 Yu-Feng Wang
Yu-Feng Wang Vladimir Parpura
Vladimir Parpura- 1Department of Physiology, School of Basic Medical Sciences, Harbin Medical University, Harbin, China
- 2Department of Neurobiology, University of Alabama at Birmingham, Birmingham, AL, USA
Brain edema formation and the ensuing brain damages are the major cause of high mortality and long term disability following the occurrence of ischemic stroke. In this process, oxygen and glucose deprivation and the resulting reperfusion injury play primary roles. In response to the ischemic insult, the neurovascular unit experiences both intracellular and extracellular edemas, associated with maladapted astrocytic plasticity. The astrocytic plasticity includes both morphological and functional plasticity. The former involves a reactive gliosis and the subsequent glial retraction. It relates to the capacity of astrocytes to buffer changes in extracellular chemical levels, particularly K+ and glutamate, as well as the integrity of the blood-brain barrier (BBB). The latter involves the expression and activity of a series of ion and water transport proteins. These molecules are grouped together around glial fibrillary acidic protein (GFAP) and water channel protein aquaporin 4 (AQP4) to form functional networks, regulate hydromineral balance across cell membranes and maintain the integrity of the BBB. Intense ischemic challenges can disrupt these capacities of astrocytes and result in their maladaptation. The maladapted astrocytic plasticity in ischemic stroke cannot only disrupt the hydromineral homeostasis across astrocyte membrane and the BBB, but also leads to disorders of the whole neurovascular unit. This review focuses on how the maladapted astrocytic plasticity in ischemic stroke plays the central role in the brain edema formation.
Ischemic stroke is a major cause of death and disability, largely because of brain edema formation (Lackland et al., 2014). The brain edema formation mainly results from oxygen and glucose deprivation and reperfusion injury as well as a series of secondary events (Rutkowsky et al., 2011; O’Donnell et al., 2013). These events cause disturbance of hydromineral balance in the neurovascular unit (Kempski, 2001). At the early stage of ischemic insults, injured neurons, glial and endothelial cells experience cytotoxic cell swelling due to abnormal transport of ion and water across cell membranes in the gray matter. Prolonged ischemia and reperfusion injury result in vasogenic edema in the white matter because of increased permeability and destruction of the blood-brain barrier (BBB) and hydromineral retention in extracellular space (Castillo, 2000). The edema can lead to high intracranial pressure, cerebral herniation and death (Khanna et al., 2014) and thus becomes a focus of studies on ischemic stroke.
Accumulating evidence suggests that brain edema is a continuous process modulated by the plastic changes in astrocyte structures and functions that are associated with or can be directly attributed to GFAP, water channel protein AQP4 and their associated ion-transport proteins. This review focuses on the causal relationship between maladapted astrocytic plasticity and brain edema formation during ischemic stroke.
Astrocytic Morphological Plasticity and Brain Edema Formation
Astrocytic endfeet cover more than 90% of brain capillaries to participate in blood-brain barrier (BBB) formation and wrap around neurons to modulate neuronal activity (Jukkola and Gu, 2015). This spatial structural feature allows astrocytes to mediate the communication and volume transmission between brain parenchyma and the blood (Vargová and Syková, 2014). Thus, when astrocyte processes expand or retract from neurons and blood vessels, neural activity and brain volume change dramatically (Nico and Ribatti, 2012), disorders of which constitute a basis of brain edema formation.
Reactive Gliosis
In ischemic stroke, astrocytes undergo a dual morphological plasticity with strong temporal and spatial features. Reactive astrogliosis or elongation of astrocyte processes occurs under hypoxia in primary culture of astrocytes and in ischemic rat brains (Yang et al., 2011b). The reactive gliosis occurs at the initial stage of ischemic stroke or in the penumbra of infarction in severe infarction (Günther et al., 2005; Yang et al., 2011a), and is the main component of cytotoxic edema (Mori et al., 2002). On the one hand, gliosis can buffer damaging effects of ischemia by increasing astrocyte absorption of glutamate, K+ and inflammatory cytokines (Pekny et al., 2014) and separate the necrotic and healthy brain tissues clearly at the penumbra to prevent the expansion of infarct zone (Claus et al., 2013). On the other hand, uncontrolled gliosis can trigger maladaptation of astrocytic plasticity.
Glial Retraction
As ischemia progresses, glial retraction occurs as indicated by the fragmentation of glial fibrillary acidic protein (GFAP) in the infarct zone in parallel with gliosis at the penumbra area or following the initially adaptive gliosis (Abrahám et al., 2003). This dual morphologic plasticity is in agreement with the finding that cerebral cytotoxic edema in prolonged ischemia becomes less severe than that at the initial stage of the middle cerebral artery occlusion in rats (Lu et al., 2011). This reaction can disrupt the structural integrity of the BBB directly and worsen the ischemic stress (Frydenlund et al., 2006; Steiner et al., 2012) by the following approaches: (1) In the lesion core, the swollen astrocytes can release K+, Cl−, and organic osmolytes including glutamate during a regulatory volume decrease (RVD; Quesada et al., 1998; Cardin et al., 1999; Ernest et al., 2005); (2) The RVD also reduces the absorption of excess K+ and glutamate produced during ischemia-evoked cortical spreading depolarization (Dreier, 2011; Seidel et al., 2016); and (3) Moreover, the RVD can cause cellular degeneration and metabolic silence (van der Zijden et al., 2008) as well as disruption of BBB integrity and leakage of blood components into the brain (Mdzinarishvili et al., 2012). As a result, extracellular osmolality increases, more water moves into the extracellular space from both blood and intracellular space, and brain edema forms. Clearly, ischemic challenge can elicit time- and space-dependent maladaptation of astrocytic morphological plasticity and contributes to the brain edema formation. Figure 1 is a diagrammatic drawing of the spatiotemporal features of astrocytic plasticity in ischemic stroke.
