Transient Potassium Channels: Therapeutic Targets for Brain Disorders
 Wonjun Noh
Wonjun Noh Sojeong Pak
Sojeong Pak Geunho Choi
Geunho Choi Sungchil Yang
Sungchil Yang Sunggu Yang
Sunggu Yang- 1Department of Nano-Bioengineering, Incheon National University, Incheon, South Korea
- 2Department of Biomedical Sciences, City University of Hong Kong, Kowloon, Hong Kong
- 3Department of Computer Science and Engineering, Incheon National University, Incheon, South Korea
Transient potassium current channels (IA channels), which are expressed in most brain areas, have a central role in modulating feedforward and feedback inhibition along the dendroaxonic axis. Loss of the modulatory channels is tightly associated with a number of brain diseases such as Alzheimer’s disease, epilepsy, fragile X syndrome (FXS), Parkinson’s disease, chronic pain, tinnitus, and ataxia. However, the functional significance of IA channels in these diseases has so far been underestimated. In this review, we discuss the distribution and function of IA channels. Particularly, we posit that downregulation of IA channels results in neuronal (mostly dendritic) hyperexcitability accompanied by the imbalanced excitation and inhibition ratio in the brain’s networks, eventually causing the brain diseases. Finally, we propose a potential therapeutic target: the enhanced action of IA channels to counteract Ca2+-permeable channels including NMDA receptors could be harnessed to restore dendritic excitability, leading to a balanced neuronal state.
Introduction
Sequential synaptic inputs often cause dendritic spikes. Fast-acting membrane depolarization in the dendritic tree is converted into axo-somatic action potentials (APs) that do not only propagate down the axon but also back into the dendritic tree (Golding et al., 2001; Bender and Trussell, 2012). With distance from the soma, the amplitude of back-propagation action potentials (bAPs) is gradually attenuated by the inhibitory action of A-type transient potassium channels, IA for short (Hoffman et al., 1997; Frick et al., 2003). It is because the density of these channels gradually increases from the soma to distal dendrites. In addition to the role of IA on feedback propagation, the channels play a critical role in feedforward, pre-to-post-synaptic transmission and integration by suppressing dendritic spikes (Cai et al., 2004; Kim et al., 2007; Jung et al., 2008; Kim and Hoffman, 2008; Yang et al., 2015). Therefore, a dysfunction of IA channels can lead to loss of control over neuronal excitability, the hallmark of several diseases in the brain. In this review, we discuss (1) the distribution (mostly in human) and function (in animals and human) of IA channels in the brain, (2) the common down-regulation of IA channels in several brain diseases and (3) a potential IA channel-based therapeutic target.
Subtypes of IA Channels
The potassium channels form a large and diverse family of ion channels that are involved in establishing the resting membrane potential, determining the action potential waveform, regulating neurotransmitter release, and modulating rhythmic firing patterns (Llinas, 1988; Joho and Hurlock, 2009). There are four major classes of potassium channels in the brain: Calcium-activated, inwardly rectifying, leak, and voltage-gated potassium channel. Particularly, IA channels, one type of voltage-gated potassium channels, are characterized by closed, opened, and inactivated channel states depending on the membrane voltage. IA channels form a large macromolecular complex comprising four ion-conducting alpha-subunits and beta-subunits (either being cytoplasmic or transmembrane) with auxiliary regulatory and supporting proteins (Leicher et al., 1998; Grizel et al., 2014). This voltage-gated potassium channels usually have a homotetrameric structure (with all alpha-subunits being identical), but some of them are heterotetrameric (with two or more non-identical alpha-subunits) (Sokolova, 2004; Grizel et al., 2014). The alpha-subunits (Kv1.4, Kv3.3, Kv3.4, Kv4.1, Kv4.2, and Kv4.3) that are divided into discrete families on the basis of sequence similarity form an ion pore and infrastructure of the channel. The alpha-subunits determine the fast kinetic property of IA channels, rapidly activating and inactivating (Coetzee et al., 1999; Covarrubias et al., 2008). The alpha-subunits are major components on which pharmacological agents target. Most generally used 4-aminopyridine (4-AP) not only blocks the subtypes of IA channels such as Kv1.4 and Kv4.x, but also other subtypes of voltage-gated potassium channels (Gutman et al., 2005). Alternatively, biotoxins isolated from venoms of the tarantula spider (PaTx1 and 2, HmTx1, HpTx1 and 2), sea anemone (BDS I and II) and scorpion (alpha-KTx15 subfamily) have relatively higher selectivity on the specific subtypes of IA channels (Sanguinetti et al., 1997; Diochot et al., 1999; Escoubas and Rash, 2004; Yeung et al., 2005; Prestipino et al., 2009). Plus, some components (Diclofenac and BmP02) have been identified to activate IA channels (Liu et al., 2005; Wu et al., 2016a,b).
Meanwhile, the beta-subunits and other auxiliary subunits of IA channels are known for modulating the biophysical properties and functions of IA channels (Hoffman and Johnston, 1998; Coetzee et al., 1999; An et al., 2000; Anderson et al., 2000; Varga et al., 2000; Beck et al., 2002; Yuan et al., 2002; Nadal et al., 2003; Gebauer et al., 2004). The alpha-subunit in the Kv1 complex interacts with the amino-terminal tetramerization domain of Kv beta-proteins which regulates the gating of the channels (Wulff et al., 2009). The Kv3 complex contains potassium voltage-gated channel subfamily E regulatory subunit 3 (KCNE3) which is known to carry a fast inactivating current (Wulff et al., 2009). The Kv4 complex has been associated with various ancillary subunits or scaffolding proteins including Kv beta-subunits (Aimond et al., 2005), dipeptidyl peptidase (DPP) family members (DPP6 and DPP10) (Nadal et al., 2003; Jerng et al., 2005) and K+ channel-interacting proteins (KChIP1, KChIP2, KChIP3, and KChIP4) (An et al., 2000; Morohashi et al., 2002). In particular, the KChIPs are required for function and formation of the Kv4 complex (Marionneau et al., 2009) while DPPs and beta-subunits contribute to the alteration of Kv4 currents (Yang et al., 2001; Jerng et al., 2005).
The activity and expression of IA channels can be modulated by certain post-translational modifications of phosphorylation and palmitoylation (Wulff et al., 2009) with various protein kinases such as Ca2+/calmodulin-dependent protein kinase II (CaMKII), cAMP-dependent protein kinase (PKA), protein kinase C (PKC), and mitogen-activated protein kinase (MAPK) (Jerng et al., 2004). CaMKII phosphorylation of Kv1.4 is reported to regulate the inactivation gate of IA channels arising from Kv1.4 (Roeper et al., 1997; Varga et al., 2004). Similarly, CaMKII phosphorylated Kv4.2 upregulates both the expression level of Kv4.2 proteins and the peak current of Kv4.2 (Roeper et al., 1997; Varga et al., 2004). Meanwhile, either PKA or PKC activation decreases the opening of IA channels encoded mostly by Kv4.2; accordingly, their activation increases the amplitude of bAPs in distal dendrites (Hoffman and Johnston, 1998). In other studies, PKC reduces inactivation of IA channels encoded by Kv3.3 and Kv3.4 (Covarrubias et al., 1994). Thus, the complex structure of IA channels likely determines their class and functions.
In this review, an attention is also made to (1) the microscopic expression pattern and (2) kinetics of Kv1.4 (kcna4), Kv3.3 (kcnc3), Kv3.4 (kcnc4), Kv4.1 (kcnd1), Kv4.2 (kcnd2), and Kv4.3 (kcnd3) which are the most common alpha-subunits of IA channels (Coetzee et al., 1999; Covarrubias et al., 2008). Firstly, as for the IA expression pattern in a neuron, pioneering works for characteristic IA channels on the dendrites show that 4-AP (a non-selective IA blocker)-sensitive IA channels are distributed along the apical dendritic truck and tuft of L5B pyramidal neurons in mice (Bekkers, 2000; Korngreen and Sakmann, 2000; Harnett et al., 2013). Specifically, Kv1.4 and Kv3.4 channels are largely localized in axons and dendrites (Veh et al., 1995; Serodio and Rudy, 1998) while Kv3.3 is mostly located in distal dendrites (Veh et al., 1995; Kim et al., 2005; Chang et al., 2007). Also, Kv4 family such as Kv4.1, Kv4.2, Kv4.3 is primarily found in somatodendritic membrane although Kv4.2 channels are highly concentrated in dendrites (Coetzee et al., 1999; Rudy and McBain, 2001; Song, 2002; Kim et al., 2005; Kerti et al., 2012). Secondly, Kv3 subfamily (encoding Kv3.1, Kv3.2, Kv3.3, and Kv3.4) has distinct functional properties; they activate at high thresholds (-10 mV) with rapid kinetics (Rudy and McBain, 2001; Zhao et al., 2013). In fact, homomeric Kv3.1 and Kv3.2 channels have slower inactivation kinetics than heteromultimeric Kv3.1/Kv3.4 and Kv3.2/Kv3.4 channels carrying a fast inactivating component (Rudy and McBain, 2001). Also, Kv4 channels show the fast recovery from inactivation state, a hallmark of somatodendritic IA channels. In general, IA channels respond transiently to stimuli, exhibiting a rapidly activating and inactivating kinetic. They affect the excitability and firing properties of neurons via regulatory actions of their pore-forming alpha-subunits, each of which has slightly different sensitivity to voltage changes (Locke and Nerbonne, 1997; Carrasquillo et al., 2012).
