Mammalian Resilience Revealed by a Comparison of Human Diseases and Mouse Models Associated With DNA Helicase Deficiencies
 Masaoki Kohzaki
Masaoki Kohzaki- Department of Radiobiology and Hygiene Management, Institute of Industrial Ecological Sciences, University of Occupational and Environmental Health Japan, Kitakyushu, Japan
Maintaining genomic integrity is critical for sustaining individual animals and passing on the genome to subsequent generations. Several enzymes, such as DNA helicases and DNA polymerases, are involved in maintaining genomic integrity by unwinding and synthesizing the genome, respectively. Indeed, several human diseases that arise caused by deficiencies in these enzymes have long been known. In this review, the author presents the DNA helicases associated with human diseases discovered to date using recent analyses, including exome sequences. Since several mouse models that reflect these human diseases have been developed and reported, this study also summarizes the current knowledge regarding the outcomes of DNA helicase deficiencies in humans and mice and discusses possible mechanisms by which DNA helicases maintain genomic integrity in mammals. It also highlights specific diseases that demonstrate mammalian resilience, in which, despite the presence of genomic instability, patients and mouse models have lifespans comparable to those of the general population if they do not develop cancers; finally, this study discusses future directions for therapeutic applications in humans that can be explored using these mouse models.
Role of DNA Helicases in Maintaining Genome Integrity
DNA helicase was first discovered in 1970 (Abdel-Monem et al., 1976; Mackay and Linn, 1976) as an enzyme that can move along a nucleic acid as a motor protein that unwinds and unpacks the condensed genomic DNA compacted by chromatin in an ATP-dependent manner (Lusser and Kadonaga, 2003; Clapier et al., 2017). In addition, the ATPases associated with various cellular activities’ (AAA+) domain can drive the conformational changes at the interface of neighboring subunits mediated by ATP binding and hydrolysis, sometimes forming complexes, for efficient remodeling or translocation of target substrates (Singleton et al., 2007). These unwinding and translocation activities of DNA helicase are important for many aspects of genomic metabolism, including DNA recombination, replication, repair, and transcription (Kogoma, 1997), and are indispensable for maintaining genomic integrity, which is tightly related to cancer progression and aging mechanisms (Mohaghegh and Hickson, 2001; Hoeijmakers, 2009). The human genome encodes over 31 DNA helicases and 64 RNA helicases (Umate et al., 2011). DEAD (Asp-Glu-Ala-Asp) box and related DExD/H RNA helicases and DNA–RNA helicases are involved in transcription, resolving transcription–replication conflicts, and G-quadruplex structures (Tanner and Linder, 2001). There are excellent reviews on RNA helicases (Bourgeois et al., 2016), which are not addressed in this review. Human DNA helicases are classified into six superfamilies (SF1–SF6) based on their shared sequence motifs (Gorbalenya et al., 1989; Singleton et al., 2007).
Human Diseases Associated With Hereditary Mutations in DNA Helicases and Mouse Models
It has been reported that hereditary mutations in DNA helicases can cause human diseases. The RECQ helicase family is one of the most well-known diseases associated with DNA helicase deficiencies (Croteau et al., 2014). The DNA helicase BRCA1-associated C-terminal helicase/BRCA1-interacting protein 1 (BACH1/BRIP1) is inactivated in patients with Fanconi anemia (FA); BACH1 is also known as FA complementation group (FANCJ) (Bridge et al., 2005; Levitus et al., 2005; Levran et al., 2005; Litman et al., 2005). FA is characterized by bone marrow failure, cellular hypersensitivity to DNA interstrand cross-linkers (ICLs), multiple congenital abnormalities, and cancer predisposition (Auerbach, 2009). Xeroderma pigmentosum type B (XPB) and type D (XPD) DNA helicases are involved in transcription-coupled DNA repair (Drapkin et al., 1994; Coin et al., 2007), and mutations in XPB and XPD helicases are found in patients with Cockayne syndrome (CS), trichothiodystrophy (TTD), and xeroderma pigmentosum (XP) (Lehmann A. R, 2001; Kraemer et al., 2007). Due to the biological importance of these diseases, various animal models, mainly using mice, have been established to elucidate their pathogenic mechanisms and show various symptoms related to vital function maintenance, carcinogenesis, and aging. In the following sections, I discuss the diseases caused by deficiencies in each DNA helicase and the mouse models corresponding to these diseases. Some DNA helicases, including Ku70/XRCC6 and PCNA-associated recombination inhibitor (PARI), were omitted due to uncertainty regarding their DNA helicase activity. Representative genome alterations in DNA helicases associated with human diseases are described in Figure 1, which shows specific protein domains and interacting proteins. The relationship between human and mouse is described in Table 1 and shown in the survival curve images in Figure 2. Moreover, a simplified phylogenetic tree of the evolutionary relationships of human helicases comprising the major families SF1–6 is shown in Figure 3 to demonstrate the relative relationships among the DNA helicases presented in this review. The table and figures provide additional information about the mechanisms of diseases caused by DNA helicase deficiencies.

FIGURE 1. Comparison of DNA helicases with alterations that cause human diseases. DNA helicases are divided into six superfamilies, and they are differently colored as follows: SF1, light orange; SF2, yellow; SF4, light pink; and SF6, orange. The domains characteristics of some proteins are color-coded and the names are written below the domains. The representative interacting proteins are marked with green squares at the interaction site of the DNA helicase of interest. All of the alterations including point mutations, frameshift mutations, missense mutation, nonsense mutations, silent mutations, splice mutations, deletions, insertions, duplications, and repeat elongations are marked with asterisks above the protein structure for simplicity. It is noted that the scale and the position of alterations are not exactly drawn. Please see the references for the exact location and type of each alteration.
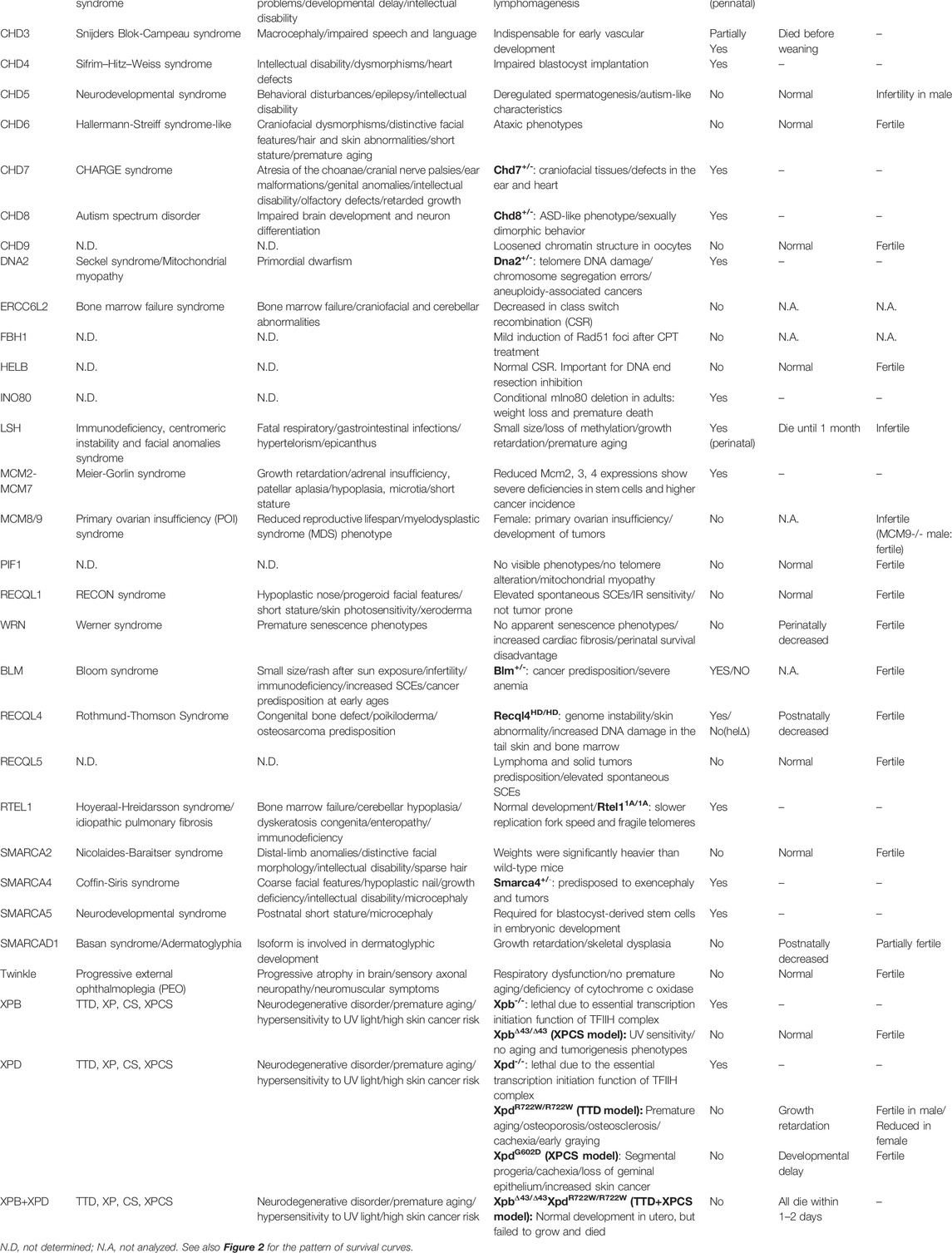
TABLE 1. Human diseases, human symptoms, and mouse phenotypes in DNA helicase deficiencies.
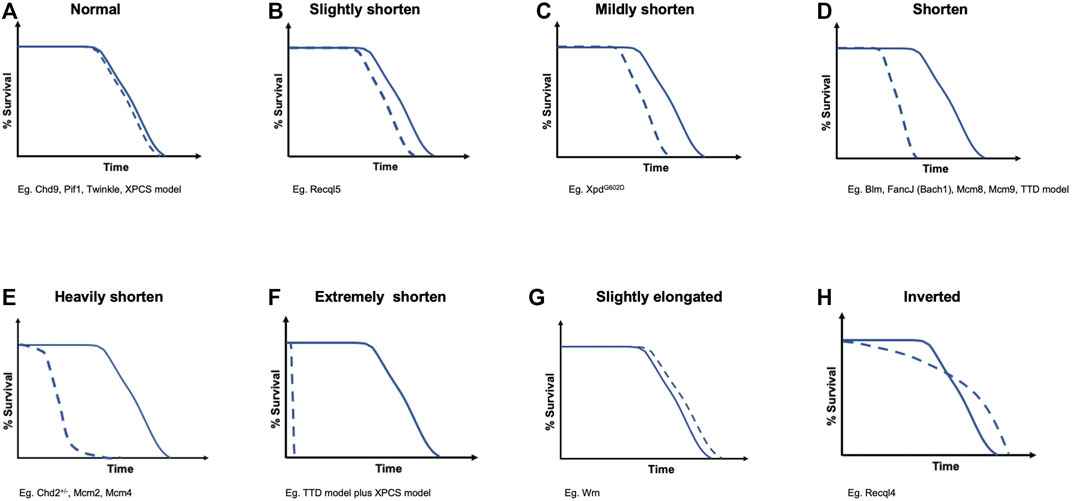
FIGURE 2. Different survival curves induced by DNA helicase deficiencies. They are categorized into eight groups as follows: (A) normal; (B) slightly shorten; (C) mildly shorten; (D) shorten; (E) heavily shorten; (F) extremely shorten; (G) slightly elongated; and (H) inverted. These categories correspond to the survival curve items in Table 1
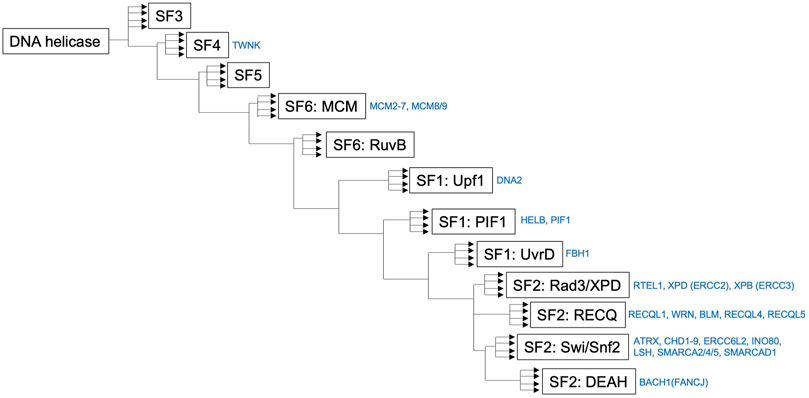
FIGURE 3. Simplified phylogenetic tree showing the evolutionary relationships of human helicases comprising the major families SF1–6. The figure was made by referring the reported figure by Jackson et al. (2014). It should be noted that a simplified phylogenetic tree does not describe the precise evolutionary time scale, and it can be used as a relative relationship for the DNA helicases presented in this review.
ATRX
ATRX belongs to the Snf2 subfamily of Swi/Snf in the superfamily 2 and shows translocation activity on dsDNA as the ATRX–DAXX complex (Xue et al., 2003; Hargreaves and Crabtree, 2011; Byrd and Raney, 2012). Both the helicase activity of ATRX and interaction with DAXX are indispensable for protection from G4-induced stress (Teng et al., 2021). ATRX shares sequences with DNMT3A, DNMT3B, and DNMT3L, which are involved in DNA methylation, and are referred to as the ATRX-DNMT3-DNMT3L (ADD) domain (Argentaro et al., 2007). Both the ADD domain and helicase domains are hotspots for mutations associated with ATR-X syndrome (Figure 1). The ADD domain interacts with PCNA (Juhász et al., 2018), and the DAXX-interacting domain (DID) interacts with death domain-associated protein (DAXX) (Tang et al., 2004), a chaperon protein. These interactions are required for DNA repair synthesis during HR and chromatin remodeling, respectively. Alpha thalassemia X-linked intellectual disability (ATR-X) syndrome shows severe psychomotor retardation, genital abnormalities, alpha-thalassemia, and characteristic facial features (Gibbons et al., 2000). The phenotypes of ATR-X syndromes might result from changes in global transcriptional regulation (Gibbons et al., 1995). ATR-X syndrome changes the DNA hypomethylation pattern at rDNA, and ATRX is required for meiotic organization in mouse oocytes (Gibbons et al., 2000; De La Fuente et al., 2004).
Atrx knockout is embryonic lethal in mice, and conditional Atrx knockout in the forebrain causes extensive hypocellularization of the hippocampus and neocortex with a marked reduction in forebrain size (Bérubé et al., 2005).
BACH1 (BRIP1 and FANCJ)
BACH1 belongs to the DEAH subfamily of superfamily 2 and shows translocation activity on ssDNA and unwind dsDNA in a 5′-to-3′ direction (Byrd and Raney, 2012). While the M299I mutant protein, a breast cancer variant, can more robustly unwind and translocate DNA than wild-type BACH1 (Sommers et al., 2009), the A349P mutant protein, which is required for DNA repair, loses ATP hydrolysis coupling for the efficient unwinding of forked structures and translocation activity on DNA (Wu et al., 2010). These results suggest that an appropriate helicase activity of BACH1 is necessary for physiological function and tumor suppression. BACH1 exists as either monomer or dimer (Wu et al., 2012). FANCJ is a protein encoded by the BRCA1-interacting protein (BRIP1) gene (Bridge et al., 2005; Levitus et al., 2005) and plays a role in DNA repair by interacting directly with BRCA1 (Cantor et al., 2001). In addition, the C-terminus of BACH1 interacts with BLM, MRE11, and TOPBP1 (Brosh and Cantor, 2014). Indeed, there are many mutations in the sites that interact with these proteins and the helicase domain (Figure 1). Monoallelic carriers of BRIP1 truncation mutations show susceptibility to breast cancer (Seal et al., 2006) and ovarian cancer (Rafnar et al., 2011), similar to those with BRCA gene mutations.
All the reported FA gene knockout mice are viable (Parmar et al., 2009) and have reduced fertility due to impaired gametogenesis (Parmar et al., 2009). Interestingly, single FA gene knockout mice do not show anemia, skeletal defects, or tumor progression (Parmar et al., 2009); however, these knockout mice exhibit accelerated epithelial tumor formation when mated with p53+/- mice (Houghtaling et al., 2005). In line with the characteristics of FA, Brip1 knockout mice are viable and fertile with very small testes, reflecting impaired spermatogonial proliferation (Sun et al., 2016). In contrast, Matsuzaki et al. independently established Fancj knockout mice that exhibit phenotypes distinct from the canonical FA phenotype, in which mitomycin C (MMC) sensitivity and lymphoma predisposition are increased (Matsuzaki et al., 2015).
CHD1–CHD9
CHD1–CHD9 belong to the Snf2 subfamily of Swi/Snf in superfamily 2 and show translocation activity on dsDNA (Hargreaves and Crabtree, 2011; Byrd and Raney, 2012). The ATPase domain of the Swi/Snf chromatin remodelers resembles the DEAD/H helicases and is required for DNA translocation activity despite no evidence of helicase activity (Hargreaves and Crabtree, 2011). SNF2 superfamily chromodomain helicase DNA-binding (Chd) family enzymes can change chromatin structure during cell metabolism for processes including DNA repair, replication, and transcription and are therefore implicated in human diseases (Marfella and Imbalzano, 2007). Proteins belonging to this family contain double chromodomains, the SNF2 N-terminal (ATPase helicase domain), and helicase C-terminal domains, which are critical for chromatin remodeling activity (Wilson et al., 2021). These CHD family proteins are divided into three subfamilies based on their additional domains, such as the DNA binding, PHD zinc finger-like, BRK (Brahma and Kismet domain), and SANT (Swi3, Ada2, N-Cor, and TFIIIB) domains (Marfella and Imbalzano, 2007) (Figure 1).
CHD1
CHD1 (chromodomain helicase DNA-binding protein 1) exists predominantly as a monomer and plays a role in ATP-dependent chromatin assembly (Lusser et al., 2005). Chd1 interacts with Rtf1, a Paf1/RNA polymerase II complex component, while Spt4-Spt5, and Spt16-Pob3 and regulate transcription elongation (Simic et al., 2003). CHD1 also regulates mouse embryonic stem cell pluripotency by opening euchromatin in mice (Gaspar-Maia et al., 2009) through histone H3 methylation (Pray-Grant et al., 2005). CHD1 plays a role in DNA repair by homologous recombination (HR) (Kari et al., 2016). In addition, CHD1 missense mutations cause neurodevelopmental phenotypes, including autism, developmental delay, facial dysmorphic features, and speech apraxia, collectively referred to as Pilarowski–Bjornsson Syndrome (Pilarowski et al., 2018).
Chd1−/− mice show embryonic lethality even in the absence of p53 mutations (Guzman-Ayala et al., 2015).
CHD2
The interaction between CHD2 and PARP1 is required for chromatin expansion and H3.3 deposition at DNA damage sites for efficient NHEJ repair (Luijsterburg et al., 2016). CHD2 is implicated in epileptic encephalopathies (Carvill et al., 2013), which are associated with self-induced seizures (Thomas et al., 2015). In addition, CHD2 haploinsufficiency can induce not only epilepsy but also behavioral problems, developmental delay, and intellectual disability (Chénier et al., 2014). CHD2 is required for neural circuit development and long-term memory (Kim et al., 2018).
Homozygous mutations in the Chd2 gene result in perinatal lethality in mice (Marfella et al., 2006). Heterozygous mutations in the Chd2 gene result in decreased neonatal viability and a shorter life span; various organs, including the kidney, are affected in Chd2+/− mice (Marfella et al., 2006). One group reported that Chd2+/− mice develop scoliosis (Kulkarni et al., 2008), and another revealed that these mice exhibit a shorter lifespan, susceptibility to lymphomas and increased extramedullary hematopoiesis (Nagarajan et al., 2009).
CHD3
Both CHD3 and CHD4 are factors of the ATP-dependent chromatin remodeling Mi-2/NuRD (nucleosome remodeling deacetylase) complex, which includes HDAC1/2, MTA1/2/3, RbAp46/48, and MDB2/3 (Xue et al., 1998). The SUMO1 interacting motif (SIM) located at the C terminal of CHD3 binds to SUMOylated KRAB-associated protein-1 (KAP-1), which mediates transcriptional regulation and DNA damage repair at specific chromatin regions. This interaction is perturbed when KAP-1 is phosphorylated at serine 824 by ATM after DNA damage for efficient DNA repair (Goodarzi et al., 2011). CHD3 mutations cause Snijders Blok–Campeau syndrome, characterized by macrocephaly and impaired language and speech (Snijders Blok et al., 2018).
Chd3 deletion causes partial embryonic lethality in mice, and all surviving Chd3-knockout mice die before weaning, Chd3 and Chd4 are indispensable for early vascular development (Xie et al., 2020).
CHD4
After oxidative damage, CHD4 interacts with DNMT1, DNMT3A, DNMT3B, PARP, PCNA, EZH2, G9a, and OGG1 (Xia et al., 2017). These interactions after oxidative damage are required for CpG island methylation and can maintain epigenetic suppression of tumor suppressor genes. Therefore, a high level of CHD4 is associated with a poor prognosis. De novo mutations in CHD4 induce intellectual disability with distinctive dysmorphisms and congenital heart defects (Sifrim et al., 2016; Weiss, et al., 2016), collectively referred to as Sifrim–Hitz–Weiss syndrome (SIHIWES).
Chd4-deficiency is embryonic lethal in mice due to impaired blastocyst implantation (O'Shaughnessy-Kirwan et al., 2015).
CHD5
CHD5 is a component protein of the nucleosome remodeling deacetylase (Mi-2/NuRD) complex (Potts et al., 2011), which is involved in ATP-dependent chromatin remodeling and histone deacetylase activities. CHD5 is expressed in the nervous system and testes (Thompson et al., 2003; Zhuang et al., 2014). Although CHD5 is a tumor suppressor gene frequently altered in many human cancers (Bagchi et al., 2007), patients with CHD5 mutations do not develop tumors (Parenti et al., 2021), suggesting that germline CHD5 alterations do not increase cancer risk. CHD5-deficient patients demonstrate dominant neurodevelopment disorders, including behavioral disturbances, epilepsy, and intellectual disability (Parenti et al., 2021).
