 Manickam Yogavel
Manickam Yogavel Jyoti Chhibber-Goel
Jyoti Chhibber-Goel Abhishek Jamwal
Abhishek Jamwal Swati Gupta
Swati Gupta Amit Sharma
Amit Sharma- Molecular Medicine - Structural Parasitology Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, India
Malaria parasite erythrocytic stages comprise of repeated bursts of parasites via cyclical invasion of host erythrocytes using dedicated receptor-ligand interactions. A family of erythrocyte-binding proteins from Plasmodium knowlesi (Pk) and Plasmodium vivax (Pv) attach to human Duffy antigen receptor for chemokines (DARC) via their Duffy binding-like domains (DBLs) for invasion. Here we provide a novel, testable and overarching interaction model that rationalizes even contradictory pieces of evidence that have so far existed in the literature on Pk/Pv-DBL/DARC binding determinants. We further address the conundrum of how parasite-encoded Pk/Pv-DBLs recognize human DARC and collate evidence for two distinct DARC integration sites on Pk/Pv-DBLs.
Introduction
Engagements of specific host receptors with parasite-encoded ligands between human erythrocytes and malaria parasite surface proteins are key events in invasion of merozoites into erythrocytes (Cowman et al., 2017). The signature Duffy binding-like domain (DBL) is present in parasite-encoded erythrocyte binding proteins (EBPs) and in protein families like Plasmodium falciparum (Pf) erythrocyte membrane protein 1 (EMP1; Cowman et al., 2017). The latter are part of the var gene family where they assist in cytoadherence—these contain several copies of DBLs in each protein, whereas the Pf-EBP named EBA-175 harbors two copies (F1 and F2) of DBLs (Mayor et al., 2005; Tolia et al., 2005). In contrast EBPs of Plasmodium knowlesi (Pk) and Plasmodium vivax (Pv) contain only single copy of DBL each. There are multiple EBPs present in the malaria parasite Pk (α, β, and γ), and it is the Pkα-DBL and Pv-DBL that mediate Duffy antigen receptor for chemokines (DARC)-dependent invasion of erythrocytes in humans (Choe et al., 2005; Hans et al., 2005; Chitnis and Sharma, 2008). Other DBL variants of Pk-EBPs: β and γ likely use alternate receptors on rhesus/human erythrocytes and may mediate invasion by Duffy antigen-independent pathways (Chitnis and Miller, 1994; Cowman et al., 2017). The Pk/Pv-DBLs (as part of their EBPs) are present on merozoite surface and are responsible for binding to the DARC receptor on reticulocytes (Figure 1) and thence mediating junction formation that is vital for the parasite invasion process (Adams et al., 1990, 1992; Cowman and Crabb, 2006; Cowman et al., 2017).
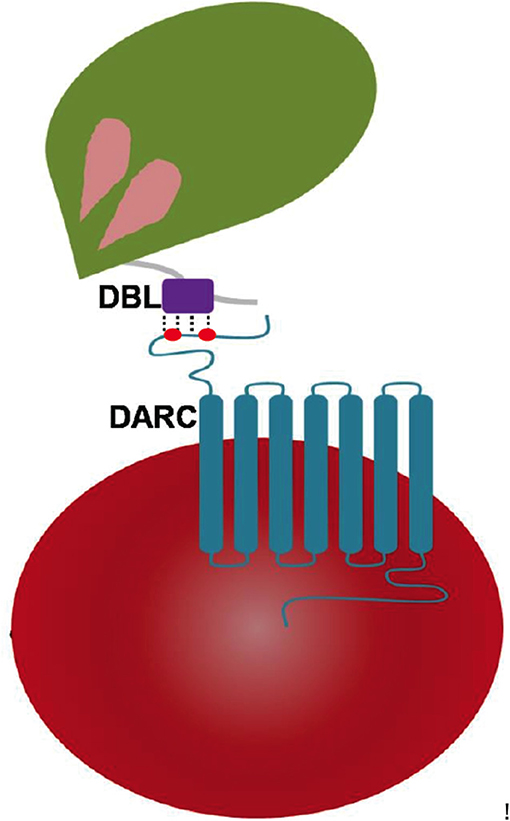
Figure 1. Pictorial representation of Plasmodium DBL domain interaction with 7 transmembrane DARC on human erythrocytes. The DBLs are part of Plasmodium-encoded membrane proteins called erythrocyte-binding proteins (EBPs) that drive receptor recognition between merozoites (green) and RBCs (red). The DARC extracellular domain of residues 1–60 contains two key tyrosyl residues at positions of 30 and 41 that are post-translationally sulfated—and are marked in red dots.
