 Arif Tasleem Jan1†‡
Arif Tasleem Jan1†‡ Mudsser Azam2‡
Mudsser Azam2‡ Safikur Rahman1
Safikur Rahman1 Angham M. S. Almigeiti1
Angham M. S. Almigeiti1 Duk Hwan Choi1Eun Ju Lee1
Duk Hwan Choi1Eun Ju Lee1 Qazi Mohd Rizwanul Haq2
Qazi Mohd Rizwanul Haq2 Inho Choi1*
Inho Choi1*- 1Department of Medical Biotechnology, Yeungnam University, Gyeongsan, South Korea
- 2Department of Biosciences, Jamia Millia Islamia, New Delhi, India
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by the progressive accumulation of β-amyloid fibrils and abnormal tau proteins in and outside of neurons. Representing a common form of dementia, aggravation of AD with age increases the morbidity rate among the elderly. Although, mutations in the ApoE4 act as potent risk factors for sporadic AD, familial AD arises through malfunctioning of APP, PSEN-1, and−2 genes. AD progresses through accumulation of amyloid plaques (Aβ) and neurofibrillary tangles (NFTs) in brain, which interfere with neuronal communication. Cellular stress that arises through mitochondrial dysfunction, endoplasmic reticulum malfunction, and autophagy contributes significantly to the pathogenesis of AD. With high accuracy in disease diagnostics, Aβ deposition and phosphorylated tau (p-tau) are useful core biomarkers in the cerebrospinal fluid (CSF) of AD patients. Although five drugs are approved for treatment in AD, their failures in achieving complete disease cure has shifted studies toward a series of molecules capable of acting against Aβ and p-tau. Failure of biologics or compounds to cross the blood-brain barrier (BBB) in most cases advocates development of an efficient drug delivery system. Though liposomes and polymeric nanoparticles are widely adopted for drug delivery modules, their use in delivering drugs across the BBB has been overtaken by exosomes, owing to their promising results in reducing disease progression.
Introduction
Alzheimer's disease (AD) is recognized as a disease of neurons and neuronal circuits. It arises as a result of progressive accumulation of β-amyloid fibrils (β-amyloid plaques) and abnormal forms of tau (tau tangles) within and outside of neurons (Jucker and Walker, 2013; Jaunmuktane et al., 2015). Approximately 46.8 million people over the age of 60 years have been diagnosed with AD worldwide (Prince et al., 2016). Though occurrence of the early onset of dementia is <1% per 4,000 individuals, the projected figure is estimated to be 131.5 million in 2050 (Prince et al., 2016). The projected increase in the prevalence of dementia is substantially higher in developing countries than in the USA and Europe, which already have a high proportion of older individuals in their populations. The Alzheimer's Association presented an estimate of 5.5 million people suffering from AD in the USA, and the incidence of AD in the USA is projected to be 7.7 millions in 2030 and 11–16 millions in 2050 (ADFF, 2016).
The pathology of AD begins well before symptom manifestation, with intracellular accumulation of neurofibrillary tangles that arise via abnormal tau protein phosphorylation and extracellular deposition of Aβ-plaques (Selkoe, 1994, 2001a,b). Interfering with neuronal communication, deposition of β-amyloid plaques causes neuronal death, while tau tangles Blocks transport of essentials into interior of the neurons. Characterized by progressive memory loss and cognitive impairment, advanced stage AD patients show symptoms ranging from neuron inflammation to neuron death. Various risk factors promote pathological changes well before the onset of clinical symptoms of AD. In addition to cardiovascular risk, studies suggest a significant contribution of lifestyle related factors such as obesity, diabetes, depression, smoking, and insufficient diet in dementia. This review presents an overview of AD with recent updates on epidemiology, factors that aggravate the disease, and a prospective insight into diagnostic markers and therapeutic options for disease treatment.
Genetic Susceptibility
AD represents one of the greatest health-care challenges of the twenty-first century. Its dependence on age and genetic background has resulted in its classification as either familial (FAD; showing early onset), which is observed in 5% of AD cases, or sporadic (SAD; showing late disease onset), which shows a high disease incidence rate. Though ApoE4 is a well-characterized risk factor in SAD, disease etiology in FAD is attributed to mutations of amyloid precursor protein (APP), presenilin1 (PS1), and presenilin 2 (PS2) genes (Goate et al., 1991; Levy-Lahad et al., 1995; Rogaev et al., 1995; Sherrington et al., 1996; Selkoe, 2001b). Accounting for 15% of total ApoE, APOE4 interferes with the clearance of Aβ from brain. Differing in amino acid substitutions at 112 and 158 positions, ApoE4 carries two arginines, while ApoE3 have two cysteines and ApoE2 has arginine and cysteine at these positions (Mahley and Huang, 2009). Attribution of APOE4 to AD is 50% in homozygous (Apo E4/E4) and 20–30% in heterozygous condition with APOE3 (Genin et al., 2011).
Over the time, substantial evidences regarding accumulation of misfolded Aβ and tau tangles (so-called seeds of pathological consequences) in the brain of patients suffering from AD have been established (Karran et al., 2011). Evidential support of Aβ and tau involvement comes from FAD studies that report mutations in APP (Aβ precursor) and PS1&2 (catalytic γ-secretase subunit; Karch and Goate, 2015; Ahmad et al., 2016). Having a critical role in the multi-causality of dementia (Boyle et al., 2013), mutations in APP enhance aggregation, while PSEN1&2 mutations cause less efficient APP processing, leading to longer and more hydrophobic Aβs (Scheuner et al., 1996; Chávez-Gutiérrez et al., 2012; Wong et al., 2013; Table 1). Though mutations in APP and PS1&2 accelerate generation of disease seeds, decreased Aβ clearance (Mawuenyega et al., 2010) and increased Aβ accumulation (Wahlster et al., 2013) enhances SAD.
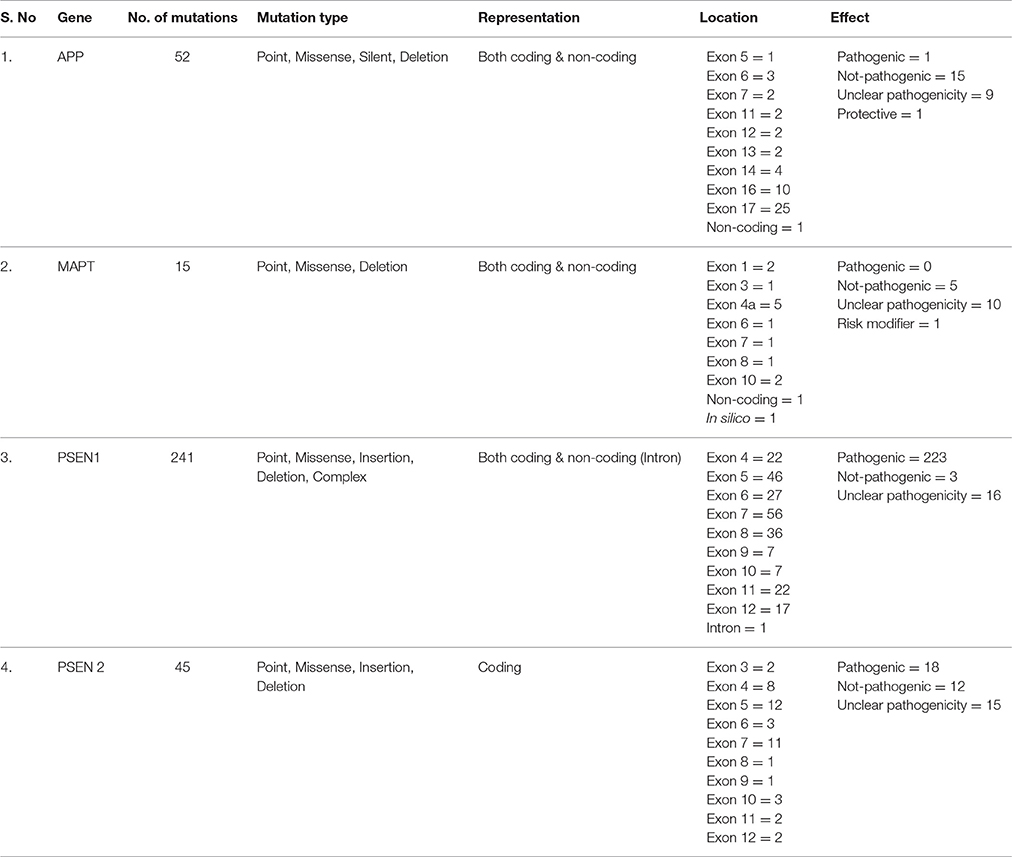
Table 1. Summary of mutations predicted in genes that have been associated with the occurrence of AD.
