Expression of mitochondrial branched-chain aminotransferase and α-keto-acid dehydrogenase in rat brain: implications for neurotransmitter metabolism
- 1Department of Neurology, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
- 2Department Physiology and Pharmacology, Wake Forest University Health Sciences, Winston-Salem, NC, USA
- 3Human Nutrition, Foods and Exercise, Virginia Tech, Blacksburg, VA, USA
In the brain, metabolism of the essential branched chain amino acids (BCAAs) leucine, isoleucine, and valine, is regulated in part by protein synthesis requirements. Excess BCAAs are catabolized or excreted. The first step in BCAA catabolism is catalyzed by the branched chain aminotransferase (BCAT) isozymes, mitochondrial BCATm and cytosolic BCATc. A product of this reaction, glutamate, is the major excitatory neurotransmitter and precursor of the major inhibitory neurotransmitter γ-aminobutyric acid (GABA). The BCATs are thought to participate in a α-keto-acid nitrogen shuttle that provides nitrogen for synthesis of glutamate from α-ketoglutarate. The branched-chain α-keto acid dehydrogenase enzyme complex (BCKDC) catalyzes the second, irreversible step in BCAA metabolism, which is oxidative decarboxylation of the branched-chain α-keto acid (BCKA) products of the BCAT reaction. Maple Syrup Urine Disease (MSUD) results from genetic defects in BCKDC, which leads to accumulation of toxic levels of BCAAs and BCKAs that result in brain swelling. Immunolocalization of BCATm and BCKDC in rats revealed that BCATm is present in astrocytes in white matter and in neuropil, while BCKDC is expressed only in neurons. BCATm appears uniformly distributed in astrocyte cell bodies throughout the brain. The segregation of BCATm to astrocytes and BCKDC to neurons provides further support for the existence of a BCAA-dependent glial-neuronal nitrogen shuttle since the data show that BCKAs produced by glial BCATm must be exported to neurons. Additionally, the neuronal localization of BCKDC suggests that MSUD is a neuronal defect involving insufficient oxidation of BCKAs, with secondary effects extending beyond the neuron.
Introduction
The three nutritionally essential branched chain amino acids (BCAAs), leucine, isoleucine, and valine, contribute nitrogen to the glutamate/glutamine cycle (Hutson et al., 2005; Yudkoff et al., 2005). These amino acids provide approximately 30–50% of the nitrogen for de novo glutamate synthesis in astrocytes (Kanamori et al., 1998). In the central nervous system (CNS), glutamate that is formed from BCAA transamination is an excitatory neurotransmitter and substrate for synthesis of the major inhibitory neurotransmitter γ-aminobutyric acid (GABA). Current theories of the function of BCAAs in brain are consistent with involvement of the glutamatergic and/or GABAergic systems in the etiology of neurological disorders (Bixel and Hamprecht, 1995; Yudkoff et al., 1996a,b; Yudkoff, 1997; Hutson et al., 1998, 2001; Kanamori et al., 1998; Sakai et al., 2004).
The first step in the catabolism of the BCAAs is reversible transamination catalyzed by the branched chain aminotransferase (BCAT) isozymes. There are two known mammalian BCAT isozymes-cytosolic (BCATc) and mitochondrial (BCATm) (Ichihara, 1985). Both BCAT enzymes reversibly transfer the α-amino group of a BCAA to an amino group acceptor, generally α-ketoglutarate. The products of the BCAT reaction are glutamate and the respective branched chain α-keto acids (BCKAs), which are α-ketoisocaproate (KIC), α-keto-β-methylvalerate (KMV), and α-ketoisovalerate (KIV). BCATc is the predominant BCAT isozyme in the CNS (Ichihara, 1985; Hall et al., 1993; Sweatt et al., 2004a,b). Previously, this laboratory has demonstrated BCATc expression in select populations of glutamatergic and GABAergic neurons (Sweatt et al., 2004b; Garcia-Espinosa et al., 2007). There is evidence that BCATm does not co-localize with BCATc, rather it is enriched in astrocytes (Bixel et al., 1997, 2001; Hutson et al., 1998; LaNoue et al., 2001), however, the detailed distribution of BCATm in the CNS has not yet been determined. It has been proposed that the BCAT isozymes participate in a nitrogen cycle that drives de novo synthesis of neurotransmitter glutamate in astrocytes (Hutson et al., 1998, 2001; LaNoue et al., 2001), facilitates nitrogen transfer for neurotransmitter glutamate in neurons and acts as a buffer to maintain glutamate levels in neurons (Yudkoff et al., 1993).
The second and irreversible step in BCAA catabolism is catalyzed by the mitochondrial branched chain α-keto acid dehydrogenase (BCKDC) enzyme complex (Harris et al., 1990). BCKDC catalyzes oxidative decarboxylation of the BCKA products of the BCAT reaction, forming NADH and the respective branched-chain acyl CoA derivative of each BCAA. Maple Syrup Urine Disease (MSUD) is an autosomal recessive disorder of this second enzyme complex. In individuals with MSUD, the oxidation of BCAAs is inhibited and, therefore, intake of BCAAs above the daily requirement for protein synthesis causes accumulation of BCAAs and their BCKAs to toxic levels (Chuang and Shih, 2001). If left untreated, most patients experience seizures, changes in muscle tone, and coma due to brain swelling. Analysis of MSUD brains by magnetic resonance diffusion imaging spectroscopy suggests impaired brain energy metabolism. Classically, there is generalized edema in brain and spinal cord, with more intense swelling in the cerebellar deep white matter, as well as edema in other areas (Lewandowski and Johnston, 1990; Sener, 2002; Jan et al., 2003; Righini et al., 2003; Ha et al., 2004; Parmar et al., 2004). Neurological disorders frequently involve disruption of the proper balance of these excitatory (glutamate) and inhibitory (GABA) neurotransmitters, which result in altered excitability.
Recent studies have focused on the key role for BCAAs in maintaining the synaptic pools of these neurotransmitters, while examining the significance of BCAAs in post-traumatic pathophysiology following traumatic brain injury. In mice there was a significant reduction in the concentrations of BCAAs in the hippocampus after a brain injury, coupled with a regionally specific alteration in both the concentration and cellular distribution of BCKDC (Cole et al., 2010). Further, BCATc, but not BCATm expression, was also diminished after a brain injury. These combined changes were associated with significantly altered excitability in the hippocampus and ultimately, impaired cognition. Impressively, offering supplemental BCAAs in the drinking water completely restored hippocampal BCAA concentrations to normal, and restored hippocampal excitability and cognitive performance in brain-injured mice. Further, similar results have been shown in clinical trials with humans, where there has been success in treating severely brain injured patients by administering IV boluses of BCAAs (Aquilani et al., 2005, 2008).
In this study immunohistochemical techniques were utilized to determine the localization of BCKDC and BCATm in adult rat brain. Understanding the distribution of these key enzymes will have a major impact on the understanding, and possibly treatment, of both traumatic brain injury and MSUD.