The Pv/Pk-DBLs specifically recognize DARC's extracellular domain and its sulfated tyrosines via intermolecular interactions (Choe et al., 2005; de Brevern et al., 2005; Chitnis and Sharma, 2008). Pv/Pk-DBLs are organized into three sub-domains and are typified (mostly) by 12-cysteine residues that are disulfide linked (Singh et al., 2003, 2006). The importance of Pv-DBL/DARC pairing is underscored by human genetic data where DARC negative individuals tend to be protected from Pv infection (Michon et al., 2001; King et al., 2011). Contrary to the established DBL-DARC invasion pathway, there is evidence for DARC-independent invasion pathway in case of Pv infections, hence calling for new perspectives in assessing the utility of DARC recognizing Pv-DBL as a Pv vaccine candidate (Ryan et al., 2006; Cavasini et al., 2007; Ménard et al., 2010; Wurtz et al., 2011; Ntumngia et al., 2016).
Duffy Antigen Receptor for Chemokines (DARC)
DARC is a seven-transmembrane protein present on surface of erythrocytes and endothelial cells (de Brevern et al., 2005). It is a promiscuous cytokine/chemokine receptor involved in pro-inflammatory processes of the immune system (Rot and von Andrian, 2004; Cowman et al., 2017). DARC is also used as an entry vehicle by malaria parasites Pk and Pv (Miller et al., 1975, 1976; Chitnis and Sharma, 2008). The binding requirements on DARC for (Pv)-DBL interaction have been previously described (Choe et al., 2005; Tournamille et al., 2005). Critical (Pv)-DBL binding residues have been mapped to an N-terminal region of DARC (Tournamille et al., 2005). Latter contains two key tyrosine residues at positions 30 and 41 whose post-translational modifications in the form of sulfations have been reported (Choe et al., 2005; Figure 2A). Sulfated Tyr41 has been considered a dominant motif and essential for (Pv/Pk)-DBL/DARC binding (Choe et al., 2005). As per report sulfated Tyr30 did not affect direct binding of (Pv)-DBL with DARC, but contributed to IL8 association with DARC (Choe et al., 2005).
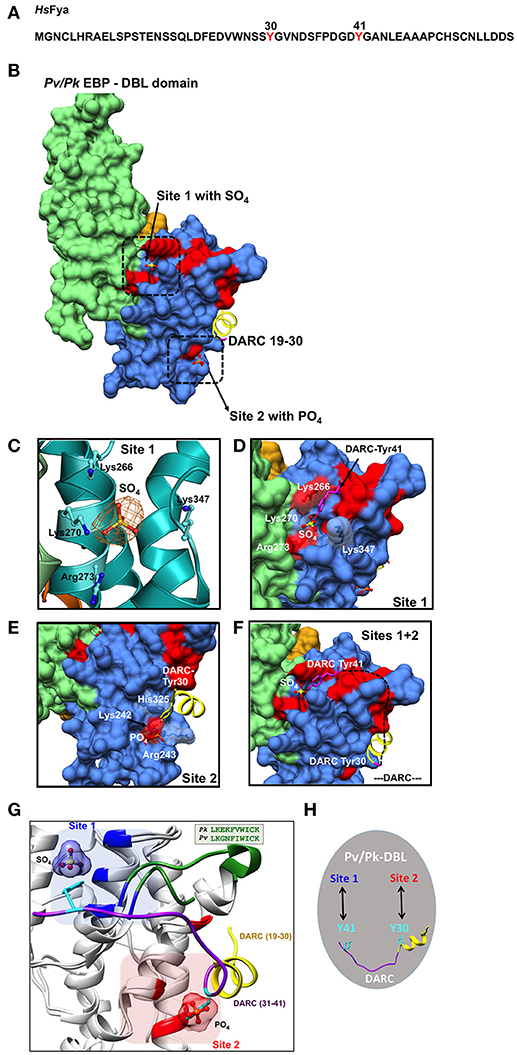
Figure 2. Binding sites on Pk/Pv-DBLs for sulfated tyrosines of DARC. (A) Sequence of human DARC (HsFya) with tyrosyl residues 30 and 41 (red) is shown. Tyr30 and Tyr41 are sulfated in human DARC and it has been proposed that sulfation of Tyr 41 is important for high affinity Pk/Pv-DBL/DARC engagement (Choe et al., 2005). (B) Pk/Pv-DBL subdomains 1 (orange), 2 (blue) and 3 (green) are shown as molecular surfaces. The bound sulfate in Pk-DBL at Site 1, phosphate in Pv-DBL at Site 2, and the N-terminal DARC peptide as ribbon (yellow) for DARC residues 19–30 are shown. Sites 1 and 2 were suggested based on mutagenesis data on Pv-DBL (VanBuskirk et al., 2004; Hans et al., 2005). The sulfate at Site 1, phosphate at Site 2 and DARC residues 19–30 at Site 2 are based on crystal structures of Pv/Pk-DBLs. The Pv-DBL residues whose mutagenesis in two different studies affects the binding of Pv-DBL with DARC are marked in red. The yellow region (19–30 of DARC) is from crystal structure of Pv-DBL/DARC complex. (C) The SA-OMIT (orange) map contoured at 3 σ level for bound SO4 in Pk-DBL (PDB: 5X6N) at Site 1 is shown. The residues Lys 266, Lys 270, Arg 273, and Lys 347 are identical in Pv and Pk-DBLs that bind DARC. Note that the corresponding residue