Mitochondrial Stress in AD
Performing vital biochemical functions, mitochondrial dysfunction significantly affects progression in the pathogenesis of AD (Swerdlow et al., 2014). Associated with the regulation of cellular metabolism, functional impairment of metabolic enzymes, in particular enzymes of the TCA cycle, causes reduction in the energy metabolism in brain (Huang et al., 2003; Bubber et al., 2005). Studies of the AD brain have revealed significant impairment in the functioning of pyruvate dehydrogenase complex (PDHC) and α-ketoglutarate dehydrogenase complex (KGDHC) enzymes, followed by impairment of isocitrate dehydrogenase (Huang et al., 2003; Bubber et al., 2005). Although, increased activity of succinate and malate dehydrogenases was observed, activities of the remaining four enzymes remains unaltered. Considering the high energy demand of neurons, a role of mitochondrial oxidative stress leading to energy imbalance appears to have a considerable effect on neurodegeneration.
Mitochondrial dysfunction, observed as altered mitochondrial DNA and increased cytochrome oxidase (COX) levels, indicates oxidative damage to the neurons of AD patients (Hirai et al., 2001; Nunomura et al., 2001; Moreira et al., 2006, 2007, 2009; Su et al., 2008). In concert with the amyloid hypothesis, altered APP processing increases Aβ deposition (Furukawa et al., 1996), whose aggregation causes oxidative stress through an increase in the production of H2O2 (Readnower et al., 2011). Inhibition of the mitochondrial electron transport chain (ETC) causes increased ROS production, which damages proteins, lipids, and nucleic acids, observed as increases in 8-hydroxy-2-deoxyguanosine (8-OHdG) and nitrotyrosine levels (Wang et al., 2005). Accumulation of Aβ in the synaptic mitochondria makes them susceptible to changes in synaptic Ca2+ as they have high levels of cyclophilin D (CypD; Sayre et al., 2005). Being a component of the mitochondrial permeability transition pore (mPTP), CypD translocation from matrix to mPTP increases interaction of CypD-mPTP with adenine nucleotide translocase resulting in opening of the pore and as such collapse of membrane potential, which ultimately leads to neuronal death (Juhaszova et al., 2004). An increase in the oxidative burden of mitochondria mediates activation of FoxO transcription factor (Kops et al., 2002; Fu and Tindall, 2008), which is associated with induction of SOD and catalase activity, as well as causing cell cycle arrest and cell death (Castellani et al., 2002). Increased ROS attenuation of the antioxidant arsenal of mitochondria alters the cellular redox state. Increased ROS levels act as an autophagy trigger and subjects mitochondria to mitophagy (Scherz-Shouval and Elazar, 2011).
Endoplasmic Reticulum (ER) Stress in AD
Acting as a site of protein synthesis, disruption in the proteostasis causes accumulation of unfolded proteins in the ER lumen (Wu and Kaufman, 2006; Ron and Walter, 2007). To reduce abnormal protein aggregation, unfolded proteins are translocated to cytoplasm for degradation; a process referred as ER-associated protein degradation (ERAD; Hetz et al., 2011; Walter and Ron, 2011). Attainment of saturation in the ER's protein-folding capacity elicits a dynamic signaling response referred as unfolded protein response (UPR; Lee, 2001; Ledoux et al., 2003). Recognition of unfolded proteins by stress sensors such as inositol requiring protein 1 (IRE1), protein kinase RNA (PKR) like ER kinase (PERK), and activating transcription factor 6 (ATF6) triggers downstream signaling via transcription factors (Hetz and Mollereau, 2014). PERK induces rapid translational attenuation by inhibition of the eukaryotic translational initiation factor 2α (eIF2α). PERK-mediated phosphorylation of eIF2α also favors translation of transcription factor ATF4, which is capable of controlling expression of genes related to amino acid metabolism, autophagy, and apoptosis. Increased phosphorylation of eIF2α in PS1 mutant knockout mice confirmed the inhibition of eIF2α phosphorylation by PS1 (Milhavet et al., 2002). Additionally, PS1-mediated abnormal processing of IRE1 disturbs UPR by interfering with the ER stress. Activation of IRE1 signaling induces splicing of X-box binding protein 1 (XBP-1), which controls the expression of lipid synthesis, ER protein translocation, protein folding, and ERAD genes (Hetz et al., 2011; Walter and Ron, 2011). Polymorphism in the promoter region of XBP-1, which reduces its transcription, is considered as a risk factor for AD (Liu et al., 2013). Expression of GPR78 and GPR94 associated with the refolding of unfolded proteins attenuated in PS2-expressed cells is attributed to impaired IRE1 phosphorylation. Though protection against Aβ toxicity through enforced Xbp1 expression in a Drosophila melanogaster AD model is attributed to reduction in the release of Ca2+ from ER (Casas-Tinto et al., 2011), a similar effect in Caenorhabditis elegans AD model is correlated with augmented stress levels and enhanced autophagy (Safra et al., 2013). Nuclear translocation of ATF6 following protease cleavage activates ERAD genes and XBP-1. Moreover, ATF4, ATF6, and XBP-1 stimulate C/EBP homologous protein (CHOP) and its target growth arrest and DNA damage inducible 34 (GADD34), as well as pro-apoptotic components of the B cell lymphoma-2 (BCL2) family of proteins (Xu et al., 2005). Neurons expressing p-tau show enhanced UPR activation (Hoozemans et al., 2009; Abisambra et al., 2013). Excessive adaptive capacity of UPR triggers pro-apoptotic events through upregulation of the cell death genes such as caspase-12 (Szegezdi et al., 2003).
Autophagy in AD
Degradation of non-essential cellular components, such as misfolded and aggregated proteins, occurs through the autophagy-lysosomal system (ALS; Li et al., 2010; Murrow and Debnath, 2013). Activated by oxidative stress, nutrient starvation, etc., clearance of unwanted entities helps in restoring substrates for cellular remodeling (Ichimura and Komatsu, 2010; Li et al., 2010; Murrow and Debnath, 2013). Abundance of growth factors and cellular nutrients activates mTOR kinase, whereas a starvation state exerts inhibitory effect on mTOR kinase activity. The mTOR kinase-mediated phosphorylation of ATG13 prevents its association to Unc-51 like kinase (ULK), and recruitment of focal adhesion kinase family interacting protein of 200 kD (FIP200) inhibits autophagy, whereas inhibition of mTOR activates phosphatases that cause dephosphorylation of ATG13 (Kundu, 2011; Lee et al., 2012), thereby promotes autophagy. Although mTOR-dependent autophagy is prominent, there are reports of mTOR-independent autophagy mediated by (1) ATG5 and ATG7 via microtubule associated light chain 3-II (LC3-II; Nishida et al., 2009); (2) autophagic proteins Beclin1, Bcl-2, and ULK1 (Nishida et al., 2009; Shimizu et al., 2010); and (3) non-canonical signaling events involving Ca2+. In addition, Ca2+ has a prominent role in both canonical and non-canonical mTOR-independent autophagy (Cárdenas and Foskett, 2012; Decuypere et al., 2013). Deletion of Beclin-1 in AD mouse models has resulted in increased Aβ accumulation, while its overexpression leads to reduction in amyloid pathology (Pickford et al., 2008; Jaeger and Wyss-Coray, 2010). Though IP3 receptor-mediated Ca2+ signaling inhibits autophagy, an increase in the cytosolic Ca2+ level promotes autophagy (Criollo et al., 2007; Wang et al., 2008; Khan and Joseph, 2010; Vingtdeux et al., 2010). Some studies have linked PS mutation in FAD with neuronal dysfunction and apoptosis via Ca2+ dysfunction (Del Prete et al., 2014; Duggan and McCarthy, 2016).
