express different phenotypes in response to particular environmental conditions. The high genome plasticity of C. perfringens can give rise to the emergence of populations carrying new toxigenic profiles by acquired virulence factors due to horizontal gene transfer (HGT) leading to rapid genetic evolution. Thus, this genomic plasticity of C. perfringens is a determinant in the adaption to different hosts, as well as in the increase of its pathogenic potential and survival in different environments (Brüggemann, 2005).
The importance of the acquisition of different virulence factors in C. perfringens is given by their adaptation within different disease niches. When exploring the virulence factors present in the C. perfringens genomes, we found that plc, colA, and nanH are present in the majority of the genomes, hence they cannot be considered markers with high host discrimination capacity (Goossens et al., 2017; Mahamat Abdelrahim et al., 2019). The identification of isolates carrying netE and netF toxins in two animal species (dogs and foals) of phylogroup I, as well as previous reports of the prevalence of these two toxins in C. perfringens isolates from dogs with acute diarrhea hemorrhagic syndrome (Sindern et al., 2019), suggest their adaptability to these specific hosts. Likewise, the presence of three sialidases (nanH, nanI, and nanJ) in isolates from the same clinical outcomes highlights the role of these enzymes in increasing the adhesion of C. perfringens to host cells (Carman, 1997; Chiarezza et al., 2009; Li and McClane, 2021) and suggests an important role in the intestinal pathogenesis in these hosts, as was previously reported (Li et al., 2015; Li and McClane, 2021). However, a better understanding of the role and specificity of these molecules in canine acute hemorrhagic diarrhea and necrotizing enteritis of foals is required.
Another virulence-related gene, pfoA, was also found in most of the genomes included here (Figure 4A), however, it was absent in toxinotype F isolates clustered in the phylogroup I, which correlates with prior studies that have revealed that some strains that produce enterotoxins and therefor cause food poisoning lack pfoA (Myers et al., 2006; Deguchi et al., 2009). This suggests that this cytolytic toxin is not the main cause of most gastrointestinal outcomes in humans, however, it could be associated with the enteritis necro-hemorrhagic or bovine enterotoxemia, as recent studies have shown (Verherstraeten et al., 2013a,b). Thus, the high number of virulence-related genes in the 372 genomes analyzed, especially in the novel isolates from Chile included in our study (Figure 4A), reveals the need to continue with the epidemiological surveillance and the molecular study of virulence factors, mainly in unexplored countries, to provide more data for a deeper knowledge of the global diversity of C. perfringens.
Another group of molecular markers of importance in health is the one associated with antibiotic resistance. We found that a high percentage of the genomes (72.8%, 270/372) harbored some type of AMR gene (Figure 4B), where a large number of isolates carried genes putatively linked to resistance to tetracycline, macrolides, and aminoglycosides. The presence of a high number of AMR genes in isolates from pigs and birds could be a consequence of the use of antibiotics as growth promoters in these animals and may be related to the appearance of resistance in zoonotic pathogens (Osman and Elhariri, 2013). Although WHO has questioned the use of additives due to the risk of antibiotic residues in meat products for human consumption, several countries continue with this practice that can be related to the increase of antibiotic resistant strains (Salvarani et al., 2012). Many factors such as the production of high concentrations of antibiotics in the global industry (Nijsingh et al., 2019; Tell et al., 2019), the indiscriminate use of antibiotics in the community (Graham et al., 2019), the contamination of natural sources by hospital waste (Larsson and Flach, 2021), the impact of intrahospital infections caused by multidrug resistant strains (Avershina et al., 2021), the use of antibiotics for animal growth, the poor management of organic waste and the use of animal excrement in the agricultural sector (Tang et al., 2017), and the antimicrobial drugs overused during the first wave of COVID-19 (Manesh and Varghese, 2021), pose a high risk for possible pathways of antimicrobial resistance (Chokshi et al., 2019).