numbers are for Pk-DBL sequence. (D) Zoomed view of Site 1 on Pv/Pk-DBLs with sulfate interacting residues (in white) that are identical between Pk/Pv-DBLs. The DARC Tyr41 (magenta) is modeled to show its proximity to the sulfate position found in crystal structure of Pk-DBL (PDB: 5X6N). The underlying red patches of Pv-DBL residues mark the path taken by those whose mutagenesis in two different studies effects binding of Pv-DBL with DARC (VanBuskirk et al., 2004; Hans et al., 2005). (E) Zoomed view of Site 2 on Pv-DBL modeled after superposition of two Pv-DBL structures (PDB IDs: 3RRC and 4NUV) with bound phosphate and unsulfated DARC peptide (residues 19–30) is shown. Note that the corresponding residue numbers are for Pv-DBL (PDB: 3RRC). The underlying red patches of Pv-DBL residues mark the path taken by the residues whose mutagenesis in two different studies effects binding of Pv-DBL with DARC (VanBuskirk et al., 2004; Hans et al., 2005). (F) Modeling of Pv-DBL with DARC region from 19 to 41 where DARC peptide (19–30, in yellow) and the bound sulfate (from Pk-DBL, in yellow) are shown. The dashed black line (DARC residues 31–40) shows the proposed trajectory of DARC peptide once its Tyr30 is bound to Site 2 and its Tyr41 is bound to Site 1. Again, note that many residues that are critical (colored red) for DARC binding, as revealed by two separate mutagenesis studies, fall along the path from Site 2 to Site 1 resulting in a wide DARC binding footprint (VanBuskirk et al., 2004; Hans et al., 2005). (G) Structural superimposition of Pk-DBL (5X6N) and Pv-DBL (4NUV) that bind DARC are shown in gray ribbons. Sulfate at Site 1 (blue) and Tyr41 (cyan) are shown along with their interacting residues (blue) from Pk-DBL (5X6N). Phosphate at Site 2 (red) and Tyr30 (cyan) are shown along with their interacting residues (red) from Pv-DBL. The loop between α6 and α7 of Pv/Pv-DBLs spanning residues 338–347 (Pk-DBL) and 369–378 (Pv-DBL) are in green. The DARC peptide spanning residues 19–41 (19–30 in yellow and 31–41 in purple) are docked on to Pk/Pv-DBLs such that it interacts via Tyr41 on Site 1 and via Tyr 30 on Site 2. (H) Proposed model on the modes of interaction between Pv/Pk-DBLs and sulfated DARC. It is proposed that soluble region of DARC peptide (from residues 19 to 41), via its sulfated Tyr41 and Tyr30 (cyan), engages at site 1 and 2 respectively on Pv/Pk-DBLs. This testable model is a dual-site mode of DARC's engagement with Pv/Pk-DBLs and can be experimentally assessed.
Binding Sites on Pk/Pv-DBLs for Sulfated Tyrosines of DARC
The Pk/Pv-DBLs and their three subdomains contain numerous disulfide linked cysteine residues that are largely conserved, and these likely contribute to DBLs' structural integrity (Singh et al., 2003, 2006; Gill et al., 2009). Conservation of hydrophobic residues and variance in solvent exposed residues within DBLs possibly allows for a parasite-specific, evolutionarily useful structural motif that can be both constant (in structural terms) and variable (in sequence). Hence, DBLs' seem to have evolved using the same principles as antibody structures, where the overall 3D core structures remain similar but sequence variation in exposed residues and loop regions allows for surface diversity that can thus engage with plethora of bio-molecular receptors.