Despite entrapping non-selective molecules, selective trapping of various molecules occurs through interaction of LC3-II with cargo-receptors such as nuclear dot protein-52 (NDP52), p62, and neighbor of BRAC1 (NRB1; Bjørkøy et al., 2005; Kirkin et al., 2009; Jo et al., 2014). Acting as an autophagic adaptor for degradation of toll/IL-1 receptor homology domain containing adaptor including IFN-β (TRIF) and tumor receptor associated factor-6 (TRAF6), a role of NDP-52 in docking of autophagosomes to T6BP, myosin VI, and optineurin for maturation has been reported (Inomata et al., 2012; Tumbarello et al., 2012). Interaction of NBR1 and p62 with ubiquitinated misfolded proteins and members of the ATG8 family makes them critical for autophagosome formation (Kabeya et al., 2000; Bjørkøy et al., 2005; Pankiv et al., 2007; Kirkin et al., 2009). PS1 mutations that cause a loss of lysosomal acidification ultimately lead to loss of lysosomal proteolytic activity (Lee et al., 2010). Disruption in the lysosomal function causes accumulation of autophagosomes containing protein aggregates, as observed in the AD brain. Studies indicate involvement of NDP-52 as a downstream facilitator of nuclear factor erythroid-2 related factor 2 (Nrf2)-mediated tau (phosphorylated) degradation (Jo et al., 2014). Reduction in the autophagy-aggravated AD pathology underlines the critical role of autophagy in the removal of Aβ aggregates (Cai et al., 2012; Di Domenico et al., 2014).
Biomarkers of AD
Cortical amyloid deposition of Aβ and phosphorylated tau (p-tau) are core biomarkers of AD in CSF (Blennow et al., 2010). With high accuracy diagnostics, specificity of the core repertoire of biomarkers is in the 80–90% range in the mild cognitive impairment stage of AD (Shaw et al., 2009; Visser et al., 2009). Despite this, assessment of the sensitivity of disease biomarkers reflects an imperfect criterion in the gold standard of clinical diagnosis of the disease because it fails to distinguish pathological changes such as plaque count between cognitively normal elderly persons and those with AD (Coart et al., 2015; Curtis et al., 2015). Changes in α-synuclein (α-syn), TDP43, and several vascular indicators, are other pathological markers in patients suffering from AD (Kovacs et al., 2013).
A step to increase the specificity and sensitivity of biomarkers has led to the screening of several different entities for use in the diagnosis of AD. One such biomarker involves Aβ oligomers (toxic forms of Aβ), which are associated with synaptic dysfunction (Overk and Masliah, 2014). Though increases in the Aβ oligomers are observed in AD, their limited occurrence in the CSF hinders their reliability in the diagnosis of AD (Hölttä et al., 2013; Yang et al., 2013). Neurogranin, a dendritic protein, is another synaptic biomarker candidate (Díez-Guerra, 2010). Increased amount of neurogranin in CSF of AD patients is correlated with the progression of mild cognitive impairment (Kvartsberg et al., 2015). Additionally, an increase in the level of presynaptic protein SNAP25 might indicate AD diagnosis (Brinkmalm et al., 2014). Of the other biomarkers, screening of CNS-specific protein candidates in blood also seems suitable; when sampling populations for AD. Combinations of proteins, lipids, and other molecules have been used in the assessment of AD (Henriksen et al., 2014; Mapstone et al., 2014). However, as they may be secreted in low amounts and are prone to degradation by plasma proteases, their suitability as biomarkers for AD is uncertain. Similarly, tau-imaging, which is commonly employed in drug trials to determine delay in the progression of AD may be another potentially suitable option for AD diagnosis.
Therapeutic Approaches to Combat AD
The fact that critical care provides patients a better quality of life family care is considered as the mainstay in the treatment of AD. At present, efforts are being made on two fronts; toward developing disease modifying therapies that can uphold progression of the disease, and; developing drugs that can act as disease pathway blockers. As Aβ and tau hyper-phosphorylation are well-recognized hallmarks of AD (Lee et al., 2005; Querfurth and Laferla, 2010; Ahmad et al., 2017), current drug approaches are aimed at:
Improved APP Processing via Inhibition and/or Activation of Enzymatic Machinery
To date, only five drugs have been approved by the USA FDA for treatment of cognitive manifestations of AD (Table 2). Of the different drug regimes, glutamate agonist memantine, and acetylcholine esterase (AchE) inhibitors such as rivastigmine, donepezil, and galantamine are employed in the treatment of the dementia phase of AD (Raina et al., 2008). Blocking the pathological stimulation of NMDA receptors, memantine protects neural cells from glutamate-mediated excitoxicity. AchE inhibitors cause temporary slowdown in cognitive function by decreasing activity of cholinesterase, leading to enhanced acetylcholine (Ach) levels and, thereby, brain functions (Godyn et al., 2016). As disruption of AchE has a direct correlation with NFT and Aβ deposits (Tavitian et al., 1993), AchE inhibitors stabilize cognitive performance and daily functioning in early dementia stages, whereas application of memantine provides benefits to patients suffering from moderate to severe dementia (Godyn et al., 2016). The fifth drug, tacrine is less prescribed due to its hepatotoxicity. Although these drugs provide symptomatic improvement in AD, benefits are generally short lived and with no effect on the pathogenic mechanism or on disease progression. Research into potential strategies is currently directed at screening bioactive compounds for their effect on enzyme inhibition, aggregation, prevention of Aβ formation, and upregulation in the removal of toxic Aβ.