Close Association of IA With Several Brain Diseases
Subtypes of IA channels are differentially distributed in the brain. The macroscopic expression pattern of IA channels is now investigated with human brain where clinical value can be highlighted. Kv1.4 is predominantly expressed in prefrontal cortex, cerebellum peduncles, pituitary and pineal gland in a ranking order; kv3.3 in prefrontal cortex, pineal gland, pituitary and cerebellum peduncles; kv3.4 in cerebellum peduncles, pituitary, prefrontal cortex and pineal gland; kv4.1 in pineal gland, prefrontal cortex, pituitary and cerebellum peduncles; kv4.2 in cerebellum peduncles, cerebellum, prefrontal cortex and hypothalamus; kv4.3 in subthalamic nucleus, pineal gland, cerebellum peduncles and prefrontal cortex (Figure 1 and Table 1). Furthermore, there are significant functional consequences of IA on brain disorders (Table 2). Interestingly, the mRNA expression of IA channels is largely downregulated in the condition of brain dysfunction (Table 3). In the following sections, we discuss the tight involvement of IA channels in brain diseases, such as Alzheimer’s disease, epilepsy, fragile X syndrome (FXS), Parkinson’s disease, chronic pain, tinnitus and ataxia.
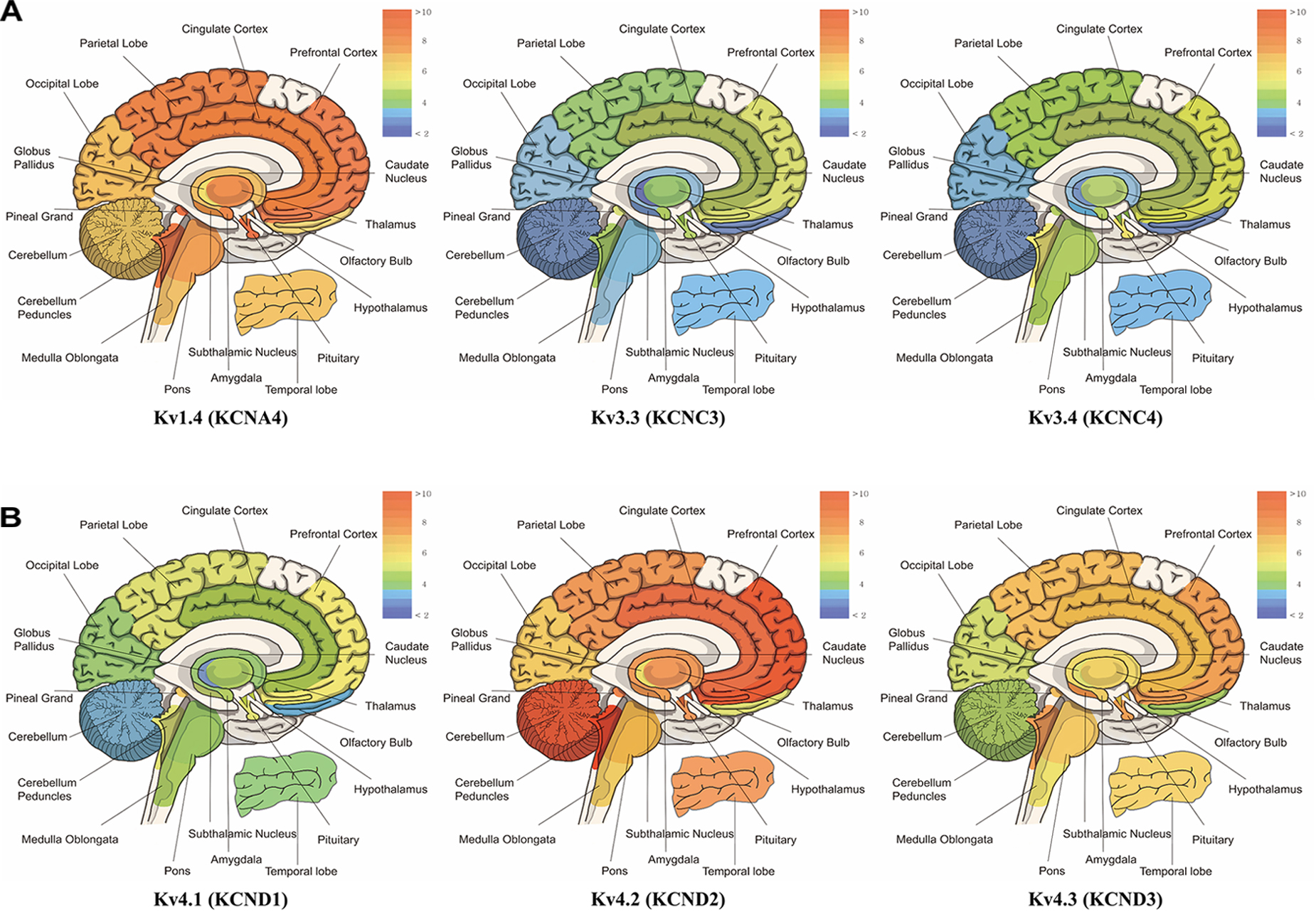
Figure 1. mRNA expression levels of panel (A) kcna4 (Kv1.4), kcnc3 (Kv3.3), and kcnc4 (Kv3.4), panel (B) kcnd1 (Kv4.1), kcnd2 (Kv4.2), and kcnd3 (Kv4.3). The microarray data have the mean value of each probe-set data obtained from BIOGPS (http://www.biogps.org), showing the tissue-specific pattern of mRNA expression according to GeneAtlas U133A, gcrma (Su et al., 2004). Note that expression of Kv1.4 is elevated in prefrontal cortex, cerebellum peduncles, pituitary and pineal gland; Kv3.3 in prefrontal cortex, pineal gland, pituitary, cerebellum peduncles; Kv3.4 in cerebellum peduncles, pituitary, prefrontal cortex and pineal gland; Kv4.1 in pineal gland, prefrontal cortex, pituitary and cerebellum peduncles; Kv4.2 in cerebellum peduncles, cerebellum, and prefrontal cortex and hypothalamus; Kv4.3 in subthalamic nucleus, pineal gland, cerebellum peduncles, pituitary and prefrontal cortex (when only top 25% of expression levels is considered).
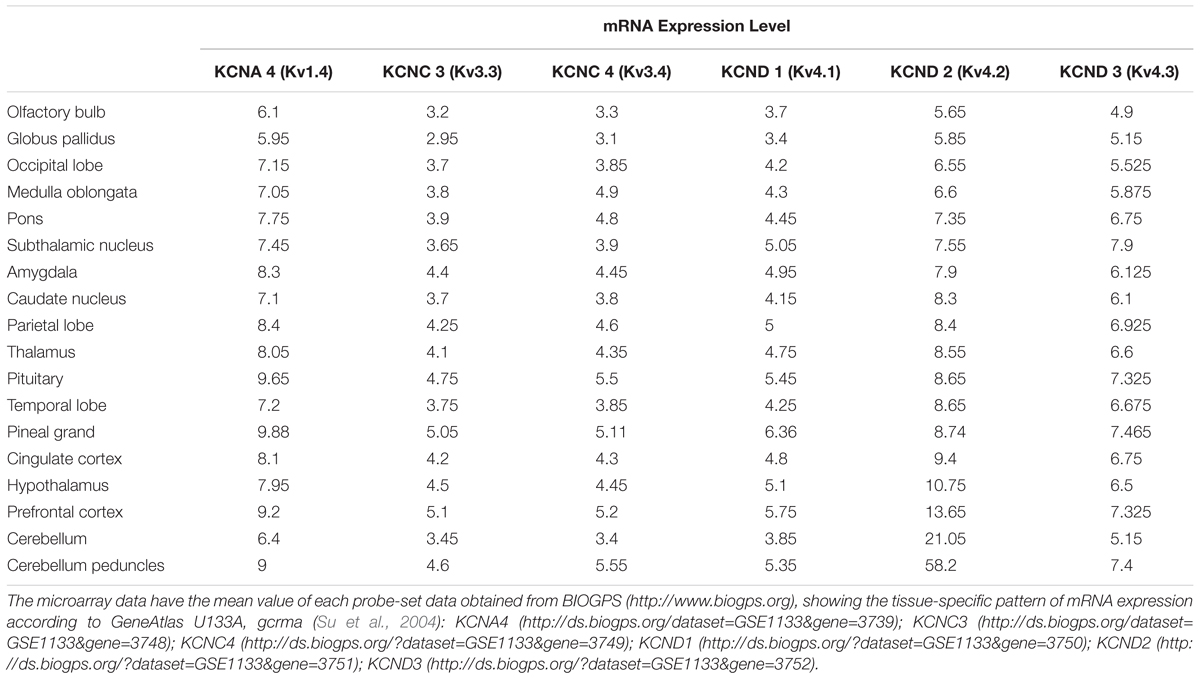
Table 1. mRNA expression levels of each IA channel subunit on the human brain.
