Identification of five novel SCN1A variants
 Baitao Zeng1,2† Haoyi Zhang3† Qing Lu1,2 Qingzi Fu1,2 Yang Yan1,2 Wan Lu1,2 Pengpeng Ma1,2 Chuanxin Feng1,2 Jiawei Qin1,2 Laipeng Luo1,2
Baitao Zeng1,2† Haoyi Zhang3† Qing Lu1,2 Qingzi Fu1,2 Yang Yan1,2 Wan Lu1,2 Pengpeng Ma1,2 Chuanxin Feng1,2 Jiawei Qin1,2 Laipeng Luo1,2  Bicheng Yang1,2*
Bicheng Yang1,2*  Yongyi Zou1,2* Yanqiu Liu1,2*
Yongyi Zou1,2* Yanqiu Liu1,2*- 1Department of Medical Genetics, Jiangxi Maternal and Child Health Hospital, Nanchang, China
- 2Jiangxi Provincial Key Laboratory of Birth Defect for Prevention and Control, Jiangxi Maternal and Child Health Hospital, Nanchang, China
- 3School of Public Health, Nanchang University, Nanchang, China
Background: Epilepsy is characterized by recurrent unprovoked seizures. Mutations in the voltage-gated sodium channel alpha subunit 1 (SCN1A) gene are the main monogenic cause of epilepsy. Type and location of variants make a huge difference in the severity of SCN1A disorder, ranging from the mild phenotype (genetic epilepsy with febrile seizures plus, GEFS+) to the severe phenotype (developmental and epileptic encephalopathies, DEEs). Dravet Syndrome (DS) is an infantile-onset DEE, characterized by drug-resistant epilepsy and temperature sensitivity or febrile seizures. Genetic test results reveal SCN1A variants are positive in 80% DS patients and DS is mainly caused by de novo variants.
Methods: Trio-whole exome sequencing (WES) was used to detect variants which were associated with clinical phenotype of five probands with epilepsy or twitching. Then, Sanger sequencing was performed to validate the five novel SCN1A variants and segregation analysis. After analyzing the location of five SCN1A variants, the pathogenic potential was assessed.
Results: In this study, we identified five novel SCN1A variants (c.4224G > C, c.3744_3752del, c.209del, c.5727_5734delTTTAAAACinsCTTAAAAAG and c.5776delT) as the causative variants. In the five novel SCN1A variants, four were de novo and the remaining one was inherited. All novel variants would be classified as “pathogenic” or “likely pathogenic.”
Conclusion: The five novel SCN1A variants will enrich the SCN1A mutations database and provide the corresponding reference data for the further genetic counseling.
1. Introduction
The voltage-gated sodium channel alpha subunit 1 (SCN1A) gene encodes the α-subunit of a voltage-gated ion channel (NaV1.1), contributing to the initiation and propagation of action potentials and the regulation of neuronal excitability (Plumereau et al., 2022). As an archetypal channelopathy, the neuronal overexcitement leads to epilepsy characterized by recurrent unprovoked epileptic seizures (Scheffer and Nabbout, 2019; Plumereau et al., 2022). The phenotypic spectrum of epilepsy varies substantially across the patients harboring pathogenic variants in SCN1A gene (Zayat et al., 2022). The mild SCN1A disorder is genetic epilepsy with febrile seizures plus (GEFS+) (Fang et al., 2022). The severe form is developmental and epileptic encephalopathies (DEEs), including epilepsy with myoclonic atonic seizures (MAE), epilepsy of infancy with migrating focal seizures (EIMFS), and Dravet Syndrome (DS) (Scheffer and Nabbout, 2019; He et al., 2022). Phenotypic heterogeneity is commonly found in GEFS+ families (Grinton et al., 2022, p. 800; Türkyılmaz et al., 2022). GEFS+ patients tend to have self-limited and drug-reactive epilepsies (Scheffer and Nabbout, 2019) but normal cognitive development (Myers et al., 2018). DS is an infantile-onset DEE (Uchino et al., 2023), often accompanied by a fever or afebrile at initial seizure onset, subsequent motor and cognitive dysfunction, and intellectual disability in adults (Chilcott et al., 2022). Interestingly, DS patients harboring SCN1A variants 90% of which are de novo and only 5% of which are inherited account for 80% of cases (Kimura et al., 2005; Guerrini et al., 2010; Guerrini, 2012; Hirose et al., 2013).
The diagnosis of SCN1A disorders mainly depends on the clinical assessment and confirmation of diagnosis is based on genetic testing (Gil-Nagel et al., 2023). Moreover, using genetic testing to shorten the individual diagnosis time maybe benefit to select the antiseizure medications and improve the long-term quality of life in patients (Makiello et al., 2011; Wolff et al., 2019; Gil-Nagel et al., 2023; Matricardi et al., 2023). As a high-throughput and fast technique for genetic testing, Whole Exome Sequencing (WES) has been widely applied to identify SCN1A variants (Zhang et al., 2020; Gowda et al., 2023). To date, the Human Gene Mutation Database Gene Locus-Specific Database (HGMD) contains 2,584 SCN1A variants in total. Among these variants, truncations and missense changes account for the vast majority proportion (Zhou et al., 2021). Some research showed that truncations causing loss of function of Nav1.1 were associated with severe epilepsy, whereas missense variants often gave rise to mild phenotypes, which indicated the genotype–phenotype correlation (Zhou et al., 2021; Chen et al., 2022). However, the genotype–phenotype correlation in epilepsy patients with SCN1A variants is not definite enough (Chen et al., 2022).