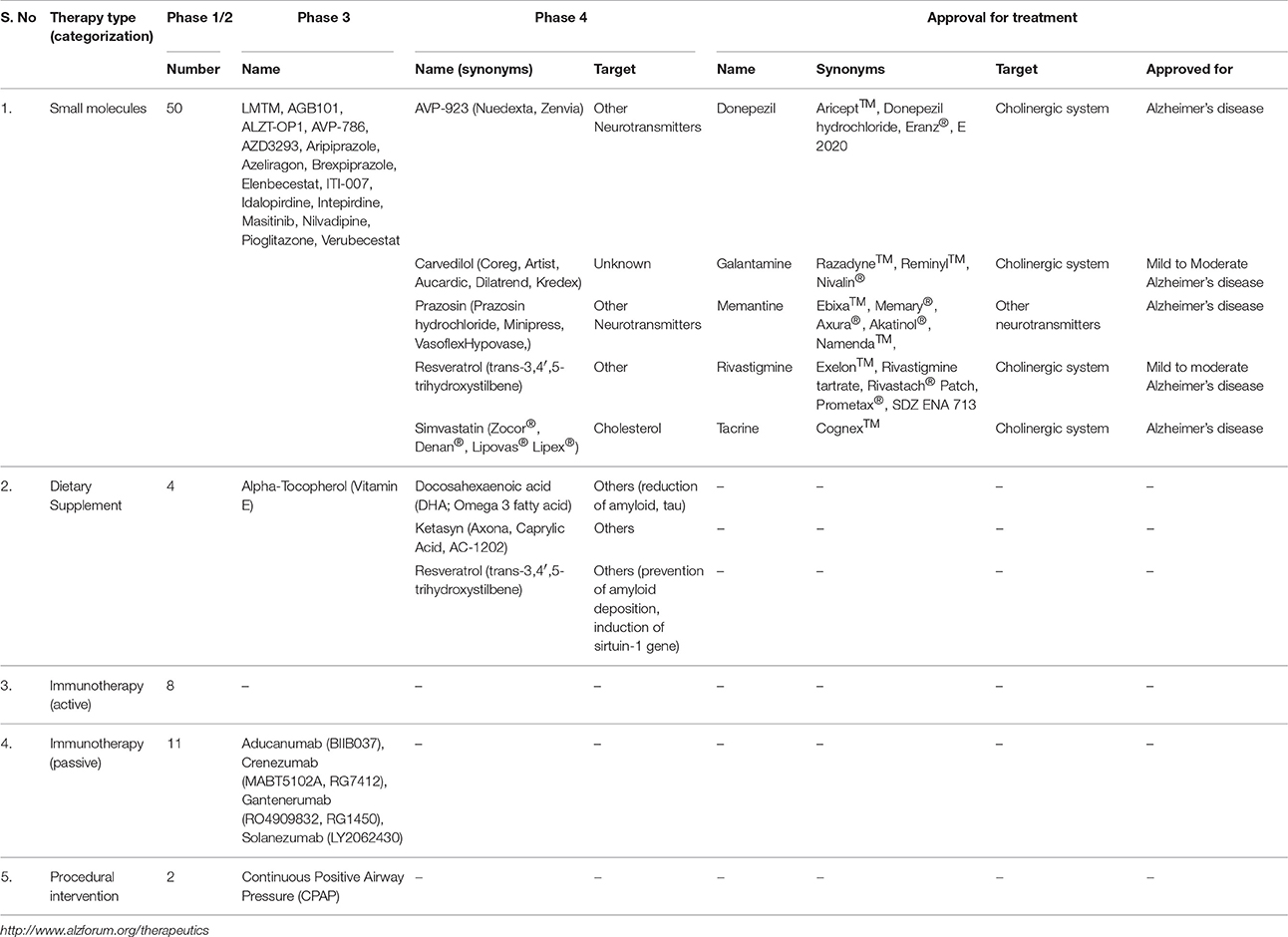
Table 2. Summary of therapeutic options available in the treatment of AD.
Immunotherapeutic Approaches to Existing Amyloids
Currently, the most attractive Aβ-directed approach, immunotherapy enlists both active (immune stimulation by vaccine for antibody production) and passive (injection of pre-prepared antibodies) immunization. Engaging both the cellular and humoral immune systems, active immunization is cost effective and ensures long-term high antibody titers. An active vaccination journey begins with AN1792, a full-length Aβ1−42 peptide injected with the immune stimulant adjuvant QS21 (Panza et al., 2014). However, on observing meningoencephalitis among 6% of the enrolled AD patients, the phase II AN1792 trial was halted in 2002. Taking strong cognizance of AN1792 trials, larger studies were directed at testing passive immunotherapy by using human anti-Aβ monoclonal antibodies. At present various 2nd generation active Aβ vaccines are undergoing clinical trials; of particular note are CAD106 (Aβ1−6; Novartis), ACC001 (Aβ1−6; Janssen and Pfizer), ACI-24, V-950, and AD-02 (Aβ1−6; Alzforum, 2016). Compared to active immunization that leads to a polyclonal antibody response, target specificity (specific against monomeric Aβ, Aβ fibrils, and their carrier and transport proteins) in passive immunization has advantages both in preventing Aβ-aggregation and in promoting Aβ-clearance in anti-Aβ immunotherapy. The first monoclonal antibody was bapineuzumab directed against Aβ1−5 (Abushouk et al., 2017; Xing et al., 2017). Although it reduced Aβ in brain during phase II trials, it failed to achieve significant benefit in a phase III trial, leading to its termination in 2012. This was followed by trials of mAb m266, which showed enhanced Aβ clearance from brain to blood (Demattos et al., 2001; Dodart et al., 2002). Using am266 precursor, Solanezumab directed against mid-domain Aβ16−24 revealed dose-dependent increases in plasma Aβ, suggesting cleavage of insoluble species from senile plaques (Farlow et al., 2012; Han and Mook-Jung, 2014). Other Abs undergoing clinical trials include gantenerumab (showing binding specificity for Aβ plaques), crenezumab IgG4 mAb (showing binding specificity for Aβ oligomers, plaques, and fibrils that inhibit aggregation), GSK933766, and BAN2401 mAb (Alzforum, 2016). Reduction in Aβ production is also achieved by using inhibitors against secretases such as NIC5-15, Bryostatin-1, AZD3293, MK8931, and E2609 (Alzforum, 2016).
Tau Centric Therapies
Tau centric therapies include the use of putative tau kinase inhibitors, microtubule stabilizers, and tau immunotherapy. Glycogen synthase kinase-3β (GSK-3β), the primary enzyme involved in tau phosphorylation, is considered the primary target for disease modification (Jaworski et al., 2011). As Aβ promotes GSK-3β activity, studies of GSK-3β inhibitors such as tideglusib and AZD1080 were pursued (King et al., 2014; Lovestone et al., 2015). Clinical trials of AZD1080 revealed nephrotoxicity (Eldar-Finkelman and Martinez, 2011), while the trials for tideglusib showed diminished clinical benefits. Microtubule stabilizers inhibit tau aggregation (Bulic et al., 2010). LMTC, a methylthionium chloride (MTC) derivative prevents tau interactions, thereby facilitating its clearance from the brain (Wischik et al., 2015). Other microtubule stabilizers include TPI207 and BMS241027 (epothilone-D). Immunotherapy for tau protein is directed toward prevention of NFT formation. Both AADvac1 (a synthetic tau derived peptide) and ACI-35 (liposome formulated based tau protein) are currently being evaluated for their effects on avoiding tau aggregation (Alzforum, 2016).
Therapies to Combat Oxidative Stress
Oxidative stress due to reactive oxygen species (ROS), being a major player in neurodegeneration strategies, are currently being devised to combat its emergence at mitochondria (Federico et al., 2012; Yan et al., 2013; Kim et al., 2015). On the forefront, ferulic acid (FA), epigallocatechin-3-gallate, and nano formulation of naturally occurring curcumin were found exerting strong antioxidant, anti-inflammatory and amyloid disintegration properties (Rezai-Zadeh et al., 2005; Yang et al., 2005; Cheng et al., 2013; Sgarbossa et al., 2015; Cascella et al., 2017). Alleviating mitochondrial dysfunction under diseased conditions, peptide based strategies complemented with different drug molecules have shown positive results in overcoming the inefficiency of low antioxidant levels (Kumar and Singh, 2015). Of them, Szeto-Schiller (SS) peptides displaying a sequence motif that directs its accumulation inside mitochondria inhibit lipid peroxidation. Accumulation of SS31 on inner mitochondrial membrane prevents release of cyt-C (Szeto, 2008; Kumar and Singh, 2015). Similarly, accumulation of drugs such as MitoQ inside mitochondria increases its potential to neutralize free radicals several 100-folds than that attributed by natural antioxidants (Tauskela, 2007; Ross et al., 2008). Probucol, a drug that helps establishment of balance of mitochondal fission-fusion processes, also maintains AD mitochondria induced extracellular signal regulated kinase (ERK) activation (Champagne et al., 2003; Gan et al., 2014).
Autophagy Enhancer Therapy
As accumulation of the aggregated proteins enhances neurodegeneration, enhancement in the degenerative capacity via, autophagy inducers seems a good therapeutic approach. Their mode of action involves prevention in the accumulation of Aβ plaques and tau tangles via degradation of the aggregates. Of the available drugs, rapamycin acting as mTOR inhibitor was found ameliorating Aβ and tau pathies in AD mouse model (Caccamo et al., 2010; Rubinsztein et al., 2012). Latrepirdine stimulation of autophagy reduces Aβ neuropathy in mouse brain (Steele and Gandy, 2013). Metformin, a PP2A agonist, inhibition of TORC1 prevents hyperphosphorylation of tau protein (Kickstein et al., 2010). Other authophagy enhancers include reservatol and its analogs, RSVA314 and RSVA405, virally packaged BECN1 and its mimetics, nicotinamide, etc (Vingtdeux et al., 2010; Shoji-Kawata et al., 2013). Though, Beclin1 deficiency increases APP and Aβ levels, its over-expression in cultured neurons cause significant reduction in Aβ accumulation (Pickford et al., 2008). Additionally, PS1 and PS2 serving as a catalytic subunit of γ-secretase was found acting as autophagy modulators (Lee et al., 2010). Associated with the substrate cleavage, their knockdown was found exhibiting inefficiency in the clearance of protein aggregates. Cells deficient in PS1 was found having reduced levels of cargo shuttle protein p62, associated with the degradation of abnormal tau (Tung et al., 2014; Caccamo et al., 2017).