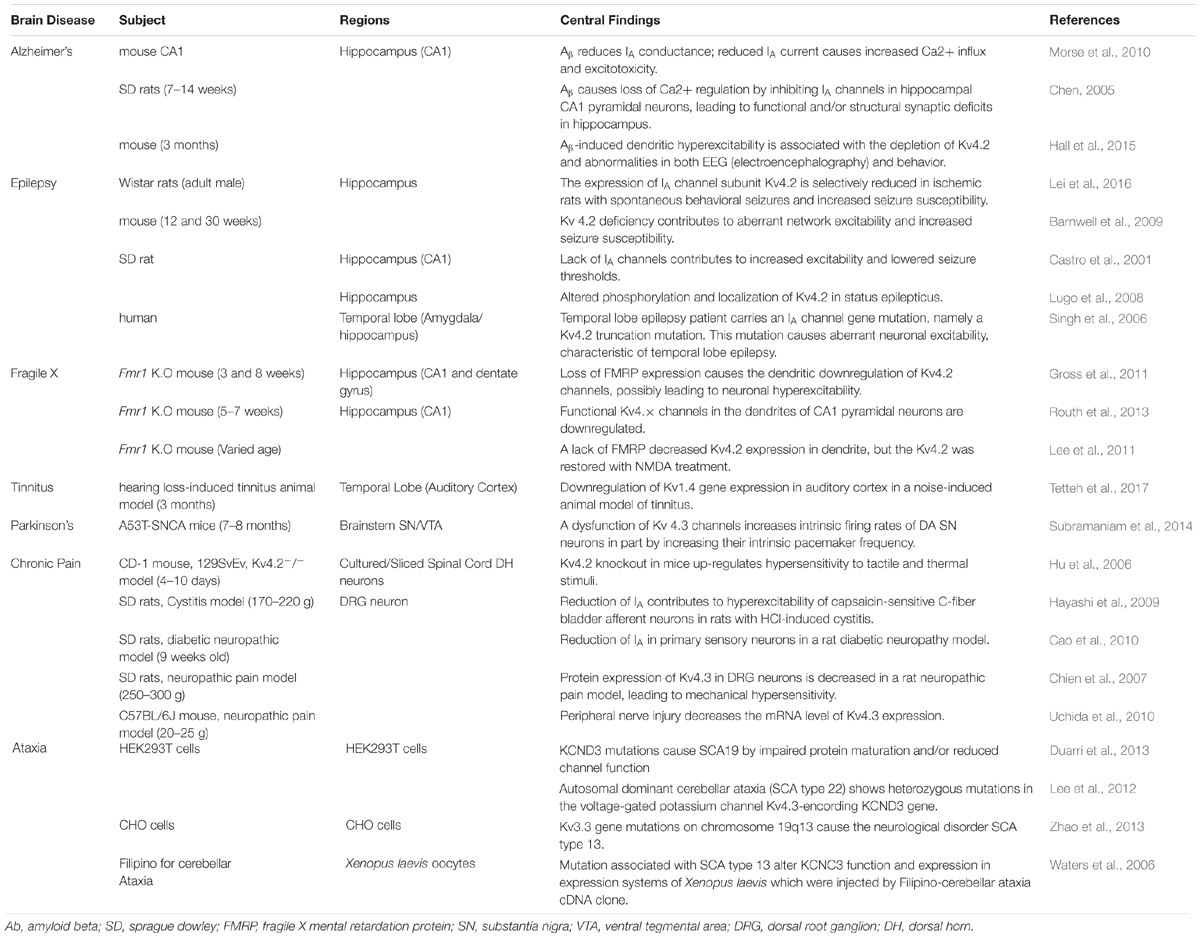
Table 2. The close association of IA channel activity and expression in brain diseases.
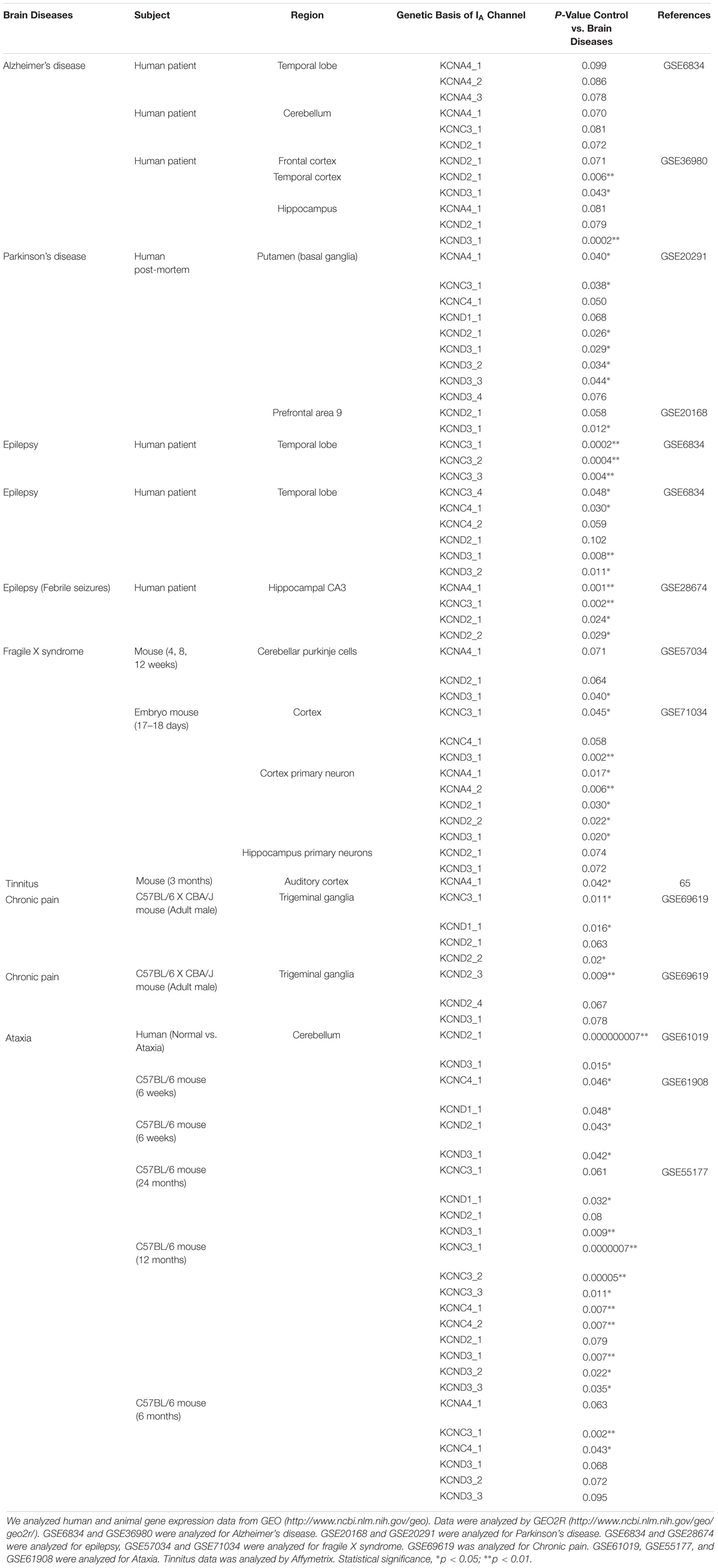
Table 3. Effects of IA channel mRNA expression in brain diseases.
Alzheimer’s Disease
Alzheimer disease (AD) is a neurodegenerative disease characterized by progressive deterioration of cognitive function. The early symptoms of AD are found in language, perception, movement, and memory. In the final phase, it causes widespread neuronal death, leading to severe memory loss, emotional disturbance and language dysfunction. Thus, patients’ independent living is impossible and relies on caregivers (Chen, 2005). One potential cause of AD appears to be the abnormal folding of the proteins Tau and/or Amyloid beta (Aβ) (Angulo et al., 2004). Interestingly, IA channels are downregulated with the plaque of Tau and/or Amyloid beta (Aβ), as reported in a study of cultured hippocampal neurons from an AD animal model (Hokama et al., 2014). This is further evidenced by other studies that Aβ-induced dendritic hyperexcitability is known to arise from a sustained increase in intracellular Ca2+ and dysfunction of IA channels in dendrites on hippocampal CA1 neurons of rodents (Chen, 2005; Morse et al., 2010; Hall et al., 2015). The loss of Ca2+ homeostasis in dendrites is attributed to abnormally enhanced back-propagation action potentials (bAPs) resulting from dysfunction of IA channels. In other words, the dysfunction of IA channels induced by Aβ misfolding and aggregation results in excessive dendritic Ca2+ influx, leading to the subsequent destruction of synapses and ultimately cell death. In contrast, a study reported that up-regulation of the kv3.4 channel (but not caused by Aβ deposition) causes alterations of the neuron activity in early AD (Angulo et al., 2004). In any cases, it is likely that the alteration of IA channels disrupts synaptic function of neurons, being involved in the clinical manifestation of AD.
Epilepsy
Epilepsy is characterized by repeated convulsive seizures. Seizures are known to be caused by excessive excitation or suppressed inhibition in neural networks (Yang and Cox, 2008, 2011; Park et al., 2018). One-third of epilepsy is a generalized seizure often accompanied by loss of consciousness, affecting the entire brain. Two-thirds of epilepsy occurs as focal seizures. Then, it often proceeds into the generalized seizures (Appleton et al., 2012). Epileptic seizure occurs as the result of environmental factors such as brain injury, stroke, brain tumors and infections, and genetic factors. Also, many of the patients are often accompanied by psychological symptoms such as anxiety and depression (Engel, 2001; Vadlamudi et al., 2003; Berkovic et al., 2006). In any case, epileptic seizures are strongly associated with the downregulation of IA channels (Tables 2, 3). In fact, the drugs that downregulate the activity of IA channels have been recognized as pro-convulsants and, therefore, inhibiting the activity and expression of IA channels is the well-established models of epilepsy (Bernard et al., 2004; Su et al., 2008). Indeed, in adult Wistar rats with spontaneous behavioral seizures, the expression of Kv4.2 is decreased in the hippocampus (Lei et al., 2016). Also, in a seizure induction study, pregnant rats were infused with a neurotoxin, MAM (methylazoxymethanol). This neurotoxin reduced DNA synthesis and caused neuronal heterotopia in the hippocampus of the rat fetuses. Postnatally, their hippocampal neurons showed IA channels (encoded by Kv4.2) dysfunctional and exhibited hyperexcitability. As a result, their seizure thresholds were lower than those in normal rats (Castro et al., 2001). With status epilepticus induced by kainic acid, the rat model is associated with the activation of extracellular signal-regulated kinase (ERK), an enzyme that causes the structural and functional impairment of dendritic Kv4.2-IA channels (Lugo et al., 2008). Apart from the expression level change of Kv4.2 in animal seizure model, the hippocampus of Kv4.2 knockout (KO) mice increases the sensitivity to convulsion-inducing stimulation compared to that in wild type mice, indicating the depletion of Kv4.2 is associated with deviant network excitability and increased seizure susceptibility (Barnwell et al., 2009). Consistent with these findings in animals, a study with surgical tissue from patients with hippocampal sclerosis-induced epilepsy showed that the dendritic regions of the hippocampus exhibit the decreased expression of Kv4.2. Similarly, the patient with one temporal lobe epilepsy carried a gene mutation of Kv4.2 channels (Singh et al., 2006; Aronica et al., 2009). Thus, these results suggest that the functional and/or structural reduction of IA channels is associated with abnormal neuronal excitability and epilepsy in the temporal lobe in both animals and humans.