Chd5 knockout mice exhibit and autism-like phenotypes (Pisansky et al., 2017) and male infertility due to decreased spermiogenesis (Zhuang et al., 2014).
CHD6
CHD6 is expressed in various mouse tissues and colocalizes with RNA polymerase II during mRNA synthesis (Lutz et al., 2006). CHD6 interacts with the PPAR complex and Nrf2, a transcription factor regulating oxidative stress response (Surapureddi et al., 2002; Nioi et al., 2005). A de novo CHD6 missense mutation was recently identified in a patient clinically presenting with Hallermann–Streiff syndrome, a premature aging disorder characterized by craniofacial and dental dysmorphisms, distinctive facial features, hair and skin abnormalities, and short stature (Kargapolova et al., 2021).
Chd6Exon12-/Exon12- mice are deficient in conserved ATPase domain expanding exons 12–19 and are viable but sterile with no obvious pathological phenotypes apart from ataxia (Lathrop et al., 2010).
CHD7
CHD7 interacts with transcription factors (Basson and van Ravenswaaij-Arts, 2015) and the BMP signaling pathway nuclear mediators SMAD1/SMAD5/SMAD8 (Liu et al., 2014). The CHD7 helicase gene has been implicated in CHARGE syndrome (Vissers et al., 2004), which is characterized by multiple symptoms, including atresia of the choanae, cranial nerve palsies, ear malformations, genital anomalies, intellectual disability, olfactory defects, and retarded growth (Sanlaville and Verloes, 2007).
Heterogeneous Chd7-mutated mice exhibit many features of CHARGE syndrome, such as craniofacial tissues and defects of the ear and heart (Bosman et al., 2005). Chd7 knockout is embryonic lethal in mice due to severe defects in the development of many tissues, including BMP signaling-regulated cardiogenic processes (Hurd et al., 2007; Liu et al., 2014).
CHD8
CHD8 is a chromatin remodeling regulator of many genes, including p53 and beta-catenin (Nishiyama et al., 2004; Nishiyama et al., 2009). CHD8 can regulate neuronal development associated with autism spectrum disorder (ASD), which is associated with chromatin modification and transcriptional regulation (Bernier et al., 2014; Sugathan et al., 2014). CHD8, KATNAL2, and SCN2A have been revealed by exome sequencing to be autism risk factors in humans (Iossifov et al., 2012; Neale et al., 2012; O'Roak et al., 2012a, 2012b; Sanders et al., 2012).
Homozygous Chd8 knockout is embryonic lethal in mice (Nishiyama et al., 2004; Nishiyama et al., 2009), but Chd8+/− mice are viable and exhibit phenotypes resembling ASD (Katayama et al., 2016). Recently, Chd8+/N2373K mice reflecting human ASD were found to show sexually dimorphic behavior changes associated with neuronal activity and gene expression (Jung et al., 2018).
CHD9/CReMM
Chd9 is highly expressed in oocytes and is associated with promoters in osteogenic cells (Shur et al., 2006).
Chd9 is involved in the chromatin loosening of growing oocytes, which is required for the acquisition of totipotency after fertilization in mice (Ooga et al., 2018). However, another group reported that Chd9 knockout mice are fertile and show no apparent phenotypes (Alendar et al., 2020).
DNA2
DNA2 possesses endonuclease activity. DNA2 belongs to the DEAD-like subfamily in superfamily 1 and can unwind dsDNA in a 5′-to-3′ direction. DNA2 forms oligomers and acts as a helicase in replication (Budd et al., 1995). DNA2 helicase/nuclease interacts with AND-1 (Ctf4) FANCD2, FEN1, PIF1, and RPA (Bae et al., 2003; Karanja et al., 2012; Sparks et al., 2020) and is indispensable for Okazaki fragment processing during DNA replication (Budd and Campbell, 1997; Bae et al., 2001). In addition, DNA2 is involved in mitochondrial DNA maintenance (Budd et al., 2006) and the double-strand break (DSB) repair pathway (Zhu et al., 2008). Human and yeast DNA2 can bind to telomeric G4 structures within ssDNA for G4 structure cleavage and unwind intermolecular G4 structures (Masuda-Sasa et al., 2008). A homogenous truncation mutation in DNA2 likely causes a form of primordial dwarfism, namely, Seckel syndrome (Shaheen et al., 2014; Tarnauskaitė et al., 2019). DNA2 mutations also induce mitochondrial myopathies (Ronchi et al., 2013).
Homozygous Dna2 knockout is embryonic lethal in mice, and heterozygous Dna2-deficient mice have increased telomere DNA damage and chromosome segregation errors that cause aneuploidy. As a result, Dna2-deficient mice develop aneuploidy-associated cancers and have dysfunctional telomeres (Lin et al., 2013).
ERCC6L2
ERCC excision repair 6 like 2 (ERCC6L2) belongs to the Snf2 subfamily of Swi/Snf in the superfamily 2 (Byrd and Raney, 2012). DNA damage induction can induce the translocation of ERCC6L2 from the cytoplasm to the mitochondria and nucleus (Tummala et al., 2014). ERCC6L2 interacts with MRI/CYREN, a cell cycle regulator of NHEJ, and regulates orientation-specific joining of broken ends during class switch recombination (CSR), which relies on the helicase activity of ERCC6L2 (Liu et al., 2020). Homozygous truncating mutations of the ERCC6L2 gene have been identified by exome sequencing in patients with bone marrow failure (BMF) and neurological dysfunction (Tummala et al., 2014; Bluteau et al., 2018). BMF is characterized by hematopoietic abnormalities and a predisposition to cancer, as typified by patients with FA (Kutler et al., 2003), and BMF patient cells with ERCC6L2 mutations show an impaired DNA repair pathway resulting from DNA-dependent protein kinase (DNA-PK) impairment and increased genomic instability similar to that observed in FANCG patient cells in response to ICLs (Tummala et al., 2018).
Ercc6l2−/− mice were viable and showed decreased nonhomologous end-joining (NHEJ)-associated CSR (Liu et al., 2020).
FBH1
The F-box DNA helicase 1 (FBH1) belongs to DNA helicase superfamily 1, and unwinds dsDNA in a 3′-to-5′ direction. The FBH1 gene is conserved from fission yeast to vertebrates (Kim et al., 2002). FBH1 interacts with PCNA via two distinct motifs (Bacquin et al., 2013). The F box domain at the N-terminus of FBH1 suggests that it is an E3 ubiquitin ligase. Although fbh1 is indispensable for viability in the absence of rqh1 (RECQ helicase in humans) or srs2 (an antirecombinase that can remove rad51 recombinase from ssDNA) DNA helicases (Krejci et al., 2003; Veaute et al., 2003), single fbh1 knockout results in mild phenotypes in yeast (Morishita et al., 2005; Osman et al., 2005). FBH1-knockout chicken B lymphocyte DT40 cells show no prominent phenotypes except mildly increased sister chromatid exchanges (SCEs), and an additive increase in SCE induction and a synergistic sensitivity to camptothecin (CPT) are observed in the absence of BLM (Kohzaki et al., 2007).
Consistently, no apparent phenotypes except mild induction of Rad51 foci after CPT treatment and micronuclei induction after topoisomerase II inhibitor treatment are observed in Fbh1 knockout mice (Laulier et al., 2010).
HELB
DNA helicase B (HELB) belongs to helicase superfamily 1B and shows translocation activity on ssDNA in a 5′-to-3′ direction (Hormeno et al., 2022). The 5′-to-3′ translocation activity of HELB is required for preventing DNA end resection in HR (Tkáč, J et al., 2016). The helicase activity of HELB is quite weak without assisting force (Hormeno et al., 2022). When HELB binds to RPA, HELB translocation activity is increased but unwinding is inhibited (Hormeno et al., 2022). HELB helicase plays a role in the early stages of replication initiation (Matsumoto et al., 1995). Indeed, HELB interacts not only with RPA but also with TOPBP1 and CDC45 (Guler et al., 2012; Gerhardt et al., 2015).
Although, mHelb knockout mice are fertile and do not show clear phenotypes under unchallenged conditions, MEF cells from Helb knockout mice show decreased replication rates after hydroxyurea treatment, suggesting that mHelb is involved in the recovery from replication stress (Tkáč, J et al., 2016).
INO80 (INOC1)
The chromatin remodeling complex inositol requiring mutant 80 (INO80) belongs to the Ino80 subfamily of Swi/Snf in superfamily 2 and shows translocation activity on dsDNA (Hargreaves and Crabtree, 2011; Byrd and Raney, 2012). INO80 is a core component of the chromatin remodeling INO80 complex, and this large INO80 complex forms a functional dimer, similar to Snf2h (Willhoft et al., 2017). In fact, INO80 has translocation activity along DNA at the H2A–H2B dimer interface for efficient H2A.Z exchange by displacing DNA (Brahma et al., 2017). In addition, the INO80 complex shows DNA helicase activity and binds Holliday junctions and replication forks in vitro (Shen et al., 2000). Therefore, INO80 is involved in DNA repair, replication, and transcriptional regulation (van Attikum and Gasser, 2009).
mIno80 knockout mice die during early embryogenesis, and conditional depletion of mIno80 in adult mice results in premature death and weight loss (Min et al., 2013).
LSH (Also Known as HELLS, PASG, or SMARCA6)
Lymphoid-specific helicase (LSH) belongs to the Snf2 subfamily of Swi/Snf in superfamily 2 and shows translocation activity on dsDNA (Hargreaves and Crabtree, 2011). The ATPase of LSH for remodeling activity can be stimulated by nucleosomes through the formation of the LSH-CDC7A complex (Jenness et al., 2018). Like CHD1-9, the ATPase domain of LSH is required for DNA translocation activity despite no evidence of helicase activity (Hargreaves and Crabtree, 2011). LSH interacts with DNMT3b, E2F3, and CDC7A and plays roles in DNA methylation (Zhu et al., 2006), transcription (von Eyss et al., 2012), and NHEJ repair (Jenness et al., 2018; Unoki et al., 2019), respectively. LSH is involved in chromatin packaging, DNA methylation, lymphoid development, and stem cell proliferation (Dennis et al., 2001). Mutations in lymphoid-specific helicase (HELLS) and cell division cycle associated 7 (CDCA7) can cause a genetically heterogeneous autosomal recessive disorder referred to as centromeric instability and facial anomalies (ICF) syndrome, which is characterized by life-threatening immunodeficiency (Thijssen et al., 2015). However, tumorigenesis induced by LSH deficiency has rarely been reported (Lee et al., 2000; Yano et al., 2004). In contrast, overexpression of LSH can promote invasive carcinoma (Yang et al., 2019).
In stark contrast to human patients, Hells-knockout mice die perinatally; thus, Hells is considered an essential gene for mouse viability (Geiman et al., 2001). Lsh-knockout mice exhibit perinatal lethality with substantial loss of genome-wide methylation (Dennis et al., 2001). Mice with Pasg mutations show growth retardation and premature aging (Sun et al., 2004).
MCM2–MCM7
Minichromosome maintenance protein complex (MCM) helicases 2–7 contain the AAA + domain and belong to superfamily 6 (Singleton et al., 2007). MCM 2–7 form a hexamer complex that plays a role in DNA replication initiation (Perkins and Diffley, 1998). Only MCM2 and MCM3 have NLS (nuclear localization signals) among the six MCM helicases (Kimura et al., 1996). The protein interaction of MCM2-7 with specific proteins may determine the specialized functions of these MCM2-7 helicases. For example, MCM3 interacts with Keap1, resulting in an antioxidant response (Tamberg et al., 2018). The interaction between STAT1 and MCM5 is required for IFNγ-induced transcriptional activation (Zhang et al., 1998). MCM6 interacts with RPA, Rb, Timeless/Tipin, and Claspin for efficient replication initiation (Komata et al., 2009; Numata et al., 2010).
Patients with partial MCM4 deficiency show growth retardation, adrenal insufficiency, and selective NK deficiency (Gineau et al., 2012). Recently, it has been reported that MCM5 is involved in Meier–Gorlin syndrome (MGS), characterized by patellar aplasia or hypoplasia, microtia, and short stature (Vetro et al., 2017). Fewer than 100 patients with MGS have been reported since the first description of the condition by Meier in 1959 and Gorlin in 1975 (de Munnik et al., 2015). Interestingly, recessive mutations in prereplication complex (pre-RC) genes, including ORC1, ORC4, ORC6, CDT1, CDC6, and CDC45, have been reported to be involved in MGS (Bicknell et al., 2011), suggesting that factors involved in DNA replication initiation play a role in suppressing MGS symptoms.
Nearly all the reported knockouts of pre-RC genes in mice are embryonic lethal (Yoshida et al., 2001; Kwon et al., 2011; Skarnes et al., 2011; Okano-Uchida et al., 2018), suggesting that replication initiation is biologically indispensable. Specifically, mice with reduced Mcm2, Mcm3, and Mcm4 expression levels show severe deficiencies in stem and progenitor cells due to replication stress and a higher cancer incidence (Pruitt et al., 2007; Shima et al., 2007; Chuang et al., 2010; Alvarez e al., 2015). In contrast, overexpression of pre-RC genes may be prognostic in several cancers (Cai et al., 2018; Li et al., 2018), suggesting that proper expression of pre-RC genes is considered necessary for homeostasis.
MCM8 and MCM9
MCM8/9 have an AAA + domain and belong to superfamily 6, similar to MCM2-7. However, MCM8 and MCM9 form an oligomeric complex that, unlike MCM2-7, is not essential for DNA replication initiation (Maiorano et al., 2005; McKinzey et al., 2021). Recently, it was reported that nonconventional NLS motifs exist at the C-terminus of MCM9 (McKinzey et al., 2021). The MCM8-MCM9 complex is required for HR induced by ICLs in chicken B lymphocyte DT40 cells (Nishimura et al., 2012). MCM8 and MCM9 interact with MCM8IP and RAD51, respectively, for efficient HR repair (Park et al., 2013; Huang et al., 2020).
Mutations in MCM8-9 can cause primary ovarian insufficiency (POI), a syndrome characterized by reduced reproductive lifespan (Desai et al., 2017). Therefore, the MCM8-MCM9 complex plays a role in meiosis mediated by HR. Recently, MCM9 mutations were found to be associated with germline predisposition to early onset inherited colorectal cancers (Goldberg et al., 2021).
MCM8-MCM9-deficient mice exhibit gametogenesis deficiency, and MCM8 knockout female mice develop ovarian tumors, although MCM8/9-knockout mice are viable (Hartford et al., 2011; Lutzmann et al., 2012). Biallelic mutations in MCM8 and MCM9 in female mice result in POI (Lutzmann et al., 2012). MCM8/9-deficient mice accumulate hematopoietic cell DNA damage with age and demonstrate an increased frequency of myeloid tumors in a p53-dependent manner (Lutzmann et al., 2019).
PIF1
PIF1 belongs to helicase superfamily 1B and is an ATP-dependent 5′–3′ DNA helicase. Like other SF1 helicases, ScPif1 exists as a monomer in solution (Lahaye et al., 1993), and ScPif1 is dimerized upon ssDNA binding (Barranco-Medina and Galletto, 2010). Interestingly, dimerized ToPif1 inhibits its helicase activity (Dai et al., 2021), suggesting a diversified helicase regulation mechanism among DNA helicases. ScPif1 interacts with PCNA (Wilson et al., 2013), Rim1, a mitochondrial ssDNA-binding protein (Ramanagoudr-Bhojappa et al., 2013), and Sub1, a G4-binding factor (Lopez et al., 2017) for recombination-coupled DNA synthesis, function in mtDNA metabolism (Ramanagoudr-Bhojappa et al., 2013) and suppressing genome instability at transcriptionally formed G4 DNA, respectively. Pif1 can resolve G-quadruplex structures (Sanders, 2010) and prevent telomere elongation (Schulz and Zakian, 1994).
In contrast, mPif1-knockout mice are viable and show no visible phenotypes and no telomere length alteration (Snow et al., 2006), suggesting that telomere protection is compensated by other DNA helicases, such as Dna2, in mice. Bannwarth et al. revealed that Pif1 knockout mice develop mitochondrial myopathy (Bannwarth et al., 2016).
RECQ Helicases
RECQ helicases belong to the RECQ-like subfamily of superfamily 2, show translocation activity on ssDNA (Kitano, 2014), and unwind dsDNA in a 3′–5′ direction (Byrd and Raney, 2012). In vertebrates, five SF2 superfamily RECQ helicases have similar helicase domains (Croteau et al., 2014). However, these proteins express different RecQ C-terminal (RQC) (Bernstein et al., 2003), helicase and RNase D–like C-terminal (HRDC) (Liu et al., 1999), exonuclease (Shen et al., 1998), sld2-like (Sangrithi et al., 2005; Matsuno et al., 2006) and Zn knuckle (Marino et al., 2013) domains, and nuclear localization sequences (NLS) (Kitao et al., 1998). Interestingly, only RECQL4 has a NLS at the N terminus of the helicase domain (Burks et al., 2007). In addition, RECQ helicases share interacting proteins, such as RAD51 and RPA, but some interactions are missing in some RECQ helicases. For example, all RECQ helicases except BLM interact with PARP1 (Croteau et al., 2014). In addition, five RECQ helicases have different binding partners involved in their function (Lu and Davis, 2021). Therefore, the outcomes and symptoms associated with deficiencies in these five RECQ helicases are very different. To date, no clear reports on patients with mutations in RECQL5 (RECQ5) have been published.
RECQ1 (RECQL1)
RECQL1 is the smallest gene in the RECQ family (Puranam and Blackshear, 1994) and forms either monomers or dimers (Vindigni and Hickson, 2009). RECQL1 helicase is related to the risk for some diseases, including breast cancer, and patient treatment outcomes (Cybulski et al., 2015; Sun et al., 2015; Viziteu et al., 2017). Recently, two patients with progeroid facial features, a hypoplastic nose, xeroderma and skin photosensitivity with short stature were identified and had the same missense mutation (Ala459er) located in the zinc-binding domain of the RECLQ1 gene, referred to as RECON (RECqlONe) (Abu-Libdeh et al., 2022).
Recql1 knockout mice show a generally normal phenotype, but MEFs obtained from Recql1 mice show genomic instability, such as chromosomal breakage, aneuploidy, SCEs (Sharma et al., 2007).
WRN (RECQL2)
Werner syndrome (WS), a rare autosomal-recessive disease, is characterized by premature senescence phenotypes in young adults (Goto et al., 1992; Friedrich et al., 2010). Among RECQ family helicases, only WRN has an exonuclease domain at the N-terminus (Huang et al., 1998). WRN forms hexamers and trimers (Perry et al., 2006). Recently, synthetic lethality between WRN and mismatch repair (MMR) has been identified by several groups (Chan et al., 2019; Kategaya et al., 2019; Lieb et al., 2019; van Wietmarschen et al., 2020), suggesting that WRN can regulate DNA repair pathway mechanisms promoting cancer cell survival.
Notably, Wrn-knockout mice show no apparent senescence-like phenotype and a normal lifespan (Lebel and Leder, 1998; Lombard et al., 2000; Ichikawa et al., 2002). In contrast, Terc−/−Wrn−/− mice exhibit premature aging phenotypes, including alopecia, cataracts, diabetes, hair graying, and osteoporosis (Chang et al., 2004; Du et al., 2004), suggesting that the abundant telomere reserve in mice results in phenotypic differences compared with that in other mammals. Wrn-deficient mice exhibit accelerated mortality in the absence of the p53 gene (Lombard et al., 2000). Wrnhel/hel mice die from increased cardiac fibrosis rather than cancers or infections (Massip et al., 2006).
BLM (RECQL3)
Like WRN, BLM forms hexamers and trimers (Karow et al., 1999). Bloom syndrome (BLM) patients are characterized by a proportional small size and a face rash that develops during Sun exposure (Rong et al., 2000; Cunniff et al., 2017). BLM syndrome patients show cancer predisposition at early ages (occurring in the second decade of life, on average), impaired fertility, and immune deficiency (Ellis et al., 1995). A significant increase in SCEs in BLM patient cells has been well recognized (Chaganti et al., 1974) and can induce mutagenesis (Carrano et al., 1978; Wilson and Thompson, 2007).
Several laboratories have established Blm-knockout mice, and some Blm-knockout mice show embryonic lethality (Chester et al., 1998; Ichikawa et al., 2002), chromosomal instability, and severe anemia (Chester et al., 1998). Viable Blm-knockout mouse strains show cancer predisposition (Luo et al., 2000), even when the knockout is heterogeneous (Goss et al., 2002).
RECQ4 (RECQL4)
Rothmund Thomson syndrome (RTS) is characterized by growth retardation with skeletal defects and congenital poikiloderma (Larizza et al., 2010). Defects in the RECLQ4 gene cause type II RTS, characterized by congenital bone defects, poikiloderma, and osteosarcoma predisposition (Kitao et al., 1999; Wang et al., 2003). RECQL4 exists as a dimer (Suzuki et al., 2009). RECQL4 is the least well-characterized among RECQ helicases in terms of function because it has an N-terminal budding yeast Sld2 domain, which is essential for DNA replication initiation (Sangrithi et al., 2005; Matsuno et al., 2006; Tanaka et al., 2007; Zegerman and Diffley, 2007), and complete RECQL4 knockout is lethal in vertebrates (Abe et al., 2011). RECQL4 knock-in human pre-B leukemia Nalm-6 cells are highly sensitive to radiation and cisplatin and show replication elongation defects after ionizing radiation (Kohzaki et al., 2012). Although RECQL4 is involved in several DNA repair pathways, such as HR (Lu et al., 2016), NHEJ (Shamanna et al., 2014), and single-strand annealing (SSA) (Kohzaki et al., 2020), a functional assay using siRNA targeting the RECQL4 helicase domain significantly reduces the expression of the Sld2 domain (Kohzaki et al., 2020), which is essential for survival and requires careful phenotyping.