Failure of biologics that target amyloids to cross the blood-brain barrier (BBB) indicates the necessity of development of an efficient drug delivery system. Liposomes and polymeric nanoparticles have shown promising results in delivering drugs and other therapeutic molecules (Ha et al., 2016); however, their use as a means to deliver drugs across the BBB has encountered roadblocks associated with biocompatibility and long-term safety. Persistent problems with low biocompatibility, restricted immune escape, circulation stability, and toxicity have led to a gradual shift of research toward the use of exosomes. Their long circulatory half-life, host biocompatibility, target-specific drug delivery ability, and low toxicity have increased the interest in using exosomes to deliver drugs in several neurodegenerative diseases (Lai and Breakefield, 2012; Liu et al., 2016). As drug delivery through exosomes has shown promising results in the ongoing studies, their utilization in reducing disease progression may indicate their suitability in reducing progression of AD.
Conclusion
AD progresses via aggregation and accumulation in the extracellular milieu of amyloid plaques and intraneuronal neurofibrillary tangles produced by p-tau. Although malfunctioning APP, PS-1 &-2 genes are considered the main culprits behind AD, mitochondrial dysfunction, ER stress and mitophagy significantly increase progression of the disease. Current research provides useful information on new targets and their utilization in designing novel inhibitors or drugs as part of attempts to achieve successful treatment of AD. It also studies different entities for employment as potent biomarkers in disease diagnosis and provides information on therapeutic suitability in the treatment of AD. Further studies on the use of exosomes as drug delivery vehicles are needed in order to reveal and reduce safety and ethical concerns. With increases in newly identified targets, studies pertaining to the design of drugs and other potent therapeutic molecules are needed in order to combat the progression of AD.
Author Contributions
IC, QH, and AJ: conceived the idea; AJ and MA: contributed to writing of the manuscript; AJ, SR, AA, DC, and EL: contributed to upgrading the contents and preparing the tables.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors extend their thanks to colleagues for their criticism, which helped to improve the quality of this paper's contents and broaden its perspective to reach a broader audience. This work was supported by the Creative Economy Leading Technology Development Program through the Gyeongsangbuk-Do and Gyeongbuk Science & Technology Promotion Center of Korea (SF316001A).
References
Abisambra, J. F., Jinwal, U. K., Blair, L. J., O'leary, J. C., Li, Q., Brady, S., et al. (2013). Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 33, 9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013
Abushouk, A. I., Elmaraezy, A., Aglan, A., Salama, R., Fouda, S., Fouda, R., et al. (2017). Bapineuzumab for mild to moderate Alzheimer's disease: a meta-analysis of randomized controlled trials. BMC Neurol 17:66. doi: 10.1186/s12883-017-0850-1
ADFF (2016). Alzheimer's disease facts and figures. Alzheimers Dement. 12, 459–509. doi: 10.1016/j.jalz.2016.03.001
Ahmad, K., Baig, M. H., Gupta, G. K., Kamal, M. A., Pathak, N., and Choi, I. (2016). Identification of common therapeutic targets for selected neurodegenerative disorders: an in silico approach. J. Comput. Sci. 17, 292–306. doi: 10.1016/j.jocs.2016.03.007
Ahmad, K., Baig, M. H., Mushtaq, G., Kamal, M. A., Greig, N. H., and Choi, I. (2017). Commonalities in biological pathways, genetics, and cellular mechanism between Alzheimer Disease and other neurodegenerative diseases: an in silico-updated overview. Curr Alzheimer Res. 14, 1190–1197. doi: 10.2174/1567205014666170203141151
Alzforum (2016). Therapeutics for Alzheimers Disease. Available online at: http://www.alzforum.org/therapeutics.
Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., et al. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell. Biol. 171, 603–614. doi: 10.1083/jcb.200507002
Blennow, K., Hampel, H., Weiner, M., and Zetterberg, H. (2010). Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 6, 131–144. doi: 10.1038/nrneurol.2010.4
Boyle, P. A., Wilson, R. S., Yu, L., Barr, A. M., Honer, W. G., Schneider, J. A., et al. (2013). Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann. Neurol. 74, 478–489. doi: 10.1002/ana.23964
Brinkmalm, A., Brinkmalm, G., Honer, W. G., Frölich, L., Hausner, L., Minthon, L., et al. (2014). SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer's disease. Mol. Neurodegener. 9:53. doi: 10.1186/1750-1326-9-53
Bubber, P., Haroutunian, V., Fisch, G., Blass, J. P., and Gibson, G. E. (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 57, 695–703. doi: 10.1002/ana.20474
Bulic, B., Pickhardt, M., Mandelkow, E. M., and Mandelkow, E. (2010). Tau protein and tau aggregation inhibitors. Neuropharmacology 59, 276–289. doi: 10.1016/j.neuropharm.2010.01.016
Caccamo, A., Ferreira, E., Branca, C., and Oddo, S. (2017). p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 22, 865–873. doi: 10.1038/mp.2016.139
Caccamo, A., Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2010). Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120. doi: 10.1074/jbc.M110.100420
Cai, Z., Zhao, B., Li, K., Zhang, L., Li, C., Quazi, S. H., et al. (2012). Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer's disease? J. Neurosci. Res. 90, 1105–1118. doi: 10.1002/jnr.23011
Cárdenas, C., and Foskett, J. K. (2012). Mitochondrial Ca(2+) signals in autophagy. Cell Calcium 52, 44–51. doi: 10.1016/j.ceca.2012.03.001
Casas-Tinto, S., Zhang, Y., Sanchez-Garcia, J., Gomez-Velazquez, M., Rincon-Limas, D. E., and Fernandez-Funez, P. (2011). The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 20, 2144–2160. doi: 10.1093/hmg/ddr100
Cascella, M., Bimonte, S., Muzio, M. R., Schiavone, V., and Cuomo, A. (2017). The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer's disease: an overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agent Cancer 12:36. doi: 10.1186/s13027-017-0145-6
Castellani, R., Hirai, K., Aliev, G., Drew, K. L., Nunomura, A., Takeda, A., et al. (2002). Role of mitochondrial dysfunction in Alzheimer's disease. J. Neurosci. Res. 70, 357–360. doi: 10.1002/jnr.10389
Champagne, D., Pearson, D., Dea, D., Rochford, J., and Poirier, J. (2003). The cholesterol-lowering drug Probucol increases apolipoprotein e production in the hippocampus of aged rats: implications for Alzheimer's disease. Neuroscience 121, 99–110. doi: 10.1016/S0306-4522(03)00361-0
Chávez-Gutiérrez, L., Bammens, L., Benilova, I., Vandersteen, A., Benurwar, M., Borgers, M., et al. (2012). The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274. doi: 10.1038/emboj.2012.79
Cheng, K. K., Yeung, C. F., Ho, S. W., Chow, S. F., Chow, A. H., and Baum, L. (2013). Highly stabilized curcumin nanoparticles tested in an in vitro blood-brain barrier model and in Alzheimer's disease Tg2576 mice. AAPS J. 15, 324–336. doi: 10.1208/s12248-012-9444-4
Coart, E., Barrado, L. G., Duits, F. H., Scheltens, P., van der Flier, W. M., Teunissen, C. E., et al. (2015). Correcting for the absence of a gold standard improves diagnostic accuracy of biomarkers in Alzheimer's Disease. J Alzheimers Dis. 46, 889–899. doi: 10.3233/JAD-142886
Criollo, A., Maiuri, M. C., Tasdemir, E., Vitale, I., Fiebig, A. A., Andrews, D., et al. (2007). Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 14, 1029–1039. doi: 10.1038/sj.cdd.4402099
Curtis, C., Gamez, J. E., Singh, U., Sadowsky, C. H., Villena, T., Sabbagh, M. N., et al. (2015). Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 72, 287–294. doi: 10.1001/jamaneurol.2014.4144
Decuypere, J. P., Kindt, D., Luyten, T., Welkenhuyzen, K., Missiaen, L., De Smedt, H., et al. (2013). mTOR-controlled autophagy requires intracellular Ca(2+) signaling. PLoS ONE 8:e61020. doi: 10.1371/journal.pone.0061020
Del Prete, D., Checler, F., and Chami, M. (2014). Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 9, 21–29. doi: 10.1186/1750-1326-9-21