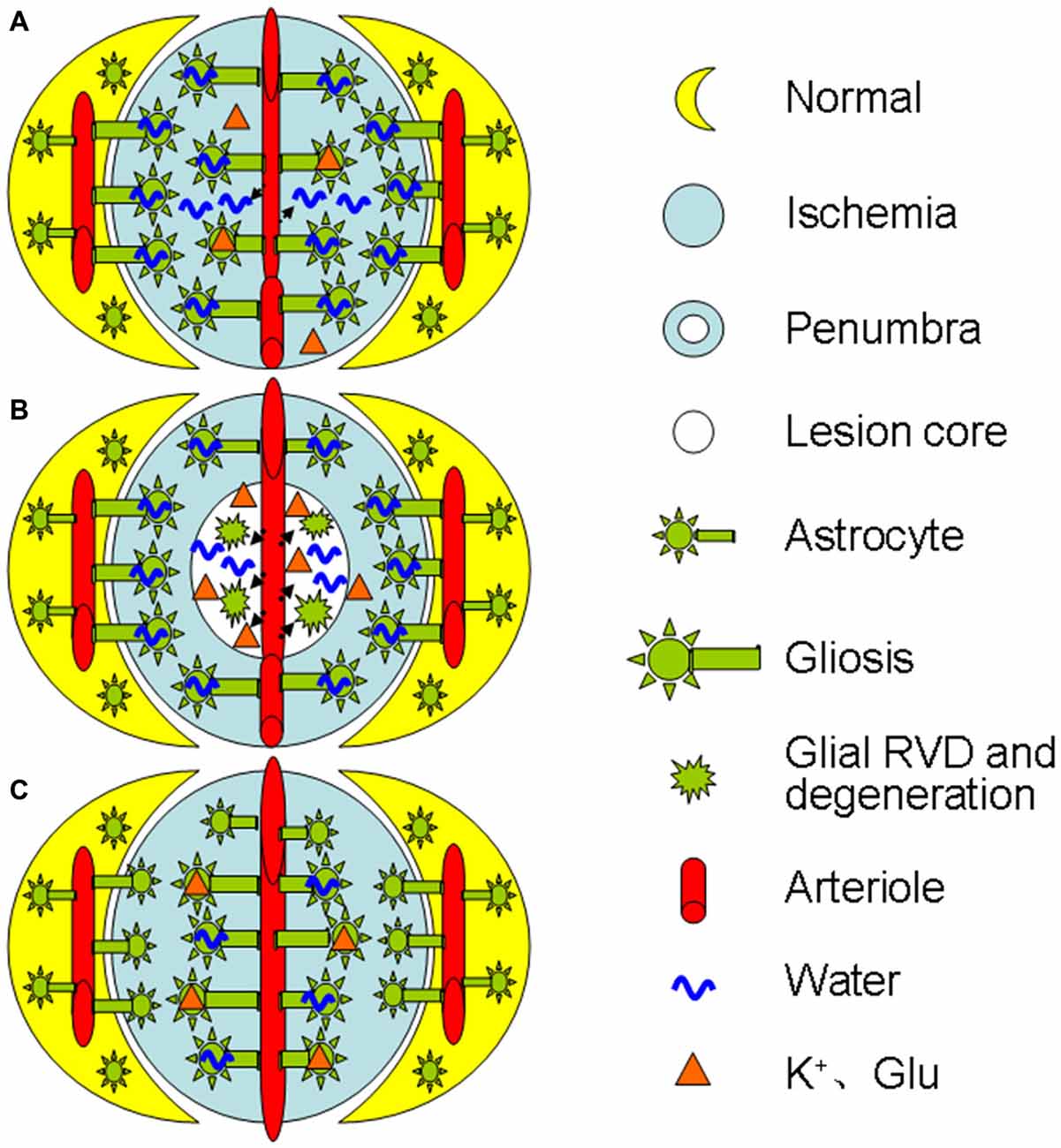
Figure 1. Diagrammatic drawing of the spatiotemporal features of astrocytic plasticity in ischemic stroke (A–C). Spatiotemporal features of astrocytic involvement in ischemic stroke in the early stage (0–24 h) of a mild stroke (A), middle stage (>24–72 h, B) and the late stage (C), respectively. The abbreviations are, Glu, glutamate; RVD, regulatory volume decrease.
Astrocytic Functional Plasticity and Edema Formation
Along with morphological plasticity, astrocytes also possess functional plasticity in brain hydromineral homeostasis by changing the expression of ion transport molecules. The molecules that are involved in this astrocytic functional plasticity in the edema formation include GFAP (Abrahám et al., 2003), AQP4 (Wang and Hatton, 2009), Na+, K+, 2Cl− and water cotransporter (NKCC)1 (Hertz et al., 2014), sulfonylurea receptor 1-transient receptor potential melastatin4 channel (Karschin et al., 1998), sodium pump (Illarionova et al., 2010), glutamine synthetase (Wang et al., 2013b), glutamate-aspartate transporter (Sullivan et al., 2007; Gottipati et al., 2015), and glutamate transporter-1 (GLT-1) (Afadlal et al., 2014; Mogoanta et al., 2014) in addition to those that are involved in GABA (Wang et al., 2013a) and glutamate metabolism (Wang et al., 2013b). Among these molecules, GFAP is the leading molecule that influences the spatial localization of other molecules.
GFAP and Edema
GFAP has long been known as the major cytoskeletal element of astrocytes, and assembling of its monomers or disassembling its filaments can largely determine if astrocyte processes expand or retract under environmental challenges (Barreto et al., 2012; Hol and Pekny, 2015). A scaffolding role of GFAP in astrocytic plasticity has been extensively explored in studying the effects of dehydration and lactation on supraoptic astrocyte activity as previously reviewed (Wang and Zhu, 2014). Consistently, single-walled carbon nanotubes cause an increase in astrocyte uptake of extracellular glutamate by increasing glutamate-aspartate transporter on cell surface, which results from an increase in GFAP filaments (Gottipati et al., 2015). Thus, the expansion of GFAP filaments in astrocyte processes pulls these associated molecules into the vicinity of neurons as shown in the distribution of AQP4 and glutamine synthetase in rats (Wang and Hatton, 2009). Consequently, astrocytes can more efficiently buffer neurochemical changes in the extracellular space around neurons.
Under ischemic challenges, the expression pattern of GFAP changes dramatically during reactive gliosis or retraction. Accumulation of GFAP in reactive astrocytes is a characteristic pathological feature of ischemic brain injury (Kalaivani et al., 2014). Following global cerebral ischemia, a tendency towards GFAP elevation was noticed after 24 h and a significant increase was observed 7 days later (Sulkowski et al., 2002). Moreover, the timing and magnitude of GFAP expression have dramatic regional variations relative to the infarct core (Abrahám et al., 2003). In contrast to the strong expression of GFAP within the penumbral tissues, GFAP staining in the lesion core area is significantly lower (Shen et al., 2010; Bazan et al., 2012), suggesting glial retraction. Correspondingly, the expression of other molecules also exhibit similar time- and region-specific features as exemplified in glutamate metabolism and ion transport activity.