The analysis of 372 genomes conducted here, is the largest effort to snapshot the global genomic diversity of C. perfringens to date. The genomic plasticity of this microorganism due to its low GC content (~ 28%; Uzal et al., 2014), its short doubling time (~ 7 min; Maiden, 2006), and a high percentage of horizontally transferred toxin encoding genes (Xiao et al., 2012; Uzal et al., 2014) contributes to the spread of existing toxinotypes to new hosts, as well as to the increase of food poisoning outbreaks and the growth of AMR. The use of MLST and WGS in a “One Health” framework that connects the health of humans and animals in a shared environment represents an optimal approach to advance knowledge of the global genetic diversity of C. perfringens. Our findings emphasize the need for further studies using a larger number of isolates from different ecological niches to elucidate the genetic characteristics, diversity, and zoonotic potential of C. perfringens and to improve strategies to reduce the growing threat to public health by this pathogen.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
AC, EG-A, and MM designed the study and performed the data analyses. AC and MM performed methodology, formal analysis, data curation, and visualization and wrote the original draft preparation. JDR, DP-S, CR, SC, LV, and MC-A validated the results and revised and edited the manuscript. JDR and MM supervised the study and contributed to review and editing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was financially supported by Dirección de Investigación e Innovación from Universidad del Rosario, Bogotá D.C. and the Universidad de Boyacá, Tunja, Colombia. In addition, this work mas supported by start-up funds from Texas A&M University and by ANID, Millennium Science Initiative Program–NCN17_093 to DP-S.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the Universidad de Boyacá and the Universidad del Rosario in Colombia for their support.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.952081/full#supplementary-material
Footnotes
1. ^https://www.ebi.ac.uk/ena/browser/home
2. ^http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
3. ^https://github.com/widdowquinn/pyani
4. ^https://github.com/EnzoAndree/FastMLST
References
Abdel-Glil, M. Y., Thomas, P., Linde, J., Busch, A., Wieler, L. H., Neubauer, H., et al. (2021). Comparative in silico genome analysis of Clostridium perfringens unravels stable phylogroups with different genome characteristics and pathogenic potential. Sci. Rep. 11:6756. doi: 10.1038/s41598-021-86148-8
Al-Shukri, M. S., Hmood, A. M., and Al-Charrakh, A. H. (2021). Sequencing of Clostridium perfringens toxin genes (cpa, etx, iap) from Iraqi hospitals and detection by PCR of the genes encoding resistance to metronidazole, tetracycline, and clindamycin. Indian J. Med. Microbiol. 39, 289–294. doi: 10.1016/j.ijmmb.2021.03.017
Avershina, E., Shapovalova, V., and Shipulin, G. J. (2021). Fighting antibiotic resistance in hospital-acquired infections: current state and emerging technologies in disease prevention, diagnostics and therapy. Front. Microbiol. 12:707330. doi: 10.3389/fmicb.2021.707330
Awad, M. M., Bryant, A. E., Stevens, D. L., and Rood, J. I. (1995). Virulence studies on chromosomal α-toxin and Θ-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of α-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 15, 191–202. doi: 10.1111/j.1365-2958.1995.tb02234.x
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bintsis, T. J. (2017). Foodborne pathogens. AIMS Microbiol. 3, 529–563. doi: 10.3934/microbiol.2017.3.529
Bolger, A. M., Lohse, M., and Usadel, B. J. B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brüggemann, H. (2005). Genomics of clostridial pathogens: implication of extrachromosomal elements in pathogenicity. Curr. Opin. Microbio. 8, 601–605. doi: 10.1016/j.mib.2005.08.006