Demattos, R. B., Bales, K. R., Cummins, D. J., Dodart, J. C., Paul, S. M., and Holtzman, D. M. (2001). Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 98, 8850–8855. doi: 10.1073/pnas.151261398
Di Domenico, F., Head, E., Butterfield, D. A., and Perluigi, M. (2014). Oxidative stress and proteostasis network: culprit and casualty of Alzheimer's- like Neurodegeneration. Adv. Geriatr. 2014:14. doi: 10.1155/2014/527518
Díez-Guerra, F. J. (2010). Neurogranin, a link between calcium/calmodulin and protein kinase C signaling in synaptic plasticity. IUBMB Life 62, 597–606. doi: 10.1002/iub.357
Dodart, J. C., Bales, K. R., Gannon, K. S., Greene, S. J., Demattos, R. B., Mathis, C., et al. (2002). Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat. Neurosci. 5, 452–457. doi: 10.1038/nn842
Duggan, S. P., and McCarthy, J. V. (2016). Beyond gamma-secretase activity: the multifunctional nature of presenilins in cell signalling pathways. Cell Signal 28, 1–11. doi: 10.1016/j.cellsig.2015.10.006
Eldar-Finkelman, H., and Martinez, A. (2011). GSK-3 Inhibitors: preclinical and clinical focus on CNS. Front. Mol. Neurosci. 4:32. doi: 10.3389/fnmol.2011.00032
Farlow, M., Arnold, S. E., Van Dyck, C. H., Aisen, P. S., Snider, B. J., Porsteinsson, A. P., et al. (2012). Safety and biomarker effects of solanezumab in patients with Alzheimer's disease. Alzheimers Dement. 8, 261–271. doi: 10.1016/j.jalz.2011.09.224
Federico, A., Cardaioli, E., Da Pozzo, P., Formichi, P., Gallus, G. N., and Radi, E. (2012). Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 322, 254–262. doi: 10.1016/j.jns.2012.05.030
Fu, Z., and Tindall, D. J. (2008). FOXOs, cancer and regulation of apoptosis. Oncogene 27, 2312–2319. doi: 10.1038/onc.2008.24
Furukawa, K., Sopher, B. L., Rydel, R. E., Begley, J. G., Pham, D. G., Martin, G. M., et al. (1996). Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J. Neurochem. 67, 1882–1896. doi: 10.1046/j.1471-4159.1996.67051882.x
Gan, X., Huang, S., Wu, L., Wang, Y., Hu, G., Li, G., et al. (2014). Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer's disease cybrid cell. Biochim. Biophys. Acta 1842, 220–231. doi: 10.1016/j.bbadis.2013.11.009
Genin, E., Hannequin, D., Wallon, D., Sleegers, K., Hiltunen, M., Combarros, O., et al. (2011). APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907. doi: 10.1038/mp.2011.52
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706. doi: 10.1038/349704a0
Godyn, J., Jonczyk, J., Panek, D., and Malawska, B. (2016). Therapeutic strategies for Alzheimer's disease in clinical trials. Pharmacol. Rep. 68, 127–138. doi: 10.1016/j.pharep.2015.07.006
Ha, D., Yang, N., and Nadithe, V. (2016). Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm. Sin. B 6, 287–296. doi: 10.1016/j.apsb.2016.02.001
Han, S. H., and Mook-Jung, I. (2014). Diverse molecular targets for therapeutic strategies in Alzheimer's disease. J. Korean Med. Sci. 29, 893–902. doi: 10.3346/jkms.2014.29.7.893
Henriksen, K., O'Bryant, S. E., Hampel, H., Trojanowski, J. Q., Montine, T. J., Jeromin, A., et al. (2014). The future of blood-based biomarkers for Alzheimer's disease. Alzheimers Dement. 10, 115–131. doi: 10.1016/j.jalz.2013.01.013
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hetz, C., Martinon, F., Rodriguez, D., and Glimcher, L. H. (2011). The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 91, 1219–1243. doi: 10.1152/physrev.00001.2011
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 21, 3017–3023.
Hölttä, M., Hansson, O., Andreasson, U., Hertze, J., Minthon, L., Nägga, K., et al. (2013). Evaluating amyloid-beta oligomers in cerebrospinal fluid as a biomarker for Alzheimer's disease. PLoS ONE 8:e66381. doi: 10.1371/journal.pone.0066381
Hoozemans, J. J., Van Haastert, E. S., Nijholt, D. A., Rozemuller, A. J., Eikelenboom, P., and Scheper, W. (2009). The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am. J. Pathol. 174, 1241–1251. doi: 10.2353/ajpath.2009.080814
Huang, H. M., Ou, H. C., Xu, H., Chen, H. L., Fowler, C., and Gibson, G. E. (2003). Inhibition of alpha-ketoglutarate dehydrogenase complex promotes cytochrome c release from mitochondria, caspase-3 activation, and necrotic cell death. J. Neurosci. Res. 74, 309–317. doi: 10.1002/jnr.10756
Ichimura, Y., and Komatsu, M. (2010). Selective degradation of p62 by autophagy. Semin. Immunopathol. 32, 431–436. doi: 10.1007/s00281-010-0220-1
Inomata, M., Niida, S., Shibata, K., and Into, T. (2012). Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell. Mol. Life Sci. 69, 963–979. doi: 10.1007/s00018-011-0819-y
Jaeger, P. A., and Wyss-Coray, T. (2010). Beclin 1 complex in autophagy and Alzheimer disease. Arch. Neurol. 67, 1181–1184. doi: 10.1001/archneurol.2010.258
Jaunmuktane, Z., Mead, S., Ellis, M., Wadsworth, J. D., Nicoll, A. J., Kenny, J., et al. (2015). Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature 525, 247–250. doi: 10.1038/nature15369
Jaworski, T., Dewachter, I., Lechat, B., Gees, M., Kremer, A., Demedts, D., et al. (2011). GSK-3alpha/beta kinases and amyloid production in vivo. Nature 480, E4–E5; discussion: E6. doi: 10.1038/nature10615
Jo, C., Gundemir, S., Pritchard, S., Jin, Y. N., Rahman, I., and Johnson, G. V. (2014). Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 5:3496. doi: 10.1038/ncomms4496
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Juhaszova, M., Zorov, D. B., Kim, S. H., Pepe, S., Fu, Q., Fishbein, K. W., et al. (2004). Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 113, 1535–1549. doi: 10.1172/JCI19906
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi: 10.1093/emboj/19.21.5720
Karch, C. M., and Goate, A. M. (2015). Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Karran, E., Mercken, M., and De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712. doi: 10.1038/nrd3505
Khan, M. T., and Joseph, S. K. (2010). Role of inositol trisphosphate receptors in autophagy in DT40 cells. J. Biol. Chem. 285, 16912–16920. doi: 10.1074/jbc.M110.114207
Kickstein, E., Krauss, S., Thornhill, P., Rutschow, D., Zeller, R., Sharkey, J., et al. (2010). Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 21830–21835. doi: 10.1073/pnas.0912793107
Kim, G. H., Kim, J. E., Rhie, S. J., and Yoon, S. (2015). The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340. doi: 10.5607/en.2015.24.4.325
King, M. K., Pardo, M., Cheng, Y., Downey, K., Jope, R. S., and Beurel, E. (2014). Glycogen synthase kinase-3 inhibitors: rescuers of cognitive impairments. Pharmacol. Ther. 141, 1–12. doi: 10.1016/j.pharmthera.2013.07.010
Kirkin, V., Lamark, T., Sou, Y. S., Bjørkøy, G., Nunn, J. L., Bruun, J. A., et al. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516. doi: 10.1016/j.molcel.2009.01.020
Kops, G. J., Dansen, T. B., Polderman, P. E., Saarloos, I., Wirtz, K. W., Coffer, P. J., et al. (2002). Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321. doi: 10.1038/nature01036
Kovacs, G. G., Milenkovic, I., Wöhrer, A., Höftberger, R., Gelpi, E., Haberler, C., et al. (2013). Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol. 126, 365–384. doi: 10.1007/s00401-013-1157-y
Kumar, A., and Singh, A. (2015). A review on mitochondrial restorative mechanism of antioxidants in Alzheimer's disease and other neurological conditions. Front. Pharmacol. 6:206. doi: 10.3389/fphar.2015.00206
Kundu, M. (2011). ULK1, mammalian target of rapamycin, and mitochondria: linking nutrient availability and autophagy. Antioxid. Redox Signal. 14, 1953–1958. doi: 10.1089/ars.2010.3809
Kvartsberg, H., Duits, F. H., Ingelsson, M., Andreasen, N., Ohrfelt, A., Andersson, K., et al. (2015). Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement. 11, 1180–1190. doi: 10.1016/j.jalz.2014.10.009
Lai, C., and Breakefield, X. (2012). Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front. Physiol. 3:228. doi: 10.3389/fphys.2012.00228
Ledoux, S., Yang, R., Friedlander, G., and Laouari, D. (2003). Glucose depletion enhances P-glycoprotein expression in hepatoma cells: role of endoplasmic reticulum stress response. Cancer Res. 63, 7284–7290.