Glutamate Transport and Edema
Glutamate uptake by astrocytes prevents elevation of excitotoxic glutamate in the brain’s extracellular space and is a critical determinant of neuronal survival and cytotoxic edema in the ischemic penumbra (Hansson et al., 2000; Uckermann et al., 2004). In parallel with the change in GFAP expression, glutamate transport proteins also exhibit dual expression in a similar spatiotemporal order. For instance, the decreased GLT-1 experssion in the infarct zone during acute ischemic phase (Rebel et al., 2005; Ketheeswaranathan et al., 2011) can partially account for the increased glutamate levels (Yang et al., 2010) and the metabolic silence in the lesion core (van der Zijden et al., 2008), a result of glutamate excitotoxicity. In contrast, 3–7 days after the ligation of bilateral common carotid arteries in rats, GLT-1 expression is increased significantly (Yatomi et al., 2013). This change is coincident with astrocyte/GFAP extension into the lesion core (Shen et al., 2010), which meets the demand of removing excess extracellular glutamate, but at the expense of glial scar formation (Yang et al., 2010).
Ion Transport and the Edema
Cytotoxic astrocyte swelling largely depends on osmotic gradients built by changes in ion transport activity, which involves NKCC1, inward rectifier K+ channel (Kir) 4.1 and sodium pump. Astrocyte NKCC1 is activated by exposure to high levels of extracellular K+ and to the regulatory volume increase in cells shrunken in response to hypertonic challenge (Larsen et al., 2014), an extracellular environment existing in the lesion core (van der Zijden et al., 2008; Yang et al., 2010). Increases in extracellular K+ levels, known to increase GFAP expression (Wang and Hatton, 2009), also increase Ca2+ influx and intracellular cAMP levels during stroke. The Ca2+ influx induces an increase in intracellular Ca2+ levels and the activation of NKCC1 and Kir4.1 (Song et al., 2015); increased cAMP level causes AQP4 phosphorylation and increases membrane water permeability (Song and Gunnarson, 2012). Thus, synergistic changes in the activity of diverse ion transport proteins with GFAP expression build the osmotic gradients.
Pivotal Roles of AQP4 in Ischemia-Elicited Astrocyte Plasticity
Among many GFAP-associated molecules, AQP4 is distinguished not only by its water permeability but also by its role of a “cotransporter” of osmolytes. AQP4 is colocalized or assembled with the transient receptor potential vanilloid channel 4 (Jo et al., 2015), gap junction protein Cx43 (Nicchia et al., 2005), Kir4.1/Kir5.1 (Lichter-Konecki et al., 2008), GLT-1 (Mogoanta et al., 2014), metabotropic glutamate receptor 5, and sodium pump (Illarionova et al., 2010). AQP4 and these ion transport proteins are mainly colocalized to astrocytic endfeet and exhibit strong functional correlation. For instance, deletion of AQP4 interferes K+ (Strohschein et al., 2011) and glutamate uptake (Nagelhus and Ottersen, 2013) while significantly decreasing GFAP expression in perivascular processes of astrocytes (Zhou et al., 2008). This molecular association likely reflects the demands of maintaining intracellular osmotic homeostasis via synergistically changing osmotic pressure and water transport.
Temporal Features
Following the occurrence of stroke, AQP4 expression presents two peaks despite the time variations among individual observations. The first peak occurs at the initial stage of stroke; the second peak appears days after reperfusion and during recovery. As shown in mice (Ribeiro Mde et al., 2006) and rats (Fang et al., 2016), there are two peaks of maximal hemispheric swelling that respectively occur at 1 h and at 48 h after the onset of ischemia and are accompanied with synergistic increases in AQP4 expression (Ribeiro Mde et al., 2006). Moreover, AQP4 at the infarct zone is found to be lower at 24 h and higher at 72 h than that at the non-occluded areas (Zeng et al., 2012). This biphasic AQP4 expression is consistent with the internalization and decomposition of AQP4 at 3 h after the onset of ischemia (Huang et al., 2013). Clearly, AQP4 expression is temporally synergistic with GFAP/astrocytic morphological plasticity.
Spatial Features
In mice with 1 h transient focal brain ischemia, AQP4 expression is significantly increased at astrocyte endfeet in the lesion core initially and it increases in whole astrocytes at the border of lesion at 48 h (Ribeiro Mde et al., 2006). Moreover, the most affected part of the cortex loses perivascular AQP4 but shows a partial recovery toward 72 h of reperfusion; however, the cortical border zone differs from the lesion core by showing no loss of perivascular AQP4 at 24 h but rather a slight increase (Frydenlund et al., 2006). These region-specific changes indicate that AQP4 plays different role, depending on the regions, consistent with the general alteration in GFAP expression and astrocytic morphological plasticity.
Further studies reveal that the spatial distribution rather than the expression level of AQP4 is closely associated with its functions. The loss of perivascular AQP4 is associated with the disappearance of the perivascular astrocyte membrane (Frydenlund et al., 2006), indicating that AQP4 is removed from its functioning sites along with the disassembly of GFAP filaments and glial retraction. Following cerebral ischemia, cortical astrocytes exhibit reduced perivascular AQP4 and unchanged AQP4 protein abundance (Stokum et al., 2015). The loss of AQP4 polarization and the disruption of laminins of the basement membrane disrupt water efflux from the brain while increasing BBB permeability (Steiner et al., 2012). Moreover, in mild focal brain ischemia, these changes occur only in the area of lesion core but not in the penumbra (Steiner et al., 2012). These facts allow us to conclude that AQP4 expression and localization in stroke are correlated with the extent and stage of brain edema formation, which allows different groups of astrocytes to respond to ischemic insults differentially. Figure 2 is a diagrammatic drawing of the astrocytic functional plasticity in ischemic stroke.
