Whole genome sequencing of Malaysian colorectal cancer patients reveals specific druggable somatic mutations
 Ryia Illani Mohd Yunos1
Ryia Illani Mohd Yunos1  Nurul-Syakima Ab Mutalib1*
Nurul-Syakima Ab Mutalib1*  Jia-shiun Khoo2 Sazuita Saidin1
Jia-shiun Khoo2 Sazuita Saidin1  Muhiddin Ishak1
Muhiddin Ishak1  Saiful Effendi Syafruddin1
Saiful Effendi Syafruddin1  Francis Yew Fu Tieng1
Francis Yew Fu Tieng1  Najwa Farhah Md Yusof1 Mohd Ridhwan Abd Razak1 Norshahidah Mahamad Nadzir1
Najwa Farhah Md Yusof1 Mohd Ridhwan Abd Razak1 Norshahidah Mahamad Nadzir1  Nadiah Abu1 Isa Md Rose3 Ismail Sagap4 Luqman Mazlan4
Nadiah Abu1 Isa Md Rose3 Ismail Sagap4 Luqman Mazlan4  Rahman Jamal1*
Rahman Jamal1*- 1UKM Medical Molecular Biology Institute (UMBI), Kuala Lumpur, Malaysia
- 2Codon Genomics SB, Seri Kembangan, Selangor, Malaysia
- 3Department of Pathology, Faculty of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
- 4Department of Surgery, Faculty of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
The incidences of colorectal cancer (CRC) are continuously increasing in some areas of the world, including Malaysia. In this study, we aimed to characterize the landscape of somatic mutations using the whole-genome sequencing approach and identify druggable somatic mutations specific to Malaysian patients. Whole-genome sequencing was performed on the genomic DNA obtained from 50 Malaysian CRC patients’ tissues. We discovered the top significantly mutated genes were APC, TP53, KRAS, TCF7L2 and ACVR2A. Four novel, non-synonymous variants were identified in three genes, which were KDM4E, MUC16 and POTED. At least one druggable somatic alteration was identified in 88% of our patients. Among them were two frameshift mutations in RNF43 (G156fs and P192fs) predicted to have responsive effects against the Wnt pathway inhibitor. We found that the exogenous expression of this RNF43 mutation in CRC cells resulted in increased cell proliferation and sensitivity against LGK974 drug treatment and G1 cell cycle arrest. In conclusion, this study uncovered our local CRC patients’ genomic landscape and druggable alterations. It also highlighted the role of specific RNF43 frameshift mutations, which unveil the potential of an alternative treatment targeting the Wnt/β-Catenin signalling pathway and could be beneficial, especially to Malaysian CRC patients.
1 Introduction
Colorectal cancer (CRC) is among the top three most common cancer worldwide, with 1.93 million new cases and 900,00 deaths reported in 2020 (Sung et al., 2021; Xi and Xu, 2021). Asian countries have also experienced a significant hike in CRC incidences in these past 20 years, especially with the changes in lifestyle and diet (Arnold et al., 2017). In Malaysia, CRC is the most common cancer among men and the second most common among women (Hashimah et al., 2019). According to the National Cancer Patient Registry-Colorectal Cancer, from 2008 to 2013, 4,501 cases of CRC were reported, most of which were Chinese, followed by Malays and Indians (Abu Hassan et al., 2016).
Substantial efforts have been made to understand the basic molecular mechanisms of CRC through profiling of somatic mutations, including the International Cancer Genome Consortium (Hudson et al., 2010), The Cancer Genome Atlas (TCGA) (The Cancer Genome Atlas Network, 2012) and the Pan-Cancer Analysis of Whole Genomes (PCAWG) (Campbell et al., 2020). However, there is still a lack of understanding in using the publicly available information to treat CRC patients effectively and unfortunately, their potential clinical significances are largely unexplored. Prognostication and treatment decision-making have been improved by numerous biomarkers discovered through comprehensive molecular profiling (Sveen et al., 2020). Several biomarkers and prognostic values, such as KRAS and EGFR, have been widely studied (Grady and Pritchard, 2014). Despite many studies on KRAS as a biomarker, the Ras protein has not yielded any therapeutic intervention due to the absence of a suitable site to which drugs could bind (McCormick, 2015; Liu et al., 2019). Alternatively, studies have been focused on blocking the pathways downstream of RAS, especially the RAF-MAPK pathway and the PI3 kinase pathways, to provide clinical benefit for patients with Ras-associated cancer (McCormick, 2015). Thus, by exploring the landscape of the alterations in cancer patients, new possible therapeutic targets and clinically relevant somatic mutations may be identified.
One of the most important signalling pathways implicated in CRC pathogenesis is the Wnt/β-catenin signalling pathway (Cheng et al., 2019) which is involves in various physiological and developmental processes such as proliferation, differentiation, apoptosis, migration, invasion and tissue homeostasis (Clevers and Nusse, 2012; Ng et al., 2019). Dysregulation of the pathway may contribute to the development and progression of specific solid tumours and haematological malignancies. (Cheng et al., 2019; Zhang and Wang, 2020). There has been increasing evidence supporting the potential relevance of the Wnt/β-catenin signalling pathway as a therapeutic target in cancer treatment (Blagodatski et al., 2014; Zhang and Wang, 2020). One of the components of this pathway is RNF43 (E3 ubiquitin-protein ligase RNF43), a type of ubiquitin ligase located in the transmembrane region (Zebisch and Jones, 2015). In cancer cells, Wnt signalling is activated through loss of function of RNF43 via mutations, leading to a decrease in the degradation of Frizzled (Serra and Chetty, 2018). Studies have shown that RNF43 mutations can have dual roles, either as a negative or positive regulator of the Wnt/β-catenin signalling pathway, depending on the type and location of the mutations in the gene (Yu et al., 2020; Cho et al., 2022; Fang et al., 2022). Somatic mutations in RNF43 have been associated with increased sensitivity to compounds that target the Wnt pathway, such as the porcupine (PORCN) inhibitor LGK974. LGK974 impairs the PORCN protein that will subsequently suppress the post-translational acylation of Wnt-ligands and inhibit their secretion. Consequently, it prevents the activation of Wnt ligands, dysregulates the Wnt-mediated signalling, and inhibits cell growth in Wnt-driven tumours (Liu et al., 2013). Therefore, as the PORCN inhibitors and other upstream inhibitors advance into clinical trials, it is essential to identify the suitable patients to be treated with these Wnt inhibitors. Hence, a comprehensive map of druggable mutations is required.
Whole-genome sequencing (WGS) can provide insight into the mutational spectra of cancers across the entire genome. In the past decades, several new promising therapeutic targets have been discovered through this approach. Extensive reviews and studies on how germline and somatically derived variants can guide therapeutic decisions have been carried out, which highlighted the importance of genome profiling of cancer (Jia et al., 2014; Ab Mutalib et al., 2017; Chan et al., 2019; Yang H. et al., 2019). Moreover, personal genome sequencing may become essential for diagnosing, preventing, and treating human diseases, particularly cancer (Cragun et al., 2016). Patient care can also be improved by transforming genomic research into personalized medicine applications by developing new and better genomics-based diagnostic tests. In this study, we employed WGS to characterize the landscape of somatic alterations in 50 Malaysian CRC patients, identify somatic alterations suitable for anticancer drug treatment, and predict the drug response. In addition, we functionally characterized two novel RNF43 variants and demonstrated that these are potentially clinically relevant variants worth exploring in future studies.
2 Materials and methods
2.1 Clinical materials
A total of 50 Malaysian CRC patients were enrolled from 2010 to 2018. All the individuals gave their written informed consent, and the study was approved under UKM PPI/111/8/JEP-2017–583. All the patients were categorized according to clinicopathological characteristics such as the age of diagnosis, ethnicity, gender, TNM classification, metastasis status, differentiation, tumour localization and survival status. Fifty paired colorectal carcinoma and their corresponding blood DNA or adjacent normal tissues were collected. The collected tissues were subjected to H&E staining and only tissues with 80% of tumour cells, confirmed by the pathologist, were selected to be used in the present study. DNA extraction was performed using AllPrep DNA/RNA/miRNA universal Kit (Qiagen, Germany) according to the manufacturer’s protocol. The quantity of the extracted DNA was assessed using Qubit Fluorometer (Thermo Fisher Scientific, United States). The quality of the extracted DNA was evaluated by agarose gel electrophoresis and NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, United States). To confirm the identity and to avoid contamination of each tumour and blood or normal tissue paired samples, we profiled the DNA based on 15 polymorphic STR markers using Investigator® IDplex Plus (Qiagen, Germany). The microsatellite status of each patient was determined using MSI Analysis System, Version 1.2 (Promega Corporation, United States) according to the manufacturer’s protocol. The amplified fragments were detected on the 3130xl Genetic Analyzer (Applied Biosystem, United States).
2.2 Library construction and Whole Genome sequencing
Libraries for WGS were constructed using TruSeq® Nano DNA HT Library Prep Kit (Illumina, United States) according to the manufacturer’s protocol. One μg (1 μg) of genomic DNA was randomly fragmented by the S220 Covaris instrument (Covaris, country, United Kingdom) following the manufacturer’s protocol. The fragmented DNA was viewed using gel electrophoresis and purified using AxyPrep Mag PCR clean-up kit (Thermo Fisher Scientific, United States) and then underwent end-repairing, phosphorylation and A-tailing reactions. WGS was performed as 150 bp paired-end, with the average coverage of at least 30X, on Illumina HiSeq X-Ten (Illumina, United States).
2.3 Bioinformatic analysis
We utilized FASTQC v0.10.01 software to perform adapter trimming and removal of low quality (less than Q30), short and ambiguous reads (Andrews, 2010). The resulting clean reads were then aligned to the reference human genome (UCSC hg19; http://genome.ucsc.edu/) (International Human Genome Consortium et al., 2001) using the Burrows-Wheeler Aligner (BWA) MEM (Li and Durbin, 2010). Picard tools and Genome Analysis Tool Kit (GATK) IndelRealigner and BaseRecalibrator were adopted to remove duplicate reads, base quality score recalibration and indel realignment. Somatic single nucleotide variants (SNVs) and insertion deletions (Indels) calling were carried out for each pair of tumour-normal samples using MuTect2 (Cibulskis et al., 2013). Functional annotation of the variants identified was performed on 1st February 2016 using ANNOVAR (Wang et al., 2010). Annotations, against Ensembl database, for mutation function (including frameshift insertion/deletion, non-frameshift insertion/deletion, synonymous SNV, non-synonymous SNV, stopgain and stoploss), mutation location (including exonic, intronic, splicing, upstream, downstream, 3′untranslated region (UTR) and 5′UTR), amino acid changes, allele frequencies (based on 1000 Genomes Project, Exome Aggregation Consortium (ExAC), Exome Sequencing Project v.6500 (ESP6500) data) and dbSNP (version 144), as well as COSMIC (v70-14th August 2014), were performed. Finally, all detected variants were manually reviewed using the Integrative Genomics Viewer (IGV) (Thorvaldsdóttir et al., 2013). cBioPortal was utilized to visualize the location of the identified mutations in the gene compared to other publicly available datasets (https://www.cbioportal.org/mutation_mapper) (Cerami et al., 2012; Gao et al., 2013).