Materials and Methods
Animals and Tissues
BCATm
Because the BCATm antibody exhibits non-specific staining with paraformaldehyde-fixed tissue, three adult male Sprague–Dawley rats (250–300 g) were deeply anesthetized with sodium pentobarbital (30 mg/kg) or ketamine/xylazine (90 and 10 mg/kg, respectively) then sacrificed. Freshly dissected brains were divided into three equal-sized blocks along the rostral-caudal axis, and cryo-embedded without fixation. Serial coronal sections (15 μM thickness) were collected on glass slides. Prior to immunolabeling, the sections were immersed in acetone (20 min) and lyophilized overnight. Cresyl violet (Nissl) staining of selected sections provided anatomical reference points, identified with the aid of a rat brain atlas (Paxinos and Watson, 1998).
BCKDC and BCATc
For localization of BCKDC, three anesthetized rats were sequentially perfused via the left ventricle with 150 mL phosphate buffered saline (PBS) and 300 mL of 0.1 M sodium phosphate buffer (pH 7.4) containing 4% paraformaldehyde. Brains were removed and placed in the paraformaldehyde fixative for 24 h. For frozen sections, the tissues were cryoprotected by sequential immersion (24 h each) in 10, 20, and 30% sucrose/PBS solutions, embedded in OCT compound (Sakura Finetek, Torrance, CA) and frozen on dry ice. Some fixed brains were not frozen, but were sectioned with a vibratome. Serial coronal sections (34 μM thickness) were obtained from six brains and stored as floating sections in PBS at 4°C or in antifreeze media (25% glycerol, 25% ethylene glycol in 50 mM phosphate buffer, pH 7.4) at −20°C. Experimental procedures involving animals conformed to NIH guidelines and were approved by the Institutional Animal Care and Use Committee of the Wake Forest University Health Sciences.
Antibodies
Polyclonal antibodies were raised in rabbits against either purified recombinant human BCATm or BCATc, as previously described and characterized in brain (Lieth et al., 2001). For BCATm and BCATc, purified recombinant human BCATm and purified N-terminal rat BCATc were used as antigens to raise antisera in rabbits. The BCATc peptide contained the first 62 residues in the BCATc sequence. For BCKDC, antibodies were raised in rabbits immunized with a keyhole limpet hemocyanin-conjugate of a peptide consisting of amino acids 32–50 of the E1α subunit of the enzyme complex. Anti-E1α-peptide 32–50 recognizes a band of ~45 kDa on immunoblots of protein from whole brain, and recognizes a band of similar mass on immunoblots of protein from purified rat liver mitochondria (data not shown). All antibodies were affinity-purified from IgG fractions of serum by passage over columns of sepharose-conjugated immunogen (purified BCATm, BCATc, or E1α-peptide), followed by elution with low pH buffer and dialysis against 50% glycerol/water (Lieth et al., 2001).
In order to determine the types of cells in which BCKDC-E1α is expressed in the brain, colocalization with antibodies directed against proteins characteristically expressed by various classes of cells was performed. These marker proteins included: glial fibrillary acidic protein (GFAP, an astrocyte marker, mouse monoclonal 610566 from Becton Dickinson, Franklin Lakes NJ, used at 2.5 μg/ml), S100 (an intermediate filament protein, expressed by astrocytes, mouse monoclonal S2532 from Sigma, used at 6.4 μg protein/ml); glutamic acid dehydrogenase (GAD67, marker for GABAergic neurons, mouse monoclonal MAB 5406 from Chemicon/Millipore, Temecula CA, used at 10 μg/ml), myelin basic protein (MBP, a marker for oligodendrocytes and white matter, goat polyclonal sc-13194 from Santa Cruz Biotechnology, Santa Cruz CA, used at 0.1 μg/ml). Appropriate secondary antibodies, conjugated to horseradish peroxidase or to fluorophores (rhodamine, FITC, or Cy3), were obtained from Jackson Immunoresearch Laboratories, West Grove PA, and used at 1–3 μg/ml. All products were “Multilabel”-antibodies that had been affinity-purified and immunoadsorbed to remove cross-reactive immunoglobulins.
Immunohistochemistry
For localization of BCATm, slide-mounted brain sections were rinsed with PBS, and then incubated for 60 min in PBS containing 5% goat serum and 0.5% Triton X-100 to block nonspecific IgG binding sites. The BCATm antibody (1:50) was applied for 2 h in a humidified chamber at 37°C. Sections were rinsed with PBS and then incubated for 60 min with goat anti-rabbit IgG conjugated to horseradish peroxidase. After rinsing with PBS, color was developed with diaminobenzidine HCl/H2O2 (maximum exposure-10 min). For the immunofluorescence study, goat serum was used as the blocking agent, and anti-BCATm and anti-GFAP as primary antibodies were used. These were followed by donkey anti-rabbit IgG-rhodamine and donkey anti-mouse IgG-FITC.
For localization of BCKDC-E1α, floating sections were rinsed and blocked as described above, using 5% donkey serum. Sections were incubated with rabbit anti-E1α-peptide (1:1000) either alone or with one of the mouse or goat antibodies directed against the marker proteins listed above. Incubations were carried out overnight at 4°C, followed by 1 h at 25°C. Sections were rinsed three times for 10 min each in PBS, and then incubated for 1 h with appropriate fluorophore-conjugated secondary antibodies. After PBS rinses, the sections were mounted on slides, and cover slips were applied using Vectashield hard-set mounting medium (Vector Laboratories Inc, Burlingame CA).
For localization of BCATc, floating sections were rinsed and blocked as described above, using 5% donkey serum. Sections were incubated with rabbit anti-BCATc (1:200). Incubations were carried out for 1 h at 25°C, then overnight at 4°C, followed by 1 h at 25°C. Sections were rinsed three times for 10 min each in PBS, and then incubated for 1 h with appropriate fluorophore-conjugated secondary antibodies. After PBS rinses, the sections were mounted on slides, and cover slips were applied using Vectashield hard-set mounting medium (Vector Laboratories Inc, Burlingame CA).
Peroxidase-labeled sections were viewed with a Zeiss Axioplan 2 microscope and images obtained via an AxioCam digital camera and AxioVision imaging software (Carl Zeiss USA, Thornwood, NY). Immunofluorescent images were acquired with a Zeiss laser scanning confocal microscope (LSM 510) with associated software. Images were converted and assembled using Adobe Photoshop 6.0 or CS2 (Adobe Systems Incorporated, San Jose CA).