Brynestad, S., Synstad, B., and Granum, P. E. J. M. (1997). The Clostridium perfringens enterotoxin gene is on a transposable element in type A human food poisoning strains. Microbiology 143, 2109–2115. doi: 10.1099/00221287-143-7-2109
Bueschel, D. M., Jost, B. H., Billington, S. J., Trinh, H. T., and Songer, J. G. (2013). Prevalence of cpb2, encoding beta2 toxin, in Clostridium perfringens field isolates: correlation of genotype with phenotype. Vet. Microbiol. 94, 121–129. doi: 10.1016/s0378-1135(03)00081-6
Canard, B., Garnier, T., Saint-Joanis, B., and Cole, S. T. (1994). Molecular genetic analysis of the nagH gene encoding a hyaluronidase of Clostridium perfringens. Mol. Gen. Genet. 243, 215–224. doi: 10.1007/BF00280319
Carman, R. J. (1997). Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microbiol. 8:S46. doi: 10.1097/00013542-199712001-00024
Chalmers, G., Bruce, H., Hunter, D., Parreira, V., Kulkarni, R., Jiang, Y.-F., et al. (2008). Multilocus sequence typing analysis of Clostridium perfringens isolates from necrotic enteritis outbreaks in broiler chicken populations. J. Clin. Microbiol. 46, 3957–3964. doi: 10.1128/JCM.01548-08
Chen, J., and McClane, B. A. (2015). Characterization of Clostridium perfringens TpeL toxin gene carriage, production, cytotoxic contributions, and trypsin sensitivity. Infect. Inmun. 83, 2369–2381. doi: 10.1128/IAI.03136-14
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Chiarezza, M., Lyras, D., Pidot, S. J., Flores-Díaz, M., Awad, M. M., Kennedy, C. L., et al. (2009). The NanI and NanJ sialidases of Clostridium perfringens are not essential for virulence. Infect. Immun. 77, 4421–4428. doi: 10.1128/IAI.00548-09
Chokshi, A., Sifri, Z., Cennimo, D., and Horng, H. (2019). Global contributors to antibiotic resistance. J. Glob. Infect. Dis. 11, 36–42. doi: 10.4103/jgid.jgid_110_18
Coursodon, C., Glock, R., Moore, K., Cooper, K., and Songer, J. J. A. (2012). TpeL-producing strains of Clostridium perfringens type A are highly virulent for broiler chicks. Anaerobe 18, 117–121. doi: 10.1016/j.anaerobe.2011.10.001
Deguchi, A., Miyamoto, K., Kuwahara, T., Miki, Y., Kaneko, I., Li, J., et al. (2009). Genetic characterization of type A enterotoxigenic Clostridium perfringens strains. PLoS One 4:5598. doi: 10.1371/journal.pone.0005598
Feil, E. J., and Enright, M. C. (2004). Analyses of clonality and the evolution of bacterial pathogens. Curr. Opin. Microbiol. 7, 308–313. doi: 10.1016/j.mib.2004.04.002
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Francisco, A. P., Vaz, C., Monteiro, P. T., Melo-Cristino, J., Ramirez, M., and Carriço, J. A. (2012). PHYLOViZ: phylogenetic inference and data visualization for sequence-based typing methods. BMC Bioinform. 13, 1–10. doi: 10.1186/1471-2105-13-87
Freedman, J. C., Theoret, J. R., Wisniewski, J. A., Uzal, F. A., Rood, J. I., and McClane, B. (2015). Clostridium perfringens type A–E toxin plasmids. Res. Microbiol. 166, 264–279. doi: 10.1016/j.resmic.2014.09.004
Garcia, J., Adams, V., Beingesser, J., Hughes, M. L., Poon, R., Lyras, D., et al. (2013). Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect. Inmun. 81, 2405–2414. doi: 10.1128/IAI.00238-13
Geier, R. R., Rehberger, T. G., and Smith, A. H. (2021). Comparative genomics of Clostridium perfringens reveals patterns of host-associated phylogenetic clades and virulence factors. Front. Microbiol. 12:649953. doi: 10.3389/fmicb.2021.649953
Goossens, E., Valgaeren, B. R., Pardon, B., Haesebrouck, F., Ducatelle, R., Deprez, P. R., et al. (2017). Rethinking the role of alpha toxin in Clostridium perfringens-associated enteric diseases: a review on bovine necro-haemorrhagic enteritis. Vet. Res. 48, 9–17. doi: 10.1186/s13567-017-0413-x
Graham, D. W., Giesen, M. J., and Bunce, J. (2019). Strategic approach for prioritising local and regional sanitation interventions for reducing global antibiotic resistance. Water 11:27. doi: 10.3390/w11010027