2.4 Single nucleotide variant and indels variants prioritization
Variants with a quality score above Q30 were considered for further analysis. Frequent variants were removed based on a minimal allele frequency (MAF) threshold of more than 5% from 1000 Genomes Project, ExAC and ESP6500 databases. Besides, variants not resulting in amino acid changes and/or identified in unannotated genes (unknown) and non-exonic regions (based on Ensembl) were also removed. Somatic variants were identified by excluding those specified in both tumour and normal samples and considered a true novel if the variant has not been reported in both dbSNP and COSMIC databases. We ensured that the corresponding normal sample has at least ten reads covering the position with zero variant reads for each of the novel somatic mutation candidates identified. For the resulting candidate of somatic mutations, the alignment of each sample was manually examined for possible mapping ambiguities and sequencing artefacts using Intergrative Genomics Viewer (IGV). Finally, we assessed the potential functional effects of each identified somatic variant based on protein impact prediction tools, SIFT and PolyPhen2.
2.5 Druggable and tumor driver alterations
We employed Cancer Genome Interpreter (Tamborero et al., 2018) to assess the relevance of the shortlisted somatic alterations as biomarkers of drug response and identify possible tumour driver alterations. Colorectal adenocarcinoma (COREAD) was selected as a cancer type for annotation.
2.6 Variants validation by sanger sequencing
All the shortlisted SNVs were validated using the Sanger sequencing method on both tumour and matched blood samples. Primers corresponding to the selected locations were designed using PrimerQuest (Integrated DNA Technologies, United States). PCR products were generated and purified using QIAquick PCR Purification Kit (Qiagen, Germany) and cycle sequencing was performed using the BigDye™ Terminator V3.1 reagent (Applied Biosystem, United States). The cycle sequencing products were then processed using ethanol precipitation, and sequencing was carried out using the 3130xl Genetic Analyzer (Applied Biosystem, United States). The results were analyzed using the Sequence Scanner software (Applied Biosystem, United States).
2.7 Lentiviral vectors construction
The RNF43 coding sequences harbouring the mutations of interest, G156Afs and p.P192Gfs, were purchased from Origene Technologies (United States). The wild-type RNF43 coding sequence was purchased from Genscript (United States). These wild-type and mutant RNF43 coding sequences were amplified to incorporate the 3x FLAG upstream of the start codon, and the EcoRI and XbaI restriction sites at the 5′ and 3′ ends, respectively, for sub-cloning purpose into the expression vector pLVX-Puro (Clontech Laboratories Inc., United States). The RNF43 coding sequences were ligated into the expression vector using T4 Ligase (New England Biolabs) according to the manufacturer’s recommendation and transformed into the chemically competent DH5α E.coli.
2.8 Cell lines, lentiviral transduction and transient transfection
HEK293T cells were used for lentivirus production and cultured in DMEM (Nacalai Tesque, Japan), supplemented with 10% (v/v) fetal bovine serum (FBS) (Nacalai Tesque, Japan) and 1% (v/v) penicillin-streptomycin mixed solution (Nacalai Tesque, Japan). HEK293T cells were seeded at 1.2 × 106 cells/well in six wells plate a day before transfection. The RNF43-expressing plasmid was co-transfected into the HEK293T cells along with packaging plasmids psPAX2 (Addgene, United States) and pMD2.G (Addgene, United States) using Lipofectamine 2000 according to the manufacturer’srecommendation. The media containing the lentiviral were collected 72 h post-transfection and filtered through a 0.45 μM PVDF sterile filter (Merck Milipore, Germany). For the lentiviral transduction, the SW48 cells were seeded into the six wells plate at a density of 2.5 × 106 cells/well, and transduced with the collected supernatant on the following day in the presence of 8 μg/ml polybrene (Merck Millipore, Germany).
2.9 Gene expression and protein analysis
Total RNA was extracted from the cell line using the AllPrep DNA/RNA mini kit (Qiagen, Germany) according to the manufacturer’s protocol and cDNA was synthesized using the iScript cDNA synthesis kit (Bio-rad Laboratories Inc., United States). Quantitative PCR (qPCR) was performed using Sso Advanced™ universal SYBR® green mastermix (Bio-Rad Laboratories Inc., United States) and run on CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, United States). GAPDH and β-actin were used as standardization controls, and fold change was calculated based on 2−∆∆Ct (Schmittgen and Livak, 2008).
Total protein from cell cultures was extracted using RIPA buffer and resolved on 10% acrylamide gel. The target protein was analyzed against the following primary antibody: Anti Flag M2 (Sigma Aldrich, United States, 1:1,000) and the secondary antibody was Rabbit anti-mouse IgG/HRP conjugated (Dako, Denmark, 1:1,000).
2.10 Ki67 proliferation assay
Muse® Ki67 Proliferation kit (Merck Milipore, Germany) was used to determine the percentage of proliferating cells based on Ki67 expression according to the manufacturer’s protocol. The stained cells were analyzed on the Muse® Cell Analyzer (Merck Milipore, Germany), followed by data analysis using Muse 1.7 Analysis software (Merck Milipore, Germany).
2.11 LGK974 drug sensitivity assay
The SW48 cells expressing wild type (Flagged-RNF43WT) and mutant RNF43 proteins (Flagged-RNF43p.G156fs and Flagged-RNF43p.P192fs) were treated with different concentrations of LGK974 ranging from 10 to 100 µM for 48 h. The control groups, cells treated with 0.1 and 1% (v/v) of DMSO, were included in each experiment. The cell viability was analyzed by using the XTT Cell Viability Assay Kit (Biotium, Germany) based on the manufacturer’s user guide to assess the sensitivity of the cells against the drug treatment.
2.12 Cell cycle assay
Cell cycle analysis was performed on SW48 cells expressing wild type (Flagged-RNF43WT) and mutant RNF43 proteins (Flagged-RNF43p.G156fs and Flagged-RNF43p.P192fs) treated with 50 µM LGK974 for 48 h. The harvested cells were stained with propidium iodide provided in BD Cycletest™ Plus DNA Reagent Kit (BD Biosciences, US) according to the manufacturer’s instructions. The DNA content of at least 10,000 cells was analyzed by FACS Aria II flow cytometry (BD Biosciences, United States) for each experiment before the data was analyzed using ModFit LT 5.0 (Verity Software House, United States).
3 Results
3.1 Patients characteristic
The characteristics of all 50 patients are listed in Table 1. Most patients were in stage 3, 70% (n = 35). The average age of patients was approximately 64 years old (range 30 – 89 years old). The samples comprised an equal number of well-differentiated adenocarcinomas and moderately differentiated adenocarcinoma. Of these 50 patients, 36% (n = 18) have died and 54% (n = 27) are still alive (as when the data were collected in 2018).

TABLE 1. Clinicopathological data of 50 colorectal cancer patients.
3.2 Whole Genome sequencing analysis and coverage
A total of 100 (50 pairs) tumour and normal samples were sequenced using the Illumina HiSeq X Ten platform. At least 730 million reads were generated for each sample, producing approximately 30× to 50× sequencing depth. The reference mapping of the data against human genome hg19 and alignment refinement were carried out based on GATK Best Practices. On average, ∼99% of the reads were found to align to hg19, with at least 83% achieving more than 20× coverage (Table 2).
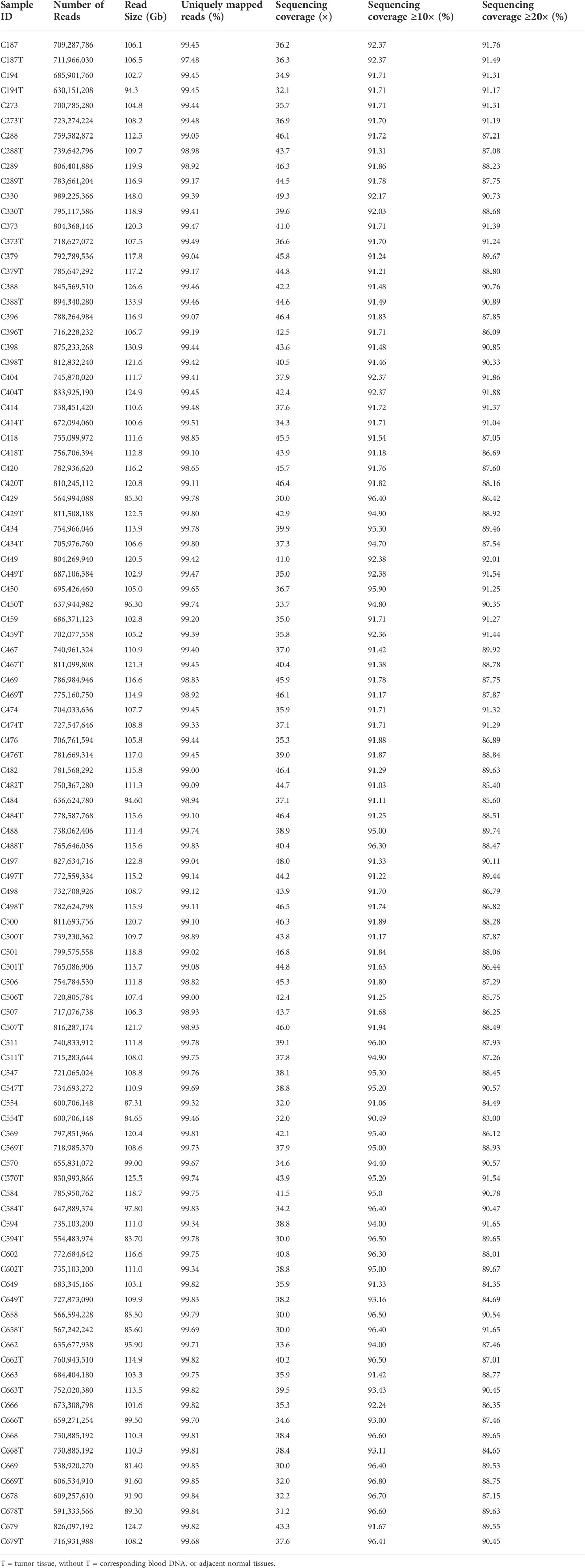
TABLE 2. Summary of sequencing statistics in each sample.