Western Blots
Western blots were performed using standard laboratory techniques. Briefly, tissue samples were stored at −20°C in 2× Laemmli buffer with each vial prepared to contain an equal concentration of protein (2 mg/ml). After thawing and boiling the samples for 4 min, 20–40 μg of protein were loaded per lane on 4–12% Tris-glycine gradient gels (Invitrogen, Carlsbad, CA) and subsequently transferred to PVDF membranes (Invitrogen, Carlsbad, CA) at 24 V for 2 h at 4°C. After transfer, the membrane was incubated for 1 h at room temperature with PBS-Tween (PBS-T) containing 2.5% non-fat dairy milk. The membrane was then incubated overnight with BCKD antibody. Following this, the membrane was washed three times (5 min each) with PBS-T, and then incubated with anti-rabbit secondary antibodies (Cell Signaling, Danvers, MA) for 1 h. Enhanced chemiluminescence detection (Millipore, Billerica, MA) was used to detect the bands. The films were then scanned into a computer for generation of the Figure 1, which demonstrates the specificity of the antibody.
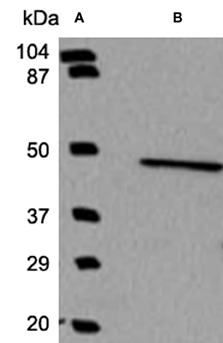
Figure 1. Western blot analysis demonstrates the specificity of the BCKD antibody. Using a molecular weight marker in lane A, a single band was observed in lane B indicating that the antibody generated to identify BCKD does recognize a band of the appropriate size and is specific to this protein and does not recognize other antigens.
Results
BCATm in the Cerebellum and Hippocampal Formation
In the cerebellum, immunolabeling for BCATm was most prominent in astrocytes of the white matter tracts and in the distal processes of the Bergmann glia of the cerbellar cortex (Figure 2). There was some labeling of astrocytes of the granule cell layer. Astrocytes in the deep cerebellar nuclei were also labeled for BCATm (data not shown). Purkinje cells and granule cells were not labeled. Co-localization of BCATm and GFAP (Figures 2C–E) in astrocytes of the cerebellar white matter confirmed the astrocytic expression of BCATm.
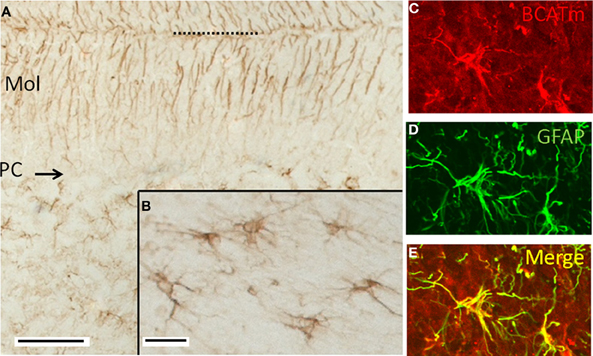
Figure 2. Immunolocalization of BCATm in the cerebellar cortex. (A) Peroxidase immunolabel for BCATm. Label is detected in the molecular (Mol) and granular layers (GL), but not in the Purkinje cell layer (PC). Dotted line indicates the pial surface. (B) Higher resolution image of a portion of the cerebellar white matter, showing label in cells that appear to be astrocytes. (C) Rhodamine-immunolabel of BCATm in the cerebellar white matter. (D) FITC-immunolabel of GFAP in the same section as in panel C. (E) Merged image of panels C and D, demonstrating label of BCATm in several astrocytes. Scale bars: (A) 100 μm, (B–E) 25 μm.
In the hippocampal formation, immunoreactivity for BCATm was restricted to astrocytes. Labeling for BCATm was seen in astrocytes in the molecular, granule cell, and polymorphic layers of the dentate gyrus (Figures 3A,B). Labeled astrocytes in the polymorphic layer typically had a stellate morphology, while those in the molecular layer usually had fewer and more elongated processes, the longest of which were generally oriented perpendicular to the granular layer (Figure 3C). In the granule cell layer, the most obvious labeling was of long processes of astrocytes at the inner margin (Figure 3B). Labeling of the smaller, stellate astrocytes extended throughout the stratum radiatum of the CA1 and CA2 hippocampal fields and into the fimbria of the hippocampus (data not shown). BCATm-labeled astrocytic processes often surrounded blood vessels, were concentrated along the hippocampal fissure and in the subpial region, and extended to the pial surface (data not shown).
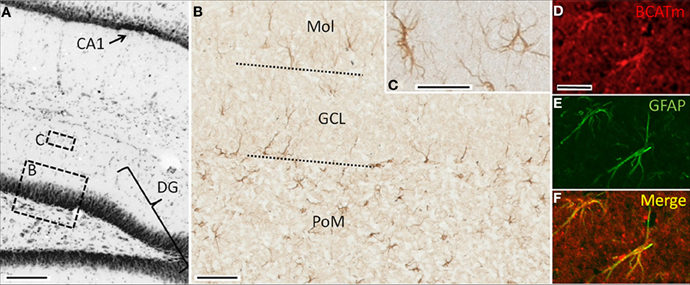
Figure 3. Immunolocalization of BCATm in the hippocampal formation. (A) Nissl-stained coronal section showing portions of the dentate gyrus (DG) and field CA1. Regions similar to those enclosed by boxes B and C are shown at higher resolution in panels B and C. (B) Peroxidase-visualized immunolabel of BCATm in the dentate gyrus. Approximate boundaries of the granule cell layer (GCL) are shown by the dotted lines. (C) Higher resolution image of BCATm-immunolabeled astrocytes of the molecular layer (MOL) of the dentate gyrus. (D) Rhodamine-immunofluorescent labeling for BCATm in the molecular layer of the dentate gyrus. (E) FITC-immunofluorescent labeling for GFAP in the same section as shown in panel D, showing GFAP in the proximal processes and cell bodies of two astrocytes. (F) Merged image of panels D and E demonstrating co-localization of BCATm and GFAP. Scale bars: (A) 200 μm; (B) 25 μm; (C) 10 μm; (D–F) 20 μm.
Double immunolabeling of the hippocampal region for BCATm and GFAP, an astrocyte specific protein, confirmed the astrocyte specificity of BCATm expression. Figures 3D,E show prominent immunoreactivity of BCATm and GFAP in cells of the polymorphic layer. Merging the two images (Figure 3F) shows the virtually complete co-localization of GFAP and BCATm, confirming that BCATm is expressed strictly in astroglia.
BCATm in the Spinal Cord
There was striking labeling for BCATm (Figure 4A) in radially oriented structures in the spinal cord white matter. The labeling pattern was coincident with that for GFAP (Figure 4B), indicating that the astrocytic processes were BCATm-positive (Figure 4C). Many, but not all, BCATm-labeled cell bodies in the white matter were clearly associated with GFAP-labeled processes (Figure 4D). However, this could be attributed to the intracellular distribution of BCATm and GFAP, with BCATm localizing in mitochondria in the cell bodies, while GFAP strongly labels astrocytic processes.