Ichikawa et al. first confirmed RECQL4 lethality (Ichikawa et al., 2002). Recql4 knockout mice established by another group demonstrated embryonic lethality (Hoki et al., 2003), confirming the essential role of the Sld2 domain at the N-terminus of the Recql4 gene. Viable Recql4-deficient (Recql4HD/HD) mice exhibit genomic instability, skin abnormalities and increased intestinal adenomas when adenomatous polyposis coli (Apc) is heterogeneously absent (Mann et al., 2005). Recql4 HD/HD mice exhibit senescence with increased DNA damage in tail fibroblasts, sparser tail hair, and significantly fewer blood cells in the bone marrow (Lu et al., 2014). In contrast, we did not observe such a significant reduction in blood cells in Recql4 HD/HD mice, and the function of Recql4 in hematopoiesis should be carefully tested in the future. Interestingly, viable Recql4 HD/HD mice show a pleiotropic phenotype in which more than half show a dramatically reduced risk of age-related mortality (approximately seven-fold) compared to that of wild-type mice throughout their lifespan (Kohzaki et al., 2021).
RECQL5 (RECQ5)
RECQL5 can interact with RNAPII (Aygün et al., 2008; Izumikawa et al., 2008) and harbors two RNAPII interaction domains for different phenotypes (Islam et al., 2010). RECQL5 forms a monomer (Garcia et al., 2004). RECQL5 inhibits transcription and affects transcription elongation to suppress genomic rearrangements and transcription stress (Aygün et al., 2009; Saponaro et al., 2014). Despite these important biological features, no human patients with RECQL5 mutations have been identified to date.
Recql5-knockout mice show a predisposition to various types of cancer, including lymphoma and solid tumors (Hu et al., 2007), and exhibit elevated levels of SCEs (Hu et al., 2005), suggesting that RECQL5 is a tumor suppressor gene.
RTEL1
Regulator of telomere elongation helicase 1 (RTEL1), a human novel helicase-like (NHL) gene, belongs to the Rad3/XPD subfamily of helicase superfamily 2 (Byrd and Raney, 2012), and oligomerized RTEL1 unwinds dsDNA in a 5′ to 3′ direction (Ghisays et al., 2021). RTEL1 helicase interacts with PCNA through the PCNA interacting protein (PIP) motif and regulates telomere length and HR to maintain genomic integrity (Bai et al., 2000; Ding et al., 2004; Vannier et al., 2013). Furthermore, the telomeric G4 unwinding activity of RTEL1 depends on the RTEL1-PCNA interaction, suggesting that RTEL1 can resolve the telomeric G4 structure of replication forks during DNA replication (Vannier et al., 2013). Biallelic RTEL1 mutations have been observed in patients with congenital dyskeratosis (Ballew et al., 2013; Walne et al., 2013). Heterozygous RTEL1 mutations are associated with idiopathic pulmonary fibrosis with telomere shortening (Kannengiesser et al., 2015; Stuart et al., 2015; Newton et al., 2016). Several RTEL1 mutations have also been identified in patients with Hoyeraal–Hreidarsson syndrome symptoms, including cerebellar hypoplasia, enteropathy, hypocellular bone marrow failure, B-/NK-cell lymphopenia, and immunodeficiency (Speckmann et al., 2017).
Homozygous Rtel1 knockout is embryonic lethal in mice (Ding et al., 2004). Mice with Rtel1 mutations (Rtel1IA/IA) in the IA PIP box, which is required for PCNA interaction, are viable and show normal development and morphology (Vannier et al., 2013). However, Rtel1 IA/IA mice demonstrate accelerated tumorigenesis in the absence of p53 (Vannier et al., 2013), suggesting that Rtel1 is a potent tumor suppressor gene, which has been previously revealed by a genome-wide association study that identified it as a glioma susceptibility locus (Shete et al., 2009).
SMARCA2 (BRM)
Multi-subunit chromatin remodeling complex Switch/Sucrose nonfermentable (SWI/SNF) consists of helicase/ATPase catalytic subunits, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamilies A2 (SMARCA2) and A4 (SMARCA4) (Wilson and Roberts, 2011). Therefore, SMARCA2 and SMARCA4 belong to the Swi/Snf subfamily in superfamily 2 and show translocation activity on dsDNA (Hargreaves and Crabtree, 2011; Byrd and Raney, 2012). Like other SWI/SNF family members, the ATPase domain of SMARCA2/4/5/SMARCAD1 are required for DNA translocation activity despite no evidence of helicase activity (Hargreaves and Crabtree, 2011). Heterozygous missense mutations in SWI/SNF-related matrix associated, actin-dependent regulator of chromatin, subfamily A, member 2 (SMARCA2) cause Nicolaides–Baraitser syndrome, characterized by distal-limb anomalies, distinctive facial morphology, intellectual disability, and sparse hair (Van Houdt et al., 1997). Although it has recently been reported that mutations in SMARCA2 outside of the helicase domain can also cause a new recognizable syndrome with intellectual disability and blepharophimosis distinct from Nicolaides–Baraitser syndrome (Cappuccio et al., 2020), it remains unclear whether these mutations affect ATP-dependent helicase activity.
Mice lacking Smarca2 are viable, fertile and significantly heavier than wild-type mice (Reyes et al., 1998). Smarca2 plays a greater role than Smarca4 in cell proliferation, although Smarca2 and Smarca4 are partially redundant (Reyes et al., 1998).
SMARCA4 (BRG1)
Along with SMARCB1, ARID1A, and ARID1B mutations, SMARCA4 mutations are associated with Coffin–Siris syndrome, a rare congenital anomaly syndrome characterized by coarse facial features, hypoplastic nail of the fifth finger and/or toe, growth deficiency, intellectual disability, and microcephaly with an autosomal dominant inheritance mechanism (Tsurusaki et al., 2012). Recently, mutations in SMARCA4 have been identified in a group of undifferentiated thoracic malignancies, which are distinct from lung carcinomas and transcriptionally related to BAF-deficient sarcomas (Le Loarer et al., 2015).
In stark contrast to Smarca2, Smarca4 knockout is early embryonic lethal in mice, and Smarca4 heterozygotes are predisposed to exencephaly and tumors (Bultman et al., 2000).
SMARCA5 (SNF2H)
SMARCA5 (SNF2h) is an ATPase-dependent helicase subunit of the Imitation SWItch (ISWI) complex and regulates DNA accessibility by sliding the histone octamer (Fan et al., 2003). Thus, SMARCA5 belongs to the Snf2 subfamily of Swi/Snf in superfamily 2 and shows translocation activity on dsDNA (Hargreaves and Crabtree, 2011; Byrd and Raney, 2012). Only a minor fraction of SMARCA5 can bind to chromatin and translocate the post-translational modified nucleosome, while the majority of SMARCA5 is highly mobile (Erdel et al., 2010). De novo or rare heterozygous variants of SMARCA5 in 12 patients were found to cause a neurodevelopmental syndrome with recurrent dysmorphic features, such as postnatal short stature associated with mild developmental delay and microcephaly (Li et al., 2021).
Homozygous Smarca5 knockout is early embryonic lethal in mice (Stopka and Skoultchi, 2003).
SMARCAD1 (ETL1)
SMARCAD1 (ETL1) is a member of the SNF2 family, belongs to the Snf2 subfamily of Swi/Snf in superfamily 2 and shows translocation activity on dsDNA (Korn et al., 1992). SMARCAD1 is as chromatin remodeler recruited to DSBs for efficient resection for subsequent HR repair (Costelloe et al., 2012). A skin-specific SMARCAD isoform regulates dermatoglyphic development (Nousbeck et al., 2011). Recently, two patients with SMARCAD1 variants showed symptoms of Basan Syndrome characterized by absent or reduced sweating, congenital adermatoglyphia, neonatal acral bullae, and transient congenital facial milia (Elhaji et al., 2021).
Smarcad1 knockout mice are viable but exhibit decreased viability, impaired fertility, and skeletal dysplasia (Schoor et a., 1999).
Twinkle (TWNK) Mitochondrial DNA Helicase
TWNK belongs to helicase superfamily 4 and unwinds DNA in a 5′-to-3′ direction. Replication of mitochondrial DNA (mtDNA) depends on the replisome containing DNA helicase TWNK, mitochondrial ssDNA-binding protein (mtSSB), and DNA polymerase G (POLG). TWNK forms stable hexamers for helicase loading (Farge et al., 2008). TWNK is indispensable for mitochondrial helicase activity required for the synthesis of D-loop strands to complete mtDNA replication (Milenkovic et al., 2013). Some patients with TWINKLE mutations develop adult-onset progressive external ophthalmoplegia (PEO), progressive atrophy of the cerebellum, brain stem, spinal cord, sensory axonal neuropathy, and neuromuscular symptoms (Suomalainen et al., 1997; Spelbrink et al., 2001; Goffart et al., 2009).
Mice with Twinkle mutations demonstrate key features observed in the muscles of PEO patients, including progressive respiratory dysfunction and cytochrome C oxidase deficiency in distinct neuronal populations, at 1 year of age. Interestingly, these mice do not show premature aging, suggesting that progressive respiratory chain dysfunction and mtDNA deletions are not strong aging accelerators (Tyynismaa et al., 2005).
XPD (ERCC2)/XPB (ERCC3)
XPD belongs to the Rad3/XPD subfamily in superfamily 2, shows translocation activity on ssDNA and unwinds dsDNA in a 5′-to-3′ direction (Fan et al., 2008; Byrd and Raney, 2012). In contrast, XPB also belongs to superfamily 2 but can unwind DNA in a 3′–5′ direction. These opposite unwinding directions are indispensable for nucleotide excision repair (NER). Both XPB and XPD are components of the TFIIH transcription complex in NER repair. CS caused by mutations in CSA or CSB is a rare autosomal recessive neurodegenerative disorder characterized by an impaired nervous system, growth failure, lipodystrophy, photosensitivity without skin cancer predisposition, and a very short (<20 years) life expectancy due to premature aging (Calmels et al., 2018). Both CSA and CSB are involved in base excision repair (BER) and transcription-coupled nucleotide excision repair (TC-NER) (van der Horst et al., 1997; Stevnsner et al., 2002).
XP is an autosomal recessive disorder characterized by a deficiency in NER and hypersensitivity to UV light (Lehmann et al., 2011). As a result, XP patients are extremely sensitive to sunlight and have a dramatically increased skin cancer risk, and their life expectancy is approximately 30 years less than that of the general population.
TTD is an autosomal recessive disorder characterized by brittle hair, a Sun-sensitive form of severe neurodevelopment, developmental abnormalities, photosensitivity, skin abnormality, maternal pregnancy complications, and reduced life span with high mortality rates at a young age (Faghri et al., 2008). Mutations in the XPB (ERCC3) and XPD (ERCC2) genes can lead to TTD, XP, CS, and XP combined with CS (XPCS) (Coin et al., 1999; Dubaele et al., 2003; Oh et al., 2006; Oksenych and Coin, 2010).
There are several mouse models of XPD and XPB. XPD is essential for cell viability. Deletion of XPD in mice is embryonic lethal (de Boer et al., 1998b). Mice with Xpd mutated at amino acid 722 (XpdR722W/R722W) reflected TTD patients (de Boer et al., 1998a). TTD model mice have compromised transcription and show many symptoms, including premature aging, osteoporosis, osteosclerosis, cachexia, early graying, sterility, and a shortened lifespan (de Boer et al., 2002). TTD mice show prominent premature aging phenotypes and a shortened lifespan without mutation accumulation in the kidney and liver (Dollé, et al., 2006), suggesting that increased genomic instability induced by DNA repair-deficiency is not a prerequisite for lifespan shortening in mice. Mice with the Xpd mutation (G602D) are considered xeroderma pigmentosum/Cockayne syndrome (XPCS) model mice and show segmental progeria, cachexia, progressive loss of germinal epithelium, and skin cancer-predisposition due to unusual NER dysfunction (Andressoo et al., 2006). The phenotypes of these XPCS-disorder mice are similar to those of TCR-deficient Csa−/− mice, which show white matter microglial activation (Jaarsma et al., 2011), suggesting that NER plays a role in neuronal maintenance.
Xpb knockout is embryonic lethal in mice (Andressoo et al., 2009). Xpb mice with the last 43 amino acids deleted (XpdΔ43/Δ43) were established as a model for XPCS, and XpdΔ43/Δ43 mice are fertile with no overt phenotypes such as growth retardation, premature aging, and cancer predisposition, while XpdΔ43/Δ43 mice are sensitive to UVB irradiation of the skin (Andressoo et al., 2009). Interestingly, when a second TFIIH helicase mutation is present, Xpd knockout mice are viable but die during the neonatal period and exhibit increased DNA damage (Andressoo et al., 2009), suggesting that the basal TFIIH transcription function during development in utero is sufficient without these two helicases but insufficient for postnatal development.
Mechanisms of DNA Helicases that Cause Human Diseases
DNA helicases can act on various substrates at different biological steps through different interacting proteins (Figure 1). In addition, DNA helicases have functional preferences in DNA repair, recombination, replication, and transcription (Figure 4). Therefore, the outcomes induced by DNA helicase deficiencies result in variable phenotypes in humans and mice (Table 1). Common mechanisms associated with DNA helicase deficiency involve DNA metabolism, including DNA repair, recombination, replication, repair, and transcription. For example, patients with progeroid syndromes caused by DNA helicase deficiencies, including FA, CS, TTD, and WRN, often also exhibit neurodegenerative phenotypes, suggesting that some DNA helicases are required for neuronal development and/or homeostasis (Maynard et al., 2015). DNA helicases that result in embryonic lethality when deficient must be involved in the critical steps of DNA metabolism (Table 1). Considering the case of Ino80-knockout mice, it is likely that the INO80 helicase is also required for human embryonic development but has never been recognized as a human syndrome. In contrast, some DNA helicase-deficient mice demonstrate a nearly normal phenotype, and some DNA helicases have not been associated with human diseases. Notably, mice deficient in DNA helicases such as Chd9, Fbh1, and Pif1, which are not associated with human syndromes, are viable, fertile, and show normal postnatal growth (Table 1), suggesting that deficiencies in these DNA helicases are easily compensated by other DNA helicases in vivo; thus, it is challenging to detect mutations associated with human disease. Alternatively, a human disease may not relate to the overlap of helicase functions and may simply be due to the relatively rare frequency of heterozygous mutations in the general population. Deficiencies in some DNA helicases, such as RECQL5, are not linked to human diseases. It is tempting to speculate that populations with RECQL5 mutations may have mild, if any, phenotypes, and the mutations are less likely to be detected than other hereditary diseases associated with easily detectable symptoms, including BLM and WRN. Thus, the population with RECQL5 mutations may have a lifespan similar to that of the general population. As a result, it is expected that most patients who have germline mutations with unnoticed phenotypes may be missed until they present with cancers or other age-related diseases and undergo precise genetic testing.
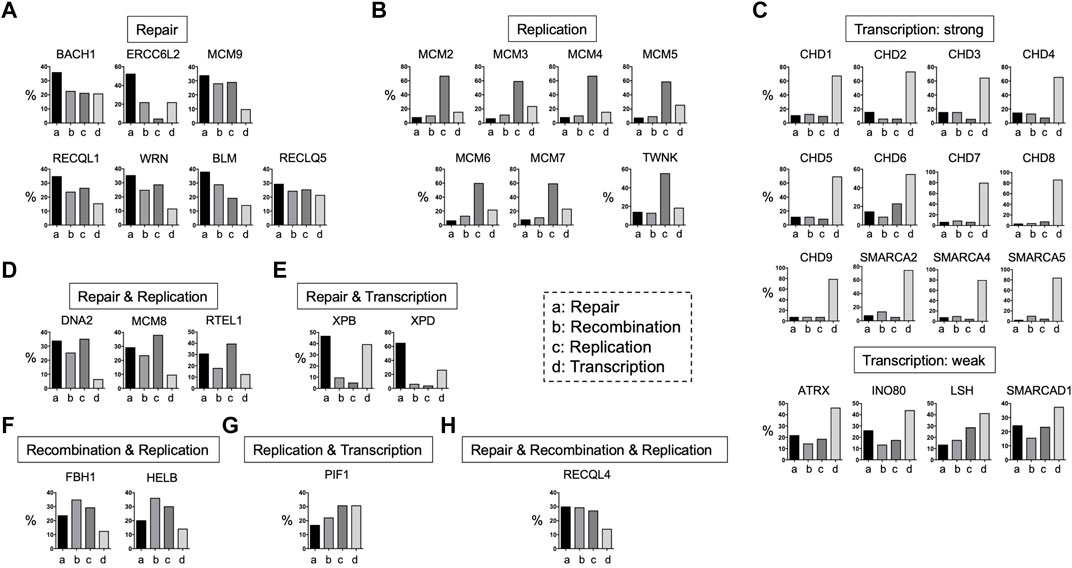
FIGURE 4. Functions of DNA helicases. DNA helicases have four main functions: DNA repair, DNA recombination, DNA replication, and transcription based on the PubMed search in June of 2022. They are divided into several categories based on the ratios of these four functions. (A): Repair, (B): replication, (C): transcription, (D): repair and replication, repair and transcription, (E): repair and transcription, (F): recombination and replication, (G): replication and transcription, and (H): repair and recombination and replication.
DNA mutations as a consequence of DNA metabolism errors resulting from DNA helicases deficiencies are now accurately determined using sequencing technologies (Schumacher et al., 2021; Cagan et al., 2022). These studies revealed that somatic mutations accumulate with age in all mammals. Therefore, either germline mutations or spontaneous somatic mutations occurring with aging resulting from DNA helicase deficiencies may affect complex physiological systems, including epigenetic regulation, gene regulatory networks, and noncoding region regulation, leading to diverse phenotypes (Sen et al., 2016; Anastasiadou et al., 2018; Vijg and Dong, 2020).
Possible Reasons for the Difference Between Human Diseases and Mouse Models
Experimental mouse models are indispensable tools for understanding human diseases due to their uniform genetic background, short lifespan, and physiologic characteristics similar to those of other mammalian species. However, there are several limitations to using mice as model animals. First, it is debatable to what extent mice with a uniform genetic background can reflect the fairly heterogeneous human population. Second, mice and humans may have different physiological circadian rhythm functions due to the reversal of the active phase between day and night. In addition, mice are susceptible to experimental conditions and the rearing environment, including pathogen-free conditions, temperature, and the microbiome (Ngyen et al., 2015). Third, different mouse strains have different phenotypes, and an appropriate mouse strain should be selected. Forth, neutrophils are the predominant white blood cells in humans, while lymphocytes are the predominant white blood cells in mice in the hematopoietic system (Kohnken et al., 2017). As a result, a high proportion of lymphomagenesis often masks tumor induction in mouse models, such as models of breast cancer (Evers and Jonkers, 2006). Fifth, there are some intrinsic differences in the gut microbiota between mice and humans, and the gut microbiota is associated with some human diseases (Nguyen et al., 2015). Last, differences between the innate and adaptive immune systems and inflammatory responses in humans and mice should be considered (Mestas and Hughes, 2004; Seok et al., 2013). However, Takao and Miyakawa have argued that the inflammatory response data between humans and mice was highly correlated when the data used by Seok et al. was analyzed by sophisticated and commonly used (Seok et al., 2013; Takao and Miyakawa, 2015), suggesting that care should be taken when choosing analytical methods to assess mouse phenotypes.
Due to these limitations, some mutations in DNA helicases, such as ATRX and CDH genes, in humans that result in viable individuals with clinical features are embryonic lethal in mice. On the other hand, the characteristics of FA patients, including cancer predisposition and anemia, are absent in FA gene-knockout mice (Parmar et al., 2009). Generally, p53 gene deficiency combined with DNA helicase knockout results in severe phenotypes and enhanced cancer progression in mice (Lombard et al., 2000; Houghtaling et al., 2005; Mann et al., 2005; Vannier et al., 2013; Lutzmann et al., 2019), apart from in Chd1-knockout mice (Guzman-Ayala et al., 2015). Thus, in the absence of DNA helicases in the mouse, a short-lived laboratory animal, p53 is essential for maintaining genomic integrity and preventing cancers.
Comprehensive whole-genome sequencing using 16 mammalian species revealed that aging-associated somatic mutation accumulation is mainly attributed to the deamination of 5-methylcytosine in humans, while somatic mutation accumulation in mice is highly induced by oxidative DNA damage (Cagan et al., 2022). Indeed, mouse cells are more sensitive to oxidative DNA damage than are human cells (Coppé et al., 2010). Such a difference in mutation accumulation between humans and mice may be one of the reasons for the phenotypic differences. In addition, DNA helicase deficiency may alter germline and/or somatic integrity, affecting complicated gene regulatory networks or noncoding networks (Anastasiadou et al., 2018; Vijg and Dong, 2020); the response of these networks may differ among species.
Mammalian Resilience in DNA-Metabolizing Enzyme Disorders
When we consider the relationship between aging and lifespan, we can refer to several aging hypotheses, such as the “mutation accumulation” hypothesis (Medawar, 1952), “antagonistic pleiotropy” hypothesis (Williams, 1957), and “disposable soma” hypothesis (Kirkwood, 2005), which explain the accumulation rate of multiple types of damage in complex networks with maintenance and repair functions in a stochastic and plastic manner. These hypotheses suggest that it is important for long-lived mammals to minimize the damage accumulation associated with tumorigenesis induced by DNA metabolism deficiency (Marmot et al., 2013). Recently, an aging model in which deleterious parental alleles were negatively selected to achieve early life mortality revealed that overall mortality throughout the lifespan was affected (Kinzina et al., 2019). This early life mortality can also be considered a bottleneck period in mouse development. In general, the more severe the bottleneck postnatal growth retardation in transgenic mice is, the higher the probability of lifetime risk of death in mouse experiments will be (Figures 2B–F) (Andressoo et al., 2009). Moreover, the developmental origins of health and disease (DOHaD) concept has been proposed, in which environmental influences during early development can affect the risks of physiological processes, including chronic diseases and noncommunicable diseases (NCDs), later in life (Hanson and Gluckman, 2014). Interestingly, Recql4-deficient mice have a higher postnatal mortality rate than wild-type mice, but the risk of death decreases dramatically with age, and the Kaplan–Meier survival curve is reversed later in life (Figure 2H) (Kohzaki et al., 2021). This suggests that resilience against accumulating mutations and genomic instability with aging in mammals may exist under certain physiological conditions.