3.3 Mutation rate and microsatellite status
In this study, we defined a high mutation rate or hypermutated as >12 mutations/Mb, in concordant as reported in TCGA 2012. The somatic mutation rates varied among the samples. The average mutation rate was 13.11 per Mb, with a range of 0.83–243.97 mutations per Mb. Of the 50 patients tested for microsatellite instability, 10% (n = 5) were classified as microsatellite instability-high (MSI-H), 10% (n = 5) as microsatellite instability low (MSI-L) and the remaining 80% (n = 40) were microsatellite stable (MSS). The median mutation rate in MSI-H, MSI-L and MSS groups were 57.7/Mb, 4.1/Mb and 3.8/Mb, respectively. The median mutation rate was significantly higher in MSI-H patients (p < 0.05) compared to MSI-L. Nearly all (4/5, 80%) MSI-H patients, except one (C289T), were classified as hypermutated and the association of MSI-H status with a high mutation rate was statistically significant (p < 0.001). Besides, one of the microsatellite stable (MSS) tumours, patient C569T, was hypermutated with a mutation rate of 243.97/Mb (Figure 1).
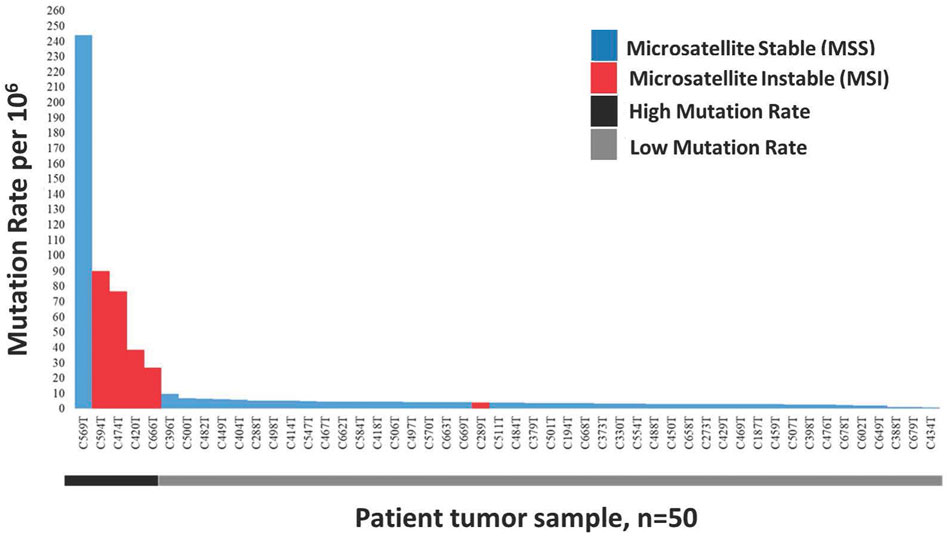
FIGURE 1. Distribution of 50 Microsatellite Instable (MSI) and Microsatellite Stable (MSS) CRCs sorted by decreasing mutation rate. Blue represents non MSI-High tumours consist of MSI-Low and MSS, while red represent MSI-High tumours.
3.4 Somatic mutations landscape in malaysian colorectal cancer
The total somatic variants detected in each CRC patient ranged between 2,587 and 756,750. Single nucleotide variants (SNVs) (Figure 2A) was the most variant type detected, with missense mutations being the highest variant class (Figure 2B). We analyzed the mutational signature underlying the development of our local CRC patients and three signatures similar to the COSMIC signature 6, 10 and 1 with cosine similarity of 0.949, 0.904 and 0.83, respectively, were discovered (Figure 3). The COSMIC signature six is related to defective DNA mismatch. COSMIC signature 10 is associated with defects in polymerase POLE, while signature one pertains to spontaneous deamination of 5-methylcytosine.
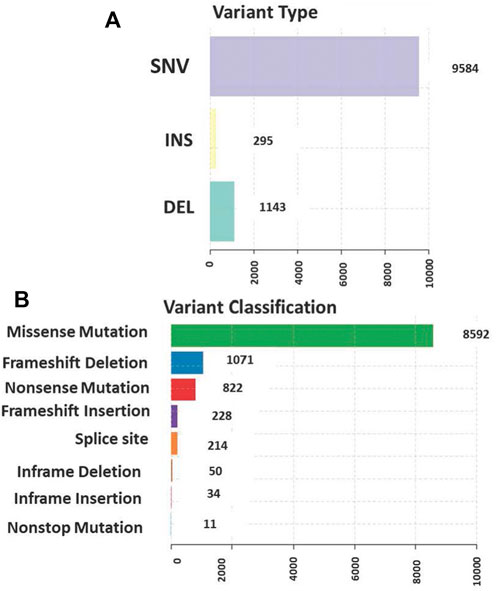
FIGURE 2. (A) Single nucleotide variants (SNVs) was the most common variant type detected (B) The distribution of the variants in genomic regions and types of exonic variants detected with missense mutation being the highest among all classes.
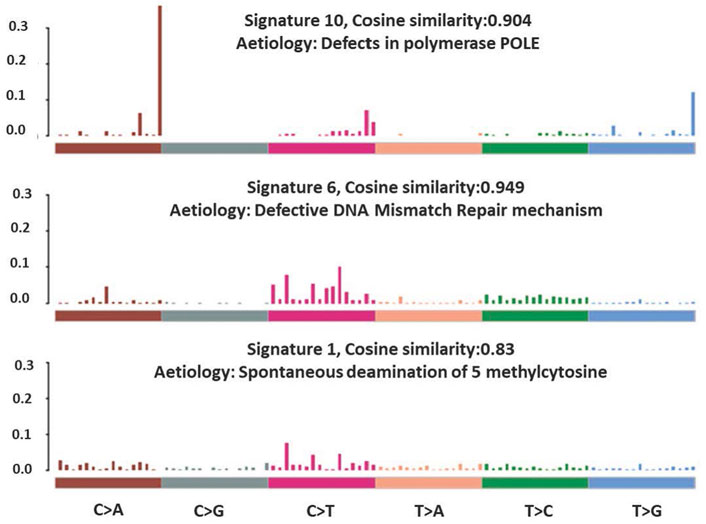
FIGURE 3. Mutational signatures identified in the CRC patients.
The top ten most frequently mutated genes are APC, TP53, KRAS, MUC4, TCF7L2, CCDC168, FAT3, KMT2C, LRP1B, PCLO, SCN1A and SPEG. Mutations in three well-established CRC genes, APC, TP53 and KRAS, were present in 70%, 66% and 34% of the patients, respectively (Figure 4). Using MutSigCV, we identified significantly mutated genes and from this analysis, the number of significantly mutated genes with p-values less than 0.001, 0.01, and 0.05 were 15, 25, and 113, respectively. Among these, the top significantly mutated genes (p < 0.001 and q < 1.0) are APC, TP53, KRAS, TCF7L2 and ACVR2A. These genes, APC, KRAS and TP53, were mutated in more than 30% of the patients. The remaining two were mutated in less than 20%, namely TCF7L2 (20%, p < 0.0001, q = 0.02) and ACVR2A (8%, p < 0.0001, q = 0.63). The significantly mutated genes (SMGs) identified (p < 0.01 and q < 1.0) are summarized in Table 3.
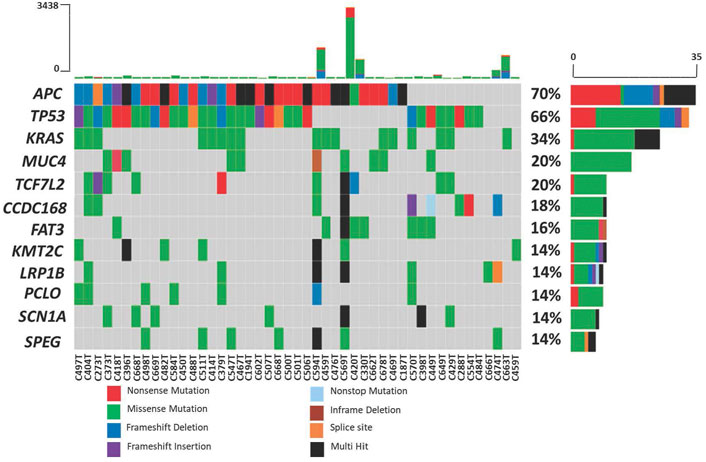
FIGURE 4. Mutation frequency in 50 Malaysian colorectal cancers. Each color in the boxes represent the mutation types.

TABLE 3. List of five top significantly mutated genes with p < 0.001 and q < 1.0.
Upon variants prioritization, 64 were identified as recurrent in 54 genes of two to six patients. As expected, most of the recurrent variants were presented in well-established CRC genes such as KRAS, APC and TP53, for which nearly all (92%) are known variants reported in the dbSNP or COSMIC database. In 12% of the patients (6/50 patients), KRAS G12D was observed to be the most frequent variant, followed by ACVR2A K435fs (8%, 4/50 patients) and TP53 R175H (8%, 4/50 patients).
Eleven clinically significant variants, classified as pathogenic, were identified in five genes, which were KRAS (rs121913529, rs112445441, rs121913529), APC (rs587781392, rs587782518, rs121913332), TP53 (rs28934576, rs121912651), PIK3CA (rs104886003) and BRAF (rs113488022). All of the mentioned variants are listed in Table 4. We identified 20 candidate driver genes using the oncodrive function in maftool v2.0.16. However, only two genes were significantly mutated (FDR<0.1), which were KRAS (G12D) and ACVR2A (K435fs). Remarkably, even with less than 20% of frequency, the ACVR2A gene was discovered to be one of the driver genes and was significantly mutated among other genes, suggesting its possible role in tumorigenesis of CRC in our local patients. The list of identified cancer driver genes is shown in Table 5.