Lee, A. S. (2001). The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 26, 504–510. doi: 10.1016/S0968-0004(01)01908-9
Lee, H. G., Perry, G., Moreira, P. I., Garrett, M. R., Liu, Q., Zhu, X., et al. (2005). Tau phosphorylation in Alzheimer's disease: pathogen or protector? Trends Mol. Med. 11, 164–169. doi: 10.1016/j.molmed.2005.02.008
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Lee, J., Giordano, S., and Zhang, J. (2012). Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem. J. 441, 523–540. doi: 10.1042/BJ20111451
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H., et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269, 973–977. doi: 10.1126/science.7638622
Li, L., Zhang, X., and Le, W. (2010). Autophagy dysfunction in Alzheimer's disease. Neurodegener. Dis. 7, 265–271. doi: 10.1159/000276710
Liu, D., Yang, F., Xiong, F., and Gu, N. (2016). The smart drug delivery system and its clinical potential. Theranostics 6, 1306–1323. doi: 10.7150/thno.14858
Liu, S. Y., Wang, W., Cai, Z. Y., Yao, L. F., Chen, Z. W., Wang, C. Y., et al. (2013). Polymorphism−116C/G of human X-box-binding protein 1 promoter is associated with risk of Alzheimer's disease. CNS Neurosci. Ther. 19, 229–234. doi: 10.1111/cns.12064
Lovestone, S., Boada, M., Dubois, B., Hull, M., Rinne, J. O., Huppertz, H. J., et al. (2015). A phase II trial of tideglusib in Alzheimer's disease. J. Alzheimers Dis. 45, 75–88. doi: 10.3233/JAD-141959
Mahley, R. W., and Huang, Y. (2009). Alzheimer disease: multiple causes, multiple effects of apolipoprotein E4, and multiple therapeutic approaches. Ann. Neurol. 65, 623–625. doi: 10.1002/ana.21736
Mapstone, M., Cheema, A. K., Fiandaca, M. S., Zhong, X., Mhyre, T. R., Macarthur, L. H., et al. (2014). Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 20, 415–418. doi: 10.1038/nm.3466
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 330:1774. doi: 10.1126/science.1197623
Milhavet, O., Martindale, J. L., Camandola, S., Chan, S. L., Gary, D. S., Cheng, A., et al. (2002). Involvement of Gadd153 in the pathogenic action of presenilin-1 mutations. J. Neurochem. 83, 673–681. doi: 10.1046/j.1471-4159.2002.01165.x
Moreira, P. I., Cardoso, S. M., Santos, M. S., and Oliveira, C. R. (2006). The key role of mitochondria in Alzheimer's disease. J. Alzheimers Dis. 9, 101–110. doi: 10.3233/JAD-2006-9202
Moreira, P. I., Duarte, A. I., Santos, M. S., Rego, A. C., and Oliveira, C. R. (2009). An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer's disease. J. Alzheimers Dis. 16, 741–761. doi: 10.3233/JAD-2009-0972
Moreira, P. I., Santos, M. S., and Oliveira, C. R. (2007). Alzheimer's disease: a lesson from mitochondrial dysfunction. Antioxid. Redox Signal. 9, 1621–1630. doi: 10.1089/ars.2007.1703
Murrow, L., and Debnath, J. (2013). Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu. Rev. Pathol. 8, 105–137. doi: 10.1146/annurev-pathol-020712-163918
Nishida, Y., Arakawa, S., Fujitani, K., Yamaguchi, H., Mizuta, T., Kanaseki, T., et al. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. doi: 10.1038/nature08455
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/jnen/60.8.759
Overk, C. R., and Masliah, E. (2014). Pathogenesis of synaptic degeneration in Alzheimer's disease and Lewy body disease. Biochem. Pharmacol. 88, 508–516. doi: 10.1016/j.bcp.2014.01.015
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145. doi: 10.1074/jbc.M702824200
Panza, F., Logroscino, G., Imbimbo, B. P., and Solfrizzi, V. (2014). Is there still any hope for amyloid-based immunotherapy for Alzheimer's disease? Curr. Opin. Psychiatry 27, 128–137. doi: 10.1097/YCO.0000000000000041
Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P. A., et al. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 118, 2190–2199. doi: 10.1172/JCI33585
Prince, M., Comas-Herrera, A., Knapp, M., Guerchet, M., and Karagiannidou, M. (2016). World Alzheimer Report. Alzheimers Disease International.
Querfurth, H. W., and Laferla, F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Raina, P., Santaguida, P., Ismaila, A., Patterson, C., Cowan, D., Levine, M., et al. (2008). Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann. Intern. Med. 148, 379–397. doi: 10.7326/0003-4819-148-5-200803040-00009
Readnower, R. D., Sauerbeck, A. D., and Sullivan, P. G. (2011). Mitochondria, amyloid beta, and Alzheimer's Disease. Int. J. Alzheimers Dis. 2011:104545. doi: 10.4061/2011/104545
Rezai-Zadeh, K., Shytle, D., Sun, N., Mori, T., Hou, H., Jeanniton, D., et al. (2005). Green Tea Epigallocatechin-3-Gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in alzheimer transgenic mice. J. Neurosci. 25:8807. doi: 10.1523/JNEUROSCI.1521-05.2005
Rogaev, E. I., Sherrington, R., Rogaeva, E. A., Levesque, G., Ikeda, M., Liang, Y., et al. (1995). Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376, 775–778. doi: 10.1038/376775a0
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Ross, M. F., Prime, T. A., Abakumova, I., James, A. M., Porteous, C. M., Smith, R. A., et al. (2008). Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem. J. 411, 633–645. doi: 10.1042/BJ20080063
Rubinsztein, D. C., Codogno, P., and Levine, B. (2012). Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 11, 709–730. doi: 10.1038/nrd3802
Safra, M., Ben-Hamo, S., Kenyon, C., and Henis-Korenblit, S. (2013). The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C. elegans. J. Cell. Sci. 126, 4136–4146. doi: 10.1242/jcs.123000
Sayre, L. M., Moreira, P. I., Smith, M. A., and Perry, G. (2005). Metal ions and oxidative protein modification in neurological disease. Ann. Ist. Super. Sanita 41, 143–164.