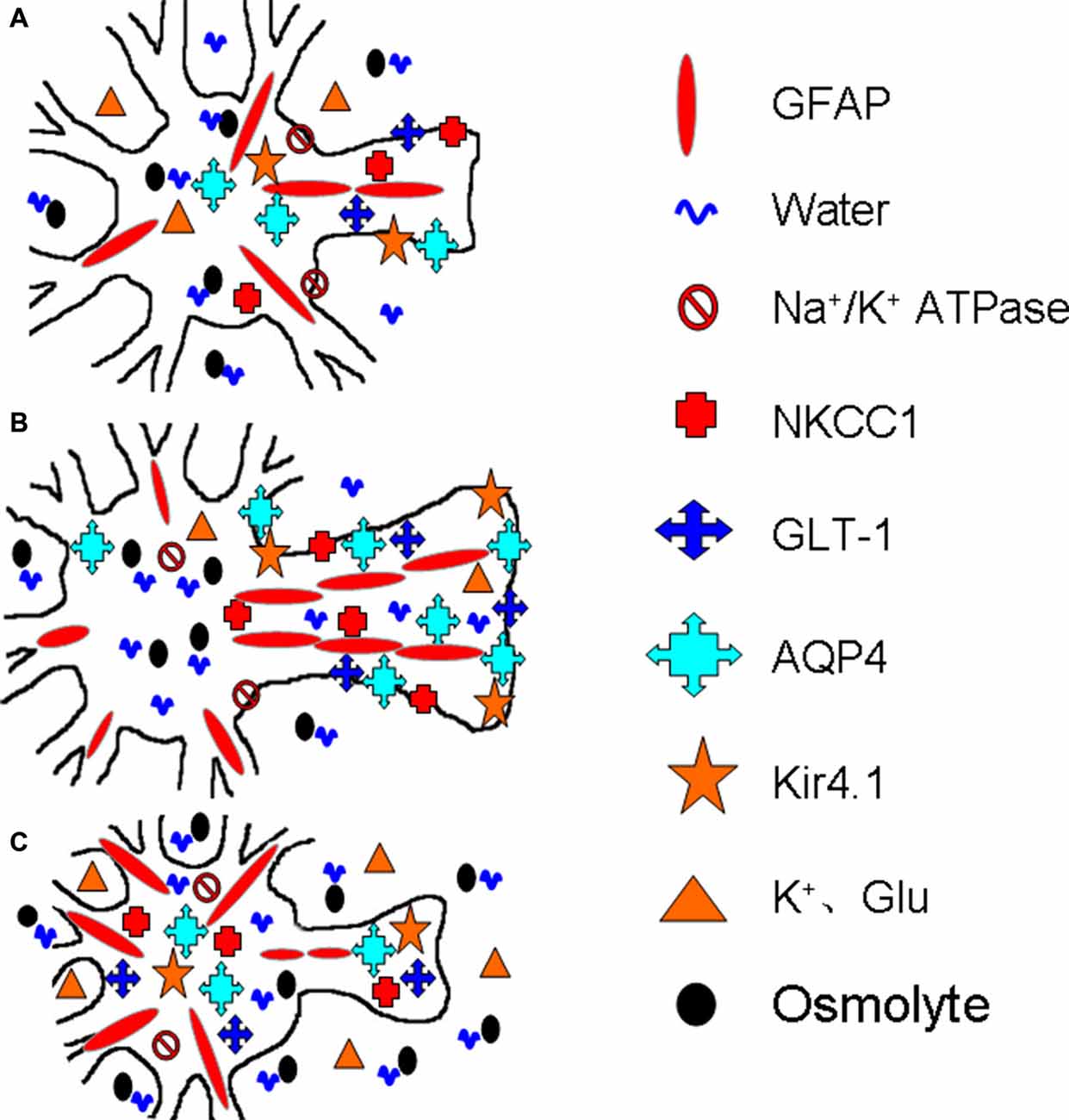
Figure 2. Diagrammatic drawing of the astrocytic functional plasticity in ischemic stroke (A–C). The functional plasticity at different loci relative to the infarct core: normally irrigated tissues (A), the penumbra (B) and the lesion core (C), respectively. The abbreviations are, AQP4, Aquaporin-4; GFAP, glial fibrillary acidic protein; GLT-1, glutamate transporter-1; Kir4.1, inward rectifier K+ channel 4.1; NKCC1, Na+, K+, 2Cl− and water co-transporter. Others refer to Figure 1.
Central Role of Astrocytic Plasticity in Brain Edema Formation
As stated above, astrocytes are not only the major cell type exhibiting cytotoxic edema during ischemia and reperfusion injury, but also a main factor that disrupts BBB integrity. Further analysis reveals that the astrocytic plasticity plays a central role in brain edema formation as detailed below.
Determining Factor of Ischemic Brain Edema
Astrocytes can extensively modulate the activities of other components of the neurovascular units (Theodosis et al., 2008), and maladaptive alterations of astrocytic plasticity are the major cause of brain edema formation in stroke. Reactive gliosis protects the brain from excessive damage caused by swelling by stabilizing hydromineral balance through absorption of increased extracellular K+ (Strohschein et al., 2011) and glutamate (Nagelhus and Ottersen, 2013; Seidel et al., 2016), reducing NH4+ toxicity and oxidative stress by synthesizing glutamine, antagonizing glutamate toxicity by releasing inhibitory adenosine, taurine and β-alanine (Morán et al., 2001; Parpura et al., 2004), alleviating vasogenic edema by repairing injured BBB, limiting the spreading of inflammation from the infarct core into penumbra, and releasing neuroprotectants (Kawano et al., 2012). In addition, the increased AQP4 level can reduce the osmotic fragility of astrocytes (Gu et al., 2003). However, during prolonged ischemic challenge, astrocytes lose their powers of being an energy provider (e.g., providing metabolic substrates to neurons), a buffer of extracellular chemical and an inhibitor of neuron activity (Marrif and Juurlink, 1999), leading to the malfunction and damage of the entire neurovascular unit. Importantly, inhibiting the activity of AQP4 (Bhattacharya et al., 2013; Yao et al., 2015) and NKCC1 (Su et al., 2002; Yan et al., 2003) significantly reduces ischemic brain edema and the ensuing brain injury while inactivation of GLT-1 worsens damage to the brain (Namura et al., 2002). These facts validate the central role of astrocytes in ischemic brain edema formation.