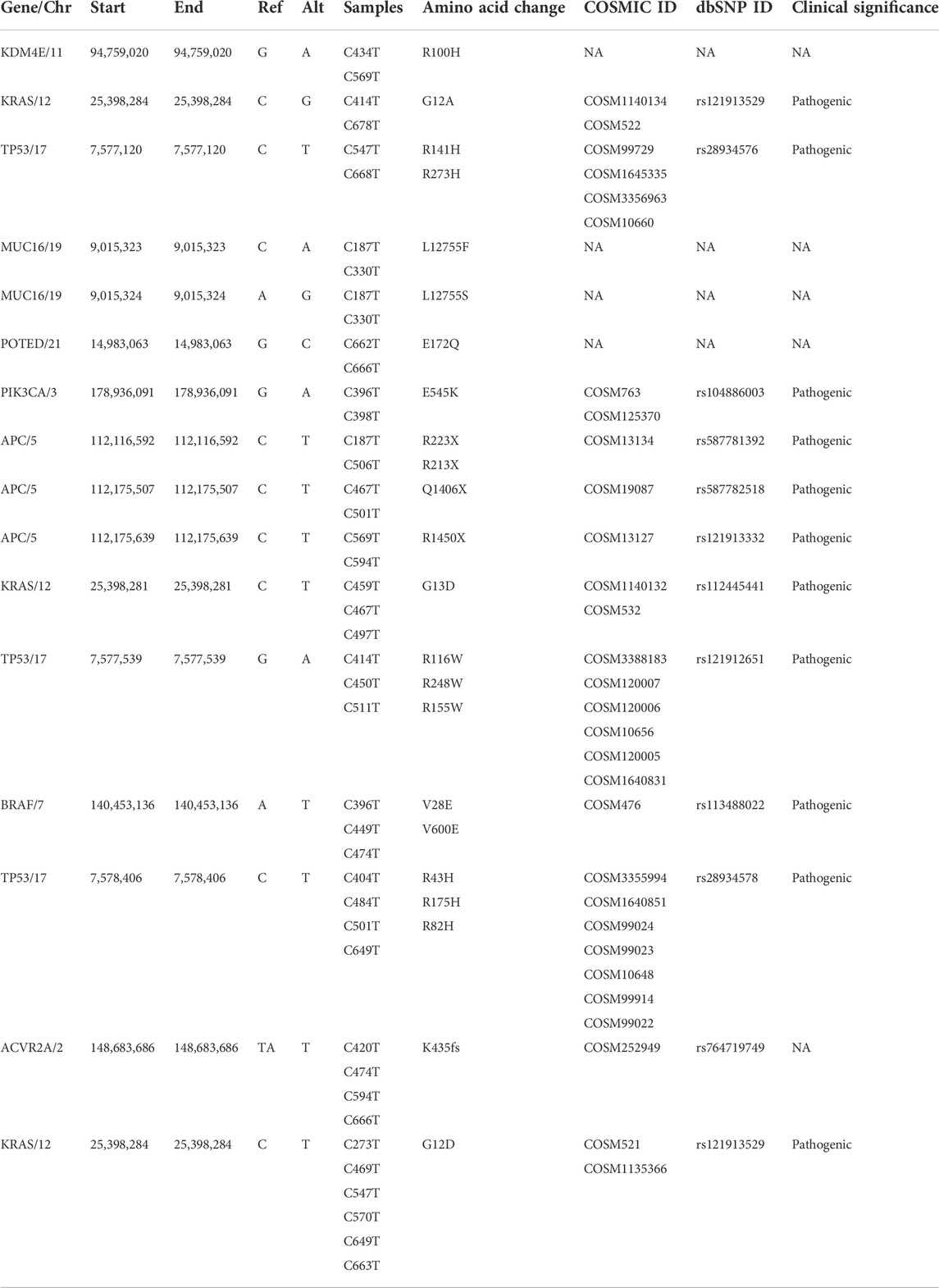
TABLE 4. List of identified known and novel recurrent somatic variants with their clinical significance in CRC genomes.
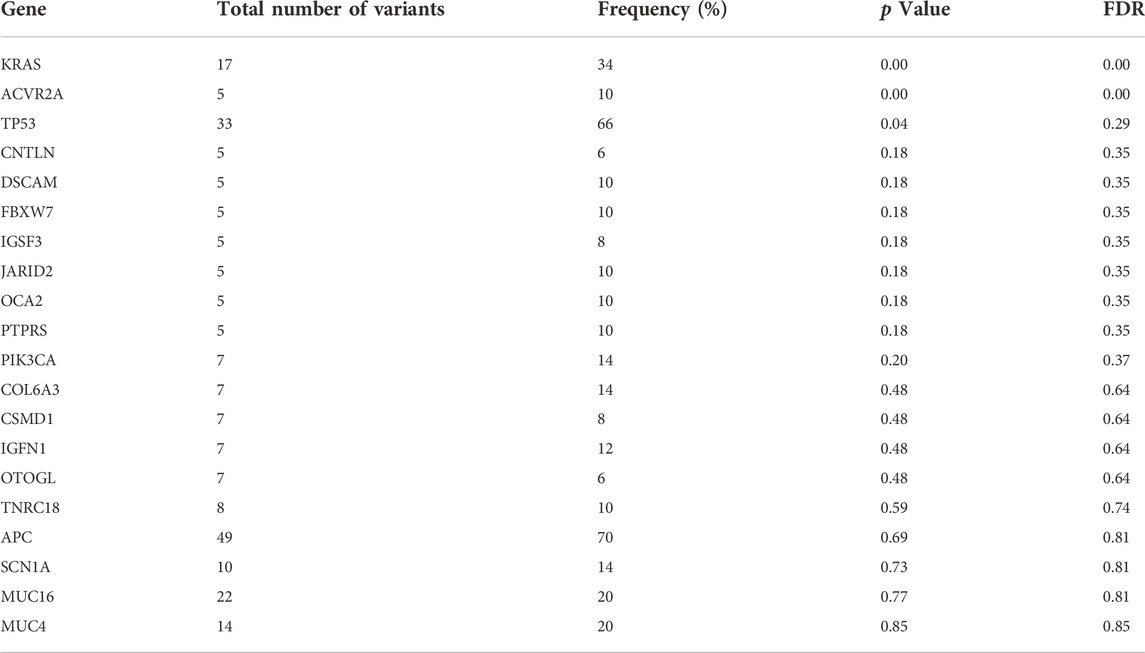
TABLE 5. List of cancer driver genes analyzed using oncodrive function in maftools. FDR<0.1 indicates the most significantly mutated driver genes.
3.5 Distribution of KDM4E, MUC16 and POTED hotspot and novel mutations
Four novel, non-synonymous variants, were identified in three genes; KDM4E R100H, MUC16 L12755F and L12755S, and POTED E172Q. At the time of analysis, these variants had not been previously reported in neither COSMIC or dbSNP. The mutation hotspots of the genes were analyzed using the cBioPortal web-based tool (https://www.cbioportal.org/) (Cerami et al., 2012; Gao et al., 2013). The lollipop plot in Figures 5A–C shows the distribution and classes of hotspot mutations in these three genes across eight different CRC datasets (n = 5,323) (assessed on 30.08.2022). The red arrow indicates the location of the identified novel variant across protein domains of the genes.
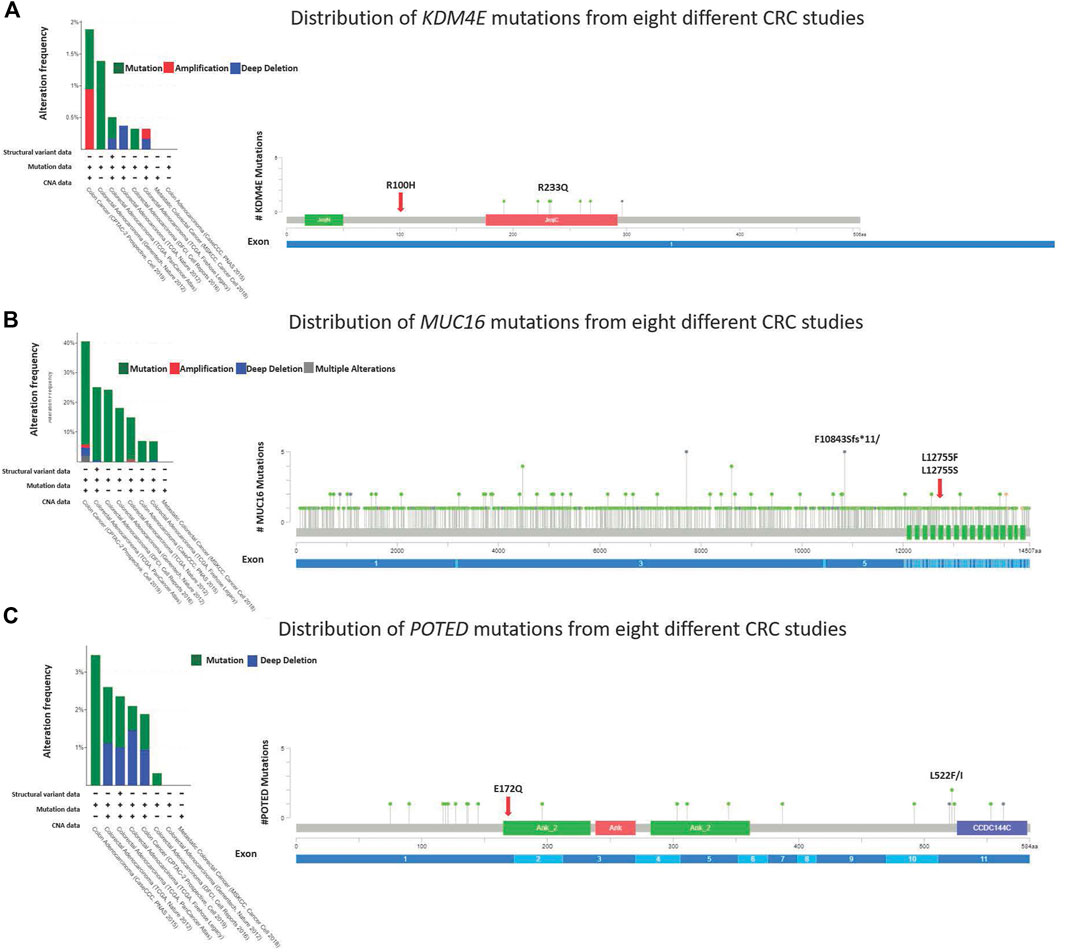
FIGURE 5. Lollipop diagram corresponding colorectal cancer studies selected in cBioportal and the frequency and types of changes occur.
3.6 Druggable somatic alterations
Based on the clinical annotation using Cancer Genome Interpreter, 88% (44/50) of the patients harboured at least one (range from 1 to 16) predicted candidate of druggable alterations. These alterations were either the targets of existing therapies (FDA guidelines or NCCN guidelines) or are currently being investigated in clinical trials (case reports, early trials, late trials and pre-clinical). Among them were various APC variants, detected in 72% (36/50) of the patients and predicted to respond against tankyrase inhibitors at the pre-clinical level. KRAS G12D was detected in 12% (6/50) of the patients and these patients were predicted to be resistant to several EGFR monoclonal antibody inhibitors (Panitumumab and Cetuximab) and ERBB2 monoclonal antibody inhibitor (Trastuzumab and Lapatinib). Six other KRAS variants were also observed in 10 (20%) different patients, which were predicted to be responsive to the combination of monoclonal antibody inhibitors such as MEK and PIK3 pathway inhibitors and MEK and MEK BCL-XL inhibitors.