In efforts to understand the structural underpinnings of Pk/Pv-DBL/DARC complex, the resolved crystal structure of Pk-DBL had suggested a region on its subdomain 2 that could accommodate the DARC's sulfated tyrosine (Singh et al., 2006), and serve as its recognition site (here referred to as Site 1, Figures 2B,C). The Pk-DBL subdomain 2 presents a remarkably surface exposed region of highly conserved residues that arrange into distinct regions lying adjacent to each other—formed by positively charged residues (Lys96, Lys100, Arg103, and Lys177) and non-polars (Tyr94, Leu168, and Ile175; Singh et al., 2006). These two dual-charge/hydrophobicity character surfaces on Pk-DBL were proposed to engage with DARC's sulfated tyrosyl based on structural considerations, sequence conservation patterns, atomic properties, and experimental data emanating from collation of mutagenesis experiments from two distinct groups at the time (VanBuskirk et al., 2004; Hans et al., 2005). Further, the proposed residues on Pk-DBL were invariant in the DARC binding Pv-DBL, hence lending support to the proposal of their essential role in acting as a recognition site for DARC's sulfated tyrosine (VanBuskirk et al., 2004; Choe et al., 2005; Hans et al., 2005; Singh et al., 2006). These data, coupled with the observation that the sulfated Tyr41 of DARC allows strong binding to Pk/Pv-DBLs (Choe et al., 2005) led to a model of DARC's docking onto Pk/Pv-DBLs via the identified site (here labeled as Site 1, Figures 2C,D). The crystal structures of Pv-DBLs (PDBs: 3RRC, 4NUU, and 4NUV) later identified another site for DARC binding, hereafter labeled Site 2 (Figure 2B), where based on (a) the binding of crystallization liquor phosphates (possibly mimicking the sulfate of DARC's sulfated Tyr30), and (b) DARC peptide spanning residues 19–30, a key site for DARC binding was proposed (Batchelor et al., 2011, 2014). In these above studies, it is to be noted that the Pk/Pv-DBLs were bacterially produced, and the DARC peptide used for co-crystallization was evidently not sulfated (see PDBs 4NUU and 4NUV). The Pv-DBL/DARC engagement was further proposed to lead to dimerization of Pv-DBL (Batchelor et al., 2011, 2014). The existence of this DARC binding site (Site 2 in this review) was proposed based on the evidence of bound phosphate (PDB: 3RRC) at the (proposed) dimer interface of Pv-DBL (Batchelor et al., 2011), and then in a latter study of Pv-DBL/DARC complex, several DARC residues (numbers 19–30) were found in proximity to Site 2 (Figures 2B,E)—although the bound Tyr30 was not sulfated (Batchelor et al., 2011, 2014). The above sets of structural and biochemical data led to a conundrum on the location of the binding site for DARC's sulfated Tyr41 on Pk/Pv-DBLs, and its significance in light of a previous report (Choe et al., 2005) that had suggested that sulphation of Tyr41 was an essential factor for coupling of DARC with Pk/Pv-DBLs (Choe et al., 2005). We shall return to this in a later section.
It has been also proposed that Pv-DBL engagement with DARC extracellular domain drives dimerization of Pv-DBLs (PDB IDs: 3RRC, 4NUU, and 4NUV; Batchelor et al., 2011, 2014). Extensive analysis using PISA (http://www.ebi.ac.uk/pdbe/pisa/) and EPPIC (http://www.eppic-web.org/), two software that analyze the oligomeric states of biomolecular structures deposited in the public database PDB (Krissinel and Henrick, 2007; Duarte et al., 2012; Baskaran et al., 2014), does not strongly support dimerization of Pv-DBL when bound to phosphate or to the DARC peptide spanning residues 19–30 (we encourage readers to assess this using online PISA and EPPIC software and the PDBs: 3RRC, 4NUU, and 4NUV). However, we do not preclude the oligomerization of Pv-DBLs when bound to DARC in vivo, and/or in the context of full-length proteins of each when they interact on their respective cell surfaces. Yet from the available structural data in PDB, it can be construed that the Pv/Pk-DBL monomer contains most vital structural elements required for engagement with two sites on sulfated DARC. Indications of dimeric states gathered from in solution studies like SAXS, ITC and analytical ultracentrifugation do not necessarily reveal the exact binding interfaces in a biomolecular complex. The structural resolution of Pk/Pv-EBPs bound to fully sulfated DARC will eventually reveal the modes of complex formation between these receptor ligand pairs.