Fragile X Syndrome
Fragile X syndrome (FXS) is the most common heritable mental disability. In general, the person with FXS has a long, narrow face, big ears and flexible fingers, often having autistic behaviors. FXS is associated with the dysfunction of the protein fragile X mental retardation protein (FMRP) encoded by a gene FMR1 on the X chromosome, a regulator of protein synthesis in axons and dendrites (Deng et al., 2011). FMRP possessing multiple RNA binding domains binds to polyribosome complexes and regulates protein synthesis (Ashley et al., 1993; Siomi et al., 1993; Brown et al., 2001; Greenough et al., 2001; Li et al., 2001; Todd et al., 2003; Darnell et al., 2005). Loss of FMRP causes a variety of symptoms such as sensory hypersensitivity and repetitive/excessive behavior which are hallmarks of FXS (Musumeci et al., 1999; Bureau et al., 2008; Bhakar et al., 2012; Yang et al., 2014b; Zhang et al., 2014). Recent findings show that FMRP is associated with the expression and activation of Kv4.2 channels in the mouse hippocampus (Table 2). There are two different conclusions on the interactional relation between FMRP and Kv4.2 channels. On one hand, in an fmr1 KO mouse model, the dendritic protein level of Kv4.2 in the hippocampus was found to be reduced, possibly causing neural hyperexcitability which underlies a plausible mechanism of FXS (and its associated epilepsy) (Gross et al., 2011). Also, the finding is supported by a following study with fmr1 KO mice that Kv4.x-mediated currents are reduced in dendrites, but not soma, of hippocampal CA1 pyramidal neurons, and accordingly the amplitude of back-propagation action potentials (bAPs) is increased in distal dendrites (Routh et al., 2013). On the sharp contrary, the local translation of Kv4.2 in CA1 dendrites increases in fmr1 KO mice, indicating that FMRP is a negative controller of dendritic Kv4.2 (Lee et al., 2011). In any cases, dendritic IA channels have been found important in FXS. Further studies are required to explore the dynamic role of dendritic IA channels in FXS.
Parkinson’s Disease
Parkinson’s disease (PD) is one of the most common neurodegenerative disorders characterized by an impairment of dopaminergic systems. Patients who suffer from PD have problems with voluntary behavior and thinking. Dementia and emotional problems such as depression and anxiety can be gradually developed over time. The cause of PD has several candidates: (1) environmental factors such as exposure to pesticides and a history of head injury and (2) genetic factors related to certain genes including SNCA, LRRK2, GNA, PRKN, PINK1, PARK7, VPS35, EIF4G1, DNAJC13, and CHCHD2 (Kalia and Lang, 2015). The pathological hallmark of PD is known to be the loss of dopaminergic neurons in the substantia nigra (SN). Another pathological feature of PD is the formation of protein aggregates called Lewy bodies (Davie, 2008). Furthermore, PD is also associated with dysfunction of IA channels (Lawson, 2000). For instance, the inhibition of Kv4.3 channels increases the spontaneous activity of nigral dopamine (DA) neurons and disrupts DA release, ultimately inducing the onset of PD (Subramaniam et al., 2014). In the research, the enhanced neuronal firing was observed only in DA neurons of the SN, but not in DA neurons of the ventral tegmental area. These animal studies are consistent with findings that IA expression is altered in PD patients, as shown in Table 3.
Chronic Pain
Chronic pain severely affects most daily activities, disrupting social life with financial burden (Breivik et al., 2006; Bouhassira et al., 2008; Henschke et al., 2015). Two symptoms dominate the disease: (1) inflammatory pain caused by a tissue reaction to injury or infection and (2) neuropathic pain from nerve injury, which prevails after the primary damage has healed (Woolf and Salter, 2000). At the root of chronic pain lies the activity of sensory neurons in the dorsal root ganglion (DRG) and spinal dorsal horn (DH). These neurons are responsible for transmitting nociceptive information from the periphery to the brain. Chronic pain is largely associated with neuronal hyperexcitability of the DRGs and DH neurons. Interestingly, several lines of evidence indicate that these neurons’ hyperexcitability is caused by reduced inhibitory currents and/or expression of IA channels in the DRGs and DH neurons (see Table 2). For example, in a rat diabetic neuropathy model, the reduction of 4-aminopyridine (4-AP)-sensitive IA currents and the decreased mRNA expression level of IA alpha-subunits; Kv1.4, Kv4.2, and Kv4.3 in the medium and large-diameter DRG neurons leads to the hyperexcitability of the neurons through BDNF activity on TrkB receptor (Cao et al., 2010). In addition, by peripheral nerve injury resulting in neuropathic pain, hypoacetylated histone H4 at the location of Kv4.2-NRSE is associated with the decreased mRNA expression of Kv4.3 in mouse DRG neurons, and the reduction of Kv4.3 is implicated in neuronal hyperexcitability (Kim et al., 2002; Uchida et al., 2010). Finally, DRGs in a neuropathic rat model show reduced Kv4.3 protein expression after spinal nerve ligation to DRGs, which drives mechanical hypersensitivity (Chien et al., 2007). Apart from DRGs, neurons in the DH of animals with formalin-induced inflammatory chronic pain are Kv4.2-deficient and exhibit increased excitability, resulting in increased sensitivity to tactile and thermal stimuli (Hu et al., 2006). Moreover, in a rat model of cystitis with visceral hypersensitivity, the decrease in 4-aminopyridine (4-AP)-sensitive IA current as well as the decreased expression of Kv1.4 channels are observed in DH neurons, leading to neuronal hyperexcitability (Hayashi et al., 2009). In fact, the role of IA channels in chronic pain has so far been studied mostly in the DRGs and DH neurons. It would be of great interest to investigate the role of IA channels along the somatosensory pathway.
Tinnitus
Tinnitus is a ringing in the ear that can be perceived even in the absence of external acoustic stimuli. Tinnitus is induced mainly by environmental factors such as traumatic noise, aging and ear infection. Environmental damage to cochlear hair cells alters the activity of the neural networks along the ascending auditory pathway, increasing the ratio of excitation to inhibition in central auditory neurons. Our group previously reported that auditory neurons in animals with noise-induced tinnitus exhibit intrinsic hyperexcitability. Also, in our preliminary data, noise-induced tinnitus is probably associated with the downregulation of Kv1.4 channels in rat auditory neurons (Yang et al., 2012; Tetteh et al., 2017). Thus, we believe this hyperexcitability to be due to the abnormal expression and/or function of IA channels (Table 2).
Ataxia
Ataxia is a neurodegenerative disorder caused by neural atrophy along the cerebellum-spine axis. Patients suffer from movement discoordination such as progressive ataxic gait and limb movements as well as difficulties with speech and eye movements (Duarri et al., 2013). Ataxia has three types depending on the parts of the dysfunction: (1) cerebellar ataxia due to dysfunction of the cerebellum; (2) sensory ataxia due to dysfunction of the sensory system including dorsal columns of the spinal cord, thalamus and parietal lobes; (3) vestibular ataxia due to dysfunction of the vestibular system. The main causes of ataxia are the focal lesion of corresponding CNS region, the exogenous substance like ethanol and vitamin b12 deficiency. Besides, genetic factors affect the degeneration of the cerebellum and/or the spine; 22 genes related to spinocerebellar ataxia (SCA) have been identified so far (Durr, 2010; Sailer and Houlden, 2012). The mRNA of Kv4.3 channels is highly expressed in the cerebellum of humans (Figure 1). Indeed, in animals and human patients with ataxia (Table 3), Kv4.3 channel mRNA expression is altered. For example, SCA type 19 and 22 are caused by mutation of Kv4.3 gene (Duarri et al., 2012, 2015; Lee et al., 2012; Table 2). In addition to Kv4.2 channels, Kv3.3 is the causative gene of SCA type 13, an autosomal dominant neurological disorder (Zhao et al., 2013). There are lines of evidence that SCA type 13 is caused by point mutations in the coding region of the Kv3.3 gene on chromosome 19q13 (Herman-Bert et al., 2000; Waters et al., 2005, 2006; Zhao et al., 2013). Moreover, the expression and function of Kv3.3 channels are altered in CHO cells transfected with mutated SCA13 as well as mRNA expression of Kv3.3 in animal with cerebellar ataxia (Herman-Bert et al., 2000; Waters et al., 2005, 2006; Zhao et al., 2013); Table 3). Taken all together, the alteration of IA channels is deeply involved in several types of ataxia.