Grass, J. E., Gould, L. H., and Mahon, B. E. (2013). Epidemiology of foodborne disease outbreaks caused by Clostridium perfringens, United States, 1998–2010. Foodborne Pathog. Dis. 10, 131–136. doi: 10.1089/fpd.2012.1316
Guerrero-Araya, E., Munoz, M., Rodríguez, C., and Paredes-Sabja, D. J. (2020). FastMLST: a multi-core tool for multilocus sequence typing of draft genome assemblies. Bioinform. Biol. Insights 15. doi: 10.1177/11779322211059238
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. J. B. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hassan, K. A., Elbourne, L. D., Tetu, S. G., Melville, S. B., Rood, J. I., and Paulsen, I. T. (2015). Genomic analyses of Clostridium perfringens isolates from five toxinotypes. Res. Mirobiol. 166, 255–263. doi: 10.1016/j.resmic.10.003
Hassani, S., Pakbin, B., Brück, W. M., Mahmoudi, R., and Mousavi, S. (2022). Prevalence, antibiotic resistance, toxin-typing and genotyping of Clostridium perfringens in raw beef meats obtained from Qazvin City Iran. Antibiotics 11:340. doi: 10.3390/antibiotics11030340
Huson, D. H. J. B. (1998). SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14, 68–73. doi: 10.1093/bioinformatics/14.1.68
Huson, D. H., and Bryant, D. J. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Immerseel, F. V., Buck, J. D., Pasmans, F., Huyghebaert, G., Haesebrouck, F., and Ducatelle, R. (2004). Clostridium perfringens in poultry: an emerging threat for animal and public health. Climate Change Responses 33, 537–549. doi: 10.1186/s40665-016-0025-0
Jolley, K. A., Bray, J. E., and Maiden, M. C. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST. Org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Keyburn, A. L., Boyce, J. D., Vaz, P., Bannam, T. L., Ford, M. E., Parker, D., et al. (2008). NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 4:e26. doi: 10.1371/journal.ppat.0040026
Kiu, R., Brown, J., Bedwell, H., Leclaire, C., Caim, S., Pickard, D., et al. (2019a). Genomic analysis on broiler-associated Clostridium perfringens strains and exploratory caecal microbiome investigation reveals key factors linked to poultry necrotic enteritis. Anim. Microbiome. 1:12. doi: 10.1186/s42523-019-0015-1
Kiu, R., Caim, S., Alexander, S., Pachori, P., and Hall, L. J. (2017). Probing genomic aspects of the multi-host pathogen Clostridium perfringens reveals significant pangenome diversity, and a diverse array of virulence factors. Front. Microbiol. 8:2485. doi: 10.3389/fmicb.2017.02485
Kiu, R., and Lindsay, J. H. (2018). An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 7, 1–15. doi: 10.1038/s41426-018-0144-8
Kiu, R., Sim, K., Shaw, A., Cornwell, E., Pickard, D., Kroll, J. S., et al. (2019b). Genomic analysis of Clostridium perfringens BEC/CPILE-positive, Toxinotype D and E strains isolated from healthy children. Toxins 11:543. doi: 10.3390/toxins11090543
Knight, D. R., Imwattana, K., Kullin, B., Guerrero-Araya, E., Paredes-Sabja, D., Didelot, X., et al. (2021). Major genetic discontinuity and novel toxigenic species in Clostridioides difficile taxonomy. Elife 10:10. doi: 10.7554/eLife.64325
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Larsson, D., and Flach, C. (2021). Antibiotic resistance in the environment. Nat. Rev. Microbiol. 20, 257–269. doi: 10.1038/s41579-021-00649-x
Layana, J. E., Miyakawa, M. E. F., and Uzal, F. (2006). Evaluation of different fluids for detection of Clostridium perfringens type D epsilon toxin in sheep with experimental enterotoxemia. Anaerobe 12, 204–206. doi: 10.1016/j.anaerobe.2006.05.001
Letunic, I., and Bork, P. (2021). Interactive tree Of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, J., and BA, M. C. (2014). The sialidases of Clostridium perfringens type D strain CN3718 differ in their properties and sensitivities to inhibitors. Appl. Environ. Microbiol. 80, 1701–1709. doi: 10.1128/AEM.03440-13