A New Overarching Model for Coupling of Pk/Pv-DBLs With DARC
The Pv-DBL (PDB: 3RRC) has two bound phosphates near/at Site 2, where later DARC peptide residues (19–30) were also found (PDBs: 4NUU, 4NUV). During process of routine structure re-refinement of deposited crystal structures, as recommended (Read et al., 2011) due to the more robust convergence routines and higher quality of resulting stereochemical models for proteins, we observed a bound sulfate at Site 1 in the Pk-DBL crystal (PDB: 5X6N, Figures 2B,C). The bound sulfate site overlaps with the earlier proposed sulfa-tyrosine recognition region (Singh et al., 2006), nestled by highly conserved positively charged residues—most of which were earlier mapped based on mutagenesis screens (VanBuskirk et al., 2004; Choe et al., 2005; Hans et al., 2005; Singh et al., 2006). Indeed, both Pv/Pk-DBLs have other phosphate/sulfate binding sites in their crystal structures but we have taken into account only those that are proximal to the regions where mutagenesis and biochemical data together provide strong evidence for DARC engagement (VanBuskirk et al., 2004; Hans et al., 2005; Sampath et al., 2013). The Pk-DBL protein migrates as a monomer on gel filtration column in the absence of detergent, and in the crystal structure (PDB: 5X6N) it is also an unambiguous monomer—even when bound to a SO4 in Site 1 (PDB ID: 5X6N and kindly assess via PISA/EPPIC). The likely irrelevant lone detergent binding site in Pk-DBL (PDB: 5X6N) is distal from both Sites 1 and 2. The presence of bound sulfate at Site 1 in Pk-DBL is clearly a striking observation, and it immediately opens the possibility of reassessing and reinterpreting the engagement rules for DARC recognition and binding by Pk/Pv-DBLs.
Therefore, based on (a) Pk-DBL-sulfate complex (Site 1, Figures 2B–D), (b) Pv-DBL-phosphate complex (Site 2, Figures 2B,E), (c) Pv-DBL-DARC peptide complex (Site 2, Figures 2B,E) and available mutagenesis and biochemical data on Pv-DBL/DARC interactions (VanBuskirk et al., 2004; Hans et al., 2005; Sampath et al., 2013), we are able to propose a simple, novel, testable and largely rationalized resolution to the conundrum of Pk/Pv-DBL/DARC coupling (Figures 2F,G). We envisage that the DARC peptide 1–60 may dock on to Pk/Pv-DBLs via its sulfated Tyr41 on Site 1 (Figures 2B,F,H; PDB: 5X6N) and on to Site 2 via the DARC peptide containing residues 19 to 30 (Figures 2F,G; PDB 4NUV). Structural, molecular size, and stereochemical considerations will allow the DARC peptide (spanning residues 19–41) to traverse from Site 2 (where Tyr 30 is) to Site 1 (where Tyr 41 is) on Pk/Pv-DBLs. Site 1 on Pk/Pv-DBLs is located on N-cap region of α2 helix and the loop between α6 and α7 helices, while Site 2 is rests on α1, α5, and α6 helices (Figure 2G). The loop (residues: 338–347 in Pk-DBL, 369–378 in Pv-DBL) between α6 and α7 is highly conserved in Pk/Pv-DBLs (Figure 2G). It is noteworthy that this loop is partly disordered in Pv-DBL (4NUV), but is ordered in sulfate bound crystal structure of Pk-DBL (5X6N).
A dual-site DARC hooking on to Pk/Pv-DBLs offers a more complete binding footprint of DARC (Figure 2G). Indeed, the molecular size of a sulfated tyrosine (from its C-α to sulfate) is ~14Å (Figure 2C). Structural modeling of the atomic space between Pk-DBL's Tyr94 (Site 1 residue, i.e., Tyr 264, based on full length Pk-DBL, as in Singh et al., 2006) and the bound sulfate shows that a sulfated tyrosine will fit snugly into it (Figure 2C). Further, the side chains in Pk/Pv-DBLs that constitute Site 1 are identical (4/4), or mostly (2/3) conserved for Site 2 (Figures 2D,E). Invariance in Site 1 residues of Pk/Pv-DBLs: Lys96, Lys100, Arg103, and Lys 177 (Pk-DBL numbering, as in Singh et al., 2006) that nestle (via charge complementarity) the bound sulfate moiety (Figures 2C,D), and its potential as a sulfa-tyrosine recognition site based on earlier mutagenesis experiments together provide very strong support for the significance of Site 1 in Pk/Pv-DBLs/DARC interactions. The above analyses hence propose that both Sites 1 and 2 are part of the overall binding footprint of DARC on Pv/Pk-DBLs, and that Sites 1 and 2 likely represent the regions of engagement where sulfated Tyr41 and Tyr30 (respectively) are recognized by the Pv/Pk-DBLs (Figures 2F,G). The intervening regions of Pv/Pk-DBL and DARC will also likely make contact with one another, and therefore the mutagenesis footprint on DBLs will be larger than that alone for Sites 1 and 2—as indeed seems to be the case, and shall be discussed below (VanBuskirk et al., 2004; Hans et al., 2005).