Spatiotemporal Correlation
In ischemia, the extent of spatiotemporally differentiated reactive gliosis and the associated RVD is highly correlated with the severity of brain swelling, excitotoxicity, oxidative stress, metabolic disorders, and disruption of the BBB. On the one hand, reactive gliosis, indicated by increasing GFAP and AQP4 expression, is a sign of astrocyte swelling at the early stage of mild stroke and in the penumbra of severe stroke. This gliosis is likely an evolutionary wise selection. It allows the brain, in a situation of a substantial energy deficit in ischemic stroke, to rebalance extracellular K+ and glutamate levels through siphoning K+ and glutamate uptake, to re-establish neuronal membrane potential and minimize excitotoxic impact at the cost of least energy. However, uncontrolled gliosis can be harmful because it produces high levels of bioactive compounds that are noxious for neuronal cell functions, such as the bioactive free radical nitric oxide (Ghasemi and Fatemi, 2014). In addition, excess gliosis also serves as a trigger of the RVD that could bring additional injury to the neurovascular unit. Thus, despite the protective capacity of gliosis, over-activated astrocytes can worsen the brain edema.
On the other hand, breakdown of this gliosis can lead to degeneration of neural tissues in the lesion core. The reduced volume during the RVD can avoid overload of osmolytes and water in astrocytes, and thus preserve structural integrity of astrocytes. However, the reduced absorption of ion, glutamate and water, decreased volume transmission into the blood in infarct areas as well as the destruction of BBB integrity can seriously disrupt the homeostasis of the whole neurovascular unit, leading to its apoptosis, autophagic cell death and necrosis (Puyal et al., 2013).
Continuous Involvement
Following the onset of stroke, astrocytes keep changing their structure and function as indicated by the expression of GFAP and AQP4, which determine a conversion of the intracellular edema into extracellular edema in the lesion core while forming a shell of swollen astrocytes at the penumbra. Moreover, overly increased astrocyte volume triggers the RVD wherein the reduced AQP4 could disrupt the adaptive plastic change and dispersion of intracellular volume through the junctional coupling among astrocytes (Nicchia et al., 2005; Mühlfeld and Richter, 2006), thereby leading to the degeneration and even death of astrocytes in the lesion core, promoting vasogenic edema. During the recovery phase, expanded astrocyte processes and newly differentiated glial cells invade the infarct areas to rebuild the neural tissue by cleaning the necrotic tissue, filling in the space and releasing many growth factors (Wasielewski et al., 2012). Thus, astrocytes are continuously involved in the pathogenesis of ischemic brain edema.
Conclusion
Together with the view presented previously (Verkhratsky et al., 2015), we propose that astrocytes are not only involved in the formation of cytotoxic edema and vasogenic edema but also serve as the central player of the disturbed neurovascular unit in the ischemic brain during edema formation. In this process, astrocytic plasticity exhibits strong spatiotemporal features and continuous involvement in brain swelling, in which, GFAP and AQP4 are the keys to determining the structural and functional plasticity. Further clarification of astrocytic plasticity in ischemic brain edema formation will help to define a therapeutic window of differentially modulating astrocytic plasticity and achieve better prognosis of ischemic brain injury.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank for the advice of Dr. Bao-Feng Yang. This work is sponsored by the National Natural Science Foundation of China (Grant No. 31471113, Y-FW), National Institutes of Health (The Eunice Kennedy Shriver National Institute of Child Health and Human Development award, HD078678, VP), and the higher education talents funds of Heilongjiang province (Grant No. 002000154, Y-FW).
References
Abrahám, H., Somogyvári-Vigh, A., Maderdrut, J. L., Vigh, S., and Arimura, A. (2003). Rapidly activated microglial cells in the preoptic area may play a role in the generation of hyperthermia following occlusion of the middle cerebral artery in the rat. Exp. Brain Res. 153, 84–91. doi: 10.1007/s00221-003-1572-8
Afadlal, S., Labetoulle, R., and Hazell, A. S. (2014). Role of astrocytes in thiamine deficiency. Metab. Brain Dis. 29, 1061–1068. doi: 10.1007/s11011-014-9571-y
Barreto, G. E., White, R. E., Xu, L., Palm, C. J., and Giffard, R. G. (2012). Effects of heat shock protein 72 (Hsp72) on evolution of astrocyte activation following stroke in the mouse. Exp. Neurol. 238, 284–296. doi: 10.1016/j.expneurol.2012.08.015
Bazan, N. G., Eady, T. N., Khoutorova, L., Atkins, K. D., Hong, S., Lu, Y., et al. (2012). Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp. Neurol. 236, 122–130. doi: 10.1016/j.expneurol.2012.04.007
Bhattacharya, P., Pandey, A. K., Paul, S., Patnaik, R., and Yavagal, D. R. (2013). Aquaporin-4 inhibition mediates piroxicam-induced neuroprotection against focal cerebral ischemia/reperfusion injury in rodents. PLoS One 8:e73481. doi: 10.1371/journal.pone.0073481
Cardin, V., Peña-Segura, C., and Pasantes-Morales, H. (1999). Activation and inactivation of taurine efflux in hyposmotic and isosmotic swelling in cortical astrocytes: role of ionic strength and cell volume decrease. J. Neurosci. Res. 56, 659–667. doi: 10.1002/(SICI)1097-4547(19990615)56:63.3.CO;2-N
Claus, H. L., Walberer, M., Simard, M. L., Emig, B., Muesken, S. M., Rueger, M. A., et al. (2013). NG2 and NG2-positive cells delineate focal cerebral infarct demarcation in rats. Neuropathology 33, 30–38. doi: 10.1111/j.1440-1789.2012.01322.x