Cellular Mechanisms and Therapeutic Targets of IA
IA channels and Ca2+-permeable channels including NMDA receptors (NMDARs) and voltage-gated calcium channels (VGCCs) are abundant in dendrites, and their interaction determines the level of dendritic excitability and synaptic plasticity (Frick et al., 2003; Cai et al., 2004; Losonczy and Magee, 2006; Kim et al., 2007; Jung et al., 2008; Losonczy et al., 2008; Wang et al., 2014; Yang et al., 2015). Ca2+ channels are kinetically slow to activate and inactivate (Figure 2A; Yang et al., 2015). Ca2+ dendritic spikes are regenerative and, thus, difficult to regulate once initiated. To efficiently suppress Ca2+-mediated hyperexcitability, an active filter is required just before it occurs. Rapid action of IA channels may play such a role as the active filter. For instance, NMDA spikes are mostly induced in distal dendrites of cortical neurons where most synaptic events occur (Larkum et al., 2009; Yang et al., 2011, 2014a, 2016). In turn, VGCCs magnify the NMDA-mediated dendritic spikes in apical dendrites, propagating the spikes to the axo-somatic sodium spikes zone for AP generation. Here, IA channels could suppress the anterograde propagation of local Na+- and Ca2+-mediated signals from dendrites as well as the backward spread of APs into dendrites (Losonczy and Magee, 2006; Losonczy et al., 2008). When IA channels are not active, the conductance of Ca2+-permeable channels can easily cause the membrane to slip toward depolarization, leading to synaptic enhancement in the axon terminals. In fact, internalization-induced reduction of IA channels enhances synaptic plasticity while increased IA channels do not display synaptic plasticity, suggesting a synaptic role of Ca2+-permeable channels heavily relying on the activity of IA channels (Kim et al., 2007; Jung et al., 2008). The activity of Ca2+-permeable channels which are inflated by inactive IA channels can often cause excessive neuronal excitability, resulting in an imbalance of excitation and inhibition in the whole network. We view this mechanism as a common cause for brain diseases such as Alzheimer’s disease, epilepsy, FXS, Parkinson’s disease, chronic pain, tinnitus and ataxia (Figure 2B).
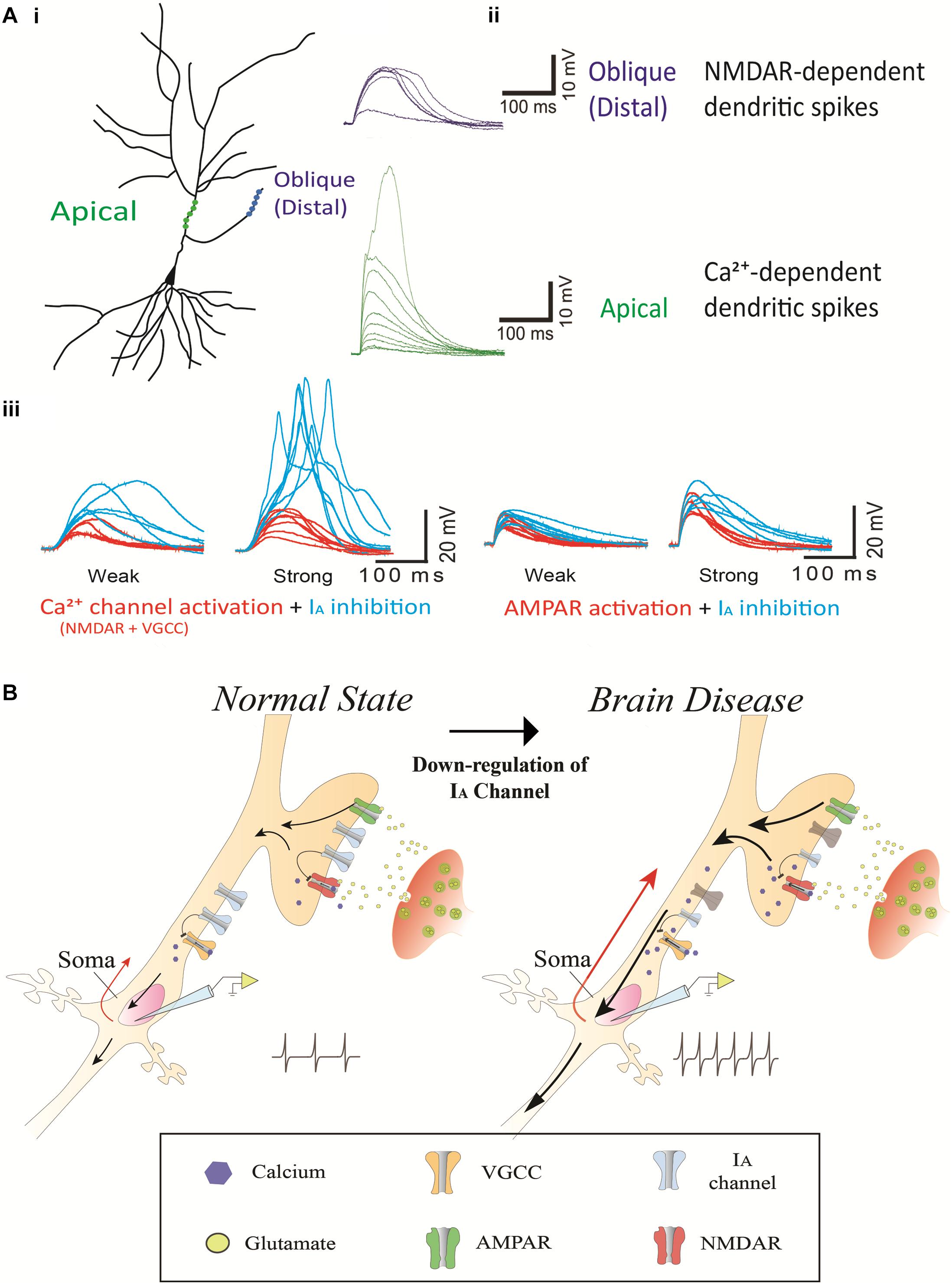
Figure 2. The contribution of IA channels on neuron excitability and brain disease. (Ai) A schematic of two photoactivated locations (distal and apical trunk) of a hippocampal neuron. (Aii) Examplar gluEPSPs (or glutamate-evoked EPSPs) in response to photoactivation in the distal and apical trunk. Note that there is a sudden increase in the response, being referred to as a NMDA-dependent dendritic spike and Ca2+-dependent dendritic spike in the distal and apical trunk, respectively. (Aiii) The blockage of 4-AP (3 mM) sensitive-IA channels potentiates the responses of NMDARs but not AMPARs. The left traces illustrate NMDAR (and VGCC)-mediated EPSPs, respectively, in a thin oblique dendrite or in an apical dendrite over an input strength; weak (averaged 6.3 μJ) and strong (averaged 11.2 μJ) energy. The right traces illustrate AMPAR-mediated EPSPs in a thin oblique dendrite over an input strength; weak (averaged 2.4 μJ) and strong (averaged 4.3 μJ) energy. (B) A putative model regarding the contribution of IA channels on neuron excitability and brain disease. It shows a molecular mechanism that dendritic impairment of IA channels can augment AP generation. The data was reused with permission from Yang et al. (2015).
Most drugs that are currently used to treat these diseases directly modulate ion channels and receptors that affect intrinsic and synaptic properties. However, as most of these ion channel-related drugs act across the whole brain, they produce a wide range of non-specific side effects, leading to the discontinuation of many potential treatments in the past. For instance, antagonists of NMDA receptors and voltage-sensitive sodium channels could in principle be used to suppress hyperexcitability. However, they also disrupt the physiological function of unintended brain areas. Furthermore, the drugs that activate GABA receptors also suppress other brain areas, often resulting in sedating and over-suppression effects that significantly reduce the quality of life. One way to circumvent these limitations of classical excitatory or inhibitory drugs would be to develop chemical compounds that “indirectly” modulate Na+ channels, NMDA or GABA receptors. A recent study reports that the application of 4-AP restores low- and high-threshold dendritic spikes in the distal and apical dendrites, respectively, which can be driven by Ca2+-permeable channels (NMDARs and/or Ni2+-sensitive voltage-gated Ca2+ channels) in rat hippocampal CA1 neurons (Yang et al., 2015). A similar study demonstrates that glutamate-mediated dendritic spikes are enhanced by 4-AP treatment and normalized by D-AP5 (an NMDA receptor antagonist) in mouse L3 pyramidal neuron (Biro et al., 2018). Thus, Ca2+-permeable channels and IA channels play a counterbalancing role in regulating neuronal excitability (Figure 2). In this respect, IA channels as an indirect modulator can be predominantly activated in a hyperexcitable condition caused by excessive activity of Ca2+-permeable channels, yet having minimal impact on the resting condition of neurons. Furthermore, given homeostatic regulation of neural networks, the lack of sensory or modulatory inputs likely causes the increment of excitation and/or decrement of inhibition in the affected neurons. These changes seem to be required for maintaining network excitability for neuron survival. In the hyperexcitable condition of the affected neurons often causing brain disorders, for example, an agonist of IA channels can function to modulate neuronal excitability by counteracting against Ca2+-permeable channels/receptors. Thus, IA channels as a therapeutic target may open an avenue for more sustainable treatments of brain disease with minimal interference in normal physiological function, leading to long-term clinical benefits. However, it is notable that there are limitations incurred when attempting to link the animal disease model to human neuropathology in the context of the expression and function of IA channels. It is currently unclear whether the suggested therapeutic mechanism of IA channels with animal disease models can apply to human diseases having a different neural network. Further research is required to delineate the precise contributions of IA channels to brain diseases in human.
Author Contributions
SgY and ScY designed, conceived, and wrote the manuscript. WN, GC, and SP collected the data, carried out data analysis, and prepared the manuscript. All authors reviewed the manuscript.
Funding
This work was supported by a grant 16172MFD340 from the Ministry of Food and Drug Safety in 2016 and the Incheon National University (International Cooperative) Research Grant for SgY, and the GRF grants (21104716 and 11102618) and the Health and Medical Research Fund (04150076) for ScY.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aimond, F., Kwak, S. P., Rhodes, K. J., and Nerbonne, J. M. (2005). Accessory Kvbeta1 subunits differentially modulate the functional expression of voltage-gated K+ channels in mouse ventricular myocytes. Circ. Res. 96, 451–458. doi: 10.1161/01.RES.0000156890.25876.63
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., Mendoza, G., et al. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556. doi: 10.1038/35000592
Anderson, A. E., Adams, J. P., Qian, Y., Cook, R. G., Pfaffinger, P. J., and Sweatt, J. D. (2000). Kv4.2 phosphorylation by cyclic AMP-dependent protein kinase. J. Biol. Chem. 275, 5337–5346.