The direct comparison between germline and somatic mutation rates in humans and mice reveals that somatic mutations have an approximately two orders of magnitude higher frequency than germline mutations (Milholland et al., 2017), suggesting that strong negative selection aggressively occurs to eliminate deleterious mutations in the germline. Moreover, somatic mutations negatively correlate with lifespan in mammals (Cagan et al., 2022). These somatic mutations that accumulate with aging are considered a consequence of DNA metabolism failure, including DNA helicases and DNA polymerases, and the DNA damage theory of aging is a convincing aging theory (Figures 2B–F) (Hoeijmakers, 2009; Freitas and de Magalhães, 2011). Indeed, the accumulation of chromosomal aberrations with aging, such as loss of the Y chromosome (LOY), is associated with a higher risk for cancer, a shorter lifespan, and other age-related diseases (Dumanski et al., 2016; Loftfield et al., 2018; Thompson et al., 2019). Consistently, it was proposed that DNA DSB repair efficacy correlates with maximum lifespan based on results obtained from 18 rodents with varying lifespans (Tian et al., 2019). Therefore, less efficient DNA repair results in genomic instability increases, which can accelerate the aging rate (Figures 2B–F). However, it remains to be determined why Recql4-deficiency is associated with resilience against aging in mice with higher levels of genomic instability and DNA damage than wild-type mice (Figures 2G,H) (Mann et al., 2005; Lu et al., 2014; Kohzaki et al., 2021).
Cancer predisposition syndromes caused by germline mutations in DNA polymerase epsilon (POLE) and delta (POLD) and the DNA glycosylase MUTYH gene, a BER factor, have recently been reported (Robinson et al., 2021a; 2021b). Patients with these mutations show a markedly increased risk for colorectal and endometrial cancer with several-fold higher mutation rates than those of the general population. Interestingly, these patients do not show overt premature aging or age-related diseases, and many patients survive until the late decades, comparable with the unaffected population. These phenotypes are reminiscent those of RTS patients, who can live for as long as the general population if they do not develop cancers (Larrizza et al., 2010). Importantly, these phenotypes in human patients are similar to those in mice with germline mutations of Pole, Pold, and homozygous knockout of Mutyh that show a normal lifespan with no apparent premature aging phenotypes in untreated conditions (Venkatesan et al., 2007; Albertson et al., 2009; Li et al., 2017). This is also the case with other DNA repair-deficient mice in which increased genomic instability is not correlated with a shortened lifespan (Figure 2A) (Dollé et al., 2006). Given that OGG1 and MUTYH are important for BER to remove accumulated endogenous oxidative DNA lesions such as 7,8-dihydro-8-oxoguanine (8-oxoG), the lack of overt phenotypes in untreated Mutyh knockout mice is surprising, particularly considering that mutations accumulate during aging. These findings indicate that the elevated mutation burdens associated with POLD/POLE and MUTYH deficiency can be mitigated by unknown factors, resulting in resistance to small insertion and deletion (ID) mutations and single-base substitution (SBS) mutations throughout life and no apparent aging-associated biological dysfunction. Many factors are involved in DNA repair pathway responses to various types of endogenously or exogenously induced DNA damage. Therefore, the loss of some DNA helicases may be efficiently compensated by other factors and may not cause an obvious phenotype such as Fbh1 and Pif1 knockout mice (Table 1). It is tempting to speculate that the system leading to the mammalian resilience system (MRS) may be activated when a certain level of mutations or certain conditions of genomic instability occur. Increased apoptosis due to DNA damage accumulation can be triggered to exclude these deleterious products in organs (Dollé et al., 2006). However, this possibility appears unlikely because programmed cell death, including apoptosis, declines with age (Tower, 2015), and p53-mediated apoptosis and/or senescence contribute to aging (Herranz and Gil, 2018). Alternatively, lifespan-limiting factors could be counteracted by factors that can elongate the lifespan. Mammalian aging and longevity processes consist of a complex variety of biological functions that are strongly influenced by environmental factors and may not be improved by a single or small number of factors or genes (Schumacher et al., 2021). However, several studies have focused on lifespan extension in mice, investigating caloric restriction and lipid metabolism and models such as dwarf and transgenic mice, including Atg5-overexpressing mice (Liang et al., 2003; Pyo et al., 2013; Johnson and Stolzing, 2019; Singh et a., 2019). These mice may be useful to identify lifespan-limiting factors to develop methods for elongating lifespan regardless of the accumulation of somatic mutations with age, which is accelerated by DNA-metabolizing enzyme disorders.
Future Perspectives
Deficiencies in some DNA helicases, such as BLM, FANCJ, RECLQ4, WRN, and XPD (Table 1) induce cancers in humans. On the other hand, overexpression of some of these DNA helicases is implicated in various types of carcinogenesis. Therefore, appropriate expression of DNA-metabolizing enzymes, including DNA helicases and DNA polymerases, is indispensable for maintaining homeostasis, particularly in later life, when physiological deficiencies are likely to be more pronounced in long-lived mammals such as humans (Hanson and Gluckman, 2014). Such balanced homeostasis can be easily affected in shot-lived mammals such as mice in the absence of p53, a guardian of genomic stability. Since accumulating somatic mutations with age and mammalian resilience are a homeostatic balance system regulated by complex and multiple physiological systems and influenced by environmental conditions, mouse strains that show unconventional lifespans or environmental settings would be useful to uncover largely unknown MRS. Understanding the mechanisms of mammalian resilience could yield a better understanding of healthy life expectancy for applied use in the future.
Author Contributions
MK conceived the concept and wrote the manuscript. MK contributed to the manuscript and approved the submitted version.
Funding
The work was supported by JSPS KAKENHI Grant numbers 15K21565 and 20K12172, the University of Occupational and Environmental Health Grants numbers 908, 1714, 031404, 904, and 909 the Takeda Science Foundation, the Kaibara Morikazu Medical Science Promotion Foundation, the Kakihara Science and Technology Foundation, the GSK Japan Research Grant, the Kurata Grants from The Hitachi Global Foundation, and the Program of the Network-type Joint Usage/Research Center for Radiation Disaster Medical Science.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The author explains that there may be a possibility for not citing the articles that should be cited due to the limitation to the manuscript frame or incomplete search. The author thanks Kenichiro Matsuzaki (Kindai University) for the helpful information about FancJ-knockout mice and appreciates the support from the members of our laboratory, the Shared-Use Research Center at the University of Occupational and Environmental Health (UOEH), the Laboratory Animal Research Center UOEH, and the Radioisotope Research Center UOEH. This work was supported by the Radiation Biology Center, Kyoto University.
References
Abdel-Monem, M., Dürwald, H., and Hoffmann-Berling, H. (1976). Enzymic Unwinding of DNA. 2. Chain Separation by an ATP-dependent DNA Unwinding Enzyme. Eur. J. Biochem. 65, 441–449. doi:10.1111/j.1432-1033.1976.tb10359.x
Abe, T., Yoshimura, A., Hosono, Y., Tada, S., Seki, M., and Enomoto, T. (2011). The N-Terminal Region of RECQL4 Lacking the Helicase Domain Is Both Essential and Sufficient for the Viability of Vertebrate Cells. Biochimica Biophysica Acta (BBA) - Mol. Cell. Res. 1813, 473–479. doi:10.1016/j.bbamcr.2011.01.001
Abu-Libdeh, B., Jhujh, S. S., Dhar, S., Sommers, J. A., Datta, A., Longo, G. M., et al. (2022). RECON Syndrome Is a Genome Instability Disorder Caused by Mutations in the DNA Helicase RECQL1. J. Clin. Investig. 132, e147301. doi:10.1172/JCI147301
Albertson, T. M., Ogawa, M., Bugni, J. M., Hays, L. E., Chen, Y., Wang, Y., et al. (2009). DNA Polymerase ε and δ Proofreading Suppress Discrete Mutator and Cancer Phenotypes in Mice. Proc. Natl. Acad. Sci. U.S.A. 106, 17101–17104. doi:10.1073/pnas.0907147106
Alendar, A., Lambooij, J. P., Bhaskaran, R., Lancini, C., Song, J. Y., van Vugt, H., et al. (2020). Gene Expression Regulation by the Chromodomain Helicase DNA-Binding Protein 9 (CHD9) Chromatin Remodeler Is Dispensable for Murine Development. PLoS One 15, e0233394. doi:10.1371/journal.pone.0233394
Alvarez, S., Díaz, M., Flach, J., Rodriguez-Acebes, S., López-Contreras, A. J., Martínez, D., et al. (2015). Replication Stress Caused by Low MCM Expression Limits Fetal Erythropoiesis and Hematopoietic Stem Cell Functionality. Nat. Commun. 6, 8548. doi:10.1038/ncomms9548
Anastasiadou, E., Jacob, L. S., and Slack, F. J. (2018). JNon-coding RNA Networks in Cancer. Nat. Rev. Cancer 18, 5–18. doi:10.1038/nrc.2017.99
Andressoo, J.-O., Mitchell, J. R., de Wit, J., Hoogstraten, D., Volker, M., Toussaint, W., et al. (2006). An Xpd Mouse Model for the Combined Xeroderma pigmentosum/Cockayne Syndrome Exhibiting Both Cancer Predisposition and Segmental Progeria. Cancer Cell. 10, 121–132. doi:10.1016/j.ccr.2006.05.027
Andressoo, J.-O., Weeda, G., de Wit, J., Mitchell, J. R., Beems, R. B., van Steeg, H., et al. (2009). An Xpb Mouse Model for Combined Xeroderma Pigmentosum and Cockayne Syndrome Reveals Progeroid Features upon Further Attenuation of DNA Repair. Mol. Cell. Biol. 29, 1276–1290. doi:10.1128/MCB.01229-08
Argentaro, A., Yang, J.-C., Chapman, L., Kowalczyk, M. S., Gibbons, R. J., Higgs, D. R., et al. (2007). Structural Consequences of Disease-Causing Mutations in the ATRX-DNMT3-Dnmt3l (ADD) Domain of the Chromatin-Associated Protein ATRX. Proc. Natl. Acad. Sci. U.S.A. 104, 11939–11944. doi:10.1073/pnas.0704057104
Auerbach, A. D. (2009). Fanconi Anemia and its Diagnosis. Mutat. Research/Fundamental Mol. Mech. Mutagen. 668, 4–10. doi:10.101610.1016/j.mrfmmm.2009.01.013
Aygün, O., Svejstrup, J., and Liu, Y. (2008). A RECQ5-RNA Polymerase II Association Identified by Targeted Proteomic Analysis of Human Chromatin. Proc. Natl. Acad. Sci. U.S.A. 105, 8580–8584. doi:10.1073/pnas.0804424105
Aygün, O., Xu, X., Liu, Y., Takahashi, H., Kong, S. E., Conaway, R. C., et al. (2009). Direct Inhibition of RNA Polymerase II Transcription by RECQL5. J. Biol. Chem. 284, 23197–23203. doi:10.1074/jbc.M109.015750
Bacquin, A., Pouvelle, C., Siaud, N., Perderiset, M., Salomé-Desnoulez, S., Tellier-Lebegue, C., et al. (2013). The Helicase FBH1 Is Tightly Regulated by PCNA via CRL4(Cdt2)-Mediated Proteolysis in Human Cells. Nucleic Acids Res. 41, 6501–6513. doi:10.1093/nar/gkt397
Bae, K.-H., Kim, H. S., Bae, S. H., Kang, H. Y., Brill, S., and Seo, Y. S. (2003). Bimodal Interaction between Replication-Protein A and Dna2 Is Critical for Dna2 Function Both In Vivo and In Vitro. Nucleic Acids Res. 31, 3006–3015. doi:10.1093/nar/gkg422
Bae, S.-H., Bae, K.-H., Kim, J.-A., and Seo, Y.-S. (2001). RPA Governs Endonuclease Switching during Processing of Okazaki Fragments in Eukaryotes. Nature 412, 456–461. doi:10.1038/3510.1038/35086609
Bagchi, A., Papazoglu, C., Wu, Y., Capurso, D., Brodt, M., Francis, D., et al. (2007). CHD5 Is a Tumor Suppressor at Human 1p36. Cell. 128, 459–475. doi:10.1016/j.cell.2006.11.052
Bai, C., Connolly, B., Metzker, M. L., Hilliard, C. A., Liu, X., Sandig, V., et al. (2000). Overexpression of M68/DcR3 in Human Gastrointestinal Tract Tumors Independent of Gene Amplification and its Location in a Four-Gene Cluster. Proc. Natl. Acad. Sci. U.S.A. 97, 1230–1235. doi:10.1073/pnas.97.3.1230
Ballew, B. J., Yeager, M., Jacobs, K., Giri, N., Boland, J., Burdett, L., et al. (2013). Germline Mutations of Regulator of Telomere Elongation Helicase 1, RTEL1, in Dyskeratosis Congenita. Hum. Genet. 132, 473–480. doi:10.1007/s00439-013-1265-8
Bannwarth, S., Berg-Alonso, L., Augé, G., Fragaki, K., Kolesar, J. E., Lespinasse, F., et al. (2016). Inactivation of Pif1 Helicase Causes a Mitochondrial Myopathy in Mice. Mitochondrion 30, 126–137. doi:10.1016/j.mito.2016.02.005
Barranco-Medina, S., and Galletto, R. (2010). DNA Binding Induces Dimerization of Saccharomyces cerevisiae Pif1. Biochemistry 49, 8445–8454. doi:10.1021/bi100984j
Basson, M. A., and van Ravenswaaij-Arts, C. (2015). Functional Insights into Chromatin Remodelling from Studies on CHARGE Syndrome. Trends Genet. 31, 600–611. doi:10.1016/j.tig.2015.05.009
Bernier, R., Golzio, C., Xiong, B., Stessman, H. A., Coe, B. P., Penn, O., et al. (2014). Disruptive CHD8 Mutations Define a Subtype of Autism Early in Development. Cell. 158, 263–276. doi:10.1016/j.cell.2014.06.017
Bernstein, D. A., Zittel, M. C., and Keck, J. L. (2003). High-resolution Structure of the E.Coli RecQ Helicase Catalytic Core. EMBO J. 22, 4910–4921. doi:10.1093/emboj/cdg500
Bérubé, N. G., Mangelsdorf, M., Jagla, M., Vanderluit, J., Garrick, D., Gibbons, R. J., et al. (2005). The Chromatin-Remodeling Protein ATRX Is Critical for Neuronal Survival during Corticogenesis. J. Clin. Investig. 115, 258–267. doi:10.1172/JCI2232910.1172/jci200522329
Bicknell, L. S., Bongers, E. M. H. F., Leitch, A., Brown, S., Schoots, J., Harley, M. E., et al. (2011). Mutations in the Pre-replication Complex Cause Meier-Gorlin Syndrome. Nat. Genet. 43, 356–359. doi:10.1038/ng.775
Bluteau, O., Sebert, M., Leblanc, T., Peffault de Latour, R., Quentin, S., Lainey, E., et al. (2018). A Landscape of Germ Line Mutations in a Cohort of Inherited Bone Marrow Failure Patients. Blood 131, 717–732. doi:10.1182/blood-2017-09-806489
Bosman, E. A., Penn, A. C., Ambrose, J. C., Kettleborough, R., Stemple, D. L., and Steel, K. P. (2005). Multiple Mutations in Mouse Chd7 Provide Models for CHARGE Syndrome. Hum. Mol. Genet. 14, 3463–3476. doi:10.1093/hmg/ddi375
Bourgeois, C. F., Mortreux, F., and Auboeuf, D. (2016). The Multiple Functions of RNA Helicases as Drivers and Regulators of Gene Expression. Nat. Rev. Mol. Cell. Biol. 17, 426–438. doi:10.1038/nrm.2016.50
Brahma, S., Udugama, M. I., Kim, J., Hada, A., Bhardwaj, S. K., Hailu, S. G., et al. (2017). INO80 Exchanges H2A.Z for H2A by Translocating on DNA Proximal to Histone Dimers. Nat. Commun. 8, 15616. doi:10.1038/ncomms15616
Bridge, W. L., Vandenberg, C. J., Franklin, R. J., and Hiom, K. (2005). The BRIP1 Helicase Functions Independently of BRCA1 in the Fanconi Anemia Pathway for DNA Crosslink Repair. Nat. Genet. 37, 953–957. doi:10.1038/ng1627
Brosh, R. M., and Cantor, S. B. (2014). Molecular and Cellular Functions of the FANCJ DNA Helicase Defective in Cancer and in Fanconi Anemia. Front. Genet. 5, 372. doi:10.3389/fgene.2014.00372
Budd, M. E., and Campbell, J. L. (1997). A Yeast Replicative Helicase, Dna2 Helicase, Interacts with Yeast FEN-1 Nuclease in Carrying Out its Essential Function. Mol. Cell. Biol. 17, 2136–2142. doi:10.1128/MCB.17.4.2136
Budd, M. E., Choe, W.-C., and Campbell, J. L. (1995). DNA2 Encodes a DNA Helicase Essential for Replication of Eukaryotic Chromosomes. J. Biol. Chem. 270, 26766–26769. doi:10.1074/jbc.270.45.26766
Budd, M. E., Reis, C. C., Smith, S., Myung, K., and Campbell, J. L. (2006). Evidence Suggesting that Pif1 Helicase Functions in DNA Replication with the Dna2 Helicase/Nuclease and DNA Polymerase δ. Mol. Cell. Biol. 26, 2490–2500. doi:10.1128/MCB.26.7.2490-2500.2006
Bultman, S., Gebuhr, T., Yee, D., La Mantia, C., Nicholson, J., Gilliam, A., et al. (2000). A Brg1 Null Mutation in the Mouse Reveals Functional Differences Among Mammalian SWI/SNF Complexes. Mol. Cell. 6, 1287–1295. doi:10.1016/s1097-2765(00)00127-1
Burks, L. M., Yin, J., and Plon, S. E. (2007). Nuclear Import and Retention Domains in the Amino Terminus of RECQL4. Gene 391, 26–38. doi:10.1016/j.gene.2006.11.019
Byrd, A. K., and Raney, K. D. (2012). Superfamily 2 Helicases. Front. Biosci. 17, 2070–2088. doi:10.2741/4038
Cagan, A., Baez-Ortega, A., Brzozowska, N., Abascal, F., Coorens, T. H. H., Sanders, M. A., et al. (2022). Somatic Mutation Rates Scale with Lifespan across Mammals. Nature 604, 517–524. doi:10.1038/s41586-022-04618-z2022
Cai, H.-Q., Cheng, Z.-J., Zhang, H.-P., Wang, P.-F., Zhang, Y., Hao, J.-J., et al. (2018). Overexpression of MCM6 Predicts Poor Survival in Patients with Glioma. Hum. Pathol. 78, 182–187. doi:10.1016/j.humpath.2018.04.024
Calmels, N., Botta, E., Jia, N., Fawcett, H., Nardo, T., Nakazawa, Y., et al. (2018). Functional and Clinical Relevance of Novel Mutations in a Large Cohort of Patients with Cockayne Syndrome. J. Med. Genet. 55, 329–343. doi:10.1136/jmedgenet-2017-104877
Cantor, S. B., Bell, D. W., Ganesan, S., Kass, E. M., Drapkin, R., Grossman, S., et al. (2001). BACH1, a Novel Helicase-like Protein, Interacts Directly with BRCA1 and Contributes to its DNA Repair Function. Cell. 105, 149–160. doi:10.1016/s0092-8674(01)00304-x
Cappuccio, G., Sayou, C., Tanno, P. L., Tisserant, E., Bruel, A.-L., Kennani, S. E., et al. (2020). De Novo SMARCA2 Variants Clustered outside the Helicase Domain Cause a New Recognizable Syndrome with Intellectual Disability and Blepharophimosis Distinct from Nicolaides-Baraitser Syndrome. Genet. Med. 22, 1838–1850. doi:10.1038/s41436-020-0898-y
Carrano, A. V., Thompson, L. H., Lindl, P. A., and Minkler, J. L. (1978). Sister Chromatid Exchange as an Indicator of Mutagenesis. Nature 271, 551–553. doi:10.1038/271551a0
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O'Roak, B. J., Cook, J., et al. (2013). Targeted Resequencing in Epileptic Encephalopathies Identifies De Novo Mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830. doi:10.1038/ng.2646
Chaganti, R. S. K., Schonberg, S., and German, J. (1974). A Manyfold Increase in Sister Chromatid Exchanges in Bloom's Syndrome Lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 71, 4508–4512. doi:10.1073/pnas.71.11.4508
Chan, E. M., Shibue, T., McFarland, J. M., Gaeta, B., Ghandi, M., Dumont, N., et al. (2019). WRN Helicase Is a Synthetic Lethal Target in Microsatellite Unstable Cancers. Nature 568, 551–556. doi:10.1038/s41586-019-1102-x
Chang, S., Multani, A. S., Cabrera, N. G., Naylor, M. L., Laud, P., Lombard, D., et al. (2004). Essential Role of Limiting Telomeres in the Pathogenesis of Werner Syndrome. Nat. Genet. 36, 877–882. doi:10.1038/ng1389
Chester, N., Kuo, F., Kozak, C., O’Hara, C. D., and Leder, P. (1998). Stage-specific Apoptosis, Developmental Delay, and Embryonic Lethality in Mice Homozygous for a Targeted Disruption in the Murine Bloom's Syndrome Gene. Genes. Dev. 12, 3382–3393. doi:10.1101/gad.12.21.3382
Chénier, S., Yoon, G., Argiropoulos, B., Lauzon, J., Laframboise, R., Ahn, J. W., et al. (2014). CHD2 Haploinsufficiency Is Associated with Developmental Delay, Intellectual Disability, Epilepsy and Neurobehavioural Problems. J. Neurodev. Disord. 6, 9. doi:10.1186/1866-1955-6-9
Chuang, C.-H., Wallace, M. D., Abratte, C., Southard, T., and Schimenti, J. C. (2010). Incremental Genetic Perturbations to MCM2-7 Expression and Subcellular Distribution Reveal Exquisite Sensitivity of Mice to DNA Replication Stress. PLoS Genet. 6, e1001110. doi:10.1371/journal10.1371/journal.pgen.1001110
Clapier, C. R., Iwasa, J., Cairns, B. R., and Peterson, C. L. (2017). Mechanisms of Action and Regulation of ATP-dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell. Biol. 18, 407–422. doi:10.1038/nrm.2017.26
Coin, F., Bergmann, E., Tremeau-Bravard, A., and Egly, J. M. (1999). Mutations in XPB and XPD Helicases Found in Xeroderma Pigmentosum Patients Impair the Transcription Function of TFIIH. EMBO J. 18, 1357–1366. doi:10.1093/emboj/18.5.1357
Coin, F., Oksenych, V., and Egly, J.-M. (2007). Distinct Roles for the XPB/p52 and XPD/p44 Subcomplexes of TFIIH in Damaged DNA Opening during Nucleotide Excision Repair. Mol. Cell. 26, 245–256. doi:10.1016/j.molcel.2007.03.009
Coppé, J.-P., Patil, C. K., Rodier, F., Krtolica, A., Beauséjour, C. M., Parrinello, S., et al. (2010). A Human-like Senescence-Associated Secretory Phenotype Is Conserved in Mouse Cells Dependent on Physiological Oxygen. PLoS One 5, e9188. doi:10.1371/journal.pone.0009188
Costelloe, T., Louge, R., Tomimatsu, N., Mukherjee, B., Martini, E., Khadaroo, B., et al. (2012). The Yeast Fun30 and Human SMARCAD1 Chromatin Remodellers Promote DNA End Resection. Nature 489, 581–584. doi:10.1038/nature11353
Croteau, D. L., Popuri, V., Opresko, P. L., and Bohr, V. A. (2014). Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 83, 519–552. doi:10.1146/10.1146/annurev-biochem-060713-035428
Cunniff, C., Bassetti, J. A., and Ellis, N. A. (2017). Bloom's Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 8, 4–23. doi:10.1159/10.1159/000452082
Cybulski, C., Carrot-Zhang, J., Kluźniak, W., Rivera, B., Kashyap, A., Wokołorczyk, D., et al. (2015). Germline RECQL Mutations Are Associated with Breast Cancer Susceptibility. Nat. Genet. 47, 643–646. doi:10.1038/ng.3284
Dai, Y.-X., Chen, W.-F., Liu, N.-N., Teng, F.-Y., Guo, H.-L., Hou, X.-M., et al. (2021). Structural and Functional Studies of SF1B Pif1 from Thermus Oshimai Reveal Dimerization-Induced Helicase Inhibition. Nucleic Acids Res. 49, 4129–4143. doi:10.1093/nar/gkab188
de Boer, J., Andressoo, J. O., de Wit, J., Huijmans, J., Beems, R. B., van Steeg, H., et al. (2002). Premature Aging in Mice Deficient in DNA Repair and Transcription. Science 296, 1276–1279. doi:10.1126/science.1070174
de Boer, J., de Wit, J., van Steeg, H., Berg, R. J. W., Morreau, H., Visser, P., et al. (1998a). A Mouse Model for the Basal Transcription/DNA Repair Syndrome Trichothiodystrophy. Mol. Cell. 1, 981–990. doi:10.1016/s1097-2765(00)80098-2
de Boer, J., Donker, I., de Wit, J., Hoeijmakers, J. H., and Weeda, G. (1998b). Disruption of the Mouse Xeroderma Pigmentosum Group D DNA Repair/basal Transcription Gene Results in Preimplantation Lethality. Cancer Res. 58, 89–94.