Dreier, J. P. (2011). The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 17, 439–447. doi: 10.1038/nm.2333
Ernest, N. J., Weaver, A. K., Van Duyn, L. B., and Sontheimer, H. W. (2005). Relative contribution of chloride channels and transporters to regulatory volume decrease in human glioma cells. Am. J. Physiol. Cell Physiol. 288, C1451–C1460. doi: 10.1152/ajpcell.00503.2004
Fang, Q., Yan, X., Li, S., Sun, Y., Xu, L., Shi, Z., et al. (2016). Methylene blue ameliorates ischemia/reperfusion-induced cerebral edema: an MRI and transmission electron microscope study. Acta Neurochir. Suppl. 121, 227–236. doi: 10.1007/978-3-319-18497-5_41
Frydenlund, D. S., Bhardwaj, A., Otsuka, T., Mylonakou, M. N., Yasumura, T., Davidson, K. G., et al. (2006). Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc. Natl. Acad. Sci. U S A 103, 13532–13536. doi: 10.1073/pnas.0605796103
Ghasemi, M., and Fatemi, A. (2014). Pathologic role of glial nitric oxide in adult and pediatric neuroinflammatory diseases. Neurosci. Biobehav. Rev. 45, 168–182. doi: 10.1016/j.neubiorev.2014.06.002
Gottipati, M. K., Bekyarova, E., Haddon, R. C., and Parpura, V. (2015). Chemically functionalized single-walled carbon nanotubes enhance the glutamate uptake characteristics of mouse cortical astrocytes. Amino Acids 47, 1379–1388. doi: 10.1007/s00726-015-1970-9
Gu, F., Hata, R., Toku, K., Yang, L., Ma, Y. J., Maeda, N., et al. (2003). Testosterone up-regulates aquaporin-4 expression in cultured astrocytes. J. Neurosci. Res. 72, 709–715. doi: 10.1002/jnr.10603
Günther, A., Küppers-Tiedt, L., Schneider, P. M., Kunert, I., Berrouschot, J., Schneider, D., et al. (2005). Reduced infarct volume and differential effects on glial cell activation after hyperbaric oxygen treatment in rat permanent focal cerebral ischaemia. Eur. J. Neurosci. 21, 3189–3194. doi: 10.1111/j.1460-9568.2005.04151.x
Hansson, E., Muyderman, H., Leonova, J., Allansson, L., Sinclair, J., Blomstrand, F., et al. (2000). Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamate-induced cell swelling and gap-junction communication. Neurochem. Int. 37, 317–329. doi: 10.1016/s0197-0186(00)00033-4
Hertz, L., Xu, J., Chen, Y., Gibbs, M. E., Du, T., Hertz, L., et al. (2014). Antagonists of the Vasopressin V1 receptor and of the β1-Adrenoceptor inhibit cytotoxic brain edema in stroke by effects on astrocytes–but the mechanisms differ. Curr. Neuropharmacol. 12, 308–323. doi: 10.2174/1570159x12666140828222723
Hol, E. M., and Pekny, M. (2015). Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 32, 121–130. doi: 10.1016/j.ceb.2015.02.004
Huang, J., Sun, S. Q., Lu, W. T., Xu, J., Gan, S. W., Chen, Z., et al. (2013). The internalization and lysosomal degradation of brain AQP4 after ischemic injury. Brain Res. 1539, 61–72. doi: 10.1016/j.brainres.2013.09.022
Illarionova, N. B., Gunnarson, E., Li, Y., Brismar, H., Bondar, A., Zelenin, S., et al. (2010). Functional and molecular interactions between aquaporins and Na,K-ATPase. Neuroscience 168, 915–925. doi: 10.1016/j.neuroscience.2009.11.062
Jo, A. O., Ryskamp, D. A., Phuong, T. T., Verkman, A. S., Yarishkin, O., MacAulay, N., et al. (2015). TRPV4 and AQP4 channels synergistically regulate cell volume and calcium homeostasis in retinal muller glia. J. Neurosci. 35, 13525–13537. doi: 10.1523/JNEUROSCI.1987-15.2015
Jukkola, P., and Gu, C. (2015). Regulation of neurovascular coupling in autoimmunity to water and ion channels. Autoimmun. Rev. 14, 258–267. doi: 10.1016/j.autrev.2014.11.010
Kalaivani, P., Ganesh, M., Sathiya, S., Ranju, V., Gayathiri, V., and Saravana Babu, C. (2014). Alteration in bioenergetic regulators, SirT1 and Parp1 expression precedes oxidative stress in rats subjected to transient cerebral focal ischemia: molecular and histopathologic evidences. J. Stroke Cerebrovasc. Dis. 23, 2753–2766. doi: 10.1016/j.jstrokecerebrovasdis.2014.06.026
Karschin, A., Brockhaus, J., and Ballanyi, K. (1998). KATP channel formation by the sulphonylurea receptors SUR1 with Kir6.2 subunits in rat dorsal vagal neurons in situ. J. Physiol. 509, 339–346. doi: 10.1111/j.1469-7793.1998.339bn.x
Kawano, H., Kimura-Kuroda, J., Komuta, Y., Yoshioka, N., Li, H. P., Kawamura, K., et al. (2012). Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res. 349, 169–180. doi: 10.1007/s00441-012-1336-5
Ketheeswaranathan, P., Turner, N. A., Spary, E. J., Batten, T. F., McColl, B. W., and Saha, S. (2011). Changes in glutamate transporter expression in mouse forebrain areas following focal ischemia. Brain Res. 1418, 93–103. doi: 10.1016/j.brainres.2011.08.029
Khanna, A., Kahle, K. T., Walcott, B. P., Gerzanich, V., and Simard, J. M. (2014). Disruption of ion homeostasis in the neurogliovascular unit underlies the pathogenesis of ischemic cerebral edema. Transl. Stroke Res. 5, 3–16. doi: 10.1007/s12975-013-0307-9
Lackland, D. T., Roccella, E. J., Deutsch, A. F., Fornage, M., George, M. G., Howard, G., et al. (2014). Factors influencing the decline in stroke mortality: a statement from the American heart association/American stroke association. Stroke 45, 315–353. doi: 10.1161/01.str.0000437068.30550.cf
Larsen, B. R., Assentoft, M., Cotrina, M. L., Hua, S. Z., Nedergaard, M., Kaila, K., et al. (2014). Contributions of the Na(+)/K(+)-ATPase, NKCC1 and Kir4.1 to hippocampal K(+) clearance and volume responses. Glia 62, 608–622. doi: 10.1002/glia.22629
Lichter-Konecki, U., Mangin, J. M., Gordish-Dressman, H., Hoffman, E. P., and Gallo, V. (2008). Gene expression profiling of astrocytes from hyperammonemic mice reveals altered pathways for water and potassium homeostasis in vivo. Glia 56, 365–377. doi: 10.1002/glia.20624
Lu, H., Hu, H., and He, Z. P. (2011). Reperfusion of the rat brain tissues following acute ischemia: the correlation among diffusion-weighted imaging, histopathology and aquaporin-4 expression. Chin. Med. J. 124, 3148–3153.