Earlier, single amino acid mutagenesis data had suggested over ~two dozen mutations that could effect binding of DARC to Pv-DBL (VanBuskirk et al., 2004; Hans et al., 2005). Based on comprehensive collation of structural and experimental data, we now propose a model that shows that majority of the 25 mutations (VanBuskirk et al., 2004; Hans et al., 2005) that diminish Pv-DBL/DARC engagement fall in a predicted trajectory that DARC peptide may take when traversing from Site 1 from Site 2 (Figures 2F,G). Two independent mutagenesis screens had earlier identified several sets of residues on Pv-DBL that altered binding to DARC—several of these residues line and nestle the sulfate in Pk-DBL at Site 1, besides being proximal to Site 2 (VanBuskirk et al., 2004; Hans et al., 2005). Further support for our model comes from a glycan masking study that was focused on Pv-DBL/DARC interactions wherein it was suggested that addition of an N-glycan site near the proposed Site 1 (our proposed model), but not in other regions of Pv-DBL, including the proposed dimer interface site (Site 2), abrogated DARC binding (Sampath et al., 2013). These data therefore additionally support our envisaged model of twin engagement of DARC via its sulfated Tyr41 at Site 1 and with DARC residues 19–30 at Site 2. Again, a comprehensive mapping of mutagenesis data support the idea that DARC residues 19–41 (via Tyr 30 and Tyr 41) traverse from Site 2 toward Site 1 along the path where numerous mutants map (Figures 2F,G; VanBuskirk et al., 2004; Hans et al., 2005).
It needs to be emphasized again that our proposed model is not discounting the importance of DARC (residues 19–30) identified earlier (Tournamille et al., 2005; Batchelor et al., 2014). Indeed, our new suggested structural framework model of Pk/Pv-DBL/DARC interactions supports, collates and integrates many previous studies (VanBuskirk et al., 2004; Choe et al., 2005; Hans et al., 2005; Singh et al., 2006; Batchelor et al., 2011, 2014). Our model is consistent with most mutagenesis and crystallography work published so far, and provides a more comprehensive explanation for DARC's binding via its sulfated Tyr30 and Tyr41 to two binding sites (Sites 1 and 2) each on Pk/Pv-DBLs (Figures 2G,H). The proposed architecture of the complex sheds new light on DBL-DARC interactions, and our overview suggests clear avenues for experimental validation of the proposal. Our model also offers scope to make new sets of site-directed mutants in Pk/Pv-DBLs that can directly address the above hypotheses using the available 3D structures.
Finally, the indicated structural framework model of Pk/Pv-DBL-DARC significantly resolves the puzzle of recognition site(s) that underpin this complex. Our analyses integrate new evidence for two distinct DARC binding sites on Pk/Pv-DBLs (called Sites 1 and 2, in the chronological order they were described in the literature) that together likely engage the extracellular domain of DARC via sulfated Tyr41 and Tyr30, respectively (Figures 2G,H). Our analyses shall be important for critical assessment of both malaria vaccine and inhibitor development efforts that are targeted at abrogating Pk/Pv-DBL-DARC interactions as a possible avenue to prevent invasion of Pk/Pv malaria parasites into human erythrocytes.
Author Contributions
MY, JC-G, AJ, and SG analyzed the data and prepared figures. MY, JC-G, and AS wrote and finalized the manuscript. MY and AS designed the study.