Angulo, E., Noe, V., Casado, V., Mallol, J., Gomez-Isla, T., Lluis, C., et al. (2004). Up-regulation of the Kv3.4 potassium channel subunit in early stages of Alzheimer’s disease. J. Neurochem. 91, 547–557. doi: 10.1111/j.1471-4159.2004.02771.x
Appleton, R. E., Freeman, A., and Cross, J. H. (2012). Diagnosis and management of the epilepsies in children: a summary of the partial update of the 2012 NICE epilepsy guideline. Arch. Dis. Child. 97, 1073–1076. doi: 10.1136/archdischild-2012-302822
Aronica, E., Boer, K., Doorn, K. J., Zurolo, E., Spliet, W. G., van Rijen, P. C., et al. (2009). Expression and localization of voltage dependent potassium channel Kv4.2 in epilepsy associated focal lesions. Neurobiol. Dis. 36, 81–95. doi: 10.1016/j.nbd.2009.06.016
Ashley, C. T. Jr., Wilkinson, K. D., Reines, D., and Warren, S. T. (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566. doi: 10.1126/science.7692601
Barnwell, L. F., Lugo, J. N., Lee, W. L., Willis, S. E., Gertz, S. J., Hrachovy, R. A., et al. (2009). Kv4.2 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia 50, 1741–1751. doi: 10.1111/j.1528-1167.2009.02086.x
Beck, E. J., Bowlby, M., An, W. F., Rhodes, K. J., and Covarrubias, M. (2002). Remodelling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein. J. Physiol. 538(Pt. 3), 691–706. doi: 10.1113/jphysiol.2001.013127
Bekkers, J. M. (2000). Properties of voltage-gated potassium currents in nucleated patches from large layer 5 cortical pyramidal neurons of the rat. J. Physiol. 525(Pt. 3), 593–609. doi: 10.1111/j.1469-7793.2000.t01-1-00593.x
Bender, K. J., and Trussell, L. O. (2012). The physiology of the axon initial segment. Annu. Rev. Neurosci. 35, 249–265. doi: 10.1146/annurev-neuro-062111-150339
Berkovic, S. F., Mulley, J. C., Scheffer, I. E., and Petrou, S. (2006). Human epilepsies: interaction of genetic and acquired factors. Trends Neurosci. 29, 391–397. doi: 10.1016/j.tins.2006.05.009
Bernard, C., Anderson, A., Becker, A., Poolos, N. P., Beck, H., and Johnston, D. (2004). Acquired dendritic channelopathy in temporal lobe epilepsy. Science 305, 532–535. doi: 10.1126/science.1097065
Bhakar, A. L., Dolen, G., and Bear, M. F. (2012). The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443. doi: 10.1146/annurev-neuro-060909-153138
Biro, A. A., Bremaud, A., Falck, J., and Ruiz, A. J. (2018). A-type K+channels impede supralinear summation of clustered glutamatergic inputs in layer 3 neocortical pyramidal neurons. Neuropharmacology 140, 86–99. doi: 10.1016/j.neuropharm.2018.07.005
Bouhassira, D., Lanteri-Minet, M., Attal, N., Laurent, B., and Touboul, C. (2008). Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 136, 380–387. doi: 10.1016/j.pain.2007.08.013
Breivik, H., Collett, B., Ventafridda, V., Cohen, R., and Gallacher, D. (2006). Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur. J. Pain 10, 287–333. doi: 10.1016/j.ejpain.2005.06.009
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A., et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. doi: 10.1016/s0092-8674(01)00568-2
Bureau, I., Shepherd, G. M., and Svoboda, K. (2008). Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J. Neurosci. 28, 5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008
Cai, X., Liang, C. W., Muralidharan, S., Kao, J. P., Tang, C. M., and Thompson, S. M. (2004). Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 44, 351–364. doi: 10.1016/j.neuron.2004.09.026
Cao, X. H., Byun, H. S., Chen, S. R., Cai, Y. Q., and Pan, H. L. (2010). Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J. Neurochem. 114, 1460–1475. doi: 10.1111/j.1471-4159.2010.06863.x
Carrasquillo, Y., Burkhalter, A., and Nerbonne, J. M. (2012). A-type K+ channels encoded by Kv4.2, Kv4.3 and Kv1.4 differentially regulate intrinsic excitability of cortical pyramidal neurons. J. Physiol. 590, 3877–3890. doi: 10.1113/jphysiol.2012.229013
Castro, P. A., Cooper, E. C., Lowenstein, D. H., and Baraban, S. C. (2001). Hippocampal heterotopia lack functional Kv4.2 potassium channels in the methylazoxymethanol model of cortical malformations and epilepsy. J. Neurosci. 21, 6626–6634. doi: 10.1523/jneurosci.21-17-06626.2001
Chang, S. Y., Zagha, E., Kwon, E. S., Ozaita, A., Bobik, M., Martone, M. E., et al. (2007). Distribution of Kv3.3 potassium channel subunits in distinct neuronal populations of mouse brain. J. Comp. Neurol. 502, 953–972. doi: 10.1002/cne.21353
Chen, C. (2005). beta-Amyloid increases dendritic Ca2+ influx by inhibiting the A-type K+ current in hippocampal CA1 pyramidal neurons. Biochem. Biophys. Res. Commun. 338, 1913–1919. doi: 10.1016/j.bbrc.2005.10.169
Chien, L. Y., Cheng, J. K., Chu, D., Cheng, C. F., and Tsaur, M. L. (2007). Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J. Neurosci. 27, 9855–9865. doi: 10.1523/JNEUROSCI.0604-07.2007
Coetzee, W. A., Amarillo, Y., Chiu, J., Chow, A., Lau, D., McCormack, T., et al. (1999). Molecular diversity of K+ channels. Ann. N. Y. Acad. Sci. 868, 233–285.
Covarrubias, M., Bhattacharji, A., De Santiago-Castillo, J. A., Dougherty, K., Kaulin, Y. A., Na-Phuket, T. R., et al. (2008). The neuronal Kv4 channel complex. Neurochem. Res. 33, 1558–1567. doi: 10.1007/s11064-008-9650-8
Covarrubias, M., Wei, A., Salkoff, L., and Vyas, T. B. (1994). Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron 13, 1403–1412. doi: 10.1016/0896-6273(94)90425-1
Darnell, J. C., Fraser, C. E., Mostovetsky, O., Stefani, G., Jones, T. A., Eddy, S. R., et al. (2005). Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 19, 903–918. doi: 10.1101/gad.1276805
Davie, C. A. (2008). A review of Parkinson’s disease. Br. Med. Bull. 86, 109–127. doi: 10.1093/bmb/ldn013
Deng, P. Y., Sojka, D., and Klyachko, V. A. (2011). Abnormal presynaptic short-term plasticity and information processing in a mouse model of fragile X syndrome. J. Neurosci. 31, 10971–10982. doi: 10.1523/JNEUROSCI.2021-11
Diochot, S., Drici, M. D., Moinier, D., Fink, M., and Lazdunski, M. (1999). Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 126, 251–263. doi: 10.1038/sj.bjp.0702283
Duarri, A., Jezierska, J., Fokkens, M., Meijer, M., Schelhaas, H. J., den Dunnen, W. F., et al. (2012). Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann. Neurol. 72, 870–880. doi: 10.1002/ana.23700
Duarri, A., Lin, M. C., Fokkens, M. R., Meijer, M., Smeets, C. J., Nibbeling, E. A., et al. (2015). Spinocerebellar ataxia type 19/22 mutations alter heterocomplex Kv4.3 channel function and gating in a dominant manner. Cell. Mol. Life Sci. 72, 3387–3399. doi: 10.1007/s00018-015-1894-2
Duarri, A., Nibbeling, E., Fokkens, M. R., Meijer, M., Boddeke, E., Lagrange, E., et al. (2013). The L450F [Corrected] mutation in KCND3 brings spinocerebellar ataxia and Brugada syndrome closer together. Neurogenetics 14, 257–258. doi: 10.1007/s10048-013-0370-0
Durr, A. (2010). Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 9, 885–894. doi: 10.1016/S1474-4422(10)70183-6
Engel, J. Jr. (2001). Mesial temporal lobe epilepsy: what have we learned? Neuroscientist 7, 340–352. doi: 10.1177/107385840100700410
Escoubas, P., and Rash, L. (2004). Tarantulas: eight-legged pharmacists and combinatorial chemists. Toxicon 43, 555–574. doi: 10.1016/j.toxicon.2004.02.007
Frick, A., Magee, J., Koester, H. J., Migliore, M., and Johnston, D. (2003). Normalization of Ca2+ signals by small oblique dendrites of CA1 pyramidal neurons. J. Neurosci. 23, 3243–3250. doi: 10.1523/jneurosci.23-08-03243.2003
Gebauer, M., Isbrandt, D., Sauter, K., Callsen, B., Nolting, A., Pongs, O., et al. (2004). N-type inactivation features of Kv4.2 channel gating. Biophys. J. 86(1 Pt. 1), 210–223. doi: 10.1016/S0006-3495(04)74097-7
Golding, N. L., Kath, W. L., and Spruston, N. (2001). Dichotomy of action-potential backpropagation in CA1 pyramidal neuron dendrites. J. Neurophysiol. 86, 2998–3010. doi: 10.1152/jn.2001.86.6.2998
Greenough, W. T., Klintsova, A. Y., Irwin, S. A., Galvez, R., Bates, K. E., and Weiler, I. J. (2001). Synaptic regulation of protein synthesis and the fragile X protein. Proc. Natl. Acad. Sci. U.S.A. 98, 7101–7106. doi: 10.1073/pnas.141145998
Grizel, A. V., Glukhov, G. S., and Sokolova, O. S. (2014). Mechanisms of activation of voltage-gated potassium channels. Acta Naturae 6, 10–26.