Marrif, H., and Juurlink, B. H. (1999). Astrocytes respond to hypoxia by increasing glycolytic capacity. J. Neurosci. Res. 57, 255–260. doi: 10.1002/(SICI)1097-4547(19990715)57:23.0.CO;2-6
Mdzinarishvili, A., Sumbria, R., Lang, D., and Klein, J. (2012). Ginkgo extract EGb761 confers neuroprotection by reduction of glutamate release in ischemic brain. J. Pharm. Pharm. Sci. 15, 94–102. doi: 10.18433/j3ps37
Mogoanta, L., Ciurea, M., Pirici, I., Margaritescu, C., Simionescu, C., Ion, D. A., et al. (2014). Different dynamics of aquaporin 4 and glutamate transporter-1 distribution in the perineuronal and perivascular compartments during ischemic stroke. Brain Pathol. 24, 475–493. doi: 10.1111/bpa.12134
Morán, J., Morales-Mulia, M., and Pasantes-Morales, H. (2001). Reduction of phospholemman expression decreases osmosensitive taurine efflux in astrocytes. Biochim. Biophys Acta. 1538, 313–320. doi: 10.1016/s0167-4889(01)00082-9
Mori, K., Miyazaki, M., Iwase, H., and Maeda, M. (2002). Temporal profile of changes in brain tissue extracellular space and extracellular ion (Na(+), K(+)) concentrations after cerebral ischemia and the effects of mild cerebral hypothermia. J. Neurotrauma 19, 1261–1270. doi: 10.1089/08977150260338047
Mühlfeld, C., and Richter, J. (2006). High-pressure freezing and freeze substitution of rat myocardium for immunogold labeling of connexin 43. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 288, 1059–1067. doi: 10.1002/ar.a.20380
Nagelhus, E. A., and Ottersen, O. P. (2013). Physiological roles of aquaporin-4 in brain. Physiol. Rev. 93, 1543–1562. doi: 10.1152/physrev.00011.2013
Namura, S., Maeno, H., Takami, S., Jiang, X. F., Kamichi, S., Wada, K., et al. (2002). Inhibition of glial glutamate transporter GLT-1 augments brain edema after transient focal cerebral ischemia in mice. Neurosci. Lett. 324, 117–120. doi: 10.1016/s0304-3940(02)00193-3
Nicchia, G. P., Srinivas, M., Li, W., Brosnan, C. F., Frigeri, A., and Spray, D. C. (2005). New possible roles for aquaporin-4 in astrocytes: cell cytoskeleton and functional relationship with connexin43. FASEB J. 19,1674–1676. doi: 10.1096/fj.04-3281fje
Nico, B., and Ribatti, D. (2012). Morphofunctional aspects of the blood-brain barrier. Curr. Drug Metab. 13, 50–60. doi: 10.2174/138920012798356970
O’Donnell, M. E., Chen, Y. J., Lam, T. I., Taylor, K. C., Walton, J. H., and Anderson, S. E. (2013). Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood-brain barrier Na/H exchanger. J. Cereb. Blood Flow Metab. 33, 225–234. doi: 10.1038/jcbfm.2012.160
Parpura, V., Scemes, E., and Spray, D. C. (2004). Mechanisms of glutamate release from astrocytes: gap junction “hemichannels”, purinergic receptors and exocytotic release. Neurochem. Int. 45, 259–264. doi: 10.1016/j.neuint.2003.12.011
Pekny, M., Wilhelmsson, U., and Pekna, M. (2014). The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 565, 30–38. doi: 10.1016/j.neulet.2013.12.071
Puyal, J., Ginet, V., and Clarke, P. G. (2013). Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog. Neurobiol. 105, 24–48. doi: 10.1016/j.pneurobio.2013.03.002
Quesada, O., González, E., Morales-Mulia, S., Bargas, J., and Pasantes-Morales, H. (1998). Effects of NaCl removal on osmolyte fluxes and regulatory volume decrease in cultured astrocytes. J. Neurosci. Res. 53, 195–202. doi: 10.1002/(sici)1097-4547(19980715)53:2<195::aid-jnr8>3.0.co;2-2
Rebel, A., Koehler, R. C., and Martin, L. J. (2005). In situ immunoradiographic method for quantification of specific proteins in normal and ischemic brain regions. J. Neurosci. Methods 143, 227–235. doi: 10.1016/j.jneumeth.2004.11.003
Ribeiro Mde, C., Hirt, L., Bogousslavsky, J., Regli, L., and Badaut, J. (2006). Time course of aquaporin expression after transient focal cerebral ischemia in mice. J. Neurosci. Res. 83, 1231–1240. doi: 10.1002/jnr.20819
Rutkowsky, J. M., Wallace, B. K., Wise, P. M., and O’Donnell, M. E. (2011). Effects of estradiol on ischemic factor-induced astrocyte swelling and AQP4 protein abundance. Am. J. Physiol. Cell Physiol. 301, C204–C212. doi: 10.1152/ajpcell.00399.2010
Seidel, J. L., Escartin, C., Ayata, C., Bonvento, G., and Shuttleworth, C. W. (2016). Multifaceted roles for astrocytes in spreading depolarization: a target for limiting spreading depolarization in acute brain injury? Glia 64, 5–20. doi: 10.1002/glia.22824
Shen, C. C., Yang, Y. C., Chiao, M. T., Cheng, W. Y., Tsuei, Y. S., and Ko, J. L. (2010). Characterization of endogenous neural progenitor cells after experimental ischemic stroke. Curr. Neurovasc. Res. 7, 6–14. doi: 10.2174/156720210790820208
Song, Y., and Gunnarson, E. (2012). Potassium dependent regulation of astrocyte water permeability is mediated by cAMP signaling. PLoS One 7:e34936. doi: 10.1371/journal.pone.0034936
Song, D., Xu, J., Hertz, L., and Peng, L. (2015). Regulatory volume increase in astrocytes exposed to hypertonic medium requires β1-adrenergic Na+/K+-ATPase stimulation and glycogenolysis. J. Neurosci. Res. 93, 130–139. doi: 10.1002/jnr.23469
Steiner, E., Enzmann, G. U., Lin, S., Ghavampour, S., Hannocks, M. J., Zuber, B., et al. (2012). Loss of astrocyte polarization upon transient focal brain ischemia as a possible mechanism to counteract early edema formation. Glia 60, 1646–1659. doi: 10.1002/glia.22383
Stokum, J. A., Mehta, R. I., Ivanova, S., Yu, E., Gerzanich, V., and Simard, J. M. (2015). Heterogeneity of aquaporin-4 localization and expression after focal cerebral ischemia underlies differences in white versus grey matter swelling. Acta Neuropathol. Commun. 3:61. doi: 10.1186/s40478-015-0239-6