Herein, taking advantage of Trio-WES, we detected five novel SCN1A variants associated with clinical phenotype in five probands with epilepsy or twitching, respectively. Then, the genetic source of five novel SCN1A variants were confirmed by the Sanger sequencing analysis. Simultaneously we analyzed their pathogenic potentials and locations. Following this, we performed prenatal diagnosis in pregnant women of family 1–3.
2. Materials and methods
2.1. Participants
Family members of five pedigrees were enrolled from the Jiangxi Maternal and Child health Hospital, Nanchang, China. General information and clinical manifestation were recorded, including gender, age, genetic relationship, and renal pathological phenomena. Peripheral blood was collected from all participants after signing written informed consent. The Clinical Research Ethics Committees of Jiangxi Maternal and Child health Hospital approved this study.
2.2. Trio-WES
Trio-WES of the proband and parents was used to detect variants which were associated with clinical phenotype. Genomic DNA was isolated from peripheral blood samples using a QIAamp DNA Mini Kit (Qiagen) and then was fragmented randomly by the ultrasonication (Covaris S220 Ultrasonicator). WES libraries were constructed, and the exons and adjacent splicing sites were amplified and sequenced on the high-throughput sequencing platforms (MGISEQ-2000, BGI) according to the manufacturer’s instructions. After generating the raw sequencing data from the sequencing platform, the sequencing adapters and low-quality sequences were trimmed. Taking advantage of BAW, all sequenced fragments were aligned and mapped to UCSC GRCh37/hg19 human reference genome and then processed for removing duplications and base quality score recalibration. Variant calling of single-nucleotide polymorphism (SNP) and indel (insertion or deletion) was carried out by GATK HaplotypeCaller. These variants were annotated and stratified for analysis using sunburst genetic analysis and interpretation platform.1 The SCN1A transcript of this study was NM_001165963.1.
Mutation nomenclature was based on the HGVS guidelines. These variants were filtered with multiple databases, such as population databases (dbSNP, 1,000 Genome, ExAC), disease databases (OMIM, HGMD, Clinvar) and biological information prediction tools (SIFT, Polyphen2, and Mutation Taster). The variants most significantly correlated with the clinical phenotypes were screened. Priority should be given to the variants which was found only in patients and do not exist in normal persons. The causative variants were classified according to the ACMG/ACG guidelines (Richards et al., 2015).
2.3. Identification of genotypes in five pedigrees
To validate the five novel variants and segregation analysis, adjacent regions variants in the SCN1A gene were amplified using the forward primer and the reverse primer designed by Primer-BLAST. Five specific primer pairs for detecting novel variants were listed in Table 1 and the PCR was performed through 2x Taq PCR Master MixII (KT211, TIANGEN). Amplification was carried out at 95°C for 5 min for initial denaturing, then 35 cycles at 95°C for 30 s, at 63°C (−0.5°C/cycles) for 30 s and at 72°C for 45 s, followed by a final extension of 8 min at 72°C in a T100 Thermal Cycler for the Classroom (BIO-RAD). The amplification products were sequenced by a sequencing provider (Tsingke, Changsha). The sequencing results alignment was completed by SeqMan Pro.

Table 1. Primers used to amplify the SCN1A gene.
3. Results
3.1. Clinical characteristics and genetic analysis
In this study, five families were recruited (Table 2). Epilepsy was found in all probands whose first seizure occurred before the age of 1 year, and incidence were often accompanied by psychomotor retardation or fever (Table 2). Through the treatment of antiseizure medications, the frequency and symptoms of epilepsy were gradually relieved (Table 2). In family 1, the proband II-1, male, was born with psychomotor retardation and epilepsy, while his parents were normal without clinical symptoms (Figure 1A; Table 2). In family 2, the female 6-month-old infant (proband II-1) was diagnosed as DS by clinic and had no family history (Figure 1B; Table 2). In family 3, the normal-appearing couple had an epileptic girl who suffered twitching once every half a month when she was born (Figure 1C; Table 2). In family 4, the normal-appearing couple had a recurrent epileptic (Figure 1D, Table 2). In family 5, clinical manifestations of the proband II-1 were twitching at the time of febrile illness (Figure 1E; Table 2). Her father was asymptomatic, while her mother had experienced the same symptoms in childhood, not now (Figure 1E; Table 2).