Gross, C., Yao, X., Pong, D. L., Jeromin, A., and Bassell, G. J. (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J. Neurosci. 31, 5693–5698. doi: 10.1523/JNEUROSCI.6661-10
Gutman, G. A., Chandy, K. G., Grissmer, S., Lazdunski, M., McKinnon, D., Pardo, L. A., et al. (2005). International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 57, 473–508. doi: 10.1124/pr.57.4.10
Hall, A. M., Throesch, B. T., Buckingham, S. C., Markwardt, S. J., Peng, Y., Wang, Q., et al. (2015). Tau-dependent Kv4. 2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J. Neurosci. 35, 6221–6230. doi: 10.1523/JNEUROSCI.2552-14.2015
Harnett, M. T., Xu, N. L., Magee, J. C., and Williams, S. R. (2013). Potassium channels control the interaction between active dendritic integration compartments in layer 5 cortical pyramidal neurons. Neuron 79, 516–529. doi: 10.1016/j.neuron.2013.06.005
Hayashi, Y., Takimoto, K., Chancellor, M. B., Erickson, K. A., Erickson, V. L., Kirimoto, T., et al. (2009). Bladder hyperactivity and increased excitability of bladder afferent neurons associated with reduced expression of Kv1.4 alpha-subunit in rats with cystitis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R1661–R1670. doi: 10.1152/ajpregu.91054.2008
Henschke, N., Kamper, S. J., and Maher, C. G. (2015). The epidemiology and economic consequences of pain. Mayo Clin. Proc. 90, 139–147. doi: 10.1016/j.mayocp.2014.09.010
Herman-Bert, A., Stevanin, G., Netter, J. C., Rascol, O., Brassat, D., Calvas, P., et al. (2000). Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3-q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation. Am. J. Hum. Genet. 67, 229–235. doi: 10.1086/302958
Hoffman, D. A., and Johnston, D. (1998). Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J. Neurosci. 18, 3521–3528. doi: 10.1523/jneurosci.18-10-03521.1998
Hoffman, D. A., Magee, J. C., Colbert, C. M., and Johnston, D. (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875. doi: 10.1038/43119
Hokama, M., Oka, S., Leon, J., Ninomiya, T., Honda, H., Sasaki, K., et al. (2014). Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb. Cortex 24, 2476–2488. doi: 10.1093/cercor/bht101
Hu, H. J., Carrasquillo, Y., Karim, F., Jung, W. E., Nerbonne, J. M., Schwarz, T. L., et al. (2006). The kv4.2 potassium channel subunit is required for pain plasticity. Neuron 50, 89–100. doi: 10.1016/j.neuron.2006.03.010
Jerng, H. H., Kunjilwar, K., and Pfaffinger, P. J. (2005). Multiprotein assembly of Kv4.2, KChIP3 and DPP10 produces ternary channel complexes with ISA-like properties. J. Physiol. 568(Pt. 3), 767–788. doi: 10.1113/jphysiol.2005.087858
Jerng, H. H., Pfaffinger, P. J., and Covarrubias, M. (2004). Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol. Cell. Neurosci. 27, 343–369. doi: 10.1016/j.mcn.2004.06.011
Joho, R. H., and Hurlock, E. C. (2009). The role of Kv3-type potassium channels in cerebellar physiology and behavior. Cerebellum 8, 323–333. doi: 10.1007/s12311-009-0098-4
Jung, S. C., Kim, J., and Hoffman, D. A. (2008). Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron 60, 657–671. doi: 10.1016/j.neuron.2008.08.029
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kerti, K., Lorincz, A., and Nusser, Z. (2012). Unique somato-dendritic distribution pattern of Kv4.2 channels on hippocampal CA1 pyramidal cells. Eur. J. Neurosci. 35, 66–75. doi: 10.1111/j.1460-9568.2011.07907.x
Kim, D. S., Choi, J. O., Rim, H. D., and Cho, H. J. (2002). Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res. Mol. Brain Res. 105, 146–152. doi: 10.1016/s0169-328x(02)00388-1
Kim, J., and Hoffman, D. A. (2008). Potassium channels: newly found players in synaptic plasticity. Neuroscientist 14, 276–286. doi: 10.1177/1073858408315041
Kim, J., Jung, S. C., Clemens, A. M., Petralia, R. S., and Hoffman, D. A. (2007). Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 54, 933–947. doi: 10.1016/j.neuron.2007.05.026
Kim, J., Wei, D. S., and Hoffman, D. A. (2005). Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J. Physiol. 569(Pt. 1), 41–57. doi: 10.1113/jphysiol.2005.095042
Korngreen, A., and Sakmann, B. (2000). Voltage-gated K+ channels in layer 5 neocortical pyramidal neurones from young rats: subtypes and gradients. J. Physiol. 525(Pt. 3), 621–639. doi: 10.1111/j.1469-7793.2000.00621.x
Larkum, M. E., Nevian, T., Sandler, M., Polsky, A., and Schiller, J. (2009). Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science 325, 756–760. doi: 10.1126/science.1171958
Lawson, K. (2000). Is there a role for potassium channel openers in neuronal ion channel disorders? Expert Opin. Investig. Drugs 9, 2269–2280. doi: 10.1517/13543784.9.10.2269
Lee, H. Y., Ge, W. P., Huang, W., He, Y., Wang, G. X., Rowson-Baldwin, A., et al. (2011). Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. doi: 10.1016/j.neuron.2011.09.033
Lee, Y. C., Durr, A., Majczenko, K., Huang, Y. H., Liu, Y. C., Lien, C. C., et al. (2012). Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 72, 859–869. doi: 10.1002/ana.23701
Lei, Z., Zhang, H., Liang, Y., and Xu, Z. C. (2016). Reduced expression of IA channels is associated with post-ischemic seizures. Epilepsy Res. 124, 40–48. doi: 10.1016/j.eplepsyres.2016.05.008
Leicher, T., Bahring, R., Isbrandt, D., and Pongs, O. (1998). Coexpression of the KCNA3B gene product with Kv1.5 leads to a novel A-type potassium channel. J. Biol. Chem. 273, 35095–35101. doi: 10.1074/jbc.273.52.35095
Li, Z., Zhang, Y., Ku, L., Wilkinson, K. D., Warren, S. T., and Feng, Y. (2001). The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 29, 2276–2283. doi: 10.1093/nar/29.11.2276
Liu, L. Y., Fei, X. W., Li, Z. M., Zhang, Z. H., and Mei, Y. A. (2005). Diclofenac, a nonsteroidal anti-inflammatory drug, activates the transient outward K+ current in rat cerebellar granule cells. Neuropharmacology 48, 918–926. doi: 10.1016/j.neuropharm.2004.12.020
Llinas, R. R. (1988). The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science 242, 1654–1664. doi: 10.1126/science.3059497
Locke, R. E., and Nerbonne, J. M. (1997). Role of voltage-gated K+ currents in mediating the regular-spiking phenotype of callosal-projecting rat visual cortical neurons. J. Neurophysiol. 78, 2321–2335. doi: 10.1152/jn.1997.78.5.2321
Losonczy, A., and Magee, J. C. (2006). Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron 50, 291–307. doi: 10.1016/j.neuron.2006.03.016
Losonczy, A., Makara, J. K., and Magee, J. C. (2008). Compartmentalized dendritic plasticity and input feature storage in neurons. Nature 452, 436–441. doi: 10.1038/nature06725
Lugo, J. N., Barnwell, L. F., Ren, Y., Lee, W. L., Johnston, L. D., Kim, R., et al. (2008). Altered phosphorylation and localization of the A-type channel, Kv4.2 in status epilepticus. J. Neurochem. 106, 1929–1940. doi: 10.1111/j.1471-4159.2008.05508.x
Marionneau, C., LeDuc, R. D., Rohrs, H. W., Link, A. J., Townsend, R. R., and Nerbonne, J. M. (2009). Proteomic analyses of native brain K(V)4.2 channel complexes. Channels 3, 284–294.
Morohashi, Y., Hatano, N., Ohya, S., Takikawa, R., Watabiki, T., Takasugi, N., et al. (2002). Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. J. Biol. Chem. 277, 14965–14975. doi: 10.1074/jbc.M200897200
Morse, T. M., Carnevale, N. T., Mutalik, P. G., Migliore, M., and Shepherd, G. M. (2010). Abnormal excitability of oblique dendrites implicated in early Alzheimer’s: a computational study. Front. Neural Circuits 4:16. doi: 10.3389/fncir.2010.00016
Musumeci, S. A., Hagerman, R. J., Ferri, R., Bosco, P., Dalla Bernardina, B., Tassinari, C. A., et al. (1999). Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 40, 1092–1099. doi: 10.1111/j.1528-1157.1999.tb00824.x
Nadal, M. S., Ozaita, A., Amarillo, Y., Vega-Saenz, de Miera, E., Ma, Y., et al. (2003). The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron 37, 449–461. doi: 10.1016/s0896-6273(02)01185-6
Park, S. W., Kim, J., Kang, M., Lee, W., Park, B. S., Kim, H., et al. (2018). Epidural electrotherapy for epilepsy. Small 14, e1801732. doi: 10.1002/smll.201801732
Prestipino, G., Corzo, G., Romeo, S., Murgia, A. R., Zanardi, I., Gurrola, G. B., et al. (2009). Scorpion toxins that block transient currents (I(A)) of rat cerebellum granular cells. Toxicol. Lett. 187, 1–9. doi: 10.1016/j.toxlet.2009.01.027
Roeper, J., Lorra, C., and Pongs, O. (1997). Frequency-dependent inactivation of mammalian A-type K+ channel KV1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J. Neurosci. 17, 3379–3391. doi: 10.1523/jneurosci.17-10-03379.1997
Routh, B. N., Johnston, D., and Brager, D. H. (2013). Loss of functional A-type potassium channels in the dendrites of CA1 pyramidal neurons from a mouse model of fragile X syndrome. J. Neurosci. 33, 19442–19450. doi: 10.1523/JNEUROSCI.3256-13.2013
Rudy, B., and McBain, C. J. (2001). Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 24, 517–526. doi: 10.1016/s0166-2236(00)01892-0
Sailer, A., and Houlden, H. (2012). Recent advances in the genetics of cerebellar ataxias. Curr. Neurol. Neurosci. Rep. 12, 227–236. doi: 10.1007/s11910-012-0267-6
Sanguinetti, M. C., Johnson, J. H., Hammerland, L. G., Kelbaugh, P. R., Volkmann, R. A., Saccomano, N. A., et al. (1997). Heteropodatoxins: peptides isolated from spider venom that block Kv4.2 potassium channels. Mol. Pharmacol. 51, 491–498.