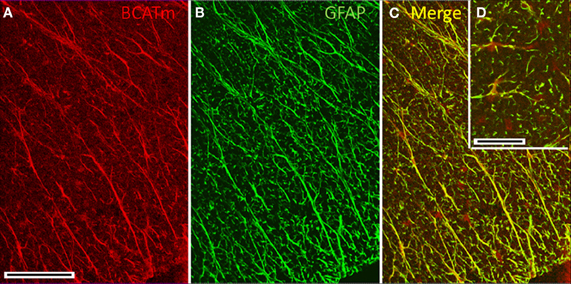
Figure 4. Immunolocalization of BCATm in the spinal cord. (A) Anti-BCATm strongly labels elongate, radially oriented elements in the white matter of the spinal cord. (B) Labeling for GFAP in the same section as shown in panel A shows that the radial elements are glial processes. (C) The merged image of panels A and B shows nearly complete colocalization of BCATm with GFAP. (D) (Inset) Cell bodies of glial cells are labeled only for BCATm. Scale bar: (A–C) 100 μm. (D) 20 μm.
BCKDC-E1α in the Cerebellum
Examination of the labeling for BCKDC-E1α in the cerebellar cortex revealed intense labeling of structures lying in the Purkinje cell layer, as well as light labeling of Purkinje cell bodies and some neurons of the molecular layer (Figure 5). The processes of astrocytes and Bergmann glia were not labeled for BCKDC-E1α. Close examination of the labeling pattern for BCKDC-E1α in the Purkinje cell layer revealed that the heavily labeled structures lie at the periphery of the Purkinje cells. The structures are clearly not labeled for GFAP, a marker for filamentous protein of astrocytes/Bergmann glia. In sections that were double immunolabeled for BCKDC-E1α and the GABAergic marker enzyme GAD67, Purkinje cell cytoplasm was clearly labeled for GAD, but the GAD label did not overlap with that of BCKDC-E1α (Figure 5 inset).
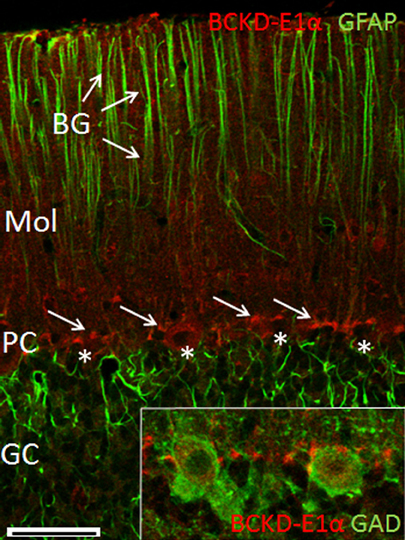
Figure 5. Immunolocalization of BCKDC-E1α in the cerebellar cortex. Co-localization of BCKDC-E1α (red) and GFAP (green) in the cerebellar cortex. Labeling for BCKDC is very prominent in structures located along the plane of Purkinje cell bodies (PC, cells marked by *; BCKDC label indicated by arrows). Label for BCKDC is also seen in some cells of the molecular layer (Mol). Immunolabel for GFAP is associated with the distal process of the Bergmann glial cells (BG, arrows) and with astrocytes in the granule cell layer (GC). (Inset) Merged image of co-labeling for BCKDC (red) and glutamate dehyrogenase (GAD), a marker for GABAergic cells. Purkinje cell bodies are clearly demarcated by anti-GAD. Strongest labeling for BCKDC is either at the far periphery of the Purkinje cells, in a distinct structure associated with surfaces of Purkinje cell bodies. Scale bars: (A) 50 μm. (B–E) 25 μm.
BCKDC-E1α immunoreactivity was also present in neurons of the deep cerebellar nuclei, where it contrasted sharply with the lack of label in the white matter (Figure 6). There was a similar paucity of label for BCKDC-E1α in the white matter of the cerebellar cortex (data not shown). This indicates that oligodendrocytes and white matter astrocytes in the cerebellum do not express BCKDC-E1α.
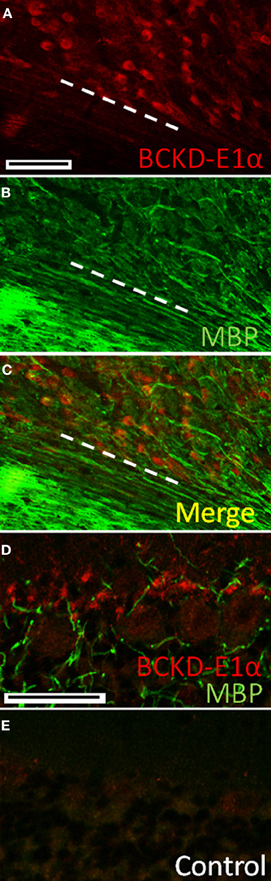
Figure 6. Immunolocalization of BCKDC in deep cerebellar nuclei and white matter. (A) Labeling for BCKDC is strong in the cell bodies of neurons of the deep cerebellar nuclei. Dashed white line marks the border between the nucleus and an unlabeled white matter tract. (B) Immunolabeling for myelin basic protein (MBP), a marker for myelin and oligodendrocytes, in the same section as in panel A. (C) Merged image of panels A and B. There is virtually no co-labeling of cells for BCKDC and MBP. (D) Co-label of the Purkinje cell layer for BCKDC and MBP. Structures labeled for BCKDC are not labeled for MBP, indicating that they are not oligodendrocytes. (E) Control section showing the Purkinje cell layer of a section incubated with secondary antibodies only. The image was obtained using the same microscope settings, and processed in the same way as the image in panel A. Scale bars: (A–C) 100 μm. (D,E) 50 μm.
BCKDC-E1α in the Hippocampal Formation
In the hippocampal formation, BCKDC-E1α immunoreactivity was generally of low-intensity, as shown in Figure 6. Labeling for BCKDC was restricted to cell bodies, most clearly to those of the granule cell layer of the dentate gyrus. Labeling for BCKDC-E1α in neurons in the hippocampus took the form of a ring of immunoreactivity surrounding an unlabeled nucleus (Figure 7A) and did not extend into the neuronal processes. In addition, co-labeling for GFAP showed that astrocytes in the hippocampus are not labeled for BCKDC-E1α (Figures 7B,C). In Figure 7, the pattern of expression of BCKDC-E1α can be contrasted with that for BCATc, which is shown for a similar section through the dentate gyrus (Figure 7D). BCATc is the neuronal isoform of the first enzyme in the BCAA catabolic pathway (Hall et al., 1993). BCATc is cytosolic, and is present mostly in the processes of neurons in the hippocampus and neocortex, as well as in the cerebellum (see Discussion).