De La Fuente, R., Viveiros, M. M., Wigglesworth, K., and Eppig, J. J. (2004). M., Wigglesworth, K., Eppig, J.JATRX, a Member of the SNF2 Family of helicase/ATPases, Is Required for Chromosome Alignment and Meiotic Spindle Organization in Metaphase II Stage Mouse Oocytes. Dev. Biol. 272, 1–14. doi:10.1016/j.ydbio.2003.12.012
de Munnik, S. A., Roukema, J., Schoots, J., Knoers, N. V., Brunner, H. G., et al. (2015). Meier-Gorlin Syndrome. Orphanet J. Rare Dis. 10, 114. doi:10.1186/s13023-015-0322-x
Dennis, K., Fan, T., Geiman, T., Yan, Q., and Muegge, K. (2001). Lsh, a Member of the SNF2 Family, Is Required for Genome-wide Methylation. Genes. Dev. 15 (15), 2940–2944. doi:10.1101/gad.929101
Desai, S., Wood-Trageser, M., Matic, J., Chipkin, J., Jiang, H., Bachelot, A., et al. (2017). MCM8 and MCM9 Nucleotide Variants in Women with Primary Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 102, 576–582. doi:10.1210/jc.2016-2565
Ding, H., Schertzer, M., Wu, X., Gertsenstein, M., Selig, S., Kammori, M., et al. (2004). Regulation of Murine Telomere Length by Rtel. Cell. 117, 873–886. doi:10.1016/j.cell.2004.05.026
Dollé, M. E. T., Busuttil, R. A., Garcia, A. M., Wijnhoven, S., van Drunen, E., Niedernhofer, L. J., et al. (2006). Increased Genomic Instability Is Not a Prerequisite for Shortened Lifespan in DNA Repair Deficient Mice. Mutat. Research/Fundamental Mol. Mech. Mutagen. 596, 22–35. doi:10.1016/j.mrfmmm.2005.11.008
Drapkin, R., Reardon, J. T., Ansari, A., Huang, J.-C., Zawel, L., Ahn, K., et al. (1994). Dual Role of TFIIH in DNA Excision Repair and in Transcription by RNA Polymerase II. Nature 368, 769–772. doi:10.1038/368769a0
Du, X., Shen, J., Kugan, N., Furth, E. E., Lombard, D. B., Cheung, C., et al. (2004). Telomere Shortening Exposes Functions for the Mouse Werner and Bloom Syndrome Genes. Mol. Cell. Biol. 24, 8437–8446. doi:10.1128/MCB.24.19.8437-8446.2004
Dubaele, S., De Santis, L. P., Bienstock, R. J., Keriel, A., Stefanini, M., Van Houten, B., et al. (2003). Basal Transcription Defect Discriminates between Xeroderma Pigmentosum and Trichothiodystrophy in XPD Patients. Mol. Cell. 11, 1635–1646. doi:10.1016/s1097-2765(03)00182-5
Dumanski, J. P., Lambert, J.-C., Rasi, C., Giedraitis, V., Davies, H., Grenier-Boley, B., et al. (2016). Mosaic Loss of Chromosome Y in Blood Is Associated with Alzheimer Disease. Am. J. Hum. Genet. 98, 1208–1219. doi:10.1016/j.ajhg.2016.05.014
Elhaji, Y., van Henten, T. M. A., Ruivenkamp, C. A. L., Nightingale, M., Santen, G. W., Vos, L. E., et al. (2021). Two SMARCAD1 Variants Causing Basan Syndrome in a Canadian and a Dutch Family. JID Innov. 1, 100022. doi:10.1016/j.xjidi.2021.100022
Ellis, N. A., Groden, J., Ye, T.-Z., Straughen, J., Lennon, D. J., Ciocci, S., et al. (1995). The Bloom's Syndrome Gene Product Is Homologous to RecQ Helicases. Cell. 83, 655–666. doi:10.1016/0092-8674(95)90105-1
Erdel, F., Schubert, T., Marth, C., Längst, G., and Rippe, K. (2010). Human ISWI Chromatin-Remodeling Complexes Sample Nucleosomes via Transient Binding Reactions and Become Immobilized at Active Sites. Proc. Natl. Acad. Sci. U.S.A. 107, 19873–19878. doi:10.1073/pnas.10010.1073/pnas.1003438107
Evers, B., and Jonkers, J. (2006). Mouse Models of BRCA1 and BRCA2 Deficiency: Past Lessons, Current Understanding and Future Prospects. Oncogene 25, 5885–5897. doi:10.1038/sj.onc10.1038/sj.onc.1209871
Faghri, S., Tamura, D., Kraemer, K. H., and Digiovanna, J. J. (2008). Trichothiodystrophy: a Systematic Review of 112 Published Cases Characterises a Wide Spectrum of Clinical Manifestations. J. Med. Genet. 45, 609–621. doi:10.1136/jmg.2008.058743
Fan, H.-Y., He, X., Kingston, R. E., and Narlikar, G. J. (2003). Distinct Strategies to Make Nucleosomal DNA Accessible. Mol. Cell. 11, 1311–1322. doi:10.1016/s1097-2765(03)00192-8
Fan, L., Fuss, J. O., Cheng, Q. J., Arvai, A. S., Hammel, M., Roberts, V. A., et al. (2008). XPD Helicase Structures and Activities: Insights into the Cancer and Aging Phenotypes from XPD Mutations. Cell 133, 789–800. doi:10.1016/j.cell.2008.04.030
Farge, G., Holmlund, T., Khvorostova, J., Rofougaran, R., Hofer, A., and Falkenberg, M. (2008). The N-Terminal Domain of TWINKLE Contributes to Single-Stranded DNA Binding and DNA Helicase Activities. Nucleic Acids Res. 36, 393–403. doi:10.1093/nar/gkm1025
Freitas, A. A., and de Magalhães, J. P. (2011). A Review and Appraisal of the DNA Damage Theory of Ageing. Mutat. Research/Reviews Mutat. Res. 728, 12–22. doi:10.1016/j.mrrev.2011.05.001
Friedrich, K., Lee, L., Leistritz, D. F., Nürnberg, G., Saha, B., Hisama, F. M., et al. (2010). WRN Mutations in Werner Syndrome Patients: Genomic Rearrangements, Unusual Intronic Mutations and Ethnic-specific Alterations. Hum. Genet. 128, 103–111. doi:10.1007/s00439-010-0832-5
Garcia, P. L., Liu, Y., Jiricny, J., West, S. C., and Janscak, P. (2004). Human RECQ5β, a Protein with DNA Helicase and Strand-Annealing Activities in a Single Polypeptide. EMBO J. 23, 2882–2891. doi:10.1038/sj.emboj.7600301
Gaspar-Maia, A., Alajem, A., Polesso, F., Sridharan, R., Mason, M. J., Heidersbach, A., et al. (2009). Chd1 Regulates Open Chromatin and Pluripotency of Embryonic Stem Cells. Nature 460, 863–868. doi:10.1038/nature08212
Geiman, T. M., Tessarollo, L., Anver, M. R., Kopp, J. B., Ward, J. M., and Muegge, K. (2001). Lsh, a SNF2 Family Member, Is Required for Normal Murine Development. Biochimica Biophysica Acta (BBA) - General Subj. 1526, 211–220. doi:10.1016/s0304-4165(01)00129-5
Gerhardt, J., Guler, G. D., and Fanning, E. (2015). Human DNA Helicase B Interacts with the Replication Initiation Protein Cdc45 and Facilitates Cdc45 Binding onto Chromatin. Exp. Cell. Res. 334 (2), 283–293. doi:10.1016/j.yexcr.2015.04.014
Ghisays, F., Garzia, A., Wang, H., Canasto-Chibuque, C., Hohl, M., Savage, S. A., et al. (2021). RTEL1 Influences the Abundance and Localization of TERRA RNA. Nat. Commun. 12, 3016. doi:10.1038/s41467-021-23299-2
Gibbons, R. J., McDowell, T. L., Raman, S., O'Rourke, D. M., Garrick, D., Ayyub, H., et al. (2000). Mutations in ATRX, Encoding a SWI/SNF-like Protein, Cause Diverse Changes in the Pattern of DNA Methylation. Nat. Genet. 24, 368–371. doi:10.1038/74191
Gibbons, R. J., Picketts, D. J., Villard, L., and Higgs, D. R. (1995). Mutations in a Putative Global Transcriptional Regulator Cause X-Linked Mental Retardation with α-thalassemia (ATR-X Syndrome). Cell. 80, 837–845. doi:10.1016/0092-8674(95)90287-2
Gineau, L., Cognet, C., Kara, N., Lach, F. P., Dunne, J., Veturi, U., et al. (2012). Partial MCM4 Deficiency in Patients with Growth Retardation, Adrenal Insufficiency, and Natural Killer Cell Deficiency. J. Clin. Investig. 122, 821–832. doi:10.1172/JCI6101410.1172/JCI61014
Goffart, S., Cooper, H. M., Tyynismaa, H., Wanrooij, S., Suomalainen, A., and Spelbrink, J. N. (2009). Twinkle Mutations Associated with Autosomal Dominant Progressive External Ophthalmoplegia Lead to Impaired Helicase Function and In Vivo mtDNA Replication Stalling. Hum. Mol. Genet. 18, 328–340. doi:10.1093/hmg/ddn359
Goldberg, Y., Aleme, O., Peled-Perets, L., Castellvi-Bel, S., Nielsen, M., and Shalev, S. A. (2021). MCM9 Is Associated with Germline Predisposition to Early-Onset Cancer-Clinical Evidence. npj Genom. Med. 6, 78. doi:10.1038/s41525-021-00242-4
Goodarzi, A. A., Kurka, T., and Jeggo, P. A. (2011). KAP-1 Phosphorylation Regulates CHD3 Nucleosome Remodeling during the DNA Double-Strand Break Response. Nat. Struct. Mol. Biol. 18, 831–839. doi:10.1038/nsmb.2077
Gorbalenya, A. E., Koonin, E. V., Donchenko, A. P., and Blinov, V. M. (1989). Two Related Superfamilies of Putative Helicases Involved in Replication, Recombination, Repair and Expression of DNA and RNA Genomes. Nucl. Acids Res. 17, 4713–4730. doi:10.1093/nar/17.12.4713
Goss, K. H., Risinger, M. A., Kordich, J. J., Sanz, M. M., Straughen, J. E., Slovek, L. E., et al. (2002). Enhanced Tumor Formation in Mice Heterozygous for Blm Mutation. Science 297, 2051–2053. doi:10.1126/science.1074340
Goto, M., Rubenstein, M., Weber, J., Woods, K., and Drayna, D. (1992). Genetic Linkage of Werner's Syndrome to Five Markers on Chromosome 8. Nature 355, 735–738. doi:10.1038/10.1038/355735a0
Guler, G. D., Liu, H., Vaithiyalingam, S., Arnett, D. R., Kremmer, E., Chazin, W. J., et al. (2012). Human DNA Helicase B (HDHB) Binds to Replication Protein A and Facilitates Cellular Recovery from Replication Stress. J. Biol. Chem. 287, 6469–6481. doi:10.1074/jbc.M111.324582
Guzman-Ayala, M., Sachs, M., Koh, F. M., Onodera, C., Bulut-Karslioglu, A., Lin, C.-J., et al. (2015). Chd1 Is Essential for the High Transcriptional Output and Rapid Growth of the Mouse Epiblast. Development 142, 118–127. doi:10.1242/dev.114843
Hanson, M. A., and Gluckman, P. D. (2014). Early Developmental Conditioning of Later Health and Disease: Physiology or Pathophysiology? Physiol. Rev. 94, 1027–1076. doi:10.1152/physrev10.1152/physrev.00029.2013
Hargreaves, D. C., and Crabtree, G. R. (2011). ATP-Dependent Chromatin Remodeling: Genetics, Genomics and Mechanisms. Cell. Res. 21, 396–420. doi:10.1038/cr.2011.32
Hartford, S. A., Luo, Y., Southard, T. L., Min, I. M., Lis, J. T., and Schimenti, J. C. (2011). Minichromosome Maintenance Helicase Paralog MCM9 Is Dispensible for DNA Replication but Functions in Germ-Line Stem Cells and Tumor Suppression. Proc. Natl. Acad. Sci. U.S.A. 108, 17702–17707. doi:10.1073/pnas.1113524108
Herranz, N., and Gil, J. (2018). Mechanisms and Functions of Cellular Senescence. J. Clin. Investig. 128, 1238–1246. doi:10.1172/JCI95148
Hoeijmakers, J. H. J. (2009). DNA Damage, Aging, and Cancer. N. Engl. J. Med. 361, 1475–1485. doi:10.1056/NEJMra0804615
Hoki, Y., Araki, R., Fujimori, A., Ohhata, T., Koseki, H., Fukumura, R., et al. (2003). Growth Retardation and Skin Abnormalities of the Recql4-Deficient Mouse. Hum. Mol. Genet. 12, 2293–2299. doi:10.1093/hmg/ddg254
Hormeno, S., Wilkinson, O. J., Aicart-Ramos, C., Kuppa, S., Antony, E., Dillingham, M. S., et al. (2022). Human HELB Is a Processive Motor Protein that Catalyzes RPA Clearance from Single-Stranded DNA. Proc. Natl. Acad. Sci. U. S. A. 119, e2112376119. doi:10.1073/pnas.2112376119
Houghtaling, S., Granville, L., Akkari, Y., Torimaru, Y., Olson, S., Finegold, M., et al. (2005). Heterozygosity for P53 (Trp53 +/−) Accelerates Epithelial Tumor Formation in Fanconi Anemia Complementation Group D2 (Fancd2) Knockout Mice. Cancer Res. 65, 85–91. doi:10.1158/0008-5472.85.65.1
Hu, Y., Lu, X., Barnes, E., Yan, M., Lou, H., Luo, G., et al. (2005). Recql5 and Blm RecQ DNA Helicases Have Nonredundant Roles in Suppressing Crossovers. Mol. Cell. Biol. 25, 3431–3442. doi:10.1128/MCB.25.9.3431-3442.2005
Hu, Y., Raynard, S., Sehorn, M. G., Lu, X., Bussen, W., Zheng, L., et al. (2007). RECQL5/Recql5 Helicase Regulates Homologous Recombination and Suppresses Tumor Formation via Disruption of Rad51 Presynaptic Filaments. Genes. Dev. 21, 3073–3084. doi:10.1101/gad.1609107
Huang, J.-W., Acharya, A., Taglialatela, A., Nambiar, T. S., Cuella-Martin, R., Leuzzi, G., et al. (2020). MCM8IP Activates the MCM8-9 Helicase to Promote DNA Synthesis and Homologous Recombination upon DNA Damage. Nat. Commun. 11, 2948. doi:10.1038/s41467-020-16718-3
Huang, S., Li, B., Gray, M. D., Oshima, J., Mian, I. S., and Campisi, J. (1998). The Premature Ageing Syndrome Protein, WRN, Is a 3′→5′ Exonuclease. Nat. Genet. 20, 114–116. doi:10.1038/10.1038/2410
Hurd, E. A., Capers, P. L., Blauwkamp, M. N., Adams, M. E., Raphael, Y., Poucher, H. K., et al. (2007). Loss of Chd7 Function in Gene-Trapped Reporter Mice Is Embryonic Lethal and Associated with Severe Defects in Multiple Developing Tissues. Mamm. Genome 18, 94–104. doi:10.1007/s00335-006-0107-6
Ichikawa, K., Noda, T., and Furuichi, Y. (2002). Preparation of the Gene Targeted Knockout Mice for Human Premature Aging Diseases, Werner Syndrome, and Rothmund-Thomson Syndrome Caused by the Mutation of DNA Helicases. Folia Pharmacol. Jpn. 119, 219–226. doi:10.1254/fpj.119.219
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De Novo gene Disruptions in Children on the Autistic Spectrum. Neuron 74, 285–299. doi:10.1016/j.neuron.2012.04.009
Islam, M. N., Fox, D., Guo, R., Enomoto, T., and Wang, W. (2010). RecQL5 Promotes Genome Stabilization through Two Parallel Mechanisms-Interacting with RNA Polymerase II and Acting as a Helicase. Mol. Cell. Biol. 30, 2460–2472. doi:10.1128/MCB.01583-09
Izumikawa, K., Yanagida, M., Hayano, T., Tachikawa, H., Komatsu, W., Shimamoto, A., et al. (2008). Association of Human DNA Helicase RecQ5β with RNA Polymerase II and its Possible Role in Transcription. Biochem. J. 413, 505–516. doi:10.1042/BJ20071392
Jaarsma, D., van der Pluijm, I., de Waard, M. C., Brandt, R., Vermeij, M., et al. (2011). Age-related Neuronal Degeneration: Complementary Roles of Nucleotide Excision Repair and Transcription-Coupled Repair in Preventing Neuropathology. PLoS Genet. 7, e1002405. doi:10.1371/journal.pgen.1002405
Jackson, R. N., Lavin, M., Carter, J., and Wiedenheft, B. (2014). Fitting CRISPR-Associated Cas3 into the Helicase Family Tree. Curr. Opin. Struct. Biol. 24, 106–114. doi:10.1016/j.sbi.2014.01.001
Jenness, C., Giunta, S., Müller, M. M., Kimura, H., Muir, T. W., and Funabiki, H. (2018). HELLS and CDCA7 Comprise a Bipartite Nucleosome Remodeling Complex Defective in ICF Syndrome. Proc. Natl. Acad. Sci. U. S. A. 115, E876–E885. doi:10.1073/pnas.1717509115
Johnson, A. A., and Stolzing, A. (2019). The Role of Lipid Metabolism in Aging, Lifespan Regulation, and Age-Related Disease. Aging Cell. 18, e13048. doi:10.1111/acel.13048
Juhász, S., Elbakry, A., Mathes, A., and Löbrich, M. (2018). ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell. 71, 11–24. doi:10.1016/j.molcel.2018.05.014
Jung, H., Park, H., Choi, Y., Kang, H., Lee, E., Kweon, H., et al. (2018). Sexually Dimorphic Behavior, Neuronal Activity, and Gene Expression in Chd8-Mutant Mice. Nat. Neurosci. 21, 1218–1228. doi:10.1038/s41593-018-0208-z
Kannengiesser, C., Borie, R., Ménard, C., Réocreux, M., Nitschké, P., Gazal, S., et al. (2015). HeterozygousRTEL1mutations Are Associated with Familial Pulmonary Fibrosis. Eur. Respir. J. 46, 474–485. doi:10.1183/09031936.00040115
Karanja, K. K., Cox, S. W., Duxin, J. P., Stewart, S. A., and Campbell, J. L. (2012). DNA2 and EXO1 in Replication-Coupled, Homology-Directed Repair and in the Interplay between HDR and the FA/BRCA Network. Cell. Cycle 11, 3983–3996. doi:10.4161/cc.22215
Kargapolova, Y., Rehimi, R., Kayserili, H., Brühl, J., Sofiadis, K., Zirkel, A., et al. (2021). Overarching Control of Autophagy and DNA Damage Response by CHD6 Revealed by Modeling a Rare Human Pathology. Nat. Commun. 12, 3014. doi:10.1038/s41467-021-23327-1
Kari, V., Mansour, W. Y., Raul, S. K., Baumgart, S. J., Mund, A., Grade, M., et al. (2016). Loss of CHD1 Causes DNA Repair Defects and Enhances Prostate Cancer Therapeutic Responsiveness. EMBO Rep. 17, 1609–1623. doi:10.15252/embr.201642352
Karow, J. K., Newman, R. H., Freemont, P. S., and Hickson, I. D. (1999). Oligomeric Ring Structure of the Bloom's Syndrome Helicase. Curr. Biol. 9, 597–600. doi:10.1016/s0960-9822(99)80264-4
Katayama, Y., Nishiyama, M., Shoji, H., Ohkawa, Y., Kawamura, A., Sato, T., et al. (2016). CHD8 Haploinsufficiency Results in Autistic-like Phenotypes in Mice. Nature 537, 675–679. doi:10.1038/nature19357
Kategaya, L., Perumal, S. K., Hager, J. H., and Belmont, L. D. (2019). Werner Syndrome Helicase Is Required for the Survival of Cancer Cells with Microsatellite Instability. iScience 13, 488–497. doi:10.1016/j.isci.2019.02.006
Kim, J., Kim, J.-H., Lee, S.-H., Kim, D.-H., Kang, H.-Y., Bae, S.-H., et al. (2002). The Novel Human DNA Helicase hFBH1 Is an F-Box Protein. J. Biol. Chem. 277, 24530–24537. doi:10.1074/jbc.M201612200
Kim, Y. J., Khoshkhoo, S., Frankowski, J. C., Zhu, B., Abbasi, S., Lee, S., et al. (2018). Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory. Neuron 100, 1180–1193. doi:10.1016/j.neuron.2018.09.049