Moreover, 14% (7/50) of CRC patients whose tumours possess PIK3CA variants were predicted to respond to the PI3K pathway inhibitor. However, these patients may not benefit from cetuximab therapy due to these variants. Four patients with different variants in the POLE gene might be suitable candidates for immunotherapy using the immune checkpoint inhibitor, PD1 antibody inhibitor. In addition, two RNF43 mutations were discovered in one of the hypermutated phenotype patients, C474T. This patient is likely to be responsive to the Wnt pathway inhibitor, also known as the porcupine inhibitor.
3.7 RNF43 G156Afs mutation promotes colorectal cancer cells proliferation
One commonly used marker for active cell proliferation is the Ki67 protein. To test the effect of harbouring the G156Afs and P192Gfs mutations on CRC cell’s proliferative capacity, we performed Ki67 FACS on the RNF43 wild type- and mutants-transduced SW48 cells. This was to compare the percentage of non-proliferating (Ki67-) and proliferating cells (Ki67+). We found that the expression of truncated RNF43 G156Afs promoted SW48 cells proliferation (78% of Ki67+cells) as compared to the SW48 cells transduced with empty vector (51.67% of Ki67+cells and wild-type RNF43 (57.4% of Ki67+cells). However, we did not observe any significant change in SW48 proliferative capacity between the cells expressing wild-type RNF43 and RNF43p.P192fs mutation (Figure 6).
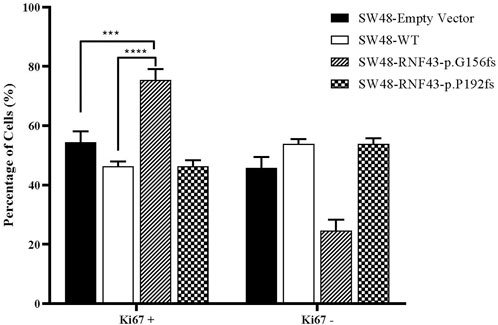
FIGURE 6. Percentage of proliferated cells assessed by Ki67 proliferation assay, 48 h post cell seeding. Percentage of Ki67+ was significantly higher in SW-RNF43-p.G156fs cells as compared to both SW48empty vector and SW48 wild type. Two-way ANOVA with Tukey′s range test, mean + SEM, n = 2, ***p < 0.005, ****p < 0.0001).
3.8 RNF43 G156Afs and P192Gfs mutation increase sensitivity against LGK974 treatment
Several studies have shown the cells that carry the inactivating RNF43 mutations are more sensitive to the porcine inhibitor LGK974 (Jiang et al., 2013; Tu et al., 2019; Zhong et al., 2019). Based on these reports, we were prompted to assess whether the expression of this RNF43 G156Afs and P192Gfs mutations would sensitize the SW48 cells to LGK974 treatment. To this end, we performed a cell viability assay upon treating the SW48 cells that expressed empty vector, wild-type RNF43 and the two RNF43 mutations with increasing concentration of LGK974 drug (10–100 µM). We found that the cells that expressed these mutations were more sensitive to a higher concentration of LGK974 (50 μM and 100 µM) as compared to the cells that expressed empty vector and wild-type RNF43 (Figure 7). We, however, did not observe any significant difference or additive effect in terms of drug sensitivity between the 50µM and 100 µM LGK974 treatments. Therefore, we used the 50 µM LGK974 in the subsequent cell cycle arrest assay.
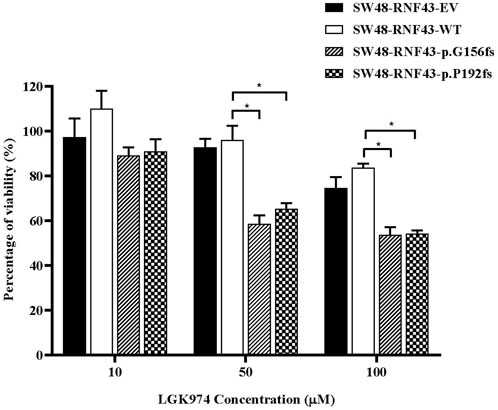
FIGURE 7. Mutants RNF43 promote reduction in cell viability. Statistical significance in all cases was measured by Two-way ANOVA with Tukey′s range test, (*p < 0.05), n = 3. Error bars represent average ± SD.
3.9 RNF43 G156Afs and P192Gfs mutation induce G1 cell cycle arrest upon LGK974 treatment
Since LGK974 is known to affect the cell cycle, we examined the effect of LGK974 treatment on the cell cycle process of each of these SW48 transduced cell lines. We treated these cells with 50 µM LGK974 for 48 h and assessed the cell cycle phases using FACS and BD Cycletest™ Plus DNA Reagent Kit (BD Biosciences, US). FACS analysis revealed a significant percentage of SW48-RNF43-p.G156fs and SW48-RNF43-p.P192s cells at the G0/G1 as compared to SW48-RNF43 wild-type cells, showing that the RNF43 mutated cells were arrested at G0/G1 phase upon the treatment. Consequently, lower percentage of cells entered S and G2/M phase due to staying in G0/G1 phase (Figure 8).
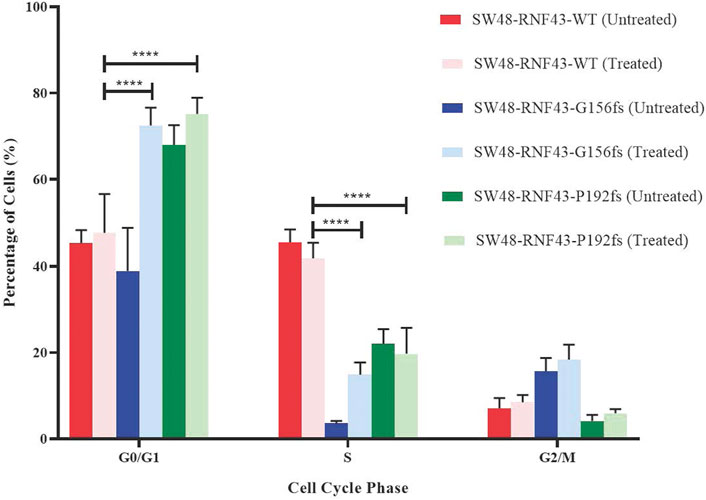
FIGURE 8. Distribution of SW48 transduced cells upon treatment with 50 μM of LGK974 throughout different cell cycle phase. Statistical significance in all cases was measured by mixed effect analysis with Tukey′s range test, (*p < 0.05), n = 4. Error bars represent average ± SD.
4 Discussion
In this present study, we performed WGS on 50 paired tumour tissues and their corresponding blood DNA or adjacent normal tissues of Malaysian CRC patients. The comprehensive analysis of the WGS data, which consisted of SNVs and Indels, resulted in the discovery of recurrent and novel variants in Malaysian CRC patients. The somatic mutation rate varied between the CRC patients. However, nearly all patients with hypermutated tumours were microsatellite instable (MSI). In the TCGA study, more than half of the hypermutated tumours had high levels of MSI (MSI-H) due to somatic mutation in mismatch repair genes, MLH1 methylation or the CpG island methylation phenotype (CIMP) (The Cancer Genome Atlas Network, 2012). The determination of MSI status is essential, especially in metastatic CRC (mCRC), because of its prognostic and therapeutic implications. MSI status has also been considered as the biomarker for the immune checkpoint inhibitor treatment response (Nojadeh et al., 2018). Two of the FDA-approved immune checkpoint inhibitors for programmed cell death-1 protein (PD-1), pembrolizumab and nivolumab, had survival benefits in patients with mCRC and MSI-H (Le et al., 2015; Overman et al., 2017). Therefore, we postulated that our C420T patient, who has a high level of MSI and mutation in the DNA polymerase epsilon (POLE) gene (R573W), might be benefited from the immune checkpoint inhibitor therapy. A recent study reported a favourable clinical response to pembrolizumab in CRC patients who have metastatic disease and are intractable to FOLFOX and FOLFIRI treatments. These patients were characterized by MSS phenotype and POLE mutation, which highlighted the importance of genomic profiling and the determination of microsatellite status for an effective therapeutic purpose (Gong et al., 2017). In addition, the POLE mutations can also serve as a prognostic marker. Patients carrying these mutations have a significantly better overall survival than those with wild type, regardless of their microsatellite status and tumour mutation burden. POLE mutations also predict a good response to the immune checkpoint inhibitor treatment. Based on this evidence, a clinical trial on toripalimab in patients with several solid tumours, including CRC, with POLE mutations and non-MSI-H, has been initiated (NCT03810339) (Wang et al., 2019). We found that our C569T patients, who has hypermutated tumour, MSS phenotype and POLE mutation, is likely to have a responsive effect against immune checkpoint inhibitor through our druggable alteration analysis.
Our genome data can be classified into three mutation signatures, signatures 1, 6 and 10, which were supported by several other studies on sporadic CRCs (Jia et al., 2014; Nagahashi et al., 2016; Tubbs and Nussenzweig, 2017). Signature one is strongly associated with an endogenous mutational process initiated by spontaneous deamination of 5-methylcytosine due to the ageing process (Tubbs and Nussenzweig, 2017). This is reflected in our patients’ age, of which 92% (n = 46) of the recruited CRC patients were above 50 years old with an average age of 64. Signature 6 and 10, on the other hand, are associated with defective MMR and defective exonuclease activity of POLE, respectively. We observed that 10% (n = 5) of the recruited patients were categorized as MSI-H, with all of them having at least one known somatic mutation in either MMR or POLE genes, which may lead to impaired MMR and exonuclease activity of POLE, respectively.
The top frequently mutated genes identified in our CRC patients cohort were APC, KRAS, TP53 and MUC4, which were also readily reported in multiple studies (The Cancer Genome Atlas Network, 2012; Abdul et al., 2017; Chang et al., 2019; Mohd Yunos et al., 2019). High mutation frequency was also observed in several other genes, including CCDC168, FAT3, KMT2C, LRP1B, PCLO, SCN1A and SPEG. Based on the MutSigCV analysis, we also identified TCF7L2 and ACVR2A among the significantly mutated genes in our CRC patients. However, these two genes were not categorized as the top ten frequently mutated genes. In MutSigCV, SMGs were defined as the genes that are mutated more often than expected by chance of given background mutation processes. Our analysis indicated that most of the top ten frequently mutated genes were not statistically significant when mutational heterogeneity was considered. Despite their high mutation frequency in CRC, these genes may not be functionally important for tumorigenesis. Compared to other studies, the mutation frequency of APC and TP53 in the Malaysian population was almost similar but much lesser than that of KRAS (The Cancer Genome Atlas Network, 2012; Abdul et al., 2017; Tanaka et al., 2017). Our previous genomic alterations profiling of Malaysian CRC patients also revealed that the APC gene was among the most frequently mutated gene, with a mutation frequency between 60% and 70% (Abdul et al., 2017; Chang et al., 2019).