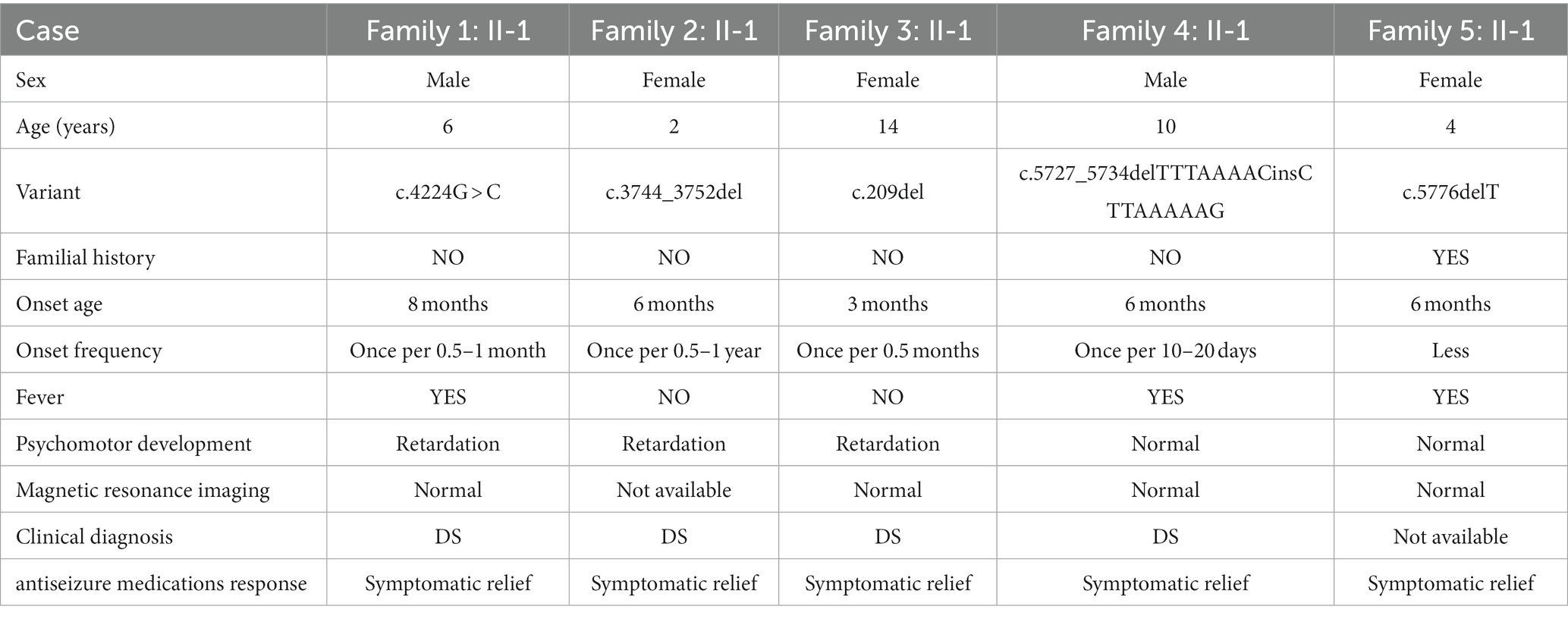
Table 2. Clinical phenotypes of five probands.
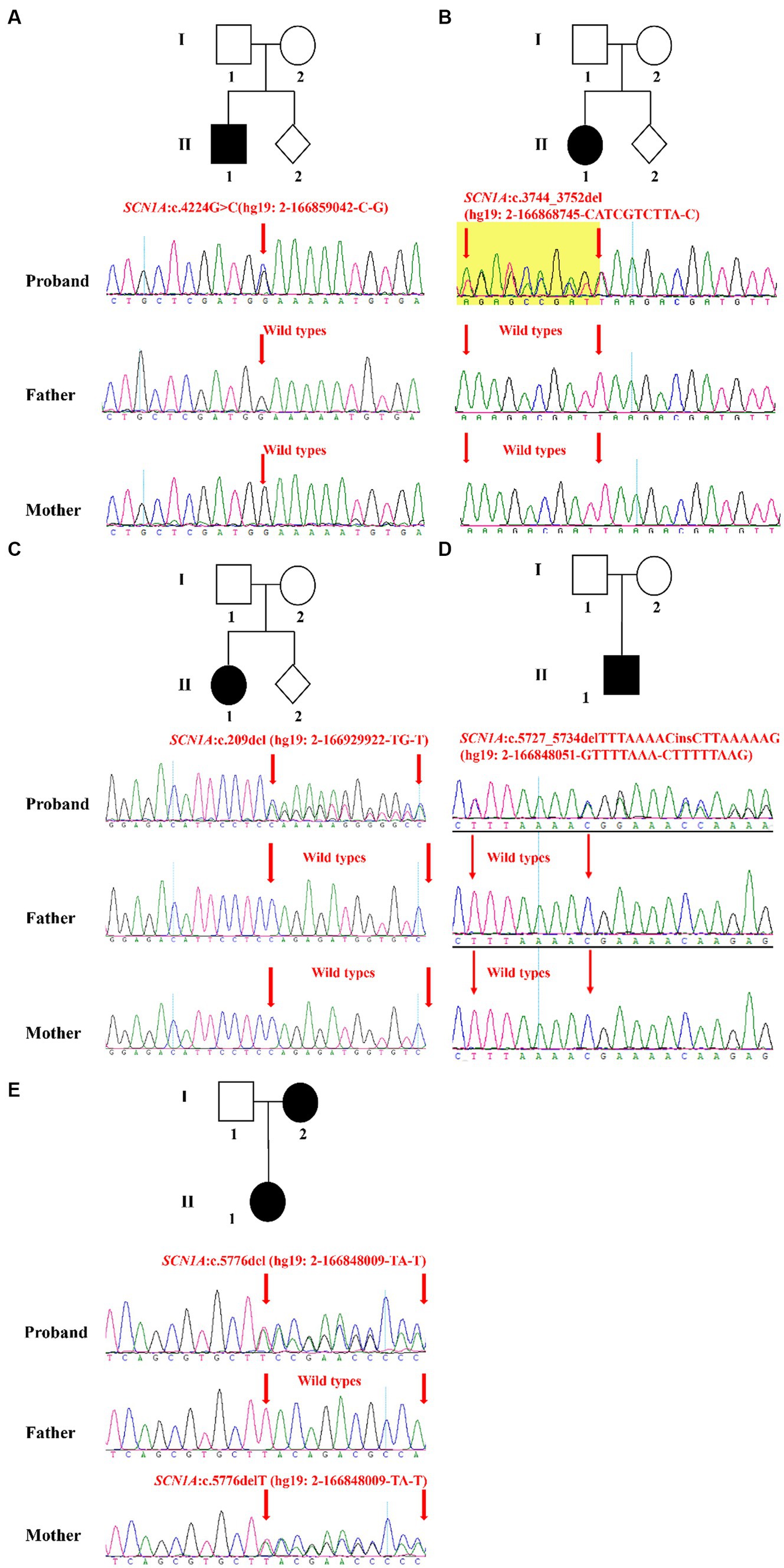
Figure 1. Validation and segregation analysis of SCN1A variants in five families. Upper: Pedigrees of family 1 (A), family 2 (B), family 3 (C), family 4 (D), and family 5 (E) with SCN1A variants; Lower: sequence chromatogram of the c.4224G > C (A), c.3744_3752del (B), c.209del (C), c.5727_5734delTTTAAAACinsCTTAAAAAG (D) and c.5776delT (E) mutations on the SCN1A gene in the proband and his/her family members.