Kimura, H., Ohtomo, T., Yamaguchi, M., Ishii, A., and Sugimoto, K. (1996). Mouse MCM Proteins: Complex Formation and Transportation to the Nucleus. Genes. Cells 1, 977–993. doi:10.1046/j.1365-2443.1996.840284.x
Kinzina, E. D., Podolskiy, D. I., Dmitriev, S. E., and Gladyshev, V. N. (2019). Patterns of Aging Biomarkers, Mortality, and Damaging Mutations Illuminate the Beginning of Aging and Causes of Early-Life Mortality. Cell. Rep. 29, 4276–4284. doi:10.1016/j.celrep.2019.11.091
Kirkwood, T. B. L. (2005). Understanding the Odd Science of Aging. Cell. 120, 437–447. doi:10.1016/j.cell.2005.01.027
Kitano, K. (2014). Structural Mechanisms of Human RecQ Helicases WRN and BLM. Front. Genet. 5, 366. doi:10.3389/fgene.2014.00366
Kitao, S., Ohsugi, I., Ichikawa, K., Goto, M., Furuichi, Y., and Shimamoto, A. (1998). Cloning of Two New Human Helicase Genes of the RecQ Family: Biological Significance of Multiple Species in Higher Eukaryotes. Genomics 54, 443–452. doi:10.1006/geno.1998.5595
Kitao, S., Shimamoto, A., Goto, M., Miller, R. W., Smithson, W. A., Lindor, N. M., et al. (1999). Mutations in RECQL4 Cause a Subset of Cases of Rothmund-Thomson Syndrome. Nat. Genet. 22, 82–84. doi:10.1038/8788
Kogoma, T. (1997). Stable DNA Replication: Interplay between DNA Replication, Homologous Recombination, and Transcription. Microbiol. Mol. Biol. Rev. 61, 212–238. doi:10.1128/mmbr10.1128/.61.2.212-238.1997
Kohnken, R., Porcu, P., and Mishra, A. (2017). Overview of the Use of Murine Models in Leukemia and Lymphoma Research. Front. Oncol. 7, 22. doi:10.3389/fonc.2017.00022
Kohzaki, M., Chiourea, M., Versini, G., Adachi, N., Takeda, S., Gagos, S., et al. (2012). The Helicase Domain and C-Terminus of Human RecQL4 Facilitate Replication Elongation on DNA Templates Damaged by Ionizing Radiation. Carcinogenesis 33, 1203–1210. doi:10.1093/carcin/bgs149
Kohzaki, M., Hatanaka, A., Sonoda, E., Yamazoe, M., Kikuchi, K., Vu Trung, N., et al. (2007). Cooperative Roles of Vertebrate Fbh1 and Blm DNA Helicases in Avoidance of Crossovers during Recombination Initiated by Replication Fork Collapse. Mol. Cell. Biol. 27, 2812–2820. doi:10.1128/MCB.02043-06
Kohzaki, M., Ootsuyama, A., Sun, L., Moritake, T., and Okazaki, R. (2020). Human RECQL4 Represses the RAD52‐mediated Single‐strand Annealing Pathway after Ionizing Radiation or Cisplatin Treatment. Int. J. Cancer 146, 3098–3113. doi:10.1002/ijc.32670
Kohzaki, M., Ootsuyama, A., Umata, T., and Okazaki, R. (2021). Comparison of the Fertility of Tumor Suppressor Gene-Deficient C57BL/6 Mouse Strains Reveals Stable Reproductive Aging and Novel Pleiotropic Gene. Sci. Rep. 11, 12357. doi:10.1038/s41598-021-91342-9
Komata, M., Bando, M., Araki, H., and Shirahige, K. (2009). The Direct Binding of Mrc1, a Checkpoint Mediator, to Mcm6, a Replication Helicase, Is Essential for the Replication Checkpoint against Methyl Methanesulfonate-Induced Stress. Mol. Cell. Biol. 29, 5008–5019. doi:10.1128/MCB.01934-08
Korn, R., Schoor, M., Neuhaus, H., Henseling, U., Soininen, R., Zachgo, J., et al. (1992). Enhancer Trap Integrations in Mouse Embryonic Stem Cells Give Rise to Staining Patterns in Chimaeric Embryos with a High Frequency and Detect Endogenous Genes. Mech. Dev. 39, 95–109. doi:10.1016/0925-4773(92)90029-j
Kraemer, K. H., Patronas, N. J., Schiffmann, R., Brooks, B. P., Tamura, D., and DiGiovanna, J. J. (2007). Xeroderma Pigmentosum, Trichothiodystrophy and Cockayne Syndrome: a Complex Genotype-Phenotype Relationship. Neuroscience 145, 1388–1396. doi:10.1016/j.neuroscience10.1016/j.neuroscience.2006.12.020
Krejci, L., Van Komen, S., Li, Y., Villemain, J., Reddy, M. S., Klein, H., et al. (2003). DNA Helicase Srs2 Disrupts the Rad51 Presynaptic Filament. Nature 423, 305–309. doi:10.1038/10.1038/nature01577
Kulkarni, S., Nagarajan, P., Wall, J., Donovan, D. J., Donell, R. L., Ligon, A. H., et al. (2008). Disruption of Chromodomain Helicase DNA Binding Protein 2 (CHD2) Causes Scoliosis. Am. J. Med. Genet. 146A, 1117–1127. doi:10.1002/ajmg.a.32178
Kutler, D. I., Singh, B., Satagopan, J., Batish, S. D., Berwick, M., Giampietro, P. F., et al. (2003). A 20-year Perspective on the International Fanconi Anemia Registry (IFAR). Blood 101, 1249–1256. doi:10.1182/blood-2002-07-2170
Kwon, H. J., Ma, S., and Huang, Z. (2011). Radial Glia Regulate Cajal-Retzius Cell Positioning in the Early Embryonic Cerebral Cortex. Dev. Biol. 351, 25–34. doi:10.1016/j.ydbio.2010.12.026
Lahaye, A., Leterme, S., and Foury, F. (1993). PIF1 DNA Helicase from Saccharomyces cerevisiae. Biochemical Characterization of the Enzyme. J. Biol. Chem. 268, 26155–26161. doi:10.1016/s0021-9258(19)74294-xPMID: 8253734
Larizza, L., Roversi, G., and Volpi, L. (2010). Rothmund-Thomson Syndrome. Orphanet J. Rare Dis. 5, 2. doi:10.1186/1750-1172-5-2
Lathrop, M. J., Chakrabarti, L., Eng, J., Harker Rhodes, C., Lutz, T., Nieto, A., et al. (2010). Deletion of the Chd6 Exon 12 Affects Motor Coordination. Mamm. Genome 21, 130–142. doi:10.1007/10.1007/s00335-010-9248-8
Laulier, C., Cheng, A., Huang, N., and Stark, J. M. (2010). Mammalian Fbh1 Is Important to Restore Normal Mitotic Progression Following Decatenation Stress. DNA Repair 9, 708–717. doi:10.1016/j.dnarep.2010.03.011
Lebel, M., and Leder, P. (1998). A Deletion within the Murine Werner Syndrome Helicase Induces Sensitivity to Inhibitors of Topoisomerase and Loss of Cellular Proliferative Capacity. Proc. Natl. Acad. Sci. U.S.A. 95, 13097–13102. doi:10.1073/pnas.95.22.13097
Lee, D. W., Zhang, K., Ning, Z. Q., Raabe, E. H., Tintner, S., Wieland, R., et al. (2000). Proliferation-associated SNF2-like Gene (PASG): a SNF2 Family Member Altered in Leukemia. Cancer Res. 60, 3612–3622.
Lehmann, A. R. (2001). The Xeroderma Pigmentosum Group D (XPD) Gene: One Gene, Two Functions, Three Diseases. Genes. Dev. 15, 15–23. doi:10.1101/gad.859501
Lehmann, A. R., McGibbon, D., and Stefanini, M. (2011). Xeroderma Pigmentosum. Orphanet J. Rare Dis. 6, 70. doi:10.1186/1750-1172-6-70
Le Loarer, F., Watson, S., Pierron, G., de Montpreville, V. T., Firmin, N., et al. (2015). SMARCA4 Inactivation Defines a Group of Undifferentiated Thoracic Malignancies Transcriptionally Related to BAF-Deficient Sarcomas. Nat. Genet. 47, 1200–1205. doi:10.1038/ng10.1038/ng.3399
Levitus, M., Waisfisz, Q., Godthelp, B. C., Vries, Y. d., Hussain, S., Wiegant, W. W., et al. (2005). The DNA Helicase BRIP1 Is Defective in Fanconi Anemia Complementation Group J. Nat. Genet. 37, 934–935. doi:10.1038/ng1625
Levran, O., Attwooll, C., Henry, R. T., Milton, K. L., Neveling, K., Rio, P., et al. (2005). The BRCA1-Interacting Helicase BRIP1 Is Deficient in Fanconi Anemia. Nat. Genet. 37, 931–933. doi:10.1038/ng1624
Li, D., Wang, Q., Gong, N. N., Kurolap, A., Feldman, H. B., Boy, N., et al. (2021). Pathogenic Variants in SMARCA5, a Chromatin Remodeler, Cause a Range of Syndromic Neurodevelopmental Features. Sci. Adv. 7, Eabf2066. doi:10.1126/sciadv.abf2066
Li, G. H., Akatsuka, S., Chew, S. H., Jiang, L., Nishiyama, T., Sakamoto, A., et al. (2017). Fenton Reaction-Induced Renal Carcinogenesis inMutyh-Deficient Mice Exhibits Less Chromosomal Aberrations Than the Rat Model. Pathol. Int. 67, 564–574. doi:10.1111/pin.125910.1111/pin.12598
Li, X., Tian, R., Gao, H., Yan, F., Ying, L., Yang, Y., et al. (2018). Identification of Significant Gene Signatures and Prognostic Biomarkers for Patients with Cervical Cancer by Integrated Bioinformatic Methods. Technol. Cancer Res. Treat. 17, 1533033818767455. doi:10.1177/10.1177/1533033818767455
Liang, H., Masoro, E. J., Nelson, J. F., Strong, R., McMahan, C. A., and Richardson, A. (2003). Genetic Mouse Models of Extended Lifespan. Exp. Gerontol. 38, 1353–1364. doi:10.1016/j.exger10.1016/j.exger.2003.10.019
Lieb, S., Blaha-Ostermann, S., Kamper, E., Rippka, J., Schwarz, C., Ehrenhöfer-Wölfer, K., et al. (2019). Werner Syndrome Helicase Is a Selective Vulnerability of Microsatellite Instability-High Tumor Cells. Elife 8, e43333. doi:10.7554/eLife.43333
Lin, W., Sampathi, S., Dai, H., Liu, C., Zhou, M., Hu, J., et al. (2013). Mammalian DNA2 Helicase/nuclease Cleaves G-Quadruplex DNA and Is Required for Telomere Integrity. EMBO J. 32 (3210), 1425–1439. doi:10.1038/emboj.2013.88
Litman, R., Peng, M., Jin, Z., Zhang, F., Zhang, J., Powell, S., et al. (2005). BACH1 Is Critical for Homologous Recombination and Appears to Be the Fanconi Anemia Gene Product FANCJ. Cancer. Cell. 8, 255–265. doi:10.1016/j.ccr.2005.08.004
Liu, X., Liu, T., Shang, Y., Dai, P., Zhang, W., Lee, B. J., et al. (2020). ERCC6L2 Promotes DNA Orientation-specific Recombination in Mammalian Cells. Cell. Res. 30, 732–744. doi:10.1038/s41422-020-0328-3
Liu, Y., Harmelink, C., Peng, Y., Chen, Y., Wang, Q., and Jiao, K. (2014). CHD7 Interacts with BMP R-SMADs to Epigenetically Regulate Cardiogenesis in Mice. Hum. Mol. Genet. 23, 2145–2156. doi:10.1093/hmg/ddt610
Liu, Z., Macias, M., Bottomley, M., Stier, G., Linge, J., Nilges, M., et al. (1999). The Three-Dimensional Structure of the HRDC Domain and Implications for the Werner and Bloom Syndrome Proteins. Structure 7, 1557–1566. doi:10.1016/s0969-2126(00)88346-x
Loftfield, E., Zhou, W., Graubard, B. I., Yeager, M., Chanock, S. J., Freedman, N. D., et al. (2018). Predictors of Mosaic Chromosome Y Loss and Associations with Mortality in the UK Biobank. Sci. Rep. 8, 12316. doi:10.1038/s41598-018-30759-1
Lombard, D. B., Beard, C., Johnson, B., Marciniak, R. A., Dausman, J., Bronson, R., et al. (2000). Mutations in the WRN Gene in Mice Accelerate Mortality in a P53-Null Background. Mol. Cell. Biol. 20, 3286–3291. doi:10.1128/MCB.20.9.3286-3291.2000
Lopez, C. R., Singh, S., Hambarde, S., Griffin, W. C., Gao, J., Chib, S., et al. (2017). Yeast Sub1 and Human PC4 Are G-Quadruplex Binding Proteins that Suppress Genome Instability at Co-transcriptionally Formed G4 DNA. Nucleic Acids Res. 45, 5850–5862. doi:10.1093/nar/gkx201
Lu, H., and Davis, A. J. (2021). Human RecQ Helicases in DNA Double-Strand Break Repair. Front. Cell. Dev. Biol. 9, 640755. doi:10.3389/fcell.2021.640755
Lu, H., Fang, E. F., Sykora, P., Kulikowicz, T., Zhang, Y., Becker, K. G., et al. (2014). Senescence Induced by RECQL4 Dysfunction Contributes to Rothmund-Thomson Syndrome Features in Mice. Cell Death Dis. 5, e1226. doi:10.1038/cddis.2014.168
Lu, H., Shamanna, R. A., Keijzers, G., Anand, R., Rasmussen, L. J., Cejka, P., et al. (2016). RECQL4 Promotes DNA End Resection in Repair of DNA Double-Strand Breaks. Cell. Rep. 16, 161–173. doi:10.1016/j.celrep.2016.05.079
Luijsterburg, M. S., de Krijger, I., Wiegant, W. W., Shah, R. G., Smeenk, G., de Groot, A. J. L., et al. (2016). PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell. 61, 547–562. doi:10.1016/j.molcel.2016.01.019
Luo, G., Santoro, I. M., McDaniel, L. D., Nishijima, I., Mills, M., Youssoufian, H., et al. (2000). Cancer Predisposition Caused by Elevated Mitotic Recombination in Bloom Mice. Nat. Genet. 26, 424–429. doi:10.1038/82548
Lusser, A., and Kadonaga, J. T. (2003). Chromatin Remodeling by ATP-dependent Molecular Machines. Bioessays 25, 1192–1200. doi:10.1002/bies.10359
Lusser, A., Urwin, D. L., and Kadonaga, J. T. (2005). Distinct Activities of CHD1 and ACF in ATP-dependent Chromatin Assembly. Nat. Struct. Mol. Biol. 12, 160–166. doi:10.1038/nsmb884
Lutz, T., Stöger, R., and Nieto, A. (2006). CHD6 Is a DNA-dependent ATPase and Localizes at Nuclear Sites of mRNA Synthesis. FEBS Lett. 580, 5851–5857. doi:10.1016/j.febslet.2006.09.049
Lutzmann, M., Bernex, F., da Costa de Jesus, C., Hodroj, D., Marty, C., Plo, I., et al. (2019). MCM8- and MCM9 Deficiencies Cause Lifelong Increased Hematopoietic DNA Damage Driving P53-dependent Myeloid Tumors. Cell. Rep. 28, 2851–2865. doi:10.1016/j.celrep10.1016/j.celrep.2019.07.095
Lutzmann, M., Grey, C., Traver, S., Ganier, O., Maya-Mendoza, A., Ranisavljevic, N., et al. (2012). MCM8- and MCM9-Deficient Mice Reveal Gametogenesis Defects and Genome Instability Due to Impaired Homologous Recombination. Mol. Cell. 47, 523–534. doi:10.1016/j.molcel.2012.05.048
Mackay, V., and Linn, S. (1976). Selective Inhibition of the Dnase Activity of the recBC Enzyme by the DNA Binding Protein from Escherichia coli. J. Biol. Chem. 251, 3716–3719. doi:10.1016/s0021-9258(17)33402-6
Maiorano, D., Cuvier, O., Danis, E., and Méchali, M. (2005). MCM8 Is an MCM2-7-Related Protein that Functions as a DNA Helicase during Replication Elongation and Not Initiation. Cell. 120, 315–328. doi:10.1016/j.cell.2004.12.010
Mann, M. B., Hodges, C. A., Barnes, E., Vogel, H., Hassold, T. J., and Luo, G. (2005). Defective Sister-Chromatid Cohesion, Aneuploidy and Cancer Predisposition in a Mouse Model of Type II Rothmund-Thomson Syndrome. Hum. Mol. Genet. 14, 813–825. doi:10.1093/hmg/ddi075
Marfella, C. G. A., and Imbalzano, A. N. (2007). The Chd Family of Chromatin Remodelers. Mutat. Research/Fundamental Mol. Mech. Mutagen. 618, 30–40. doi:10.1016/j.mrfmmm.2006.07.012
Marfella, C. G. A., Ohkawa, Y., Coles, A. H., Garlick, D. S., Jones, S. N., and Imbalzano, A. N. (2006). Mutation of the SNF2 Family Member Chd2 Affects Mouse Development and Survival. J. Cell. Physiol. 209, 162–171. doi:10.1002/jcp.20718
Marino, F., Vindigni, A., and Onesti, S. (2013). Bioinformatic Analysis of RecQ4 Helicases Reveals the Presence of a RQC Domain and a Zn Knuckle. Biophys. Chem. 177-178, 34–39. doi:10.1016/j.bpc.2013.02.009
Marmot, M. G., Altman, D. G., Altman, D. G., Cameron, D. A., Dewar, J. A., Thompson, S. G., et al. (2013). The Benefits and Harms of Breast Cancer Screening: an Independent Review. Br. J. Cancer 108, 2205–2240. doi:10.1038/bjc.2013.177
Massip, L., Garand, C., Turaga, R. V. N., Deschênes, F., Thorin, E., and Lebel, M. (2006). Increased Insulin, Triglycerides, Reactive Oxygen Species, and Cardiac Fibrosis in Mice with a Mutation in the Helicase Domain of the Werner Syndrome Gene Homologue. Exp. Gerontol. 41, 157–168. doi:10.1016/j.exger.2005.10.011
Masuda-Sasa, T., Polaczek, P., Peng, X. P., Chen, L., and Campbell, J. L. (2008). Processing of G4 DNA by Dna2 Helicase/nuclease and Replication Protein A (RPA) Provides Insights into the Mechanism of Dna2/RPA Substrate Recognition. J. Biol. Chem. 283, 24359–24373. doi:10.1074/10.1074/jbc.m802244200
Matsumoto, K., Seki, M., Masutani, C., Tada, S., Enomoto, T., and Ishimi, Y. (1995). Stimulation of DNA Synthesis by Mouse DNA Helicase B in a DNA Replication System Containing Eukaryotic Replication Origins. Biochemistry 34, 7913–7922. doi:10.1021/bi00024a016
Matsuno, K., Kumano, M., Kubota, Y., Hashimoto, Y., and Takisawa, H. (2006). The N-Terminal Noncatalytic Region of Xenopus RecQ4 Is Required for Chromatin Binding of DNA Polymerase α in the Initiation of DNA Replication. Mol. Cell. Biol. 26, 4843–4852. doi:10.1128/MCB.02267-05
Matsuzaki, K., Borel, V., Adelman, C. A., Schindler, D., and Boulton, S. J. (2015). FANCJ Suppresses Microsatellite Instability and Lymphomagenesis Independent of the Fanconi Anemia Pathway. Genes. Dev. 29, 2532–2546. doi:10.1101/gad.272740.115
Maynard, S., Fang, E. F., Scheibye-Knudsen, M., Croteau, D. L., and Bohr, V. A. (2015). DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 5, a025130. doi:10.1101/cshperspect.a025130
McKinzey, D. R., Gomathinayagam, S., Griffin, W. C., Klinzing, K. N., Jeffries, E. P., Rajkovic, A., et al. (2021). Motifs of the C-Terminal Domain of MCM9 Direct Localization to Sites of Mitomycin-C Damage for RAD51 Recruitment. J. Biol. Chem. 296, 100355. doi:10.1016/j.jbc.2021.100355
Mestas, J., and Hughes, C. C. W. (2004). Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 172, 2731–2738. doi:10.4049/jimmunol.172.5.2731
Milenkovic, D., Matic, S., Kühl, I., Ruzzenente, B., Freyer, C., Jemt, E., et al. (2013). TWINKLE Is an Essential Mitochondrial Helicase Required for Synthesis of Nascent D-Loop Strands and Complete mtDNA Replication. Hum. Mol. Genet. 22, 1983–1993. doi:10.1093/hmg/10.1093/hmg/ddt051
Milholland, B., Dong, X., Zhang, L., Hao, X., Suh, Y., and Vijg, J. (2017). Differences between Germline and Somatic Mutation Rates in Humans and Mice. Nat. Commun. 8, 15183. doi:10.1038/ncomms15183