Li, J., Freedman, J. C., and McClane, B. A. (2015). NanI sialidase, CcpA, and CodY work together to regulate epsilon toxin production by Clostridium perfringens type D strain CN3718. J. Bacteriol. 197, 3339–3353. doi: 10.1128/JB.00349-15
Li, J., and McClane, B. (2021). NanH is produced by sporulating cultures of Clostridium perfringens type F food poisoning strains and enhances the cytotoxicity of C. perfringens enterotoxin. mSphere 6. doi: 10.1128/mSphere.00176-21
Lindström, M., Heikinheimo, A., Lahti, P., and Korkeala, H. J. (2011). Novel insights into the epidemiology of Clostridium perfringens type A food poisoning. Food Microbiol. 28, 192–198. doi: 10.1016/j.fm.2010.03.020
Ma, M., Li, J., and McClane, B. A. (2012). Genotypic and phenotypic characterization of Clostridium perfringens isolates from Darmbrand cases in post-world war II Germany. Infect. Human. 80, 4354–4363. doi: 10.1128/IAI.00818-12
Mahamat Abdelrahim, A., Radomski, N., Delannoy, S., Djellal, S., Le Négrate, M., Hadjab, K., et al. (2019). Large-scale genomic analyses and toxinotyping of Clostridium perfringens implicated in foodborne outbreaks in France. Front. Microbiol. 10:777. doi: 10.3389/fmicb.2019.00777
Maiden, M. C. J. A. R. M. (2006). Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 60, 561–588. doi: 10.1146/annurev.micro.59.030804.121325
Manesh, A., and Varghese, G. M. (2021). Rising antimicrobial resistance: an evolving epidemic in a pandemic. Lancet Microbe. 2, e419–e420. doi: 10.1016/S2666-5247(21)00173-7
Mehdizadeh Gohari, I. A., Navarro, M., Li, J., Shrestha, A., Uzal, F., and McClane, B. A. (2021). Pathogenicity and virulence of Clostridium perfringens. Virulence 12, 723–753. doi: 10.1080/21505594.2021.1886777
Minh, B. Q., Nguyen, M. A. T., and von Haeseler, A. J. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Muñoz, M., Restrepo-Montoya, D., Kumar, N., Iraola, G., Herrera, G., Ríos-Chaparro, D. I., et al. (2019). Comparative genomics identifies potential virulence factors in Clostridium tertium and C. paraputrificum. Virulence 10, 657–676. doi: 10.1080/21505594.2019.1637699
Mwangi, S., Timmons, J., Fitz-Coy, S., and Parveen, S. (2019). Characterization of Clostridium perfringens recovered from broiler chicken affected by necrotic enteritis. Poult. Sci. 98, 128–135. doi: 10.3382/ps/pey332
Myers, G. S., Rasko, D. A., Cheung, J. K., Ravel, J., Seshadri, R., DeBoy, R. T., et al. (2006). Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 16, 1031–1040. doi: 10.1101/gr.5238106
Nazki, S., Wani, S. A., Parveen, R., Ahangar, S. A., Kashoo, Z. A., Hamid, S., et al. (2017). Isolation, molecular characterization and prevalence of Clostridium perfringens in sheep and goats of Kashmir Himalayas, India. Vet. World. 10, 1501–1507. doi: 10.14202/vetworld.2017.1501-1507
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. J. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nijsingh, N., Munthe, C., and Larsson, D. (2019). Managing pollution from antibiotics manufacturing: charting actors, incentives and disincentives. Environ. Health 18, 1–17. doi: 10.1186/s12940-019-0531-1
Osman, K., and Elhariri, M. (2013). Antibiotic resistance of Clostridium perfringens isolates from broiler chickens in Egypt. Rev. Sci. Tech. 32, 841–850. doi: 10.20506/rst.32.2.2212
Page, A. J., Alikhan, N.-F., Carleton, H. A., Seemann, T., Keane, J. A., and Katz, L. S. (2017). Comparison of classical multi-locus sequence typing software for next-generation sequencing data. Microb. Genom. 3:e000124. doi: 10.1099/mgen.0.000124
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pérez-Losada, M., Cabezas, P., Castro-Nallar, E., and Crandall, K. A. (2013). Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Genet. Evol. 16, 38–53. doi: 10.1016/j.meegid.2013.01.009