The result of Trio-WES confirmed maternity and paternity, and revealed that all patients harbored SCN1A heterozygous mutations. Subsequently, the Sanger sequencing analysis was performed to further validate these variants. In family 1, the c.4224G > C variant in the SCN1A gene existed in the proband and was absent in his healthy father and mother, which can explain the symptom of epilepsy (Figure 1A). In family 2, the proband carried one variant in the SCN1A gene (c.3744_3752del) associated with twitching, whereas it was not detected in parents (Figure 1B). In family 3 and 4, a definite diagnosis of epilepsy was obtained in the affected individual (I-1) by finding the c.209del variant and c.5727_5734delTTTAAAACinsCTTAAAAAG variant in the SCN1A gene, respectively (Figures 1C,D). In family 5, the detected variant in the SCN1A gene (c.5776delT) of the proband inherited from her mother (Figure 1E). Moreover, the proband and her mother were sick with the similar symptom.
3.2. Analysis of variants
Five identified variants were not recorded in HGMD and Clinvar databases in this study (Table 3). In addition, these novel variants had never been reported in previous research, two of which were located in the C-terminal domain (c.5727_5734delTTTAAAACinsCTTAAAAAG/p. Gln1914Thrfs*31, c.5776delT/p. Tyr1926Thrfs*6), one in the pore loop connecting segment 5 (S5) and segment 6 (S6) of domains 3 (D3) (c.4224G > C/p.Trp1408Cys), one in D3S1- S2(c.3744_3752del/p. Ile1248_Thr1250del), and one in N-terminal domain (c.209del/p. Pro70Glnfs*22) (Figure 2). All novel variants were not present in the population frequency databases (Table 3). After bioinformatics-based prediction, the structure or function of protein may be disrupted by these variants. Following the ACMG guideline (Richards et al., 2015), all novel variants would be classified as “pathogenic” or “likely pathogenic” (Table 3).
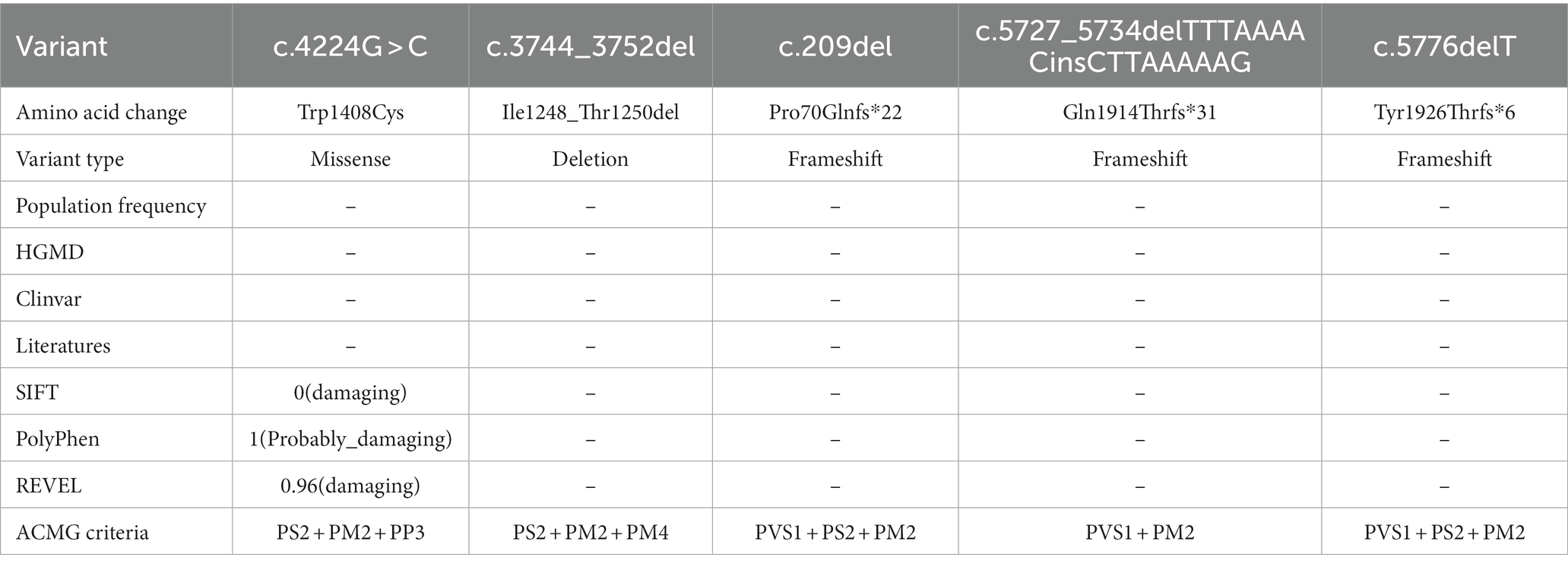
Table 3. Novel mutations identified in five families.