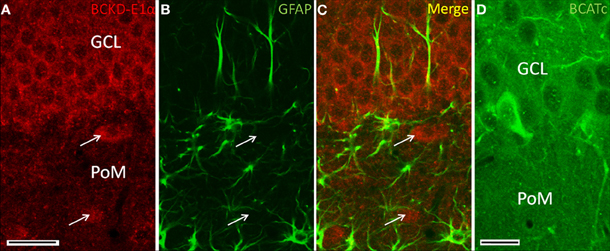
Figure 7. Immunolocalization of BCKDC in dentate gyrus. (A) BCKDC immunolabel is present in neurons of the granule cell layer (GCL). Arrows indicate specific labeling of large neurons of the polymorphic layer (PoM). (B) Immunolabel for the astrocyte marker GFAP in the same section as in panel A. Astrocyte processes are labeled in the granule cell polymorphic layers. Astrocyte cell bodies are also labeled in the polymorphic layer. (C) Merged image of panels A and B. There is no co-labeling of cells for BCKDC and GFAP. (D) Labeling of the dentate gyrus for BCATc, one of the enzymes that generates branched-chain α-keto-acids that are the substrates for BCKDC. As shown previously (Sweatt et al., 2004a), BCATc is present only in cells at the inner margin of the granule cell layer. Scale bars: (A–C) 25 μm. (D) 20 μm.
BCKDC-E1α in the Cerebral Cortex
In the cerebral cortex, the intensity of BCKDC-E1α is in clearly labeled neuronal bodies (Figure 8). In the pyramidal layers, large cell bodies are more typically immunoreactive for BCKDC-E1α, while some processes occasionally expressed BCKDC-E1α, as well. GFAP most strongly labeled astrocytes near the apical surfaces of the cortex. These labeling patterns are consistent with that observed in other regions of the brain.
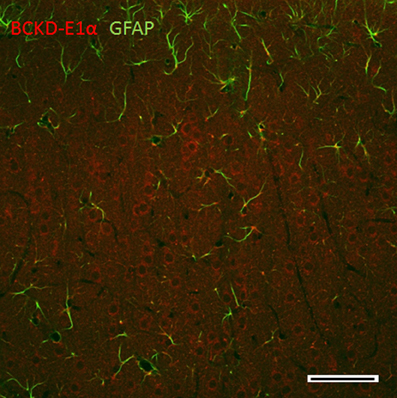
Figure 8. Immunolocalization of BCKDC in the cerebral cortex. BCKDC is clearly expressed in the perinuclear regions of neurons in the pyramidal cell bodies. GFAP immunoreactivity is observed in astrocytic processes primarily in the apical surface. Scale bars: 100 μm.
BCKDC-E1α in the Hypothalamus and other Brain Regions
Immunoreactivity for BCKDC in the hypothalamus continued the pattern of low-intensity labeling of neuronal cell bodies seen in other brain regions. The salient features are illustrated by Figure 9, in which neuronal cell bodies in the arcuate nucleus are consistently labeled. Small S100-positive cells astrocytes (Figure 9B) appear to be labeling astrocytes and tanycytes along the ventricle surface. The subependymal region, like the subpial regions elsewhere, is characterized by S100-positive processes, with few BCKDC-E1α-positive cell bodies. At the ventricular interface, the ependymal cells show strong labeling for BCKDC-E1α at their ciliary surfaces. This is especially evident in Figure 9C. At higher resolution, labeling of neurons in the arcuate nucleus for BCKDC-E1α can be compared with the distribution of glial elements (astrocytes and/or tanycytes) that form the blood-brain barrier in the median eminence and are immunoreactive for S100 (Figures 9D–F). The glial cells are not labeled for BCKDC-E1α. Elsewhere in the brain (e.g., in the thalamus and brain stem), labeling for BCKDC-E1α is similar to that shown in Figure 9 for the hypothalamus—neuronal cell bodies are generally immunoreactive (data not shown). In addition, glial cells were not labeled for BCKDC-E1α. As shown for the cerebellum (Figure 5), there was no overlap of BCKDC-E1α immunoreactivity with MBP-labeling of white matter tracts.
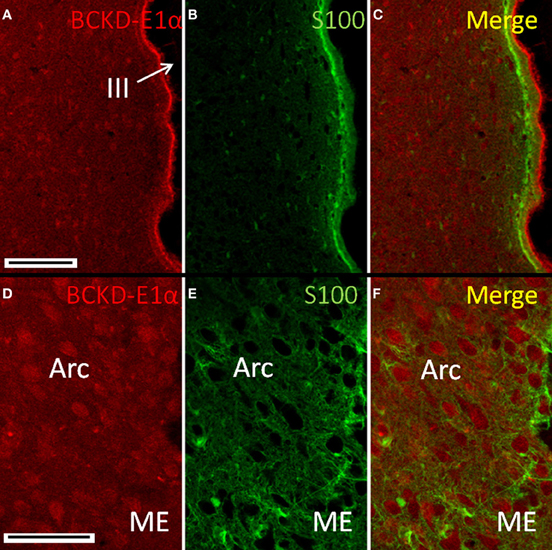
Figure 9. Immunolocalization of BCKDC in the hypothalamus. (A) Labeling for BCKDC is present in cells of hypothalamic nuclei, as well as in ependymal cells lining the third ventricle (III). (B) Immunolabel for S100 in the same section as in panel A. (C) Merged image of panels A and B. BCKDC-positive cells are not co-labeled for S100. Labeling for BCKDC in the ependymal cells clearly lies above a layer of cells that express S100. (D) Higher resolution image of the arcuate nucleus (Arc), near the transition to the median eminence (ME). (E) Immunolabel for S100 in the same section as in panel D. (F) Merged image of panels D and E. Neurons of the arcuate nucleus are labeled for BCKDC, with no overlap of labeling for S100. S100 labeling appears in occasional cells of the nucleus, as well as in processes of cells lying in the transition zone to the median eminence. Scale bars: (A–C) 100 μm. (D–F) 50 μm.
BCKDC-E1α in the Spinal Cord
Cell bodies in both the white matter and gray matter of the spinal cord were labeled for BCKDC-E1α (Figure 10). In the white matter, neuronal cell bodies that were positive for BCKDC-E1α were often in close proximity to S100-positive elements (astrocytes). Since much of the S100 labeling was located on structures that appeared to be blood vessels, the S100 was probably located in perivascular astrocytes.
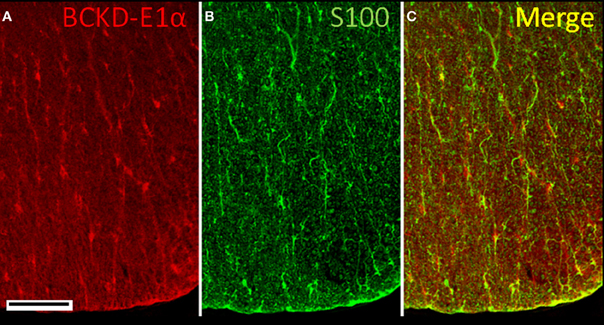
Figure 10. Immunolocalization of BCKDC in the spinal cord. (A) Labeling for BCKDC in the white matter is found on cell bodies and fine, radially oriented processes. (B) Labeling for an astrocyte marker, S100, is present over some cell bodies, but is mostly associated with serpentine structures that resemble blood vessels. (C) The merged image of panels A and B shows that BCKDC-positive cell bodies are often associated with S100-positive processes. Scale bars: (A–C) 50 μm.