Pightling, A. W., Pettengill, J. B., Luo, Y., Baugher, J. D., Rand, H., and Strain, E. (2018). Interpreting whole-genome sequence analyses of foodborne bacteria for regulatory applications and outbreak investigations. Front. Microbiol. 9:1482. doi: 10.3389/fmicb.2018.01482
Quainoo, S., Coolen, J. P., van Hijum, S. A., Huynen, M. A., Melchers, W. J., van Schaik, W., et al. (2017). Whole-genome sequencing of bacterial pathogens: the future of nosocomial outbreak analysis. BMC Infect. Dis. 20, 1015–1063. doi: 10.1186/s12879-019-4743-3
Raskin, D. M., Seshadri, R., Pukatzki, S. U., and Mekalanos, J. J. J. C. (2006). Bacterial genomics and pathogen evolution. Cell 124, 703–714. doi: 10.1016/j.cell.2006.02.002
Research UCJNa (2017). The universal protein resource (UniProt). Nucleic Acids Res. 36, D190–D195. doi: 10.1093/nar/gkm895
Revitt-Mills, S. A., Rood, J. I., and Adams, V. J. M. A. (2015). Clostridium perfringens extracellular toxins and enzymes: 20 and counting. Microbiol. Aust. 36, 114–117. doi: 10.1071/MA15039
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rood, J. I., Adams, V., Lacey, J., Lyras, D., McClane, B. A., Melville, S. B., et al. (2018). Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 53, 5–10. doi: 10.1016/j.anaerobe.2018.04.011
Sakurai, J., Nagahama, M., and Oda, M. (2004). Clostridium perfringens alpha-toxin: characterization and mode of action. J. Biochem. 136, 569–574. doi: 10.1093/jb/mvh161
Salipante, S. J., SenGupta, D. J., Cummings, L. A., Land, T. A., Hoogestraat, D. R., and Cookson, B. T. (2015). Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J. Clin. Microbiol. 53, 1072–1079. doi: 10.1128/JCM.03385-14
Salvarani, F. M., Silva, R. O. S., Pires, P. S., Cruz Júnior, E. C. C., Albefaro, I. S., Guedes, R. M. C., et al. (2012). Antimicrobial susceptibility of Clostridium perfringens isolated from piglets with or without diarrhea in Brazil. Braz. J. Microbiol. 43, 1030–1033. doi: 10.1590/S1517-838220120003000027
Sarker, M. R., Carman, R. J., and BA, M. C. (1999). Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 33, 946–958. doi: 10.1046/j.1365-2958.1999.01534.x
Sawires, Y. S., and Songer, J. G. J. A. (2006). Clostridium perfringens: insight into virulence evolution and population structure. Anaerobe 12, 23–43. doi: 10.1016/j.anaerobe.2005.10.002
Seemann, T. J. B. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shimizu, T., Ohtani, K., Hirakawa, H., Ohshima, K., Yamashita, A., Shiba, T., et al. (2002). Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. J. Proc. Natl. Acad. Sci. U. S. A. 99, 996–1001. doi: 10.1073/pnas.022493799
Siah, S. P., Merif, J., Kaur, K., Nair, J., Huntington, P. G., Karagiannis, T., et al. (2014). Improved detection of gastrointestinal pathogens using generalised sample processing and amplification panels. Pathology 46, 53–59. doi: 10.1097/PAT.0000000000000022
Silva-Andrade, C., Martin, A. J., and Garrido, D. J. M. (2022). Comparative genomics of Clostridium baratii reveals Strain-level diversity in toxin abundance. Microorganisms 10:213. doi: 10.3390/microorganisms10020213
Simpson, K. M., Callan, R. J., and Metre, V. (2018). Clostridial abomasitis and enteritis in ruminants. Vet. Clin. North Am. Food Anim. Pract. 34, 155–184. doi: 10.1016/j.cvfa.2017.10.010
Sindern, N., Suchodolski, J. S., Leutenegger, C. M., Mehdizadeh Gohari, I., Prescott, J. F., Proksch, A. L., et al. (2019). Prevalence of Clostridium perfringens netE and netF toxin genes in the feces of dogs with acute hemorrhagic diarrhea syndrome. J Vet Intern Med 33, 100–105. doi: 10.1111/jvim.15361
Songer, J. G. (2010). Clostridia as agents of zoonotic disease. Vet. Microbiol. 140, 399–404. doi: 10.1016/j.vetmic.2009.07.003
Tang, K. L., Caffrey, N. P., Nóbrega, D. B., Cork, S. C., Ronksley, P. E., Barkema, H. W., et al. (2017). Restricting the use of antibiotics in food-producing animals and its associations with antibiotic resistance in food-producing animals and human beings: a systematic review and meta-analysis. Lancet Planet Health. 1, e316–e327. doi: 10.1016/S2542-5196(17)30141-9