On top of that, we identified four novel, non-synonymous which led to amino acid substitutions in three genes; KDM4E, MUC16 and POTED. Non synonymous variant of KDM4E R100H was identified in two patients C434T and C569T. KDM4 family protein functions as histone lysine demethylases that remove methyl groups from lysine residues in the histone tail, thereby controlling the transcriptional activity of target genes (Chen et al., 2006). KDM4 proteins family consist of four paralogues, namely KDM4A- KDM4D, and two pseudogenes, KDM4E and KDM4F. While KDM4A and KDM4B are the widely-studied members of the KDM4 subfamily, the roles of KDM4E in cancers have rarely been reported (Wang et al., 2022a). Genomic alterations and overexpression of the KDM4 family are reported in different breast cancer subtypes. Several KDM4 inhibitors have already been used as anticancer drugs for breast cancers in vitro (Ye et al., 2015; Varghese et al., 2021). However, none of these drugs have undergone clinical trials yet (Varghese et al., 2021). The bioinformatics analysis demonstrated that the intronless KDM4E and KDM4F are expressed similarly to KDM4D. Because of their architecture and lack of expression, KDM4E and KDM4F are referred to as pseudogenes (Berry and Janknecht, 2013). Growing evidence that the pseudogenes have a variety of biological roles and that their dysregulation is frequently linked to human disorders like cancer signifies their potential as therapeutic targets (Prensner and Chinnaiyan, 2011; Wahlestedt, 2013; Sisu, 2021). Several genomic alterations of pseudogenes in CRC have been identified. For instance, pseudogenes DUXAP8, MST O 2P and MYLKP1 involved in supporting CRC progression and enhance cancer risk (Lynn et al., 2018; He et al., 2020; Guo and Zhang, 2022). Hence, it is worth to explore the molecular characteristic and functional relevance of the identified recurrent KDM4E R100H mutation to unravel their potential as therapeutic target in CRC.
In this present study, we have identified a recurrent, non-synonymous MUC16 L12755S mutation which was predicted to be deleterious by SIFT and PolyPhen-2 tools. Located within the tandem repeat domain, this particular mutation has not been previously reported in CRC and is worth exploring its functional relevance in future studies.The MUC16 gene encodes for a highly glycosylated protein that consists of two primary domains: a tandem repeat domain (interspersed with SEA domain) containing the CA-125 epitope and a transmembrane domain (Hattrup and Gendler, 2008; Felder et al., 2014). CA-125 is an FDA-approved serum biomarker used in monitoring cancer progression and treatment response, particularly in ovarian cancer (Bottoni and Scatena, 2015; Li et al., 2018; Charkhchi et al., 2020). MUC16 is the most frequently mutated gene in endometrial cancer (Hu and Sun, 2018), and its oncogenic properties have been investigated in several other cancers such as glioblastoma (Yang C. et al., 2019), gastric cancer (Huang et al., 2021) and colorectal cancer (Björkman et al., 2019). Meanwhile, knocking down MUC16 in CRC cells impaired their growth and metastatic capability due to the deregulation of JAK2-STAT3 signalling pathway(Liu et al., 2022). Furthermore, a significant correlation between the MUC16 mutation with tumour mutational burden and microsatellite status was shown in patients with gastric cancer (Zhang et al., 2022), colorectal cancer (Wang et al., 2020), and melanoma (Zhang et al., 2020; Wang et al., 2022b) which signifies the used of immune checkpoint inhibitor (ICI) in the treatment regimen.
To our knowledge, our study is the first to report novel POTED E172Q mutation in CRC, which were discovered in two of our patients. POTE family gene has at least ten paralogs, which encode for cancer testis antigens (CTAs) that are expressed in the germ cells of the adult testis, fetal ovary, prostate, placenta. Moreover, POTE gene family has been associated with the pathogenesis of various human cancers in which their expression is higher in cancer tissues as compared to normal tissues (Coulie et al., 2014; Sharma et al., 2019). Due to their low expression in normal tissues, POTEs are potential biomarker candidates for cancer progression and therapeutic targets (Redfield et al., 2013). POTED, also known as ANKRD21, is one of the paralogs of POTE located on chromosome 21. This gene is one of the 45 gene signatures for metastatic predictor in triple-negative breast cancer (TNBC) whereby the high expression of POTED was associated with poor prognosis (Kuo et al., 2012). Nevertheless, the mechanism regulating the POTED expression in cancer remains to be elucidated. In 2019, Shen et al. demonstrated an aberrant expression of POTEE, another paralog of POTE gene family, perturbed the SPHK1/p65 signalling axis that consequently promoted tumorigenesis by inhibiting apoptosis in CRC cells. Their study has highlighted the potential roles of POTEE as a novel biomarker for the diagnosis and intervention of CRC (Shen et al., 2019). Thus, the functional roles of other paralogs of POTE gene family, such as POTED, remain elusive and worth pursuing.
The Wnt signalling pathway is frequently activated in most CRC cases due to the loss of function mutations in the APC gene. APC mutations were discovered to be one of the potential biomarkers for sensitivity to tankyrase inhibitors in CRC. Tankyrase inhibitors enhance the degradation of β-catenin and inhibit cell proliferation in CRC cell lines that harbour APC mutations (Schatoff et al., 2019; Jang et al., 2020). In this study, we identified four previously reported pathogenic APC truncating mutations, namely the R223X, R213X, Q1406X and R1450X, which were predicted to be sensitive toward tankyrase inhibitors. We analyzed the druggability of the identified mutations, which were expected to be the target of either existing therapies or currently being investigated in clinical trials. The response of APC truncating mutations, such as Q1405X, in in vivo model was proven to be sensitive against tankyrase inhibitor, G007-LK, through WNT suppression due to tankyrase synthase inhibition (Schatoff et al., 2019). The finding demonstrates the importance of these APC mutations in CRC and an investigation into how these mutations can be translated for targeted molecular therapeutics is warranted.
Besides APC, we also identified two N-terminal truncating mutations in RNF43, specifically the G156Afs and P192Gfs. These variants were found in C474T patient who has wild-type APC, KRAS and TP53, is hypermutated, and MSI-H phenotype. From our druggable alterations analysis, those with RNF43 mutations were predicted to be responsive against the porcupine inhibitor LGK974. Even with a prevalence of less than 20% in CRC, RNF43 has been described as one of the emerging predictive markers for treatment selection, especially in those with BRAF V600E mutations and MSI-H tumors with low MLH1 expression (Jiang et al., 2013; Giannakis et al., 2014; Tu et al., 2019; Yunos et al., 2020). RNF43 gene has been functionally characterized in multiple cancers such as pancreatic (Jiang et al., 2013), gastric (Niu et al., 2015) and hepatocellular carcinoma (Xing et al., 2013). Depending on the type and position of the mutations in the gene, RNF43 mutations can function as either positive or negative regulators of the Wnt/β-catenin signalling pathway (Li et al., 2020; Yu et al., 2020). The widely reported RNF43 mutations in CRC are the R117fs and G659fs, which these mutations are commonly observed in serrated CRCs with mutated BRAF and MSI (Bond et al., 2016). The R117fs along with another RNF43 mutation, P441fs, act as positive regulators of the Wnt/β-catenin signalling pathway because the presence of these mutations resulted in FZD accumulation on the CRC cells surface. Furthermore, treatment with LGK974 decreased the Wnt/β-catenin activity induced by these mutations (Cho et al., 2022). Contrariwise, a study by Tu et al. showed that G659fs mutation does not confer any dominant-negative activities and is unlikely to play a role in supporting CRC pathogenesis (Tu et al., 2019). This finding was supported by several independent studies that show the same G659fs C-terminal truncation did not affect the Wnt/β-catenin signalling (Li et al., 2020; Cho et al., 2022). A more recent study has shed light on this observation whereby the G659fs actually promoted CRC cells growth via PI3K/mTOR instead of the Wnt signaling pathways (Fang et al., 2022). Collectively, these observations indicate that different RNF43 mutations would possess different molecular properties whereby each of these specific mutations warrant for comprehensive investigations in order to understand their roles in CRC.
Comprehensive screening of 135 RNF43 missense and frameshift mutations in multiple human cancers revealed that all of the frameshift mutations and almost all missense mutations are located in the RING domain. This resulted in the RNF43 loss of function and subsequently increased activity of Wnt/β-catenin (Yu et al., 2020). Cho et al. demonstrated that even though RNF43 R117fs could interact with FZD5, RNF43 with this specific mutation could not ubiquitinate FZD5 due to the lack of the RING domain. It suggests that the RING domain of RNF43 is vital for regulating the Wnt/β-catenin signalling pathway (Cho et al., 2022). Herein, we investigated the role of two RNF43 mutations, G156Afs and P192Gfs, identified from our WGS data in the SW48 CRC cell line. The G156fs mutation is located in the protease-associated domain (PA domain) and mutation in this domain may affect the RING domain. Our observations revealed that RNF43 G156Afs mutation, but not P192Gfs, promoted SW48 cell proliferation. Nevertheless, both mutations exhibited higher sensitivity to LGK974 treatment that was manifested via reduced cell viability and cell cycle arrest at the G0/G1 phase 48 h post-treatment. We have successfully characterized the potential roles of these truncating RNF43 mutations in CRC pathogenesis, which can be further explored for the development of novel therapeutic targets in CRC. Moreover, it is essential to further validate invidual mutation identified from any genomic profiling studies to confirm their involvement in tumorigenesis. This is because Altogether, the analysis of druggable variants from our WGS, supported by the functional characterization, enhanced our understanding of the value of genomics and translating them into precision medicine.
Data availability statement
The data presented in the study are deposited in the NCBI SRA repository, accession number PRJNA928101.
Ethics statement
The studies involving human participants were reviewed and approved by UKM. The patients/participants provided their written informed consent to participate in this study.