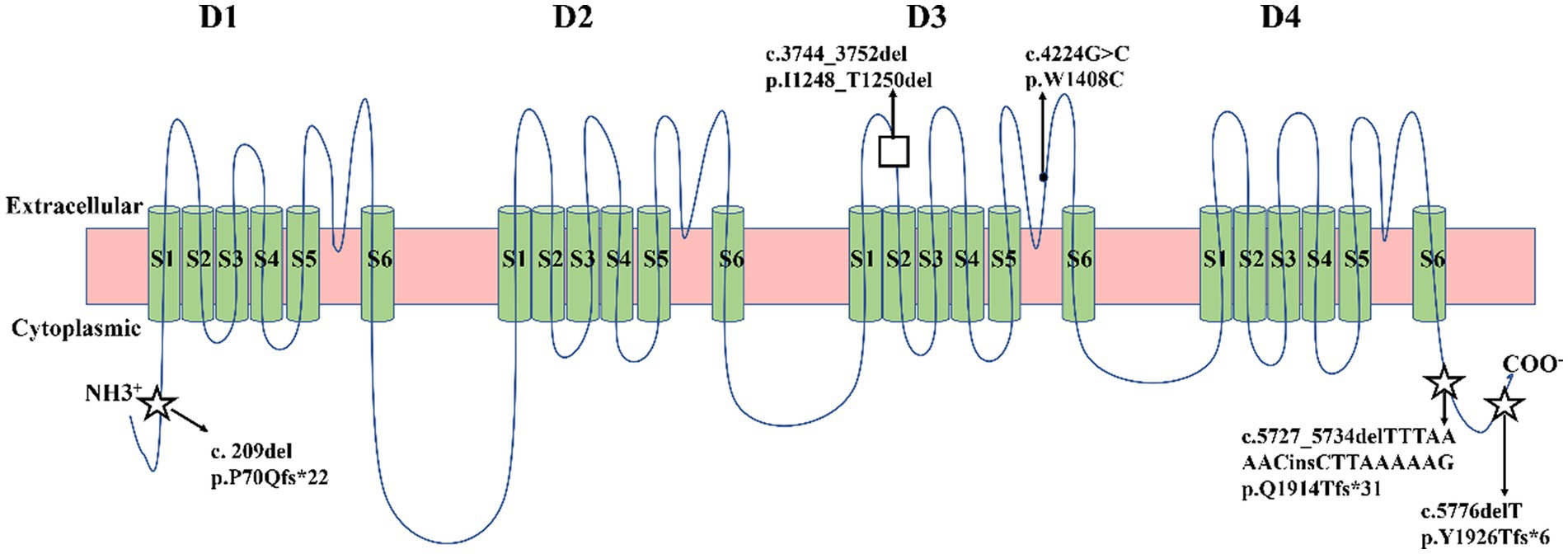
Figure 2. Schematic location of five variants in the transmembrane topology of a voltage-gated sodium channel encoded by SCN1A gene. Five green circles represented α-helical transmembrane segments (S1-S6), which can combine with a pore loop to form a homologous domain (D1-D4). Squares represented missense variants. Black dots represented in-frame deletion variants. Five-pointed stars represented frameshift variants.
4. Discussion
On clinical, genetic testing should be carried out in an infant presenting with generalized convulsions status epilepticus or recurrent febrile seizure. The most common monogenic cause of epilepsy is SCN1A variants (Brunklaus et al., 2022). Genetic test results reveal SCN1A variants were positive in 80% DS patients and 10% GEFS+ patients (Scheffer and Berkovic, 2000). Therefore, SCN1A genetic testing is mainly used for confirming the clinical diagnosis of DS and should be discouraged in GEFS+ patients (Hirose et al., 2013). In this study, five patients were born with epilepsy, twitching or febrile illness. Suspecting DS, we performed genetic testing and detected five novel SCN1A variants, respectively.
In the five variants, four were de novo and the remaining one was inherited. Previous studies reported that de novo accounted for 90% of SCN1A variants in DS patients, only 5% inherited variants (Kimura et al., 2005; Guerrini et al., 2010; Guerrini, 2012; Hirose et al., 2013). Milder GEFS+ phenotypes were observed in the family members harboring inherited variants (Hirose et al., 2013). Consistent with that reported in previous studies, the majority of detected variants were de novo in this study and the mother with inherited variants of family 5 experienced relatively mild symptoms in childhood which have disappeared now. In addition, the phenotypes varies substantially across the patients having de novo pathogenic variants in SCN1A gene, which is related to type and location of variants (de Lange et al., 2018).
Functional studies have shown missense mutations in the ion-pore and voltage-sensor regions, resulting in a lack of sodium current, caused serious clinical symptoms (Zuberi et al., 2011). Here, the patient of family 1 had psychomotor retardation and epilepsy, which were serious clinical symptoms caused by the c.4224G > C variant in the pore loop. This novel missense mutation was located at the 1408th amino acid residue where a deleterious nonsense mutation (c.4223G > A) had been reported before (Fujiwara et al., 2003, p. 1). The DS patient with c.4223G > A showed similar symptoms such as severe mental decline and childhood epilepsies (Fujiwara et al., 2003, p. 1). Here, the novel in-frame deletion (c.3744_3752del) variant in D3S1- S2 was detected in a DS patient. A recorded in-frame deletion variant (c.3740_3751del) was included in this mutation region (Wang et al., 2022). The c.3740_3751del variant was de novo and resulted in DS, according to SCN1A mutations database (Wang et al., 2022). Comparing with frameshift mutants causing premature truncation, in-frame deletion mutants lead to the loss of one or more amino acids of proteins and preserve function. However, electrophysiological analysis indicated that the SCN1A in-frame deletions will also exhibit complete loss-of-function (Wang et al., 2022). A newly discovered frameshift variant (c.209del) was in N-terminal domain. The clinical phenotype of this variant was the worst in five novel SCN1A variants. Surprisingly, in this study the patients harbored c.5727_5734delTTTAAAACinsCTTAAAAAG or c.5776del mutants in the C-terminal domain, suffered from the different severities of epilepsy and febrile convulsions. Frameshift mutants behind the two mutants (c.5741_5742delAA and c.5788delC) were both de novo and associated with DS (Harkin et al., 2007; Zuberi et al., 2011). Though the pathogenicity mechanism was haploinsufficiency for Nav1.1 (Harkin et al., 2007; Zuberi et al., 2011), the phenotypes of C-terminal variants were different, which increased the difficulty for genotype–phenotype prediction.