Discussion
The key findings of this study are the apparent restriction of BCATm expression to astrocytes, Bergmann glial cells and spinal cord radial glia, while BCKDC-E1α is expressed by neurons and ependymal cells. Most remarkable, in light of the white matter edema that occurs in MSUD, is that BCKDC-E1α, which is defective in MSUD, is not present in white matter tracts.
BCATm
For BCATm, the astrocytes in which it is heavily expressed are most often located in the neuropil and myelinated tracts, and not in gray matter. Labeling for BCATm in these astrocytes extends into the cell processes, indicating a wide intracellular distribution of the enzyme in this cell type. The intracellular pattern of BCATm expression in astrocytes follows the pattern of labeling for cytoskeletal elements (GFAP), though in spinal cord, cell bodies are also clearly labeled. This pattern may have resulted from the need to use fresh-frozen tissue to demonstrate BCATm in brain tissues. Chemical fixation would be expected to preserve mitochondria, which would exhibit a punctate labeling pattern for BCATm.
These results support the hypothesis that BCATm contributes nitrogen to the glutamate-glutamine cycle via the astrocyte. Uptake of BCAAs into the brain provides an important source of nitrogen, with BCATc and BCATm mediating the transfer of α-amino nitrogen from BCAA to glutamate which can then donate nitrogen to other brain amino acids. Net nitrogen transfer from BCAAs occurs when the BCAA carbon skeleton is lost due to oxidation (Hutson et al., 2005). Further, the rate of BCAA oxidation is greater than the rate of incorporation of BCAA into protein (Cooper and Plum, 1987). Although the distribution of these enzymes in humans has not been reported, human brain contains 20% of total body capacity to oxidize BCAAs (Suryawan et al., 1998). Current paradigms suggest that following the vesicular release of the neurotransmitter glutamate, astrocytes take up a significant portion of the glutamate to prevent excitotoxicity. The astrocyte enzyme, glutamine synthetase, converts glutamate to glutamine using free ammonia. The glutamine is then shuttled back to the neuron where it can be re-converted to glutamate and either re-used as a neurotransmitter or oxidized (Berl and Clarke, 1983). This system is not 100% efficient because glutamate is oxidized to pyruvate, therefore, a combination of de novo synthesis and vascular delivery of and/or glutamine is required to maintain glutamate homeostasis (Gamberino et al., 1997; LaNoue et al., 2007). A key finding of this study, that BCATm is astrocyte specific, reveals that this BCAT isoform is in a key position to remove α-ketoglutarate from the TCA cycle to form glutamate. Furthermore, the localization of BCKDC suggests that BCKAs produced from the BCATm reaction are oxidized outside astrocytes, which is consistent with the BCAA cycle hypothesis (Yudkoff et al., 1996b; Hutson et al., 1998).
Studies of astrocyte cultures reveal a high capacity for leucine uptake, with BCAAs being accumulated against a concentration gradient. Transfer of BCAA nitrogen into glutamate and then into glutamine occurs in primary cultures of rat astroglia (Yudkoff et al., 1983, 1994, 1996b; Hutson et al., 1998; McKenna et al., 1998). Studies of cultured neonatal cortical astrocytes support the role of BCAAs in de novo glutamate synthesis (Gamberino et al., 1997; Waagepetersen et al., 2001).
BCKDC E1α
Labeling of BCKDC E1α in neurons has a pattern consistent with mitochondrial localization, i.e., punctate labeling of the perinuclear cytoplasm. Interesting exceptions are the preferential labeling of neuronal processes in the cerebral cortex and the labeling of radially oriented glial processes in the spinal cord. The punctuate pattern of labeling in the cerebral cortical neurons suggests localization of the enzyme in mitochondria in large dendrites. In the cerebellum, the strong labeling for BCKDC-E1α in the Purkinje cell layer could represent a mitochondria-rich region either in the proximal portion of one of the large dendrites of the Purkinje cells or in structures associated with the Purkinje cell surface. These could include elements of other neurons and/or glial cells, such as Bergmann glia, which lie close to the labeled structure. Labeling for BCKDC-E1α is present in a variety of neuronal types: glutamatergic (e.g., hippocampal granule cells), GABAergic (e.g., Purkinje cells), and cholinergic (motor neurons). In addition, labeling of neurons in the arcuate nucleus indicates expression of BCKDC-E1α in neurons that produce neuropeptides that are involved in food intake regulation. Injection of leucine into the third ventricle has been reported to result in inhibition of food intake (Cota et al., 2006; Blouet et al., 2009).
The complete absence of BCKDC in astrocytes indicates BCAA oxidation is neuronal and suggests that as in the periphery, there is translocation of BCAA and BCKA between cells and separation of transamination and oxidation except in neurons that contain both BCATc and BCKDC. Depending on the specific neuron, the location of BCATc and BCKDC indicates that either reamination/transamination or oxidative decarboxylation of BCKAs occurs in neurons. The intracellular separation of the two enzymes, BCATc appearing primarily in processes and pre-synaptic varicosities while BCKDC occurs primarily in cell bodies, suggests a potential spatially regulated mechanism for determining the ultimate fate of BCKAs.
Disruptions in BCKDC result in accumulation of both BCKAs and BCAAs in neurons and astrocytes. The disease is lethal if untreated and the block at BCKDC results in accumulation of both BCAAs and BCKAs to neurologically toxic levels (Jan et al., 2003). This is accompanied by systemic metabolic decompensation. Magnetic resonance imaging, computerized tomography scans and autopsies have all demonstrated generalized brain edema, with more intense swelling in the cerebellar deep white matter with severe status spongiosus, as well as edema in the posterior brainstem, the posterior limbs of the internal capsule and the posterior aspect of the centrum semiovale (Riviello et al., 1991; Sener, 2002; Righini et al., 2003; Ha et al., 2004; Parmar et al., 2004). The affected brain regions are most notably the white matter tracts, which show almost no labeling for BCKDC. Flooding the brain with BCAAs and BCKAs may have direct effects on the white matter, especially since white matter lacks the BCKDC that might detoxify by catabolizing BCKAs. Reamination of the BCKAs by the neuronal BCATc isozyme could deplete neuronal glutamate and upset the balance of excitotoxic and inhibitory neurotransmission in areas where BCATc is expressed. The α-keto acid of leucine, KIC, is thought to be the toxic metabolite (Chuang and Shih, 2001), which is supported by the lack of MSUD symptoms found in the BCATm knockout mouse (She et al., 2007). Alternatively, the high levels of BCAAs and BCKAs in MSUD may have indirect effects on pathways that regulate the balance of fluid, ions, and small molecules in myelinated tracts. The implications of these findings for disorders of the BCAA metabolizing system are several. First, it appears that coordinated action of BCATc, BCATm, and most importantly, BCKDC in maintaining BCAA/BCKA homeostasis occurs in cell layers and neuropil, and not in the white matter tracts that undergo edema in MSUD. The only enzyme of the system that is present in white matter is BCATc, which is found in tract axons (Garcia-Espinosa et al., 2007). In MSUD, the deficit in BCKDC activity would be most severe in (or even restricted to) gray matter, among neurons and astroglia or their derivatives. Effects on white matter could result from direct action of BCAA/BCKAs on tract components or from indirect effects of the BCAA/BCKA on pathways originating in the cell body and that control white matter homeostasis. That the white matter edema of MSUD is reversible (Jan et al., 2003) might indicate a direct effect. In particular, since BCATc is distributed evenly in axons in white matter, it is tempting to speculate that this enzyme has a regulatory function (metabolic/structural) that may involve binding and or catalysis of BCAA/BCKA, and that BCATc normally operates without need of local BCKDC-E1α activity. This role may be disrupted in MSUD when the BCATc enzyme is overwhelmed by globally high levels of BCAA/BCKA.