Funding
AS is supported by JC Bose fellowship and funded by Department of Science and Technology, Government of India.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, J. H., Hudson, D. E., Torii, M., Ward, G. E., Wellems, T. E., Aikawa, M., et al. (1990). The Duffy receptor family of Plasmodium knowlesi is located within the micronemes of invasive malaria merozoites. Cell 63, 141–153. doi: 10.1016/0092-8674(90)90295-P
Adams, J. H., Sim, B. K., Dolan, S. A., Fang, X., Kaslow, D. C., and Miller, L. H. (1992). A family of erythrocyte binding proteins of malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 89, 7085–7089. doi: 10.1073/pnas.89.15.7085
Baskaran, K., Duarte, J. M., Biyani, N., Bliven, S., and Capitani, G. (2014). A PDB-wide, evolution-based assessment of protein-protein interfaces. BMC Struct. Biol. 14:22. doi: 10.1186/s12900-014-0022-0
Batchelor, J. D., Malpede, B. M., Omattage, N. S., DeKoster, G. T., Henzler-Wildman, K. A., and Tolia, N. H. (2014). Red blood cell invasion by Plasmodium vivax: structural basis for DBP engagement of DARC. PLoS Pathog. 10:e1003869. doi: 10.1371/journal.ppat.1003869
Batchelor, J. D., Zahm, J. A., and Tolia, N. H. (2011). Dimerization of Plasmodium vivax DBP is induced upon receptor binding and drives recognition of DARC. Nat. Struct. Mol. Biol. 18, 908–914. doi: 10.1038/nsmb.2088
Cavasini, C. E., Mattos, L. C., Couto, A. A., Bonini-Domingos, C. R., Valencia, S. H., Neiras, W. C., et al. (2007). Plasmodium vivax infection among Duffy antigen-negative individuals from the Brazilian Amazon region: an exception? Trans. R. Soc. Trop. Med. Hyg. 101, 1042–1044. doi: 10.1016/j.trstmh.2007.04.011
Chitnis, C. E., and Miller, L. H. (1994). Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J. Exp. Med. 180, 497–506. doi: 10.1084/jem.180.2.497
Chitnis, C. E., and Sharma, A. (2008). Targeting the Plasmodium vivax Duffy-binding protein. Trends Parasitol. 24, 29–34. doi: 10.1016/j.pt.2007.10.004
Choe, H., Moore, M. J., Owens, C. M., Wright, P. L., Vasilieva, N., Li, W., et al. (2005). Sulphated tyrosines mediate association of chemokines and Plasmodium vivax Duffy binding protein with the Duffy antigen/receptor for chemokines (DARC). Mol. Microbiol. 55, 1413–1422. doi: 10.1111/j.1365-2958.2004.04478.x
Cowman, A. F., and Crabb, B. S. (2006). Invasion of red blood cells by malaria parasites. Cell 124, 755–766. doi: 10.1016/j.cell.2006.02.006
Cowman, A. F., Tonkin, C. J., Tham, W. H., and Duraisingh, M. T. (2017). The molecular basis of erythrocyte invasion by malaria parasites. Cell Host Microbe 22, 232–245. doi: 10.1016/j.chom.2017.07.003
de Brevern, A. G., Wong, H., Tournamille, C., Colin, Y., Le Van Kim, C., and Etchebest, C. (2005). A structural model of a seven-transmembrane helix receptor: the Duffy antigen/receptor for chemokine (DARC). Biochim. Biophys. Acta 1724, 288–306. doi: 10.1016/j.bbagen.2005.05.016
Duarte, J. M., Srebniak, A., Schärer, M. A., and Capitani, G. (2012). Protein interface classification by evolutionary analysis. BMC Bioinformatics 13:334. doi: 10.1186/1471-2105-13-334
Gill, J., Chitnis, C. E., and Sharma, A. (2009). Structural insights into chondroitin sulphate a binding duffy-binding-like domains from plasmodium falciparum: implications for intervention strategies against placental malaria. Malar. J. 8:67. doi: 10.1186/1475-2875-8-67
Hans, D., Pattnaik, P., Bhattacharyya, A., Shakri, A. R., Yazdani, S. S., Sharma, M., et al. (2005). Mapping binding residues in the Plasmodium vivax domain that binds Duffy antigen during red cell invasion. Mol. Microbiol. 55, 1423–1434. doi: 10.1111/j.1365-2958.2005.04484.x
King, C. L., Adams, J. H., Xianli, J., Grimberg, B. T., McHenry, A. M., Greenberg, L. J., et al. (2011). Fy(a)/Fy(b) antigen polymorphism in human erythrocyte duffy antigen affects susceptibility to Plasmodium vivax malaria. Proc. Natl. Acad. Sci. U.S.A. 108, 20113–20118. doi: 10.1073/pnas.1109621108
Krissinel, E., and Henrick, K. (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. doi: 10.1016/j.jmb.2007.05.022
Mayor, A., Bir, N., Sawhney, N., Singh, S., Pattnaik, P., Singh, S., et al. (2005). Receptor-binding residues lie in central regions of Duffy-binding-like domains involved in red cell invasion and cytoadherence by malaria parasites. Blood 105, 2557–2563. doi: 10.1182/blood-2004-05-1722