Author contributions
RM performed the experiments, data analysis, interpretation, and manuscript drafting. KJS performed the bioinformatics analysis and visualization of the sequencing data. N-SA was involved in data interpretation, drafting the manuscript, and overseeing the experiments. SSa, MI, NM, NM and MA were heavily engaged in sample QC, DNA extraction and variants validation. SSa and FT were involved in the determination of the microsatellite status. SSy was involved in optimization in vectors construction and cell-based assay. NA gave insight into the functional analyses. LM and IS are the colorectal surgeons involved in specimen collection and IR is a pathologist. RJ was involved in the critical review of the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by the Long Research Grant Scheme (LRGS/2014/UKM-UKM/K/01) from the Ministry of Higher Education Malaysia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ab Mutalib, N.-S., Md Yusof, N. F., Abdul, S.-N., and Jamal, R. (2017). Pharmacogenomics DNA biomarkers in colorectal cancer: Current update. Front. Pharmacol. 8, 736. doi:10.3389/fphar.2017.00736
Abdul, S. N., Ab Mutalib, N. S., Sean, K. S., Syafruddin, S. E., Ishak, M., Sagap, I., et al. (2017). Molecular characterization of somatic alterations in Dukes’ B and C colorectal cancers by targeted sequencing. Front. Pharmacol. 8, 465. doi:10.3389/fphar.2017.00465
Abu Hassan, M. R., Ismail, I., Mohd Suan, M. A., Ahmad, F., Wan Khazim, W. K., Othman, Z., et al. (2016). Incidence and mortality rates of colorectal cancer in Malaysia. Epidemiol. Health 38, e2016007. doi:10.4178/epih.e2016007
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Babraham Bioinforma. http://www.bioinformatics.babraham.ac.uk/projects/.
Arnold, M., Sierra, M. S., Laversanne, M., Soerjomataram, I., Jemal, A., and Bray, F. (2017). Global patterns and trends in colorectal cancer incidence and mortality. Gut. 66, 683–691. doi:10.1136/gutjnl-2015-310912
Berry, W. L., and Janknecht, R. (2013). KDM4/JMJD2 Histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 73, 2936–2942. doi:10.1158/0008-5472.CAN-12-4300
Björkman, K., Mustonen, H., Kaprio, T., Haglund, C., and Böckelman, C. (2019). Mucin 16 and kallikrein 13 as potential prognostic factors in colon cancer: Results of an oncological 92-multiplex immunoassay. Tumour Biol. 41, 1010428319860728. doi:10.1177/1010428319860728
Blagodatski, A., Poteryaev, D., and Katanaev, V. L. (2014). Targeting the Wnt pathways for therapies. Mol. Cell. Ther. 2, 28. doi:10.1186/2052-8426-2-28
Bond, C. E., McKeone, D. M., Kalimutho, M., Bettington, M. L., Pearson, S.-A., Dumenil, T. D., et al. (2016). RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 7, 70589–70600. doi:10.18632/oncotarget.12130
Bottoni, P., and Scatena, R. (2015). The role of CA 125 as tumor marker: Biochemical and clinical aspects. Adv. Exp. Med. Biol. 867, 229–244. doi:10.1007/978-94-017-7215-0_14
Campbell, P. J., Getz, G., Korbel, J. O., Stuart, J. M., Jennings, J. L., Stein, L. D., et al. (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. doi:10.1038/s41586-020-1969-6
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. doi:10.1158/2159-8290.CD-12-0095
Chan, H. T., Chin, Y. M., and Low, S.-K. (2019). The roles of common variation and somatic mutation in cancer pharmacogenomics. Oncol. Ther. 7, 1–32. doi:10.1007/s40487-018-0090-6
Chang, Y. S., Lee, C. C., Ke, T. W., Chang, C. M., Chao, D. S., Huang, H. Y., et al. (2019). Molecular characterization of colorectal cancer using whole-exome sequencing in a Taiwanese population. Cancer Med. 8, 3738–3747. doi:10.1002/cam4.2282
Charkhchi, P., Cybulski, C., Gronwald, J., Wong, F. O., Narod, S. A., and Akbari, M. R. (2020). Ca125 and ovarian cancer: A comprehensive review. Cancers (Basel) 12 (12), 3730. doi:10.3390/cancers12123730
Chen, Z., Zang, J., Whetstine, J., Hong, X., Davrazou, F., Kutateladze, T. G., et al. (2006). Structural insights into histone demethylation by JMJD2 family members. Cell 125, 691–702. doi:10.1016/j.cell.2006.04.024
Cheng, X., Xu, X., Chen, D., Zhao, F., and Wang, W. (2019). Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 110, 473–481. doi:10.1016/j.biopha.2018.11.082
Cho, A. R., Sul, H. J., Kim, Y. J., Kim, B., and Zang, D. Y. (2022). RNF43 R117fs mutant positively regulates Wnt/β-catenin signaling by failing to internalize FZD expressed on the cell surface. Sci. Rep. 12, 7013. doi:10.1038/s41598-022-10868-8
Cibulskis, K., Lawrence, M. S., Carter, S. L., Sivachenko, A., Jaffe, D., Sougnez, C., et al. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. doi:10.1038/nbt.2514
Clevers, H., and Nusse, R. (2012). Wnt/β-catenin signaling and disease. Cell 149, 1192–1205. doi:10.1016/j.cell.2012.05.012
Coulie, P. G., Van Den Eynde, B. J., Van Der Bruggen, P., and Boon, T. (2014). Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135–146. doi:10.1038/nrc3670
Cragun, D., Lewis, C., Camperlengo, L., and Pal, T. (2016). Hereditary cancer: Example of a public health approach to ensure population health benefits of genetic medicine. Healthcare 4, 6. doi:10.3390/healthcare4010006
Fang, L., Ford-Roshon, D., Russo, M., O’Brien, C., Xiong, X., Gurjao, C., et al. (2022). RNF43 G659fs is an oncogenic colorectal cancer mutation and sensitizes tumor cells to PI3K/mTOR inhibition. Nat. Commun. 13, 3181. doi:10.1038/s41467-022-30794-7
Felder, M., Kapur, A., Gonzalez-Bosquet, J., Horibata, S., Heintz, J., Albrecht, R., et al. (2014). MUC16 (CA125): Tumor biomarker to cancer therapy, a work in progress. Mol. Cancer 13, 129. doi:10.1186/1476-4598-13-129
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1. doi:10.1126/scisignal.2004088
Giannakis, M., Hodis, E., Jasmine Mu, X., Yamauchi, M., Rosenbluh, J., Cibulskis, K., et al. (2014). RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 46, 1264–1266. doi:10.1038/ng.3127
Gong, J., Wang, C., Lee, P. P., Chu, P., and Fakih, M. (2017). Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J. Natl. Compr. Canc. Netw. 15, 142–147. doi:10.6004/jnccn.2017.0016
Grady, W. M., and Pritchard, C. C. (2014). Molecular alterations and biomarkers in colorectal cancer. Toxicol. Pathol. 42, 124–139. doi:10.1177/0192623313505155
Guo, M., and Zhang, X. (2022). LncRNA MSTO2P promotes colorectal cancer progression through epigenetically silencing CDKN1A mediated by EZH2. World J. Surg. Oncol. 20, 95–99. doi:10.1186/s12957-022-02567-5
Hashimah, B., Nirmal, K., Nabihah, A., Sukumaran, R., Balqis, B., Rahayu, O., et al. (2019). Malaysia National Cancer Registry 2012-2016.
Hattrup, C. L., and Gendler, S. J. (2008). Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 70, 431–457. doi:10.1146/annurev.physiol.70.113006.100659
He, W., Yu, Y., Huang, W., Feng, G., and Li, J. (2020). The pseudogene duxap8 promotes colorectal cancer cell proliferation, invasion, and migration by inducing epithelial-mesenchymal transition through interacting with ezh2 and h3k27me3. Onco. Targets. Ther. 13, 11059–11070. doi:10.2147/OTT.S235643
Hu, J., and Sun, J. (2018). MUC16 mutations improve patients’ prognosis by enhancing the infiltration and antitumor immunity of cytotoxic T lymphocytes in the endometrial cancer microenvironment. Oncoimmunology 7, e1487914. doi:10.1080/2162402X.2018.1487914
Huang, Y. J., Cao, Z. F., Wang, J., Yang, J., Wei, Y. J., Tang, Y. C., et al. (2021). Why MUC16 mutations lead to a better prognosis: A study based on the cancer genome Atlas gastric cancer cohort. World J. Clin. Cases 9, 4143–4158. doi:10.12998/wjcc.v9.i17.4143
Hudson, T. J., Anderson, W., Aretz, A., Barker, A. D., Bell, C., Bernabé, R. R., et al. (2010). International network of cancer genome projects. Nature 464, 993–998. doi:10.1038/nature08987
International Human Genome Consortium Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi:10.1038/35057062
Jang, M. K., Mashima, T., and Seimiya, H. (2020). Tankyrase inhibitors target colorectal cancer stem cells via axin-dependent downregulation of c-kit tyrosine kinase. Mol. Cancer Ther. 19, 765–776. doi:10.1158/1535-7163.MCT-19-0668
Jia, P., Pao, W., and Zhao, Z. (2014). Patterns and processes of somatic mutations in nine major cancers. BMC Med. Genomics 7, 11. doi:10.1186/1755-8794-7-11
Jiang, X., Hao, H.-X., Growney, J. D., Woolfenden, S., Bottiglio, C., Ng, N., et al. (2013). Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A. 110, 12649–12654. doi:10.1073/pnas.1307218110
Kuo, W. H., Chang, Y. Y., Lai, L. C., Tsai, M. H., Hsiao, C. K., Chang, K. J., et al. (2012). Molecular characteristics and metastasis predictor genes of triple-negative breast cancer: A clinical study of triple-negative breast carcinomas. PLoS One 7, e45831. doi:10.1371/journal.pone.0045831
Le, D. T., Uram, J. N., Wang, H., Bartlett, B. R., Kemberling, H., Eyring, A. D., et al. (2015). PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520. doi:10.1056/NEJMoa1500596
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi:10.1093/bioinformatics/btp698
Li, S., Lavrijsen, M., Bakker, A., Magierowski, M., Magierowska, K., Liu, P., et al. (2020). Commonly observed RNF43 mutations retain functionality in attenuating Wnt/β-catenin signaling and unlikely confer Wnt-dependency onto colorectal cancers. Oncogene 39, 3458–3472. doi:10.1038/s41388-020-1232-5
Li, X., Pasche, B., Zhang, W., and Chen, K. (2018). Association of MUC16 mutation with tumor mutation load and outcomes in patients with gastric cancer. JAMA Oncol. 4, 1691–1698. doi:10.1001/jamaoncol.2018.2805
Liu, J., Pan, S., Hsieh, M. H., Ng, N., Sun, F., Wang, T., et al. (2013). Targeting wnt-driven cancer through the inhibition of porcupine by LGK974. Proc. Natl. Acad. Sci. U. S. A. 110, 20224–20229. doi:10.1073/pnas.1314239110
Liu, P., Wang, Y., and Li, X. (2019). Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 9, 871–879. doi:10.1016/j.apsb.2019.03.002
Liu, Z., Gu, Y., Li, X., Zhou, L., Cheng, X., Jiang, H., et al. (2022). Mucin 16 promotes colorectal cancer development and progression through activation of janus kinase 2. Dig. Dis. Sci. 67, 2195–2208. doi:10.1007/s10620-021-07004-3
Lynn, H., Sun, X., Ayshiev, D., Siegler, J. H., Rizzo, A. N., Karnes, J. H., et al. (2018). Single nucleotide polymorphisms in the MYLKP1 pseudogene are associated with increased colon cancer risk in African Americans. PLoS One 13, e0200916. doi:10.1371/journal.pone.0200916
McCormick, F. (2015). KRAS as a therapeutic target. Clin. Cancer Res. 21, 1797–1801. doi:10.1158/1078-0432.CCR-14-2662
Mohd Yunos, R.-I., Ab Mutalib, N.-S., Khor, S. S., Saidin, S., Abd Razak, M. R., Mahamad Nadzir, N., et al. (2019). Whole exome sequencing identifies genomic alterations in proximal and distal colorectal cancer. Prog. Microbes Mol. Biol. 2, 1–14. doi:10.36877/pmmb.a0000036
Nagahashi, M., Wakai, T., Shimada, Y., Ichikawa, H., Kameyama, H., Kobayashi, T., et al. (2016). Genomic landscape of colorectal cancer in Japan: Clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med. 8, 136. doi:10.1186/s13073-016-0387-8
Ng, L. F., Kaur, P., Bunnag, N., Suresh, J., Sung, I. C. H., Tan, Q. H., et al. (2019). WNT signaling in disease. Cells 8, E826. doi:10.3390/cells8080826
Niu, L., Qin, H. Z., Xi, H. Q., Wei, B., Xia, S. Y., and Chen, L. (2015). RNF43 inhibits cancer cell proliferation and could be a potential prognostic factor for human gastric carcinoma. Cell. Physiol. biochem. 36, 1835–1846. doi:10.1159/000430154
Nojadeh, J. N., Sharif, S. B., and Sakhinia, E. (2018). Microsatellite instability in colorectal cancer. EXCLI J. 17, 159–168. doi:10.17179/excli2017-948
Overman, M. J., McDermott, R., Leach, J. L., Lonardi, S., Lenz, H.-J., Morse, M. A., et al. (2017). Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet. Oncol. 18, 1182–1191. doi:10.1016/S1470-2045(17)30422-9
Prensner, J. R., and Chinnaiyan, A. M. (2011). The emergence of lncRNAs in cancer biology. Cancer Discov. 1, 391–407. doi:10.1158/2159-8290.CD-11-0209
Redfield, S. M., Mao, J., Zhu, H., He, Z., Zhang, X., Bigler, S. A., et al. (2013). The C-terminal common to group 3 POTES (CtG3P): A newly discovered nucleolar marker associated with malignant progression and metastasis. Am. J. Cancer Res. 3, 278–289.