BCAAs may also be important to the injured brain where decreased expression of BCATc and BCKD has been observed, while BCATm was not affected. Disruption of these enzymes could have a profound impact on the glutamate/glutamine cycle, which may account for some of the pathophysiology following traumatic brain injury. One putative mechanism of injury is known as “glutamate excitotoxicity” in which dysregulated release of glutamate from pre-synaptic excitatory neurons floods the synaptic cleft. The astrocyte then clears the excess glutamate. Normally, some of this glutamate would be recycled, however, the decreased expression of BCATc and BCKD suggests that this may not be occurring properly. Indeed, this may result in depleted neurotransmitter pools accounting for regionally specific hypoexcitability after a brain injury.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Jeffrey T. Cole and Andrew J. Sweatt conducted the experiments, collected the data and prepared the images. Jeffrey T. Cole, Andrew J. Sweatt, and Susan M. Hutson designed the experiments, interpreted the results, and prepared the manuscript. This work was supported by NIH NS-38641 (Susan M. Hutson). Grant sponsor: National Institutes of Health/National Institute of Neurological Disorders and Stroke; Grant Number NS 38641 (Susan M. Hutson).
Disclaimer
The opinions expressed herein belong solely to the authors. They do not nor should they be interpreted as representative of or endorsed by the Uniformed Services University of the Health Sciences, US Army, US Navy, Department of Defense, or any other agency of the federal government.
References
Aquilani, R., Boselli, M., Boschi, F., Viglio, S., Iadarola, P., Dossena, M., Pastoris, O., and Verri, M. (2008). Branched-chain amino acids may improve recovery from a vegetative or minimally conscious state in patients with traumatic brain injury: a pilot study. Arch. Phys. Med. Rehabil. 89, 1642–1647.
Aquilani, R., Iadarola, P., Contardi, A., Boselli, M., Verri, M., Pastoris, O., Boschi, F., Arcidiaco, P., and Viglio, S. (2005). Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch. Phys. Med. Rehabil. 86, 1729–1735.
Berl, S., and Clarke, D. D. (1983). “The metabolic compartmentation concept,” in Glutamine, Glutamate and GABA in the Central Nervous System, eds L. Hertz, E. Kvamme, E. G. McGeer, and A. Schousboe (New York, NY: Alan R. Liss, Inc.), 205–217.
Bixel, M., Shimomura, Y., Hutson, S., and Hamprecht, B. (2001). Distribution of key enzymes of branched-chain amino acid metabolism in glial and neuronal cells in culture. J. Histochem. Cytochem. 49, 407–418.
Bixel, M. G., and Hamprecht, B. (1995). Generation of ketone bodies from leucine by cultured astroglial cells. J. Neurochem. 65, 2450–2461.
Bixel, M. G., Hutson, S. M., and Hamprecht, B. (1997). Cellular distribution of branched-chain amino acid aminotransferase isoenzymes among rat brain glial cells in culture. J. Histochem. Cytochem. 45, 685–694.
Blouet, C., Jo, Y. H., Li, X., and Schwartz, G. J. (2009). Mediobasal hypothalamic leucine sensing regulates food intake through activation of a hypothalamus-brainstem circuit. J. Neurosci. 29, 8302–8311.
Chuang, D. I., and Shih, V. E. (2001). “Disorders of branched chain amino acid and keto acid metabolism,” in The Metabolic Basis of Inherited Disease, eds C. R. Shriver, A. L. Beaudet, S. Sly, and D. Vaile (New York, NY: McGraw Hill), 1239–1277.
Cole, J. T., Mitala, C. M., Kundu, S., Verma, A., Elkind, J. A., Nissim, I., and Cohen, A. S. (2010). Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc. Natl. Acad. Sci. U.S.A. 107, 366–371.
Cooper, A. J., and Plum, F. (1987). Biochemistry and physiology of brain ammonia. Physiol. Rev. 67, 440–519.
Cota, D., Proulx, K., Smith, K. A., Kozma, S. C., Thomas, G., Woods, S. C., and Seeley, R. J. (2006). Hypothalamic mTOR signaling regulates food intake. Science 312, 927–930.
Gamberino, W. C., Berkich, D. A., Lynch, C. J., Xu, B., and LaNoue, K. F. (1997). Role of pyruvate carboxylase in facilitation of synthesis of glutamate and glutamine in cultured astrocytes. J. Neurochem. 69, 2312–2325.
Garcia-Espinosa, M. A., Wallin, R., Hutson, S. M., and Sweatt, A. J. (2007). Widespread neuronal expression of branched-chain aminotransferase in the CNS: implications for leucine/glutamate metabolism and for signaling by amino acids. J. Neurochem. 100, 1458–1468.
Ha, J. S., Kim, T. K., Eun, B. L., Lee, H. S., Lee, K. Y., Seol, H. Y., and Cha, S. H. (2004). Maple syrup urine disease encephalopathy: a follow-up study in the acute stage using diffusion-weighted MRI. Pediatr. Radiol. 34, 163–166.
Hall, T. R., Wallin, R., Reinhart, G. D., and Hutson, S. M. (1993). Branched chain aminotransferase isoenzymes. Purification and characterization of the rat brain isoenzyme. J. Biol. Chem. 268, 3092–3098.
Harris, R. A., Zhang, B., Goodwin, G. W., Kuntz, M. J., Shimomura, Y., Rougraff, P., Dexter, P., Zhao, Y., Gibson, R., and Crabb, D. W. (1990). Regulation of branched-chain α-ketoacid dehydrogenase and elucidation of a molecular basis for maple syrup urine disease. Adv. Enzyme Regul. 30, 245–263.
Hutson, S. M., Berkich, D., Drown, P., Xu, B., Aschner, M., and LaNoue, K. F. (1998). Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J. Neurochem. 71, 863–874.
Hutson, S. M., Lieth, E., and LaNoue, K. F. (2001). Function of leucine in excitatory neurotransmitter metabolism in the central nervous system. J. Nutr. 131, 846S–850S.
Hutson, S. M., Sweatt, A. J., and Lanoue, K. F. (2005). Branched-chain amino acid metabolism: implications for establishing safe intakes. J. Nutr. 135, 1557S–1564S.