Here, the mothers of family 1–3 were both pregnant again. They expressed a desire to undergo prenatal diagnosis for causative variants detection (c.4224G > C, c.3744_3752del and c.209del) in fetuses to prompt the birth risk of infants with the similar symptom of probands. The sanger sequencing results showed the likely pathogenic variant (c.4224G > C and c.3744_3752del) were not found in the fetus of family 1 and 2 (Figures 3A–C). In addition, no pathogenic variant (c.209del) was detected in the fetus of family 3 (Figure 3C). Finally, three couples were both willing to continue with the pregnancy and the three infants displayed normal phenotype.
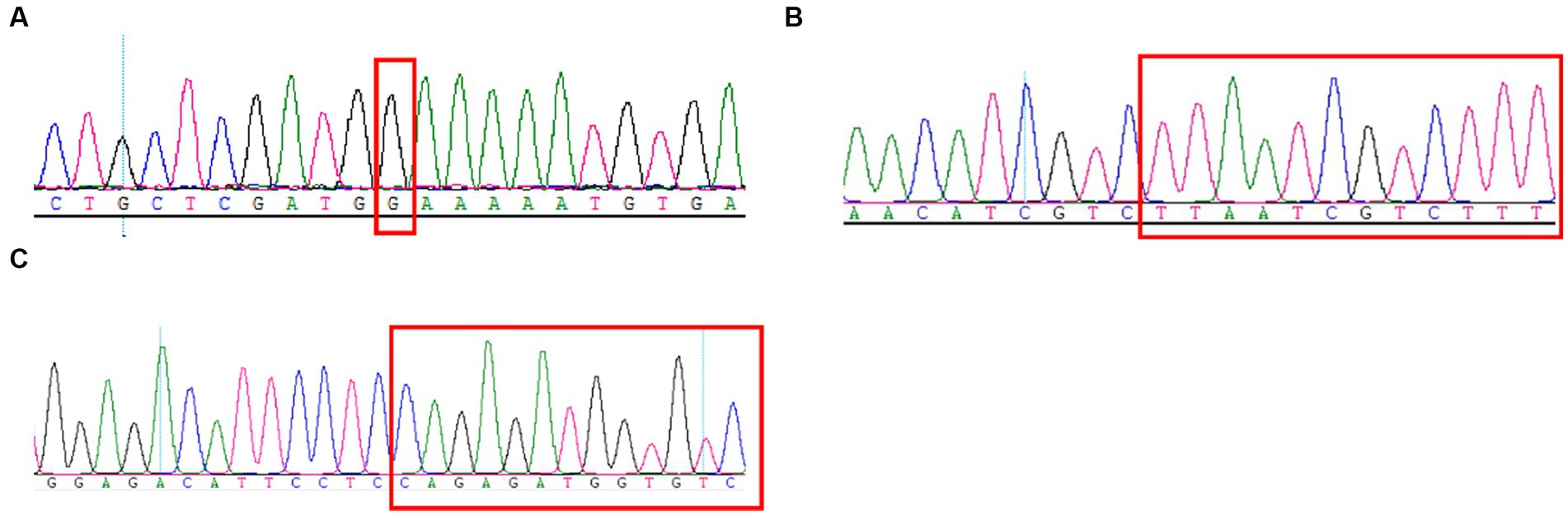
Figure 3. Sanger sequencing results of the c.4224G > C (A), c.3744_3752del (B), and c.209del (C) mutations on the SCN1A gene in the fetuses of family 1–3.
More often than not, de novo mutations occurring either in post zygotically or a single gamete are a one-off event and the risk of de novo mutations recurrence is 1% ~ 2% (Alison et al., 2023). Meanwhile, de novo mutations are constantly produced in both somatic and germ cells to form gonadal mosaicism during growth and development, which can significantly increase the recurrence risk (Acuna-Hidalgo et al., 2016). Depienne et al. observed that de novo mutations in the SCN1A gene remained a small possibility with associated recurrence risk (Depienne et al., 2006). A follow-up study showed that the percentage of mutant cells in the gonadal mosaicism was positively related to the severity of the phenotype (Depienne et al., 2010). Xu et al. found that 8.6% parents of DS children were SCN1A gene mutation mosaicism (Xu et al., 2015), which create a higher risk for family reproduction. Thus, we recommended that prenatal diagnosis was made for pregnant women of family 1–3. Sanger sequencing can detect some parental mosaicism (Xu et al., 2015), but a degree of misdetection rate still exists. Provided that a personalized risk assessment and full disclosure of all potential risk is essential for prenatal diagnosis.