Ménard, D., Barnadas, C., Bouchier, C., Henry-Halldin, C., Gray, L. R., Ratsimbasoa, A., et al. (2010). Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc. Natl. Acad. Sci. U.S.A. 107, 5967–5971. doi: 10.1073/pnas.0912496107
Michon, P., Wolley, I., Wood, E. M., Kastens, W., Zimmerman, P. A., and Adams, J. H. (2001). Duffy-null promoter heterozygosity reduces DARC expression and abrogates adhesion of the P. vivax ligand required for blood-stage infection. FEBS Lett. 495, 111–114. doi: 10.1016/S0014-5793(01)02370-5
Miller, L. H., Mason, S. J., Clyde, D. F., and McGinniss, M. H. (1976). The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 295, 302–304. doi: 10.1056/NEJM197608052950602
Miller, L. H., Mason, S. J., Dvorak, J. A., McGinniss, M. H., and Rothman, I. K. (1975). Erythrocyte receptors for (Plasmodium knowlesi) malaria: duffy blood group determinants. Science 189, 561–563. doi: 10.1126/science.1145213
Ntumngia, F. B., Thomson-Luque, R., Torres Lde, M., Gunalan, K., Carvalho, L. H., and Adams, J. H. (2016). A novel erythrocyte binding protein of Plasmodium vivax suggests an alternate invasion pathway into duffy-positive reticulocytes. MBio 7:e01261-16. doi: 10.1128/mBio.01261-16
Read, R. J., Adams, P. D., Arendall, W. B. III., Brunger, A. T., Emsley, P., Joosten, R. P., et al. (2011). A new generation of crystallographic validation tools for the protein data bank. Structure 19, 1395–1412. doi: 10.1016/j.str.2011.08.006
Rot, A., and von Andrian, U. H. (2004). Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 22, 891–928. doi: 10.1146/annurev.immunol.22.012703.104543
Ryan, J. R., Stoute, J. A., Amon, J., Dunton, R. F., Mtalib, R., Koros, J., et al. (2006). Evidence for transmission of Plasmodium vivax among a duffy antigen negative population in Western Kenya. Am. J. Trop. Med. Hyg. 75, 575–581.
Sampath, S., Carrico, C., Janes, J., Gurumoorthy, S., Gibson, C., Melcher, M., et al. (2013). Glycan masking of Plasmodium vivax duffy binding protein for probing protein binding function and vaccine development. PLoS Pathog. 9:e1003420. doi: 10.1371/journal.ppat.1003420
Singh, S. K., Hora, R., Belrhali, H., Chitnis, C. E., and Sharma, A. (2006). Structural basis for Duffy recognition by the malaria parasite Duffy-binding-like domain. Nature 439, 741–744. doi: 10.1038/nature04443
Singh, S. K., Singh, A. P., Pandey, S., Yazdani, S. S., Chitnis, C. E., and Sharma, A. (2003). Definition of structural elements in Plasmodium vivax and P. knowlesi Duffy-binding domains necessary for erythrocyte invasion. Biochem. J. 374, 193–198. doi: 10.1042/bj20030622
Tolia, N. H., Enemark, E. J., Sim, B. K., and Joshua-Tor, L. (2005). Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell 122, 183–193. doi: 10.1016/j.cell.2005.05.033
Tournamille, C., Filipe, A., Badaut, C., Riottot, M. M., Longacre, S., Cartron, J. P., et al. (2005). Fine mapping of the Duffy antigen binding site for the Plasmodium vivax Duffy-binding protein. Mol. Biochem. Parasitol. 144, 100–103. doi: 10.1016/j.molbiopara.2005.04.016
VanBuskirk, K. M., Sevova, E., and Adams, J. H. (2004). Conserved residues in the Plasmodium vivax duffy-binding protein ligand domain are critical for erythrocyte receptor recognition. Proc. Natl. Acad. Sci. U.S.A. 101, 15754–15759. doi: 10.1073/pnas.0405421101
Keywords: Duffy antigen receptor for chemokines (DARC), Duffy binding-like domains (DBLs), malaria, Plasmodium, Structure
Citation: Yogavel M, Chhibber-Goel J, Jamwal A, Gupta S and Sharma A (2018) Engagement Rules That Underpin DBL-DARC Interactions for Ingress of Plasmodium knowlesi and Plasmodium vivax Into Human Erythrocytes. Front. Mol. Biosci. 5:78. doi: 10.3389/fmolb.2018.00078
Received: 22 June 2018; Accepted: 03 August 2018;
Published: 27 August 2018.
Edited by:
Luis G. Brieba, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoReviewed by:
Rogerio R. Sotelo-Mundo, Center for Research in Food and Development (CIAD), MexicoRenato Augusto DaMatta, State University of Norte Fluminense, Brazil
Copyright © 2018 Yogavel, Chhibber-Goel, Jamwal, Gupta and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Sharma, YW1pdHBzaGFybWE2OEBnbWFpbC5jb20=