Ichihara, A. (1985). “Aminotransferases of branched-chain amino acids,” in Transaminases, eds P. Christen and D. E. Metzler (New York, NY: John Wiley and Sons), 430–438.
Jan, W., Zimmerman, R. A., Wang, Z. J., Berry, G. T., Kaplan, P. B., and Kaye, E. M. (2003). MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology 45, 393–399.
Kanamori, K., Ross, B. D., and Kondrat, R. W. (1998). Rate of glutamate synthesis from leucine in rat brain measured in vivo by 15N NMR. J. Neurochem. 70, 1304–1315.
LaNoue, K. F., Berkich, D. A., Conway, M., Barber, A. J., Hu, L. Y., Taylor, C., and Hutson, S. (2001). Role of specific aminotransferases in de novo glutamate synthesis and redox shuttling in the retina. J. Neurosci. Res. 66, 914–922.
LaNoue, K. F., Carson, V., Berkich, D. A., and Hutson, S. M. (2007). “Mitochondiral/cytosolic interactions via metabolite shuttles and transporters,” in Handbook of Neurochemistry and Molecular Neurobiology, eds G. Dienel and G. Gibson (New York, NY: Springer), 589–616.
Lewandowski, E. D., and Johnston, D. L. (1990). Reduced substrate oxidation in postischemic myocardium: 13C and 31P NMR analyses. Am. J. Physiol. Heart Circ. Physiol. 258, H1357–H1365.
Lieth, E., LaNoue, K. F., Berkich, D. A., Xu, B., Ratz, M., Taylor, C., and Hutson, S. M. (2001). Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J. Neurochem. 76, 1712–1723.
McKenna, M. C., Tildon, J. T., Stevenson, J. H., Hopkins, I. B., Huang, X., and Couto, R. (1998). Lactate transport by cortical synaptosomes from adult rat brain: characterization of kinetics and inhibitor specificity. Dev. Neurosci. 20, 300–309.
Parmar, H., Sitoh, Y. Y., and Ho, L. (2004). Maple syrup urine disease: diffusion-weighted and diffusion-tensor magnetic resonance imaging findings. J. Comput. Assist. Tomogr. 28, 93–97.
Paxinos, G., and Watson, C. (1998). The Rat Brain in Stereotaxic Coordinates, 4th Edn. San Diego, CA: Academic Press.
Righini, A., Ramenghi, L. A., Parini, R., Triulzi, F., and Mosca, F. (2003). Water apparent diffusion coefficient and T2 changes in the acute stage of maple syrup urine disease: evidence of intramyelinic and vasogenic-interstitial edema. J. Neuroimaging 13, 162–165.
Riviello, J. J. Jr. Rezvani, I., DiGeorge, A. M., and Foley, C. M. (1991). Cerebral edema causing death in children with maple syrup urine disease. J. Pediatr. 119, 42–45.
Sakai, R., Cohen, D. M., Henry, J. F., Burrin, D. G., and Reeds, P. J. (2004). Leucine-nitrogen metabolism in the brain of conscious rats: its role as a nitrogen carrier in glutamate synthesis in glial and neuronal metabolic compartments. J. Neurochem. 88, 612–622.
Sener, R. N. (2002). Diffusion magnetic resonance imaging in intermediate form of maple syrup urine disease. J. Neuroimaging 12, 368–370.
She, P., Reid, T. M., Bronson, S. K., Vary, T. C., Hajnal, A., Lynch, C. J., and Hutson, S. M. (2007). Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 6, 181–194.
Suryawan, A., Hawes, J. W., Harris, R. A., Shimomura, Y., Jenkins, A. E., and Hutson, S. M. (1998). A molecular model of human branched-chain amino acid metabolism. Am. J. Clin. Nutr. 68, 72–81.
Sweatt, A., Wood, M., Suryawan, A., Wallin, R., Willingham, M. C., and Hutson, S. M. (2004a). Branched-chain amino acid catabolism: unique segregation of pathway enzymes in organ systems and peripheral nerves. Am. J. Physiol. 286, E64–E76.
Sweatt, A. J., Garcia-Espinosa, M. A., Wallin, R., and Hutson, S. M. (2004b). Branched-chain amino acids and neurotransmitter metabolism: expression of cytosolic branched-chain aminotransferase (BCATc) in the cerebellum and hippocampus. J. Comp. Neurol. 477, 360–370.
Waagepetersen, H. S., Sonnewald, U., Larsson, O. M., and Schousboe, A. (2001). Multiple compartments with different metabolic characteristics are involved in biosynthesis of intracellular and released glutamine and citrate in astrocytes. Glia 35, 246–252.
Yudkoff, M., Daikhin, Y., Nelson, D., Nissim, I., and Erecinska, M. (1996a). Neuronal metabolism of branched-chain amino acids: flux through the aminotransferase pathway in synaptosomes. J. Neurochem. 66, 2136–2145.
Yudkoff, M., Daikhin, Y., Grunstein, L., Nissim, I., Stern, J., and Pleasure, D. (1996b). Astrocyte leucine metabolism: significance of branched-chain amino acid transamination. J. Neurochem. 66, 378–385.
Yudkoff, M., Daikhin, Y., Lin, Z.-P., Nissim, I., Stern, J., and Pleasure, D. (1994). Interrelationships of leucine and glutamate metabolism in cultured astrocytes. J. Neurochem. 62, 1192–1202.
Yudkoff, M., Daikhin, Y., Nissim, I., Horyn, O., Luhovyy, B., Lazarow, A., and Nissim, I. (2005). Brain amino acid requirements and toxicity: the example of leucine. J. Nutr. 135(Suppl. 6), 1531S–1538S.
Yudkoff, M., Nissim, I., Daikhin, Y., Lin, Z. P., Nelson, D., Pleasure, D., and Erecinska, M. (1993). Brain glutamate metabolism: neuronal-astroglial relationships. Dev. Neurosci. 15, 343–350.
Keywords: glutamate, gamma-amino butyric acid, MSUD, branched chain aminotransferase, traumatic brain injury, branched chain amino acid
Citation: Cole JT, Sweatt AJ and Hutson SM (2012) Expression of mitochondrial branched-chain aminotransferase and α-keto-acid dehydrogenase in rat brain: implications for neurotransmitter metabolism. Front. Neuroanat. 6:18. doi: 10.3389/fnana.2012.00018
Received: 01 March 2012; Paper pending published: 30 March 2012;
Accepted: 10 May 2012; Published online: 28 May 2012.
Edited by:
Alfonso Fairén, University Miguel Hernandez, SpainReviewed by:
Fiorenzo Conti, Universita Politecnica delle Marche, ItalyJianzheng Zheng, University of New Mexico, USA
Copyright: © 2012 Cole, Sweatt and Hutson. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jeffrey T. Cole, Department of Neurology, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, B-3012, USUHS, Bethesda, MD 20814, USA. e-mail: jeffrey.cole@usuhs.edu