Strohschein, S., Hüttmann, K., Gabriel, S., Binder, D. K., Heinemann, U., and Steinhauser, C. (2011). Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia 59, 973–980. doi: 10.1002/glia.21169
Su, G., Kintner, D. B., Flagella, M., Shull, G. E., and Sun, D. (2002). Astrocytes from Na(+)-K(+)-Cl(+) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am. J. Physiol. Cell Physiol. 282, C1147–C1160. doi: 10.1152/ajpcell.00538.2001
Sulkowski, G., Bubko, I., Struzynska, L., Januszewski, S., Walski, M., and Rafalowska, U. (2002). Astrocytic response in the rodent model of global cerebral ischemia and during reperfusion. Exp. Toxicol. Pathol. 54, 31–38. doi: 10.1078/0940-2993-00229
Sullivan, S. M., Lee, A., Bjorkman, S. T., Miller, S. M., Sullivan, R. K., Poronnik, P., et al. (2007). Cytoskeletal anchoring of GLAST determines susceptibility to brain damage: an identified role for GFAP. J. Biol. Chem. 282, 29414–29423. doi: 10.1074/jbc.m704152200
Theodosis, D. T., Poulain, D. A., and Oliet, S. H. (2008). Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol. Rev. 88, 983–1008. doi: 10.1152/physrev.00036.2007
Uckermann, O., Vargová, L., Ulbricht, E., Klaus, C., Weick, M., Rillich, K., et al. (2004). Glutamate-evoked alterations of glial and neuronal cell morphology in the guinea pig retina. J. Neurosci. 24, 10149–10158. doi: 10.1523/jneurosci.3203-04.2004
van der Zijden, J. P., van Eijsden, P., de Graaf, R. A., and Dijkhuizen, R. M. (2008). 1H/13C MR spectroscopic imaging of regionally specific metabolic alterations after experimental stroke. Brain 131, 2209–2219. doi: 10.1093/brain/awn139
Vargová, L., and Syková, E. (2014). Astrocytes and extracellular matrix in extrasynaptic volume transmission. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130608. doi: 10.1098/rstb.2013.0608
Verkhratsky, A., Steardo, L., Parpura, V., and Montana, V. (2015). Translational potential of astrocytes in brain disorders. Prog. Neurobiol. doi: 10.1016/j.pneurobio.2015.09.003 [Epub ahead of print].
Wang, Y. F., and Hatton, G. I. (2009). Astrocytic plasticity and patterned oxytocin neuronal activity: dynamic interactions. J. Neurosci. 29, 1743–1754. doi: 10.1523/JNEUROSCI.4669-08.2009
Wang, Y. F., Sun, M. Y., Hou, Q., and Hamilton, K. A. (2013a). GABAergic inhibition through synergistic astrocytic neuronal interaction transiently decreases vasopressin neuronal activity during hypoosmotic challenge. Eur. J. Neurosci. 37, 1260–1269. doi: 10.1111/ejn.12137
Wang, Y. F., Sun, M. Y., Hou, Q., and Parpura, V. (2013b). Hyposmolality differentially and spatiotemporally modulates levels of glutamine synthetase and serine racemase in rat supraoptic nucleus. Glia 61, 529–538. doi: 10.1002/glia.22453
Wang, Y. F., and Zhu, H. (2014). Mechanisms underlying astrocyte regulation of hypothalamic neuroendocrine neuron activity. Sheng Li Ke Xue Jin Zhan 45, 177–184.
Wasielewski, B., Jensen, A., Roth-Harer, A., Dermietzel, R., and Meier, C. (2012). Neuroglial activation and Cx43 expression are reduced upon transplantation of human umbilical cord blood cells after perinatal hypoxic-ischemic injury. Brain Res. 1487, 39–53. doi: 10.1016/j.brainres.2012.05.066
Yan, Y., Dempsey, R. J., Flemmer, A., Forbush, B., and Sun, D. (2003). Inhibition of Na(+)-K(+)-Cl(+) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 961, 22–31. doi: 10.1016/s0006-8993(02)03832-5
Yang, J., Lunde, L. K., Nuntagij, P., Oguchi, T., Camassa, L. M., Nilsson, L. N., et al. (2011a). Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J. Alzheimers Dis. 27, 711–722. doi: 10.3233/JAD-2011-110725
Yang, Q., Wang, E. Y., Huang, X. J., Qu, W. S., Zhang, L., Xu, J. Z., et al. (2011b). Blocking epidermal growth factor receptor attenuates reactive astrogliosis through inhibiting cell cycle progression and protects against ischemic brain injury in rats. J. Neurochem. 119, 644–653. doi: 10.1111/j.1471-4159.2011.07446.x
Yang, Z. J., Xie, Y., Bosco, G. M., Chen, C., and Camporesi, E. M. (2010). Hyperbaric oxygenation alleviates MCAO-induced brain injury and reduces hydroxyl radical formation and glutamate release. Eur. J. Appl. Physiol. 108, 513–522. doi: 10.1007/s00421-009-1229-9
Yao, X., Derugin, N., Manley, G. T., and Verkman, A. S. (2015). Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci. Lett. 584, 368–372. doi: 10.1016/j.neulet.2014.10.040
Yatomi, Y., Tanaka, R., Shimura, H., Miyamoto, N., Yamashiro, K., Takanashi, M., et al. (2013). Chronic brain ischemia induces the expression of glial glutamate transporter EAAT2 in subcortical white matter. Neuroscience 244, 113–121. doi: 10.1016/j.neuroscience.2013.04.018
Zeng, X. N., Xie, L. L., Liang, R., Sun, X. L., Fan, Y., and Hu, G. (2012). AQP4 knockout aggravates ischemia/reperfusion injury in mice. CNS Neurosci. Ther. 18, 388–394. doi: 10.1111/j.1755-5949.2012.00308.x
Keywords: aquaporin-4, astrocytes, brain edema formation, glial fibrillary acidic protein, functional plasticity, hydromineral balance, ischemic stroke, morphological plasticity
Citation: Wang Y-F and Parpura V (2016) Central Role of Maladapted Astrocytic Plasticity in Ischemic Brain Edema Formation. Front. Cell. Neurosci. 10:129. doi: 10.3389/fncel.2016.00129
Received: 16 February 2016; Accepted: 29 April 2016;
Published: 13 May 2016.
Edited by:
Qi Yuan, Memorial University, CanadaReviewed by:
James Donald Lechleiter, University of Texas Health Science Center, USASelva Baltan, Cleveland Clinic, USA
Copyright © 2016 Wang and Parpura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Feng Wang, eXVmZW5nd2FuZ0BlbXMuaHJibXUuZWR1LmNu;
Vladimir Parpura, dmxhZEB1YWIuZWR1