Min, J.-N., Tian, Y., Xiao, Y., Wu, L., Li, L., and Chang, S. (2013). The mINO80 Chromatin Remodeling Complex Is Required for Efficient Telomere Replication and Maintenance of Genome Stability. Cell. Res. 23, 1396–1413. doi:10.1038/cr.2013.113
Mohaghegh, P., and Hickson, I. D. (2001). DNA Helicase Deficiencies Associated with Cancer Predisposition and Premature Ageing Disorders. Hum. Mol. Genet. 10, 741–746. doi:10.1093/hmg/10.7.741
Morishita, T., Furukawa, F., Sakaguchi, C., Toda, T., Carr, A. M., Iwasaki, H., et al. (2005). Role of the Schizosaccharomyces pombe F-Box DNA Helicase in Processing Recombination Intermediates. Mol. Cell. Biol. 25, 8074–8083. doi:10.1128/MCB.25.18.8074-8083.2005
Nagarajan, P., Onami, T. M., Rajagopalan, S., Kania, S., Donnell, R., and Venkatachalam, S. (2009). Role of Chromodomain Helicase DNA-Binding Protein 2 in DNA Damage Response Signaling and Tumorigenesis. Oncogene 28, 1053–1062. doi:10.1038/onc.2008.440
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and Rates of Exonic De Novo Mutations in Autism Spectrum Disorders. Nature 485, 242–245. doi:10.1038/nature11011
Newton, C. A., Batra, K., Torrealba, J., Kozlitina, J., Glazer, C. S., Aravena, C., et al. (2016). Telomere-related Lung Fibrosis Is Diagnostically Heterogeneous but Uniformly Progressive. Eur. Respir. J. 48, 1710–1720. doi:10.1183/13993003.00308-2016
Nguyen, T. L. A., Vieira-Silva, S., Liston, A., and Raes, J. (2015). How Informative Is the Mouse for Human Gut Microbiota Research? Dis. Model. Mech. 8, 1–16. doi:10.1242/dmm.017400
Nioi, P., Nguyen, T., Sherratt, P. J., and Pickett, C. B. (2005). The Carboxy-Terminal Neh3 Domain of Nrf2 Is Required for Transcriptional Activation. Mol. Cell. Biol. 25, 10895–10906. doi:10.1128/MCB.25.24.10895-10906.2005
Nishimura, K., Ishiai, M., Horikawa, K., Fukagawa, T., Takata, M., Takisawa, H., et al. (2012). Mcm8 and Mcm9 Form a Complex that Functions in Homologous Recombination Repair Induced by DNA Interstrand Crosslinks. Mol. Cell. 47, 511–522. doi:10.1016/j.molcel10.1016/j.molcel.2012.05.047
Nishiyama, M., Nakayama, K., Tsunematsu, R., Tsukiyama, T., Kikuchi, A., and Nakayama, K. I. (2004). Early Embryonic Death in Mice Lacking the β-Catenin-Binding Protein Duplin. Mol. Cell. Biol. 24, 8386–8394. doi:10.1128/MCB.24.19.8386-8394.2004
Nishiyama, M., Oshikawa, K., Tsukada, Y.-i., Nakagawa, T., Iemura, S.-i., Natsume, T., et al. (2009). CHD8 Suppresses P53-Mediated Apoptosis through Histone H1 Recruitment during Early Embryogenesis. Nat. Cell. Biol. 11, 172–182. doi:10.1038/ncb1831
Nousbeck, J., Burger, B., Fuchs-Telem, D., Pavlovsky, M., Fenig, S., Sarig, O., et al. (2011). A Mutation in a Skin-specific Isoform of SMARCAD1 Causes Autosomal-Dominant Adermatoglyphia. Am. J. Hum. Genet. 89, 302–307. doi:10.1016/j.ajhg.2011.07.004
Numata, Y., Ishihara, S., Hasegawa, N., Nozaki, N., and Ishimi, Y. (2010). Interaction of Human MCM2-7 Proteins with TIM, TIPIN and Rb. J. Biochem. 147, 917–927. doi:10.1093/jb/mvq028
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012b). Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 338, 1619–1622. doi:10.1126/science.1227764
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012a). Sporadic Autism Exomes Reveal a Highly Interconnected Protein Network of De Novo Mutations. Nature 485, 246–250. doi:10.1038/nature10989
O'Shaughnessy-Kirwan, A., Signolet, J., Costello, I., Gharbi, S., and Hendrich, B. (2015). Constraint of Gene Expression by the Chromatin Remodelling Protein CHD4 Facilitates Lineage Specification. Development 142, 2586–2597. doi:10.1242/dev.125450
Oh, K.-S., Khan, S. G., Jaspers, N. G. J., Raams, A., Ueda, T., Lehmann, A., et al. (2006). Phenotypic Heterogeneity in the XPB DNA Helicase Gene (ERCC3): Xeroderma Pigmentosum without and with Cockayne Syndrome. Hum. Mutat. 27, 1092–1103. doi:10.1002/humu.20392
Okano-Uchida, T., Kent, L. N., Ouseph, M. M., McCarty, B., Frank, J. J., Kladney, R., et al. (2018). Endoreduplication of the Mouse Genome in the Absence of ORC1. Genes. Dev. 32, 978–990. doi:10.1101/gad.311910.118
Oksenych, V., and Coin, F. (2010). The Long Unwinding Road: XPB and XPD Helicases in Damaged DNA Opening. Cell. Cycle 9, 90–96. doi:10.4161/cc.9.1.10267
Ooga, M., Funaya, S., Hashioka, Y., Fujii, W., Naito, K., Suzuki, M. G., et al. (2018). Chd9 Mediates Highly Loosened Chromatin Structure in Growing Mouse Oocytes. Biochem. Biophysical Res. Commun. 500, 583–588. doi:10.1016/j.bbrc.2018.04.105
Osman, F., Dixon, J., Barr, A. R., and Whitby, M. C. (2005). The F-Box DNA Helicase Fbh1 Prevents Rhp51-dependent Recombination without Mediator Proteins. Mol. Cell. Biol. 25, 8084–8096. doi:10.1128/MCB.25.18.8084-8096.2005
Parenti, I., Lehalle, D., Nava, C., Torti, E., Leitão, E., Person, R., et al. (2021). Missense and Truncating Variants in CHD5 in a Dominant Neurodevelopmental Disorder with Intellectual Disability, Behavioral Disturbances, and Epilepsy. Hum. Genet. 140, 1109–1120. doi:10.1007/s10.1007/s00439-021-02283-2
Park, J., Long, D. T., Lee, K. Y., Abbas, T., Shibata, E., Negishi, M., et al. (2013). The MCM8-MCM9 Complex Promotes RAD51 Recruitment at DNA Damage Sites to Facilitate Homologous Recombination. Mol. Cell. Biol. 33, 1632–1644. doi:10.1128/MCB.01503-12
Parmar, K., D’Andrea, A., and Niedernhofer, L. J. (2009). Mouse Models of Fanconi Anemia. Mutat. Research/Fundamental Mol. Mech. Mutagen. 668, 133–140. doi:10.1016/j.mrfmmm.2009.03.015
Perkins, G., and Diffley, J. F. X. (1998). Nucleotide-dependent Prereplicative Complex Assembly by Cdc6p, a Homolog of Eukaryotic and Prokaryotic Clamp-Loaders. Mol. Cell. 2, 23–32. doi:10.1016/s1097-2765(00)80110-0
Perry, J. J. P., Yannone, S. M., Holden, L. G., Hitomi, C., Asaithamby, A., Han, S., et al. (2006). WRN Exonuclease Structure and Molecular Mechanism Imply an Editing Role in DNA End Processing. Nat. Struct. Mol. Biol. 13, 414–422. doi:10.1038/nsmb1088
Pilarowski, G. O., Vernon, H. J., Applegate, C. D., Boukas, L., Cho, M. T., Gurnett, C. A., et al. (2018). Missense Variants in the Chromatin Remodeler CHD1 Are Associated with Neurodevelopmental Disability. J. Med. Genet. 55, 561–566. doi:10.1136/jmedgenet-2017-104759
Pisansky, M. T., Young, A. E., O'Connor, M. B., Gottesman, I. I., Bagchi, A., and Gewirtz, J. C. (2017). Mice Lacking the Chromodomain Helicase DNA-Binding 5 Chromatin Remodeler Display Autism-like Characteristics. Transl. Psychiatry 7, e1152. doi:10.1038/tp.2017.111
Potts, R. C., Zhang, P., Wurster, A. L., Precht, P., Mughal, M. R., Wood, W. H., et al. (2011). CHD5, a Brain-specific Paralog of Mi2 Chromatin Remodeling Enzymes, Regulates Expression of Neuronal Genes. PLoS One 6, e24515. doi:10.1371/journal.pone.0024515
Pray-Grant, M. G., Daniel, J. A., Schieltz, D., Yates, J. R., and Grant, P. A. (2005). Chd1 Chromodomain Links Histone H3 Methylation with SAGA- and SLIK-dependent Acetylation. Nature 433, 434–438. doi:10.1038/nature03242
Pruitt, S. C., Bailey, K. J., and Freeland, A. (2007). Reduced Mcm2 Expression Results in Severe Stem/progenitor Cell Deficiency and Cancer. Stem Cells 25, 3121–3132. doi:10.1634/stemcells10.1634/stemcells.2007-0483
Puranam, K. L., and Blackshear, P. J. (1994). Cloning and Characterization of RECQL, a Potential Human Homologue of the Escherichia coli DNA Helicase RecQ. J. Biol. Chem. 269, 29838–29845. doi:10.1016/S0021-9258(18)43957-9
Pyo, J. O., Yoo, S. M., Ahn, H. H., Nah, J., Hong, S. H., Kam, T. I., et al. (2013). Overexpression of Atg5 in Mice Activates Autophagy and Extends Lifespan. Nat. Commun. 4 (4), 2300. doi:10.1038/ncomms3300
Rafnar, T., Gudbjartsson, D. F., Sulem, P., Jonasdottir, A., Sigurdsson, A., Jonasdottir, A., et al. (2011). Mutations in BRIP1 Confer High Risk of Ovarian Cancer. Nat. Genet. 43, 1104–1107. doi:10.1038/ng.955
Ramanagoudr-Bhojappa, R., Blair, L. P., Tackett, A. J., and Raney, K. D. (2013). Physical and Functional Interaction between Yeast Pif1 Helicase and Rim1 Single-Stranded DNA Binding Protein. Nucleic Acids Res. 41, 1029–1046. doi:10.1093/nar/gks1088
Reyes, J. C., Barra, J., Muchardt, C., Camus, A., Babinet, C., and Yaniv, M. (1998). Altered Control of Cellular Proliferation in the Absence of Mammalian Brahma (SNF2α). EMBO J. 17, 6979–6991. doi:10.1093/emboj/17.23.6979
Robinson, P. S., Coorens, T. H. H., Palles, C., Mitchell, E., Abascal, F., Olafsson, S., et al. (2021a). Increased Somatic Mutation Burdens in Normal Human Cells Due to Defective DNA Polymerases. Nat. Genet. 53, 1434–1442. doi:10.1038/s41588-021-00930-y
Robinson, P. S., Thomas, L. E., Abascal, F., Jung, H., Harvey, L. M. R., West, H. D., et al. (2021b). Inherited MUTYH Mutations Cause Elevated Somatic Mutation Rates and Distinctive Mutational Signatures in Normal Human Cells. Nat. Commun. 13, 3949. doi:10.1038/s41467-022-31341-0
Ronchi, D., Di Fonzo, A., Lin, W., Bordoni, A., Liu, C., Fassone, E., et al. (2013). Mutations in DNA2 Link Progressive Myopathy to Mitochondrial DNA Instability. Am. J. Hum. Genet. 92, 293–300. doi:10.1016/j.ajhg.2012.12.014
Rong, S.-B., Väliaho, J., and Vihinen, M. (2000). Structural Basis of Bloom Syndrome (BS) Causing Mutations in the BLM Helicase Domain. Mol. Med. 6, 155–164. doi:10.1007/bf03402111
Sanders, C. M. (2010). Human Pif1 Helicase Is a G-Quadruplex DNA-Binding Protein with G-Quadruplex DNA-Unwinding Activity. Biochem. J. 430, 119–128. doi:10.1042/BJ20100612
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De Novo mutations Revealed by Whole-Exome Sequencing Are Strongly Associated with Autism. Nature 485, 237–241. doi:10.1038/nature10945
Sangrithi, M. N., Bernal, J. A., Madine, M., Philpott, A., Lee, J., Dunphy, W. G., et al. (2005). Initiation of DNA Replication Requires the RECQL4 Protein Mutated in Rothmund-Thomson Syndrome. Cell. 121, 887–898. doi:10.1016/j.cell.2005.05.015
Sanlaville, D., and Verloes, A. (2007). CHARGE Syndrome: an Update. Eur. J. Hum. Genet. 15, 389–399. doi:10.1038/sj.ejhg.5201778
Saponaro, M., Kantidakis, T., Mitter, R., Kelly, G. P., Heron, M., Williams, H., et al. (2014). RECQL5 Controls Transcript Elongation and Suppresses Genome Instability Associated with Transcription Stress. Cell. 157, 1037–1049. doi:10.1016/j.cell.2014.03.048
Schoor, M., Schuster-Gossler, K., Roopenian, D., and Gossler, A. (1999). Skeletal Dysplasias, Growth Retardation, Reduced Postnatal Survival, and Impaired Fertility in Mice Lacking the SNF2/SWI2 Family Member ETL1. Mech. Dev. 85, 73–83. doi:10.1016/s0925-4773(99)00090-8
Schulz, V. P., and Zakian, V. A. (1994). The saccharomyces PIF1 DNA Helicase Inhibits Telomere Elongation and De Novo Telomere Formation. Cell. 76, 145–155. doi:10.1016/0092-8674(94)9010.1016/0092-8674(94)90179-1
Schumacher, B., Pothof, J., Vijg, J., and Hoeijmakers, J. H. J. (2021). The Central Role of DNA Damage in the Ageing Process. Nature 592, 695–703. doi:10.1038/s41586-021-03307-7
Seal, S., Thompson, D., Thompson, D., Renwick, A., Elliott, A., Kelly, P., et al. (2006). Truncating Mutations in the Fanconi Anemia J Gene BRIP1 Are Low-Penetrance Breast Cancer Susceptibility Alleles. Nat. Genet. 38, 1239–1241. doi:10.1038/ng1902
Sen, P., Shah, P. P., Nativio, R., and Berger, S. L. (2016). Epigenetic Mechanisms of Longevity and Aging. Cell. 166, 822–839. doi:10.1016/j.cell.2016.07.050
Seok, J., Warren, H. S., Cuenca, A. G., Mindrinos, M. N., Baker, H. V., Xu, W., et al. (2013). Genomic Responses in Mouse Models Poorly Mimic Human Inflammatory Diseases. Proc. Natl. Acad. Sci. U.S.A. 110, 3507–3512. doi:10.1073/pnas.1222878110
Shaheen, R., Faqeih, E., Ansari, S., Abdel-Salam, G., Al-Hassnan, Z. N., Al-Shidi, T., et al. (2014). Genomic Analysis of Primordial Dwarfism Reveals Novel Disease Genes. Genome Res. 24, 291–299. doi:10.1101/gr.160572.113
Shamanna, R. A., Singh, D. K., Lu, H., Mirey, G., Keijzers, G., Salles, B., et al. (2014). RECQ Helicase RECQL4 Participates in Non-homologous End Joining and Interacts with the Ku Complex. Carcinogenesis 35, 2415–2424. doi:10.1093/carcin/bgu137
Sharma, S., Stumpo, D. J., Balajee, A. S., Bock, C. B., Lansdorp, P. M., Brosh, R. M., et al. (2007). RECQL, a Member of the RecQ Family of DNA Helicases, Suppresses Chromosomal Instability. Mol. Cell. Biol. 27, 1784–1794. doi:10.1128/MCB.01620-06
Shen, J.-C., Gray, M. D., Oshima, J., Kamath-Loeb, A. S., Fry, M., and Loeb, L. A. (1998). Werner Syndrome Protein. J. Biol. Chem. 273, 34139–34144. doi:10.1074/jbc.273.51.34139
Shen, X., Mizuguchi, G., Hamiche, A., and Wu, C. (2000). A Chromatin Remodelling Complex Involved in Transcription and DNA Processing. Nature 406, 541–544. doi:10.1038/35020123
Shete, S., Hosking, F. J., Robertson, L. B., Dobbins, S. E., Sanson, M., Malmer, B., et al. (2009). Genome-wide Association Study Identifies Five Susceptibility Loci for Glioma. Nat. Genet. 41, 899–904. doi:10.1038/ng.407
Shima, N., Alcaraz, A., Liachko, I., Buske, T. R., Andrews, C. A., Munroe, R. J., et al. (2007). A Viable Allele of Mcm4 Causes Chromosome Instability and Mammary Adenocarcinomas in Mice. Nat. Genet. 39, 93–98. doi:10.1038/ng1936
Shur, I., Socher, R., and Benayahu, D. (2006). In Vivo association of CReMM/CHD9 with Promoters in Osteogenic Cells. J. Cell. Physiol. 207, 374–378. doi:10.1002/jcp.20586
Sifrim, A., Hitz, M. P., Hitz, M.-P., Wilsdon, A., Breckpot, J., Turki, S. H. A., et al. (2016). Distinct Genetic Architectures for Syndromic and Nonsyndromic Congenital Heart Defects Identified by Exome Sequencing. Nat. Genet. 48, 1060–1065. doi:10.1038/ng.3627
Simic, R., Lindstrom, D. L., Tran, H. G., Roinick, K. L., Costa, P. J., Johnson, A. D., et al. (2003). Chromatin Remodeling Protein Chd1 Interacts with Transcription Elongation Factors and Localizes to Transcribed Genes. EMBO J. 22, 1846–1856. doi:10.1093/emboj/cdg179
Singh, P. P., Demmitt, B. A., Nath, R. D., and Brunet, A. (2019). The Genetics of Aging: A Vertebrate Perspective. Cell. 177, 200–220. doi:10.1016/j.cell.2019.02.038
Singleton, M. R., Dillingham, M. S., and Wigley, D. B. (2007). Structure and Mechanism of Helicases and Nucleic Acid Translocases. Annu. Rev. Biochem. 76, 23–50. doi:10.1146/annurev.biochem.76.052305.115300
Skarnes, W. C., Rosen, B., West, A. P., Koutsourakis, M., Bushell, W., Iyer, V., et al. (2011). A Conditional Knockout Resource for the Genome-wide Study of Mouse Gene Function. Nature 474, 337–342. doi:10.1038/nature10163
Snijders Blok, L., Rousseau, J., Rousseau, J., Twist, J., Ehresmann, S., Takaku, M., et al. (2018). CHD3 Helicase Domain Mutations Cause a Neurodevelopmental Syndrome with Macrocephaly and Impaired Speech and Language. Nat. Commun. 9, 4619. doi:10.1038/s414610.1038/s41467-018-06014-6
Snow, B. E., Mateyak, M., Paderova, J., Wakeham, A., Iorio, C., Zakian, V., et al. (2006). Murine Pif1 Interacts with Telomerase and Is Dispensable for Telomere Function In Vivo. Mol. Cell. Biol. 27, 1017–1026. doi:10.1128/MCB.01866-06
Sommers, J. A., Rawtani, N., Gupta, R., Bugreev, D. V., Mazin, A. V., Cantor, S. B., et al. (2009). FANCJ Uses its Motor ATPase to Destabilize Protein-DNA Complexes, Unwind Triplexes, and Inhibit RAD51 Strand Exchange. J. Biol. Chem. 284, 7505–7517. doi:10.1074/jbc.M809019200
Sparks, M. A., Burgers, P. M., and Galletto, R. (2020). Pif1, RPA, and FEN1 Modulate the Ability of DNA Polymerase δ to Overcome Protein Barriers during DNA Synthesis. J. Biol. Chem. 295, 15883–15891. doi:10.1074/jbc.RA120.015699
Speckmann, C., Sahoo, S. S., Rizzi, M., Hirabayashi, S., Karow, A., Serwas, N. K., et al. (2017). Clinical and Molecular Heterogeneity of RTEL1 Deficiency. Front. Immunol. 8, 449. doi:10.3389/fimmu.2017.00449
Spelbrink, J. N., Li, F.-Y., Tiranti, V., Nikali, K., Yuan, Q.-P., Tariq, M., et al. (2001). Human Mitochondrial DNA Deletions Associated with Mutations in the Gene Encoding Twinkle, a Phage T7 Gene 4-like Protein Localized in Mitochondria. Nat. Genet. 28, 223–231. doi:10.1038/90058
Stevnsner, T., Nyaga, S., de Souza-Pinto, N. C., Gorgels, T. G. M. F., and Hogue, B. A. (2002). Mitochondrial Repair of 8-oxoguanine Is Deficient in Cockayne Syndrome Group B. Oncogene 21, 8675–8682. doi:10.1038/sj.onc.1205994