In conclusion, we performed gene diagnosis for five families with epilepsy or twitching by WES-trio, respectively. As a result, five novel variants in SCN1A gene were identified, confirmed, and analyzed. These variants will enrich the SCN1A mutations database and provide the corresponding reference data for the further genetic counseling or genotype–phenotype correlations.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The studies involving humans were approved by the Clinical Research Ethics Committees of Jiangxi Maternal and Child health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
BZ: Data curation, Formal analysis, Writing – original draft. HZ: Data curation, Formal analysis, Writing – original draft. QL: Investigation, Writing – original draft. QF: Investigation, Writing – original draft. YY: Investigation, Writing – original draft. WL: Investigation, Writing – original draft. PM: Investigation, Writing – original draft. CF: Investigation, Writing – original draft. JQ: Investigation, Writing – original draft. LL: Investigation, Writing – original draft. BY: Project administration, Supervision, Writing – review & editing. YZ: Funding acquisition, Project administration, Writing – review & editing. YL: Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Key Research and Development Program of Jiangxi Province (Grant No. 20202BBG73015 to YY), Youth Science Foundation of Jiangxi Province (Grant No. 20192BAB215010 to WL), Provincial Health Commission Program of Jiangxi (Grant No. SKJP202211156 to YY), National Natural Science Foundation of China (Grant No. 82160318 to YZ), Natural Science Foundation of Jiangxi Province (Grant No. 20224BAB206037 to YZ), Jiangxi Province Key Research and Development Project (Grant No. 20232BBG70023 to YZ) and the Jiangxi Provincial Key Laboratory of Birth Defect for Prevention and Control (No. 20202BCD42017 to YL).
Acknowledgments
We would like to thank the five families for participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Acuna-Hidalgo, R., Veltman, J. A., and Hoischen, A. (2016). New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 17, 1–19. doi: 10.1186/s13059-016-1110-1
Alison, C. K., Jonathan, W., Nina, H., and Anne, G. (2023). Providing recurrence risk counselling for parents after diagnosis of a serious geneticn condition caused by an apparently de novo mutation in their child: a qualitative investigation of the PREGCARE strategy with UK clinical genetics practitioners. J. Med. Genet. doi: 10.1136/jmg-2023-109183
Brunklaus, A., Pérez-Palma, E., Ghanty, I., Xinge, J., Brilstra, E., Ceulemans, B., et al. (2022). Development and validation of a prediction model for early diagnosis of SCN1A-related epilepsies. Neurology 98, e1163–e1174. doi: 10.1212/WNL.0000000000200028
Chen, C., Fang, F., Wang, X., Lv, J., Wang, X., and Jin, H. (2022). Phenotypic and genotypic characteristics of SCN1A associated seizure diseases. Front. Mol. Neurosci. 15:821012. doi: 10.3389/fnmol.2022.821012
Chilcott, E., Díaz, J. A., Bertram, C., Berti, M., and Karda, R. (2022). Genetic therapeutic advancements for Dravet syndrome. Epilepsy Behav. 132:108741. doi: 10.1016/j.yebeh.2022.108741
de Lange, I. M., Koudijs, M. J., Van’t Slot, R., Gunning, B., Sonsma, A. C. M., van Gemert, L. J. J. M., et al. (2018). Mosaicism of de novo pathogenic SCN1A variants in epilepsy is a frequent phenomenon that correlates with variable phenotypes. Epilepsia 59, 690–703. doi: 10.1111/epi.14021
Depienne, C., Arzimanoglou, A., Trouillard, O., Fedirko, E., Baulac, S., Saint-Martin, C., et al. (2006). Parental mosaicism can cause recurrent transmission of SCN1A mutations associated with severe myoclonic epilepsy of infancy. Hum. Mutat. 27:389. doi: 10.1002/humu.9419
Depienne, C., Trouillard, O., Gourfinkel-An, I., Saint-Martin, C., Bouteiller, D., Graber, D., et al. (2010). Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J. Med. Genet. 47, 404–410. doi: 10.1136/jmg.2009.074328
Fang, Z., Xie, L., Li, X., Gui, J., Yang, X., Han, Z., et al. (2022). Severe epilepsy phenotype with SCN1A missense variants located outside the sodium channel core region: relationship between functional results and clinical phenotype. Seizure 101, 109–116. doi: 10.1016/j.seizure.2022.07.018
Fujiwara, T., Sugawara, T., Mazaki-Miyazaki, E., Takahashi, Y., Fukushima, K., Watanabe, M., et al. (2003). Mutations of sodium channel α subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic–clonic seizures. Brain 126, 531–546. doi: 10.1093/brain/awg053
Gil-Nagel, A., Sánchez-Carpintero, R., and Villanueva, V. (2023). Patient profile, management, and quality of life associated with Dravet syndrome: a cross-sectional, multicentre study of 80 patients in Spain. Sci. Rep. 13:3355. doi: 10.1038/s41598-023-30273-z
Gowda, V. K., Battina, M., Vegda, H., Srinivasan, V. M., Chikara, S. K., Mishra, A., et al. (2023). Cohort of phenotype, genotype, and outcome of SCN developmental and epileptic encephalopathies from southern part of India. J. Pediatr. Genet. 12:32. doi: 10.1055/s-0041-1731020
Grinton, B. E., Robertson, E., Fearnley, L. G., Scheffer, I. E., Marson, A. G., O’Brien, T. J., et al. (2022). A founder event causing a dominant childhood epilepsy survives 800 years through weak selective pressure. Am. J. Hum. Genet. 109, 2080–2087. doi: 10.1016/j.ajhg.2022.10.004