Scherz-Shouval, R., and Elazar, Z. (2011). Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. 36, 30–38. doi: 10.1016/j.tibs.2010.07.007
Scheuner, D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., et al. (1996). Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med. 2, 864–870. doi: 10.1038/nm0896-864
Selkoe, D. J. (1994). Amyloid beta-protein precursor: new clues to the genesis of Alzheimer's disease. Curr. Opin. Neurobiol. 4, 708–716. doi: 10.1016/0959-4388(94)90014-0
Selkoe, D. J. (2001a). Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimers Dis. 3, 75–80. doi: 10.3233/JAD-2001-3111
Selkoe, D. J. (2001b). Presenilin, Notch, and the genesis and treatment of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 98, 11039–11041. doi: 10.1073/pnas.211352598
Sgarbossa, A., Giacomazza, D., and Di Carlo, M. (2015). Ferulic acid: a hope for Alzheimer's Disease therapy from plants. Nutrients 7, 5764–5782. doi: 10.3390/nu7075246
Shaw, L. M., Vanderstichele, H., Knapik-Czajka, M., Clark, C. M., Aisen, P. S., Petersen, R. C., et al. (2009). Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413. doi: 10.1002/ana.21610
Sherrington, R., Froelich, S., Sorbi, S., Campion, D., Chi, H., Rogaeva, E. A., et al. (1996). Alzheimer's disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum. Mol. Genet. 5, 985–988. doi: 10.1093/hmg/5.7.985
Shimizu, S., Arakawa, S., and Nishida, Y. (2010). Autophagy takes an alternative pathway. Autophagy 6, 290–291. doi: 10.4161/auto.6.2.11127
Shoji-Kawata, S., Sumpter, R., Leveno, M., Campbell, G. R., Zou, Z., Kinch, L., et al. (2013). Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201–206. doi: 10.1038/nature11866
Steele, J. W., and Gandy, S. (2013). Latrepirdine (Dimebon®), a potential Alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy 9, 617–618. doi: 10.4161/auto.23487
Su, B., Wang, X., Nunomura, A., Moreira, P. I., Lee, H. G., Perry, G., et al. (2008). Oxidative stress signaling in Alzheimer's disease. Curr. Alzheimer Res. 5, 525–532. doi: 10.2174/156720508786898451
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer's disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Szegezdi, E., Fitzgerald, U., and Samali, A. (2003). Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann. N.Y. Acad. Sci. 1010, 186–194. doi: 10.1196/annals.1299.032
Szeto, H. H. (2008). Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid. Redox Signal. 10, 601–619. doi: 10.1089/ars.2007.1892
Tavitian, B., Pappata, S., Bonnot-Lours, S., Prenant, C., Jobert, A., Crouzel, C., et al. (1993). Positron emission tomography study of [11C]methyl-tetrahydroaminoacridine (methyl-tacrine) in baboon brain. Eur. J. Pharmacol. 236, 229–238. doi: 10.1016/0014-2999(93)90593-7
Tumbarello, D. A., Waxse, B. J., Arden, S. D., Bright, N. A., Kendrick-Jones, J., and Buss, F. (2012). Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 14, 1024–1035. doi: 10.1038/ncb2589
Tung, Y. T., Wang, B. J., Hsu, W. M., Hu, M. K., Her, G. M., Huang, W. P., et al. (2014). Presenilin-1 regulates the expression of p62 to govern p62-dependent tau degradation. Mol. Neurobiol. 49, 10–27. doi: 10.1007/s12035-013-8482-y
Vingtdeux, V., Giliberto, L., Zhao, H., Chandakkar, P., Wu, Q., Simon, J. E., et al. (2010). AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 285, 9100–9113. doi: 10.1074/jbc.M109.060061
Visser, P. J., Verhey, F., Knol, D. L., Scheltens, P., Wahlund, L. O., Freund-Levi, Y., et al. (2009). Prevalence and prognostic value of CSF markers of Alzheimer's disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol. 8, 619–627. doi: 10.1016/S1474-4422(09)70139-5
Wahlster, L., Arimon, M., Nasser-Ghodsi, N., Post, K. L., Serrano-Pozo, A., Uemura, K., et al. (2013). Presenilin-1 adopts pathogenic conformation in normal aging and in sporadic Alzheimer's disease. Acta Neuropathol. 125, 187–199. doi: 10.1007/s00401-012-1065-6
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, J., Xiong, S., Xie, C., Markesbery, W. R., and Lovell, M. A. (2005). Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J. Neurochem. 93, 953–962. doi: 10.1111/j.1471-4159.2005.03053.x
Wang, S. H., Shih, Y. L., Ko, W. C., Wei, Y. H., and Shih, C. M. (2008). Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol. Life Sci. 65, 3640–3652. doi: 10.1007/s00018-008-8383-9
Wischik, C. M., Staff, R. T., Wischik, D. J., Bentham, P., Murray, A. D., Storey, J. M., et al. (2015). Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J. Alzheimers Dis. 44, 705–720. doi: 10.3233/JAD-142874
Wong, H. K., Veremeyko, T., Patel, N., Lemere, C. A., Walsh, D. M., Esau, C., et al. (2013). De-repression of FOXO3a death axis by microRNA-132 and−212 causes neuronal apoptosis in Alzheimer's disease. Hum. Mol. Genet. 22, 3077–3092. doi: 10.1093/hmg/ddt164
Wu, J., and Kaufman, R. J. (2006). From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 13, 374–384. doi: 10.1038/sj.cdd.4401840
Xing, H. Y., Li, B., Peng, D., Wang, C. Y., Wang, G. Y., Li, P., et al. (2017). A novel monoclonal antibody against the N-terminus of Abeta1-42 reduces plaques and improves cognition in a mouse model of Alzheimer's disease. PLoS ONE 12:e0180076. doi: 10.1371/journal.pone.0180076
Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005). Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664. doi: 10.1172/JCI26373
Yan, J. J., Jung, J. S., Kim, T. K., Hasan, A., Hong, C. W., Nam, J. S., et al. (2013). Protective effects of ferulic acid in amyloid precursor protein plus presenilin-1 transgenic mouse model of Alzheimer disease. Biol. Pharm. Bull. 36, 140–143. doi: 10.1248/bpb.b12-00798
Yang, F., Lim, G. P., Begum, A. N., Ubeda, O. J., Simmons, M. R., Ambegaokar, S. S., et al. (2005). Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 280, 5892–5901. doi: 10.1074/jbc.M404751200
Keywords: Alzheimer's disease, diagnostics, drugs, neurodegeneration, therapeutics
Citation: Jan AT, Azam M, Rahman S, Almigeiti AMS, Choi DH, Lee EJ, Haq QMR and Choi I (2017) Perspective Insights into Disease Progression, Diagnostics, and Therapeutic Approaches in Alzheimer's Disease: A Judicious Update. Front. Aging Neurosci. 9:356. doi: 10.3389/fnagi.2017.00356
Received: 19 June 2017; Accepted: 18 October 2017;
Published: 01 November 2017.
Edited by:
Ghulam Md Ashraf, King Abdulaziz University, Saudi ArabiaReviewed by:
Cláudia Fragão Pereira, University of Coimbra, PortugalVictoria Campos-Peña, Instituto Nacional de Neurología y Neurocirugía, Mexico
Copyright © 2017 Jan, Azam, Rahman, Almigeiti, Choi, Lee, Haq and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Inho Choi, aW5ob2Nob2lAeW51LmFjLmty
†Present Address: Arif Tasleem Jan, School of Biosciences and Biotechnology, Baba Ghulam Shah Badshah University, Rajouri, India
‡These authors have contributed equally to this work.