Stopka, T., and Skoultchi, A. I. (2003). The ISWI ATPase Snf2h Is Required for Early Mouse Development. Proc. Natl. Acad. Sci. U.S.A. 100, 14097–14102. doi:10.1073/pnas.2336105100
Stuart, B. D., Choi, J., Zaidi, S., Xing, C., Holohan, B., Chen, R., et al. (2015). Exome Sequencing Links Mutations in PARN and RTEL1 with Familial Pulmonary Fibrosis and Telomere Shortening. Nat. Genet. 47, 512–517. doi:10.1038/ng.3278
Sugathan, A., Biagioli, M., Golzio, C., Erdin, S., Blumenthal, I., Manavalan, P., et al. (2014). CHD8 Regulates Neurodevelopmental Pathways Associated with Autism Spectrum Disorder in Neural Progenitors. Proc. Natl. Acad. Sci. U. S. A. 111, E4468–E4477. doi:10.1073/pnas.1405266110.1073/pnas.1405266111
Sun, J., Wang, Y., Xia, Y., Xu, Y., Ouyang, T., Li, J., et al. (2015). Mutations in RECQL Gene Are Associated with Predisposition to Breast Cancer. PLoS Genet. 11, e1005228. doi:10.1371/journal.pgen.1005228
Sun, L.-Q., Lee, D. W., Zhang, Q., Xiao, W., Raabe, E. H., Meeker, A., et al. (2004). Growth Retardation and Premature Aging Phenotypes in Mice with Disruption of the SNF2-like Gene, PASG. Genes. Dev. 18, 1035–1046. doi:10.1101/gad.1176104
Sun, X., Brieño-Enríquez, M. A., Cornelius, A., Modzelewski, A. J., Maley, T. T., Campbell-Peterson, K. M., et al. (2016). FancJ (Brip1) Loss-Of-Function Allele Results in Spermatogonial Cell Depletion during Embryogenesis and Altered Processing of Crossover Sites during Meiotic Prophase I in Mice. Chromosoma 125, 237–252. doi:10.1007/s00412-015-0549-2
Suomalainen, A., Majander, A., Wallin, M., Setälä, K., Kontula, K., Leinonen, H., et al. (1997). Autosomal Dominant Progressive External Ophthalmoplegia with Multiple Deletions of mtDNA: Clinical, Biochemical, and Molecular Genetic Features of the 1Oq-Linked Disease. Neurology 48, 1244–1253. doi:10.1212/wnl.48.5.1244
Surapureddi, S., Yu, S., Bu, H., Hashimoto, T., Yeldandi, A. V., Kashireddy, P., et al. (2002). Identification of a Transcriptionally Active Peroxisome Proliferator-Activated Receptor α-interacting Cofactor Complex in Rat Liver and Characterization of PRIC285 as a Coactivator. Proc. Natl. Acad. Sci. U.S.A. 99, 11836–11841. doi:10.1073/pnas.182426699
Suzuki, T., Kohno, T., and Ishimi, Y. (2009). DNA Helicase Activity in Purified Human RECQL4 Protein. J. Biochem. 146, 327–335. doi:10.1093/jb/mvp074
Takao, K., and Miyakawa, T. (2015). Genomic Responses in Mouse Models Greatly Mimic Human Inflammatory Diseases. Proc. Natl. Acad. Sci. U.S.A. 112, 1167–1172. doi:10.1073/pnas.14019610.1073/pnas.1401965111
Tamberg, N., Tahk, S., Koit, S., Kristjuhan, K., Kasvandik, S., Kristjuhan, A., et al. (2018). Keap1-MCM3 Interaction Is a Potential Coordinator of Molecular Machineries of Antioxidant Response and Genomic DNA Replication in Metazoa. Sci. Rep. 8, 12136. doi:10.1038/s41598-018-30562-y
Tanaka, S., Umemori, T., Hirai, K., Muramatsu, S., Kamimura, Y., and Araki, H. (2007). CDK-Dependent Phosphorylation of Sld2 and Sld3 Initiates DNA Replication in Budding Yeast. Nature 445, 328–332. doi:10.1038/nature05465
Tang, J., Wu, S., Liu, H., Stratt, R., Barak, O. G., Shiekhattar, R., et al. (2004). A Novel Transcription Regulatory Complex Containing Death Domain-Associated Protein and the ATR-X Syndrome Protein. J. Biol. Chem. 279, 20369–20377. doi:10.1074/jbc.M401321200
Tanner, N. K., and Linder, P. (2001). DExD/H Box RNA Helicases. Mol. Cell. 8, 251–262. doi:10.1016/s1097-2765(01)00329-x
Tarnauskaitė, Ž., Bicknell, J. A., Murray, J. E., Parry, D. A., Logan, C. V., Bober, M. B., et al. (2019). Biallelic Variants in DNA2 Cause Microcephalic Primordial Dwarfism. Hum. Mutat. 40, 1063–1070. doi:10.1002/humu.23776
Teng, Y.-C., Sundaresan, A., O’Hara, R., Gant, V. U., Li, M., Martire, S., et al. (2021). ATRX Promotes Heterochromatin Formation to Protect Cells from G-Quadruplex DNA-Mediated Stress. Nat. Commun. 12, 3887. doi:10.1038/s41467-021-24206-5
Thijssen, P. E., Ito, Y., Grillo, G., Wang, J., Velasco, G., Nitta, H., et al. (2015). Mutations in CDCA7 and HELLS Cause Immunodeficiency-Centromeric Instability-Facial Anomalies Syndrome. Nat. Commun. 6, 7870. doi:10.1038/ncomms8870
Thomas, R. H., Zhang, L. M., Carvill, G. L., Archer, J. S., Heavin, S. B., Mandelstam, S. A., et al. (2015). CHD2 Myoclonic Encephalopathy Is Frequently Associated with Self-Induced Seizures. Neurology 84, 951–958. doi:10.1212/WNL.0000000000001305
Thompson, D. J., Genovese, G., Genovese, G., Halvardson, J., Ulirsch, J. C., Wright, D. J., et al. (2019). Genetic Predisposition to Mosaic Y Chromosome Loss in Blood. Nature 575, 652–657. doi:10.1038/s41586-019-1765-3
Thompson, P. M., Gotoh, T., Kok, M., White, P. S., and Brodeur, G. M. (2003). CHD5, a New Member of the Chromodomain Gene Family, Is Preferentially Expressed in the Nervous System. Oncogene 22, 1002–1011. doi:10.1038/sj.onc.1206211
Tian, X., Firsanov, D., Zhang, Z., Cheng, Y., Luo, L., Tombline, G., et al. (2019). SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell. 177, 622–638. doi:10.1016/j.cell.2019.03.043
Tkáč, J., Xu, G., Adhikary, H., Young, J. T. F., Gallo, D., Escribano-Díaz, C., et al. (2016). HELB Is a Feedback Inhibitor of DNA End Resection. Mol. Cell. 61, 405–418. doi:10.1016/j.molcel.2015.12.013
Tower, J. (2015). Programmed Cell Death in Aging. Ageing Res. Rev. 23, 90–100. doi:10.1016/j.arr.2015.04.002
Tsurusaki, Y., Okamoto, N., Ohashi, H., Kosho, T., Imai, Y., Hibi-Ko, Y., et al. (2012). Mutations Affecting Components of the SWI/SNF Complex Cause Coffin-Siris Syndrome. Nat. Genet. 44, 376–378. doi:10.1038/ng.2219
Tummala, H., Dokal, A. D., Walne, A., Ellison, A., Cardoso, S., Amirthasigamanipillai, S., et al. (2018). Genome Instability Is a Consequence of Transcription Deficiency in Patients with Bone Marrow Failure Harboring Biallelic ERCC6L2 Variants. Proc. Natl. Acad. Sci. U.S.A. 115, 7777–7782. doi:10.1073/pnas.1803275115
Tummala, H., Kirwan, M., Walne, A. J., Hossain, U., Jackson, N., Pondarre, C., et al. (2014). ERCC6L2 Mutations Link a Distinct Bone-Marrow-Failure Syndrome to DNA Repair and Mitochondrial Function. Am. J. Hum. Genet. 94, 246–256. doi:10.1016/j.ajhg.2014.01.007
Tyynismaa, H., Mjosund, K. P., Wanrooij, S., Lappalainen, I., Ylikallio, E., Jalanko, A., et al. (2005). Mutant Mitochondrial Helicase Twinkle Causes Multiple mtDNA Deletions and a Late-Onset Mitochondrial Disease in Mice. Proc. Natl. Acad. Sci. U.S.A. 102, 17687–17692. doi:10.1073/pnas.0505551102
Umate, P., Tuteja, N., and Tuteja, R. (2011). Genome-wide Comprehensive Analysis of Human Helicases. Commun. Integr. Biol. 4, 118–137. doi:10.4161/cib.4.1.1384410.4161/cib.13844
Unoki, M., Funabiki, H., Velasco, G., Francastel, C., and Sasaki, H. (2019). CDCA7 and HELLS Mutations Undermine Nonhomologous End Joining in Centromeric Instability Syndrome. J. Clin. Investig. 129, 78–92. doi:10.1172/JCI99751
van Attikum, H., and Gasser, S. M. (2009). Crosstalk between Histone Modifications during the DNA Damage Response. Trends Cell. Biol. 19, 207–217. doi:10.1016/j.tcb.2009.03.001
van der Horst, G. T. J., van Steeg, H., Berg, R. J. W., van Gool, A. J., and Weeda, G. (1997). Defective Transcription-Coupled Repair in Cockayne Syndrome B Mice Is Associated with Skin Cancer Predisposition. Cell. 89, 425–435. doi:10.1016/s0092-8674(00)80223-8
Van Houdt, J. K. J., Sousa, S. B., van Schaik, B. D. C., and Seuntjens, E. (2012). Heterozygous Missense Mutations in SMARCA2 Cause Nicolaides-Baraitser Syndrome. Nat. Genet. 44, 445–449. doi:10.1038/ng.1105
van Wietmarschen, N., Sridharan, S., Nathan, W. J., Tubbs, A., Chan, E. M., Callen, E., et al. (2020). Repeat Expansions Confer WRN Dependence in Microsatellite-Unstable Cancers. Nature 586, 292–298. doi:10.1038/s41586-020-2769-8
Vannier, J.-B., Sandhu, S., Wu, X., Nabi, Z., Ding, H., Boulton, S. J., et al. (2013). RTEL1 Is a Replisome-Associated Helicase that Promotes Telomere and Genome-wide Replication. Science 342, 239–242. doi:10.1126/science.1241779
Veaute, X., Jeusset, J., Soustelle, C., Kowalczykowski, S. C., Le Cam, E., and Fabre, F. (2003). The Srs2 Helicase Prevents Recombination by Disrupting Rad51 Nucleoprotein Filaments. Nature 423, 309–312. doi:10.1038/nature01585
Venkatesan, R. N., Treuting, P. M., Fuller, E. D., Goldsby, R. E., Norwood, T. H., Gooley, T. A., et al. (2007). Mutation at the Polymerase Active Site of Mouse DNA Polymerase δ Increases Genomic Instability and Accelerates Tumorigenesis. Mol. Cell. Biol. 27, 7669–7682. doi:10.1128/MCB.00002-07
Vetro, A., Savasta, S., Russo Raucci, A., Cerqua, C., Sartori, G., Limongelli, I., et al. (2017). MCM5: a New Actor in the Link between DNA Replication and Meier-Gorlin Syndrome. Eur. J. Hum. Genet. 25, 646–650. doi:10.1038/ejhg.2017.5
Vijg, J., and Dong, X. (2020). Pathogenic Mechanisms of Somatic Mutation and Genome Mosaicism in Aging. Cell. 182, 12–23. doi:10.1016/j.cell.2020.06.024
Villa, F., Simon, A. C., Ortiz Bazan, M. A., Kilkenny, M. L., Wirthensohn, D., Wightman, M., et al. (2016). Ctf4 Is a Hub in the Eukaryotic Replisome that Links Multiple CIP-Box Proteins to the CMG Helicase. Mol. Cell. 63, 385–396. doi:10.1016/j.molcel.2016.06.009
Vindigni, A., and Hickson, I. D. (2009). RecQ Helicases: Multiple Structures for Multiple Functions? HFSP J. 3, 153–164. doi:10.2976/1.3079540
Vissers, L. E. L. M., van Ravenswaaij, C. M. A., Admiraal, R., Hurst, J. A., de Vries, B. B. A., Janssen, I. M., et al. (2004). Mutations in a New Member of the Chromodomain Gene Family Cause CHARGE Syndrome. Nat. Genet. 36, 955–957. doi:10.1038/ng1407
Viziteu, E., Klein, B., Basbous, J., Lin, Y.-L., Hirtz, C., Gourzones, C., et al. (2017). RECQ1 Helicase Is Involved in Replication Stress Survival and Drug Resistance in Multiple Myeloma. Leukemia 31, 2104–2113. doi:10.1038/leu.2017.54
von Eyss, B., Maaskola, J., Memczak, S., Möllmann, K., Schuetz, A., Loddenkemper, C., et al. (2012). The SNF2-like Helicase HELLS Mediates E2F3-dependent Transcription and Cellular Transformation. EMBO J. 31, 972–985. doi:10.1038/emboj.2011.451
Walne, A. J., Vulliamy, T., Kirwan, M., Plagnol, V., and Dokal, I. (2013). Constitutional Mutations in RTEL1 Cause Severe Dyskeratosis Congenita. Am. J. Hum. Genet. 92, 448–453. doi:10.1016/j.ajhg.2013.02.001
Wang, L., Du, Y., Ward, J. M., Shimbo, T., Lackford, B., Zheng, X., et al. (2014). INO80 Facilitates Pluripotency Gene Activation in Embryonic Stem Cell Self-Renewal, Reprogramming, Andblastocyst Development. Cell Stem Cell 14, 575–591. doi:10.1016/j.stem.2014.02.013
Wang, L. L., Gannavarapu, A., Kozinetz, C. A., Levy, M. L., Lewis, R. A., Chintagumpala, M. M., et al. (2003). Association between Osteosarcoma and Deleterious Mutations in the RECQL4 Gene in Rothmund-Thomson Syndrome. JNCI J. Natl. Cancer Inst. 95, 669–674. doi:10.1093/jnci/95.9.669
Weiss, K., Terhal, P. A., Cohen, L., Bruccoleri, M., Irving, M., Martinez, A. F., et al. (2016). De Novo Mutations in CHD4 , an ATP-dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet. 99, 934–941. doi:10.1016/j.ajhg.2016.08.001
Willhoft, O., McCormack, E. A., Aramayo, R. J., Bythell-Douglas, R., Ocloo, L., Zhang, X., et al. (2017). Crosstalk within a Functional INO80 Complex Dimer Regulates Nucleosome Sliding. Elife 6, e25782. doi:10.7554/eLife.25782
Williams, G. C. (1957). Pleiotropy Natural Selection, and the Evolution of Senescence. Evolution 11, 398–411. doi:10.2307/2406060
Wilson, D. M., and Thompson, L. H. (2007). Molecular Mechanisms of Sister-Chromatid Exchange. Mutat. Research/Fundamental Mol. Mech. Mutagen. 616, 11–23. doi:10.1016/j.mrfmmm.2006.11.017
Wilson, B. G., and Roberts, C. W. M. (2011). SWI/SNF Nucleosome Remodellers and Cancer. Nat. Rev. Cancer 11, 481–492. doi:10.1038/nrc3068
Wilson, M. A., Kwon, Y., Xu, Y., Chung, W.-H., Chi, P., Niu, H., et al. (2013). Pif1 Helicase and Polδ Promote Recombination-Coupled DNA Synthesis via Bubble Migration. Nature 502, 393–396. doi:10.1038/nature12585
Wilson, M.-M., Henshall, D. C., Byrne, S. M., and Brennan, G. P. (2021). CHD2-Related CNS Pathologies. Int. J. Mol. Sci. 22, 588. doi:10.3390/ijms22020588
Wu, Y., Sommers, J. A., Loiland, J. A., Kitao, H., Kuper, J., Kisker, C., et al. (2012). The Q Motif of Fanconi Anemia Group J Protein (FANCJ) DNA Helicase Regulates its Dimerization, DNA Binding, and DNA Repair Function. J. Biol. Chem. 287, 21699–21716. doi:10.1074/jbc.M112.351338
Wu, Y., Sommers, J. A., Suhasini, A. N., Leonard, T., Deakyne, J. S., Mazin, A. V., et al. (2010). Fanconi Anemia Group J Mutation Abolishes its DNA Repair Function by Uncoupling DNA Translocation from Helicase Activity or Disruption of Protein-DNA Complexes. Blood 116, 3780–3791. doi:10.1182/blood-2009-11-256016
Xia, L., Huang, W., Bellani, M., Seidman, M. M., Wu, K., Fan, D., et al. (2017). CHD4 Has Oncogenic Functions in Initiating and Maintaining Epigenetic Suppression of Multiple Tumor Suppressor Genes. Cancer Cell. 31, 653–668. doi:10.1016/j.ccell.2017.04.005
Xie, J., Gao, S., Schafer, C., Colijn, S., Muthukumar, V., and Griffin, C. T. (2020). The chromatin-remodeling enzyme CHD3 plays a role in embryonic viability but is dispensable for early vascular development. PLoS One. 15, e0235799. doi:10.1371/journal.pone.0235799
Xue, Y., Wong, J., Moreno, G. T., Young, M. K., Côté, J., and Wang, W. (1998). NURD, a Novel Complex with Both ATP-dependent Chromatin-Remodeling and Histone Deacetylase Activities. Mol. Cell. 2 (2), 851–861. doi:10.1016/s1097-2765(00)80299-3
Xue, Y., Gibbons, R., Yan, Z., Yang, D., McDowell, T. L., Sechi, S., et al. (2003). The ATRX Syndrome Protein Forms a Chromatin-Remodeling Complex with Daxx and Localizes in Promyelocytic Leukemia Nuclear Bodies. Proc. Natl. Acad. Sci. U.S.A. 100, 10635–10640. doi:10.1073/pnas.1937626100
Yang, X., Miao, B. S., Wei, C. Y., Dong, R. Z., Gao, P. T., Zhang, X. Y., et al. (2019). Lymphoid‐specific Helicase Promotes the Growth and Invasion of Hepatocellular Carcinoma by Transcriptional Regulation of Centromere Protein F Expression. Cancer Sci. 110, 2133–2144. doi:10.1111/cas.14037
Yano, M., Ouchida, M., Shigematsu, H., Tanaka, N., Ichimura, K., Kobayashi, K., et al. (2004). Tumor-specific Exon Creation of the HELLS/SMARCA6 Gene in Non-small Cell Lung Cancer. Int. J. Cancer 112, 8–13. doi:10.1002/ijc.20407
Yoshida, K., Kuo, F., George, E. L., Sharpe, A. H., and Dutta, A. (2001). Requirement of CDC45 for Postimplantation Mouse Development. Mol. Cell. Biol. 21, 4598–4603. doi:10.1128/MCB10.1128/mcb.21.14.4598-4603.2001
Zegerman, P., and Diffley, J. F. X. (2007). Phosphorylation of Sld2 and Sld3 by Cyclin-dependent Kinases Promotes DNA Replication in Budding Yeast. Nature 445, 281–285. doi:10.1038/nature05432
Zhang, J. J., Zhao, Y., Chait, B. T., Lathem, W. W., Ritzi, M., Knippers, R., et al. (1998). Ser727-dependent Recruitment of MCM5 by Stat1α in IFN-γ-Induced Transcriptional Activation. EMBO J. 17, 6963–6971. doi:10.1093/emboj/17.23.6963
Zhu, H., Geiman, T. M., Xi, S., Jiang, Q., Schmidtmann, A., Chen, T., et al. (2006). Lsh Is Involved in De Novo Methylation of DNA. EMBO J. 25, 335–345. doi:10.1038/sj.emboj.7600925
Zhu, Z., Chung, W.-H., Shim, E. Y., Lee, S. E., and Ira, G. (2008). Sgs1 Helicase and Two Nucleases Dna2 and Exo1 Resect DNA Double-Strand Break Ends. Cell. 134, 981–994. doi:10.1016/j.cell.2008.08.037
Keywords: DNA helicase, human disease, mouse model, lifespan - longevity, resilience
Citation: Kohzaki M (2022) Mammalian Resilience Revealed by a Comparison of Human Diseases and Mouse Models Associated With DNA Helicase Deficiencies. Front. Mol. Biosci. 9:934042. doi: 10.3389/fmolb.2022.934042
Received: 02 May 2022; Accepted: 23 June 2022;
Published: 11 August 2022.
Edited by:
Huiming Lu, University of Texas Southwestern Medical Center, United StatesReviewed by:
Sue Cotterill, St George’s, University of London, United KingdomJoseph Newman, University of Oxford, United Kingdom
Copyright © 2022 Kohzaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masaoki Kohzaki, masaoki-k@med.uoeh-u.ac.jp