Tell, J., Caldwell, D. J., Häner, A., Hellstern, J., Hoeger, B., Journel, R., et al. (2019). Science-based targets for antibiotics in receiving waters from pharmaceutical manufacturing operations. Integr. Environ. Assess. Manag. 15, 312–319. doi: 10.1002/ieam.4141
Uzal, F. A., Freedman, J. C., Shrestha, A., Theoret, J. R., Garcia, J., Awad, M. M., et al. (2014). Towards an understanding of the role ofClostridium perfringenstoxins in human and animal disease. Future Microbiol. 9, 361–377. doi: 10.2217/fmb.13.168
Uzal, F. A., and Songer, J. G. J. (2008). Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J. Vet. Diagn. Invest. 20, 253–265. doi: 10.1177/104063870802000301
Verherstraeten, S., Goossens, E., Valgaeren, B., Pardon, B., Timbermont, L., Haesebrouck, F., et al. (2013a). Perfringolysin O: the underrated Clostridium perfringens toxin? Vet. Res. 44, 1702–1721. doi: 10.1186/1297-9716-44-45
Verherstraeten, S., Goossens, E., Valgaeren, B., Pardon, B., Timbermont, L., Vermeulen, K., et al. (2013b). The synergistic necrohemorrhagic action of Clostridium perfringens perfringolysin and alpha toxin in the bovine intestine and against bovine endothelial cells. Vet. Res. 44, 1–8. doi: 10.1186/1297-9716-44-45
Verma, A. K., Abdel-Glil, M. Y., Madesh, A., Gupta, S., Karunakaran, A. C., Inbaraj, S., et al. (2020). Multilocus sequence typing of Clostridium perfringens strains from neonatal calves, dairy workers and associated environment in India. Anaerobe 63:102212. doi: 10.1016/j.anaerobe.2020.102212
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genom. Biol. 15, R46–R12. doi: 10.1186/gb-2014-15-3-r46
Wróbel, B. J. (2008). Statistical measures of uncertainty for branches in phylogenetic trees inferred from molecular sequences by using model-based methods. J. Appl. Genet. 49, 49–67. doi: 10.1007/BF03195249
Xiao, Y., Wagendorp, A., Moezelaar, R., Abee, T., Wells-Bennik, M. H. J. A., and Microbiology, E. (2012). A wide variety of Clostridium perfringens type A food-borne isolates that carry a ChromosomalcpeGene belong to one multilocus sequence typing cluster. Appl. Environ. Microbiol. 78, 7060–7068. doi: 10.1128/AEM.01486-12
Xiu, L., Zhu, C., Zhong, Z., Liu, L., Chen, S., Xu, W., et al. (2022). Prevalence and multilocus sequence typing of Clostridium perfringens isolated from different stages of a duck production chain. Food Microbiol. 102:103901. doi: 10.1016/j.fm.2021.103901
Xu, W., Zhang, H., Hu, Z., Miao, Z., Zhang, Y., Wang, H. J. V. M., et al. (2021). Prevalence and multilocus sequence typing of Clostridium perfringens isolated from retail chicken products and diseased chickens in Tai'an region China. Vet. Med. Sci. 7, 2339–2347. doi: 10.1002/vms3.616
Yang, W.-Y., Lee, Y.-J., Lu, H.-Y., Branton, S. L., Chou, C.-H., and Wang, C. (2019). The netB-positive Clostridium perfringens in the experimental induction of necrotic enteritis with or without predisposing factors. Poult. Sci. 98, 5297–5306. doi: 10.3382/ps/pez311
Keywords: Clostridium perfringens, intra-species diversity, multilocus sequence typing, genomic epidemiology, toxinotypes
Citation: Camargo A, Guerrero-Araya E, Castañeda S, Vega L, Cardenas-Alvarez MX, Rodríguez C, Paredes-Sabja D, Ramírez JD and Muñoz M (2022) Intra-species diversity of Clostridium perfringens: A diverse genetic repertoire reveals its pathogenic potential. Front. Microbiol. 13:952081. doi: 10.3389/fmicb.2022.952081
Edited by:
Andrew Spiers, Abertay University, United KingdomReviewed by:
Joseph C. S. Brown, Aparon, United KingdomAnn-Katrin Llarena, Norwegian University of Life Sciences, Norway
Copyright © 2022 Camargo, Guerrero-Araya, Castañeda, Vega, Cardenas-Alvarez, Rodríguez, Paredes-Sabja, Ramírez and Muñoz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina Muñoz, claudia.munoz@urosario.edu.co