Schatoff, E. M., Goswami, S., Zafra, M. P., Foronda, M., Shusterman, M., Leach, B. I., et al. (2019). Distinct colorectal cancer–associated apc mutations dictate response to tankyrase inhibition. Cancer Discov. 9, 1358–1371. doi:10.1158/2159-8290.CD-19-0289
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108. doi:10.1038/nprot.2008.73
Serra, S., and Chetty, R. (2018). Rnf43. J. Clin. Pathol. 71, 1–6. doi:10.1136/jclinpath-2017-204763
Sharma, A., Albahrani, M., Zhang, W., Kufel, C. N., James, S. R., Odunsi, K., et al. (2019). Epigenetic activation of POTE genes in ovarian cancer. Epigenetics 14, 185–197. doi:10.1080/15592294.2019.1581590
Shen, Z., Feng, X., Fang, Y., Li, Y., Li, Z., Zhan, Y., et al. (2019). POTEE drives colorectal cancer development via regulating SPHK1/p65 signaling. Cell Death Dis. 10, 863. doi:10.1038/s41419-019-2046-7
Sisu, C. (2021). Pseudogenes as biomarkers and therapeutic targets in human cancers. Methods Mol. Biol. 2324, 319–337. doi:10.1007/978-1-0716-1503-4_20
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 71, 209–249. doi:10.3322/caac.21660
Sveen, A., Kopetz, S., and Lothe, R. A. (2020). Biomarker-guided therapy for colorectal cancer: Strength in complexity. Nat. Rev. Clin. Oncol. 17, 11–32. doi:10.1038/s41571-019-0241-1
Tamborero, D., Rubio-Perez, C., Deu-Pons, J., Schroeder, M. P., Vivancos, A., Rovira, A., et al. (2018). Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 10, 25. doi:10.1186/s13073-018-0531-8
Tanaka, N., Mashima, T., Mizutani, A., Sato, A., Aoyama, A., Gong, B., et al. (2017). APC mutations as a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer. Mol. Cancer Ther. 16, 752–762. doi:10.1158/1535-7163.MCT-16-0578
The Cancer Genome Atlas Network (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. doi:10.1038/nature11252
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi:10.1093/bib/bbs017
Tu, J., Park, S., Yu, W., Zhang, S., Wu, L., Carmon, K., et al. (2019). The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis. Sci. Rep. 9, 18557. doi:10.1038/s41598-019-54931-3
Tubbs, A., and Nussenzweig, A. (2017). Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656. doi:10.1016/j.cell.2017.01.002
Varghese, B., Del Gaudio, N., Cobellis, G., Altucci, L., and Nebbioso, A. (2021). KDM4 involvement in breast cancer and possible therapeutic approaches. Front. Oncol. 11, 750315. doi:10.3389/fonc.2021.750315
Wahlestedt, C. (2013). Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 12, 433–446. doi:10.1038/nrd4018
Wang, F., Zhao, Q., Wang, Y. N., Jin, Y., He, M. M., Liu, Z. X., et al. (2019). Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 5, 1504–1506. doi:10.1001/jamaoncol.2019.2963
Wang, K., Li, M., and Hakonarson, H. (2010). Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Wang, X., Duanmu, J., Fu, X., Li, T., and Jiang, Q. (2020). Analyzing and validating the prognostic value and mechanism of colon cancer immune microenvironment. J. Transl. Med. 18, 324. doi:10.1186/s12967-020-02491-w
Wang, Z., Cai, H., Zhao, E., and Cui, H. (2022a). The diverse roles of histone demethylase KDM4B in normal and cancer development and progression. Front. Cell Dev. Biol. 9, 790129. doi:10.3389/fcell.2021.790129
Wang, Z., Hou, H., Zhang, H., Duan, X., Li, L., and Meng, L. (2022b). Effect of MUC16 mutations on tumor mutation burden and its potential prognostic significance for cutaneous melanoma. Am. J. Transl. Res. 14, 849–862.
Xi, Y., and Xu, P. (2021). Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 14, 101174. doi:10.1016/j.tranon.2021.101174
Xing, C., Zhou, W., Ding, S., Xie, H., Zhang, W., Yang, Z., et al. (2013). Reversing effect of ring finger protein 43 inhibition on malignant phenotypes of human hepatocellular carcinoma. Mol. Cancer Ther. 12, 94–103. doi:10.1158/1535-7163.MCT-12-0672
Yang, C., Tan, Y., Li, S., Zhou, J., Wang, Q., Wang, Y., et al. (2019). Genomic landscapes by multiregion sequencing combined with circulation tumor DNA detection contribute to molecular diagnosis in glioblastomas. Aging 11, 11224–11243. doi:10.18632/aging.102526
Yang, H., Shah, R. H., Tegay, D., and Onel, K. (2019). Precision oncology: Lessons learned and challenges for the future. Cancer Manag. Res. 11, 7525–7536. doi:10.2147/CMAR.S201326
Ye, Q., Holowatyj, A., Wu, J., Liu, H., Zhang, L., Suzuki, T., et al. (2015). Abstract 98: Genetic alterations of KDM4 subfamily and therapeutic effect of novel demethylase inhibitor in breast cancer. Cancer Res. 5, 98–1530. doi:10.1158/1538-7445.am2015-98
Yu, J., Mohamed Yusoff, P. A., Woutersen, D. T. J., Goh, P., Harmston, N., Smits, R., et al. (2020). The functional landscape of patient-derived RNF43 mutations predicts sensitivity to Wnt inhibition. Cancer Res. 80, 5619–5632. doi:10.1158/0008-5472.CAN-20-0957
Yunos, R. I. M., Mutalib, N. S. A., Tieng, F. Y. F., Abu, N., and Jamal, R. (2020). Actionable potentials of less frequently mutated genes in colorectal cancer and their roles in precision medicine. Biomolecules 10, E476. doi:10.3390/biom10030476
Zebisch, M., and Jones, E. Y. (2015). ZNRF3/RNF43 - a direct linkage of extracellular recognition and E3 ligase activity to modulate cell surface signalling. Prog. Biophys. Mol. Biol. 118, 112–118. doi:10.1016/j.pbiomolbio.2015.04.006
Zhang, F., Li, X., Chen, H., Guo, J., Xiong, Z., Yin, S., et al. (2022). Mutation of MUC16 is associated with tumor mutational burden and lymph node metastasis in patients with gastric cancer. Front. Med. 9, 836892. doi:10.3389/fmed.2022.836892
Zhang, L., Han, X., and Shi, Y. (2020). Association of MUC16 mutation with response to immune checkpoint inhibitors in solid tumors. JAMA Netw. Open 3, e2013201. doi:10.1001/jamanetworkopen.2020.13201
Zhang, Y., and Wang, X. (2020). Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 13, 165. doi:10.1186/s13045-020-00990-3
Keywords: Malaysians colorectal cancer, whole genome sequencing, precision medicine, druggable alterations, RNF43, KDM4E, MUC16, POTED
Citation: Mohd Yunos RI, Ab Mutalib N-S, Khoo J-s, Saidin S, Ishak M, Syafruddin SE, Tieng FYF, Md Yusof NF, Abd Razak MR, Mahamad Nadzir N, Abu N, Rose IM, Sagap I, Mazlan L and Jamal R (2023) Whole genome sequencing of Malaysian colorectal cancer patients reveals specific druggable somatic mutations. Front. Mol. Biosci. 9:997747. doi: 10.3389/fmolb.2022.997747
Received: 19 July 2022; Accepted: 20 September 2022;
Published: 14 February 2023.
Edited by:
Lan Zhang, Southwest Jiaotong University, ChinaCopyright © 2023 Mohd Yunos, Ab Mutalib, Khoo, Saidin, Ishak, Syafruddin, Tieng, Md Yusof, Abd Razak, Mahamad Nadzir, Abu, Rose, Sagap, Mazlan and Jamal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nurul-Syakima Ab Mutalib, syakima@ppukm.ukm.edu.my; Rahman Jamal, rahmanj@ppukm.ukm.edu.my