Serodio, P., and Rudy, B. (1998). Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J. Neurophysiol. 79, 1081–1091. doi: 10.1152/jn.1998.79.2.1081
Singh, B., Ogiwara, I., Kaneda, M., Tokonami, N., Mazaki, E., Baba, K., et al. (2006). A Kv4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol. Dis. 24, 245–253. doi: 10.1016/j.nbd.2006.07.001
Siomi, H., Siomi, M. C., Nussbaum, R. L., and Dreyfuss, G. (1993). The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74, 291–298. doi: 10.1016/0092-8674(93)90420-u
Sokolova, O. (2004). Structure of cation channels, revealed by single particle electron microscopy. FEBS Lett. 564, 251–256. doi: 10.1016/S0014-5793(04)00254-6
Song, W. J. (2002). Genes responsible for native depolarization-activated K+ currents in neurons. Neurosci. Res. 42, 7–14. doi: 10.1016/s0168-0102(01)00305-4
Su, A. I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K. A., Block, D., et al. (2004). A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067. doi: 10.1073/pnas.0400782101
Su, T., Cong, W. D., Long, Y. S., Luo, A. H., Sun, W. W., Deng, W. Y., et al. (2008). Altered expression of voltage-gated potassium channel 4.2 and voltage-gated potassium channel 4-interacting protein, and changes in intracellular calcium levels following lithium-pilocarpine-induced status epilepticus. Neuroscience 157, 566–576. doi: 10.1016/j.neuroscience.2008.09.027
Subramaniam, M., Althof, D., Gispert, S., Schwenk, J., Auburger, G., Kulik, A., et al. (2014). Mutant alpha-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J. Neurosci. 34, 13586–13599. doi: 10.1523/JNEUROSCI.5069-13.2014
Tetteh, H., Lee, M., Lau, C. G., Yang, S., and Yang, S. (2017). Tinnitus: prospects for pharmacological interventions with a seesaw model. Neuroscientist 24:107385841773341. doi: 10.1177/1073858417733415
Todd, P. K., Mack, K. J., and Malter, J. S. (2003). The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc. Natl. Acad. Sci. U.S.A. 100, 14374–14378. doi: 10.1073/pnas.2336265100
Uchida, H., Sasaki, K., Ma, L., and Ueda, H. (2010). Neuron-restrictive silencer factor causes epigenetic silencing of Kv4.3 gene after peripheral nerve injury. Neuroscience 166, 1–4. doi: 10.1016/j.neuroscience.2009.12.021
Vadlamudi, L., Scheffer, I. E., and Berkovic, S. F. (2003). Genetics of temporal lobe epilepsy. J. Neurol. Neurosurg. Psychiatry 74, 1359–1361.
Varga, A. W., Anderson, A. E., Adams, J. P., Vogel, H., and Sweatt, J. D. (2000). Input-specific immunolocalization of differentially phosphorylated Kv4.2 in the mouse brain. Learn. Mem. 7, 321–332. doi: 10.1101/lm.35300
Varga, A. W., Yuan, L. L., Anderson, A. E., Schrader, L. A., Wu, G. Y., Gatchel, J. R., et al. (2004). Calcium-calmodulin-dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A-type potassium currents. J. Neurosci. 24, 3643–3654. doi: 10.1523/JNEUROSCI.0154-04.2004
Veh, R. W., Lichtinghagen, R., Sewing, S., Wunder, F., Grumbach, I. M., and Pongs, O. (1995). Immunohistochemical localization of five members of the Kv1 channel subunits: contrasting subcellular locations and neuron-specific co-localizations in rat brain. Eur. J. Neurosci. 7, 2189–2205. doi: 10.1111/j.1460-9568.1995.tb00641.x
Wang, K., Lin, M. T., Adelman, J. P., and Maylie, J. (2014). Distinct Ca2+ sources in dendritic spines of hippocampal CA1 neurons couple to SK and Kv4 channels. Neuron 81, 379–387. doi: 10.1016/j.neuron.2013.11.004
Waters, M. F., Fee, D., Figueroa, K. P., Nolte, D., Muller, U., Advincula, J., et al. (2005). An autosomal dominant ataxia maps to 19q13: allelic heterogeneity of SCA13 or novel locus? Neurology 65, 1111–1113. doi: 10.1212/01.wnl.0000177490.05162.41
Waters, M. F., Minassian, N. A., Stevanin, G., Figueroa, K. P., Bannister, J. P., Nolte, D., et al. (2006). Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 38, 447–451. doi: 10.1038/ng1758
Woolf, C. J., and Salter, M. W. (2000). Neuronal plasticity: increasing the gain in pain. Science 288, 1765–1769.
Wu, B., Wu, B. F., Feng, Y. J., Tao, J., and Ji, Y. H. (2016a). Mapping the interaction anatomy of BmP02 on Kv1.3 channel. Sci. Rep. 6:29431. doi: 10.1038/srep29431
Wu, B., Zhu, Y., Shi, J., Tao, J., and Ji, Y. (2016b). BmP02 atypically delays Kv4.2 inactivation: implication for a unique interaction between scorpion toxin and potassium channel. Toxins 8:E280. doi: 10.3390/toxins8100280
Wulff, H., Castle, N. A., and Pardo, L. A. (2009). Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 8, 982–1001. doi: 10.1038/nrd2983
Yang, E. K., Alvira, M. R., Levitan, E. S., and Takimoto, K. (2001). Kvbeta subunits increase expression of Kv4.3 channels by interacting with their C termini. J. Biol. Chem. 276, 4839–4844. doi: 10.1074/jbc.M004768200
Yang, S., and Cox, C. L. (2008). Excitatory and anti-oscillatory actions of nitric oxide in thalamus. J. Physiol. 586, 3617–3628. doi: 10.1113/jphysiol.2008.153312
Yang, S., and Cox, C. L. (2011). Attenuation of inhibitory synaptic transmission by glial dysfunction in rat thalamus. Synapse 65, 1298–1308. doi: 10.1002/syn.20964
Yang, S., Emiliani, V., and Tang, C. M. (2014a). The kinetics of multibranch integration on the dendritic arbor of CA1 pyramidal neurons. Front. Cell. Neurosci. 8:127. doi: 10.3389/fncel.2014.00127
Yang, S., Papagiakoumou, E., Guillon, M., de Sars, V., Tang, C. M., and Emiliani, V. (2011). Three-dimensional holographic photostimulation of the dendritic arbor. J. Neural Eng. 8:046002. doi: 10.1088/1741-2560/8/4/046002
Yang, S., Santos, M. D., Tang, C. M., Kim, J. G., and Yang, S. (2016). A postsynaptic role for short-term neuronal facilitation in dendritic spines. Front. Cell. Neurosci. 10:224. doi: 10.3389/fncel.2016.00224
Yang, S., Su, W., and Bao, S. (2012). Long-term, but not transient, threshold shifts alter the morphology and increase the excitability of cortical pyramidal neurons. J. Neurophysiol. 108, 1567–1574. doi: 10.1152/jn.00371.2012
Yang, S., Tang, C. M., and Yang, S. (2015). The shaping of two distinct dendritic spikes by A-type voltage-gated K+ channels. Front. Cell. Neurosci. 9:469. doi: 10.3389/fncel.2015.00469
Yang, S., Yang, S., Park, J. S., Kirkwood, A., and Bao, S. (2014b). Failed stabilization for long-term potentiation in the auditory cortex of FMR1 knockout mice. PLoS One 9:e104691. doi: 10.1371/journal.pone.0104691
Yeung, S. Y., Thompson, D., Wang, Z., Fedida, D., and Robertson, B. (2005). Modulation of Kv3 subfamily potassium currents by the sea anemone toxin BDS: significance for CNS and biophysical studies. J. Neurosci. 25, 8735–8745. doi: 10.1523/JNEUROSCI.2119-05.2005
Yuan, L. L., Adams, J. P., Swank, M., Sweatt, J. D., and Johnston, D. (2002). Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J. Neurosci. 22, 4860–4868. doi: 10.1523/jneurosci.22-12-04860.2002
Zhang, Y., Bonnan, A., Bony, G., Ferezou, I., Pietropaolo, S., Ginger, M., et al. (2014). Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1-/y mice. Nat. Neurosci. 17, 1701–1709. doi: 10.1038/nn.3864
Keywords: A-type potassium channels, NMDA receptor, VGCC, dendritic spikes, brain disorders
Citation: Noh W, Pak S, Choi G, Yang S and Yang S (2019) Transient Potassium Channels: Therapeutic Targets for Brain Disorders. Front. Cell. Neurosci. 13:265. doi: 10.3389/fncel.2019.00265
Received: 29 November 2018; Accepted: 28 May 2019;
Published: 13 June 2019.
Edited by:
Sergey M. Korogod, National Academy of Sciences of Ukraine, UkraineReviewed by:
Arnaud J. Ruiz, UCL School of Pharmacy, United KingdomDarrin Brager, The University of Texas at Austin, United States
Copyright © 2019 Noh, Pak, Choi, Yang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sungchil Yang, sungchil.yang@cityu.edu.hk, yangs70@gmail.com; Sunggu Yang, abiyang9@gmail.com
†These authors have contributed equally to this work