Guerrini, R. (2012). Dravet syndrome: the main issues. Eur. J. Paediatr. Neurol. 16, S1–S4. doi: 10.1016/j.ejpn.2012.04.006
Guerrini, R., Cellini, E., Mei, D., Metitieri, T., Petrelli, C., Pucatti, D., et al. (2010). Variable epilepsy phenotypes associated with a familial intragenic deletion of the SCN1A gene. Epilepsia 51, 2474–2477. doi: 10.1111/j.1528-1167.2010.02790.x
Harkin, L. A., McMahon, J. M., Iona, X., Dibbens, L., Pelekanos, J. T., Zuberi, S. M., et al. (2007). The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 130, 843–852. doi: 10.1093/brain/awm002
He, Z., Li, Y., Zhao, X., and Li, B. (2022). Dravet syndrome: advances in etiology, clinical presentation, and treatment. Epilepsy Res. 188:107041. doi: 10.1016/j.eplepsyres.2022.107041
Hirose, S., Scheffer, I. E., Marini, C., De Jonghe, P., Andermann, E., Goldman, A. M., et al. (2013). SCN1A testing for epilepsy: application in clinical practice. Epilepsia 54, 946–952. doi: 10.1111/epi.12168
Kimura, K., Sugawara, T., Mazaki-Miyazaki, E., Hoshino, K., Nomura, Y., Tateno, A., et al. (2005). A missense mutation in SCN1A in brothers with severe myoclonic epilepsy in infancy (SMEI) inherited from a father with febrile seizures. Brain Dev. 27, 424–430. doi: 10.1016/j.braindev.2004.11.005
Makiello, P., Feng, T., Dunwoody, B., Steckler, F., Symonds, J., Zuberi, S. M., et al. (2011). Comorbidities and predictors of health-related quality of life in Dravet syndrome: a 10-year, prospective follow-up study. Epilepsia. doi: 10.1111/epi.17531
Matricardi, S., Cestèle, S., Trivisano, M., Kassabian, B., Leroudier, N., Vittorini, R., et al. (2023). Gain of function SCN1A disease-causing variants: expanding the phenotypic spectrum and functional studies guiding the choice of effective antiseizure medication. Epilepsia 64, 1331–1347. doi: 10.1111/epi.17509
Myers, K. A., Scheffer, I. E., and Berkovic, S. F., ILAE Genetics Commission (2018). Genetic literacy series: genetic epilepsy with febrile seizures plus. Epileptic Disord. 20, 232–238. doi: 10.1684/epd.2018.0985
Plumereau, Q., Ebdalla, A., Poulin, H., Appendino, J. P., Scantlebury, M. H., Au, P. Y. B., et al. (2022). De novo Y1460C missense variant in NaV1.1 impedes the pore region and results in epileptic encephalopathy. Sci. Rep. 12:17182. doi: 10.1038/s41598-022-22208-x
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Scheffer, I. E., and Berkovic, S. F. (2000). Genetics of the epilepsies. Curr. Opin. Pediatr. 12, 536–542. doi: 10.1097/00008480-200012000-00004
Scheffer, I. E., and Nabbout, R. (2019). SCN1A-related phenotypes: epilepsy and beyond. Epilepsia 60, S17–S24. doi: 10.1111/epi.16386
Türkyılmaz, A., Tekin, E., Yaralı, O., and Çebi, A. H. (2022). Genetic landscape of SCN1A variants in a Turkish cohort with GEFS+ Spectrum and Dravet syndrome. Mol. Syndromol. 13, 270–281. doi: 10.1159/000521330
Uchino, K., Tanaka, Y., Ikezawa, W., Deshimaru, M., Kubota, K., Watanabe, T., et al. (2023). Astrocyte Ca2+ signaling is facilitated in Scn1a+/− mouse model of Dravet syndrome. Biochem. Biophys. Res. Commun. 643, 169–174. doi: 10.1016/j.bbrc.2022.12.084
Wang, J.-Y., Tang, B., Sheng, W.-X., Hua, L.-D., Zeng, Y., Fan, C.-X., et al. (2022). Clinical and functional features of epilepsy-associated in-frame deletion variants in SCN1A. Front. Mol. Neurosci. 15:828846. doi: 10.3389/fnmol.2022.828846
Wolff, M., Brunklaus, A., and Zuberi, S. M. (2019). Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia 60, S59–S67. doi: 10.1111/epi.14935
Xu, X., Yang, X., Wu, Q., Liu, A., Yang, X., Ye, A. Y., et al. (2015). Amplicon resequencing identified parental mosaicism for approximately 10% of “de novo” SCN1A mutations in children with Dravet syndrome. Hum. Mutat. 36, 861–872. doi: 10.1002/humu.22819
Zayat, V., Szlendak, R., and Hoffman-Zacharska, D. (2022). Concise review: stem cell models of SCN1A-related encephalopathies-current perspective and future therapies. Cells 11:3119. doi: 10.3390/cells11193119
Zhang, L., Gao, J., Liu, H., Tian, Y., Zhang, X., Lei, W., et al. (2020). Pathogenic variants identified by whole-exome sequencing in 43 patients with epilepsy. Hum. Genomics 14:44. doi: 10.1186/s40246-020-00294-0
Zhou, X., Xu, H., Cai, X., Tang, B., Liu, X., Shi, Y., et al. (2021). Differences in SCN1A intronic variants result in diverse aberrant splicing patterns and are related to the phenotypes of epilepsy with febrile seizures. Epilepsy Res. 176:106711. doi: 10.1016/j.eplepsyres.2021.106711
Keywords: SCN1A, epilepsy, Dravet syndrome, de novo, seizures
Citation: Zeng B, Zhang H, Lu Q, Fu Q, Yan Y, Lu W, Ma P, Feng C, Qin J, Luo L, Yang B, Zou Y and Liu Y (2023) Identification of five novel SCN1A variants. Front. Behav. Neurosci. 17:1272748. doi: 10.3389/fnbeh.2023.1272748
Edited by:
Hua-Jun Feng, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Yiran Xu, Third Affiliated Hospital of Zhengzhou University, ChinaMaria Augusta Montenegro, University of California, San Diego, United States
Copyright © 2023 Zeng, Zhang, Lu, Fu, Yan, Lu, Ma, Feng, Qin, Luo, Yang, Zou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bicheng Yang, yangbc1985@126.com; Yongyi Zou, zouyongyi@gmail.com; Yanqiu Liu, lyq0914@126.com
†These authors have contributed equally to this work