Truncated Tau Induces Mitochondrial Transport Failure Through the Impairment of TRAK2 Protein and Bioenergetics Decline in Neuronal Cells
 Rodrigo A. Quintanilla
Rodrigo A. Quintanilla Carola Tapia-Monsalves
Carola Tapia-Monsalves Erick H. Vergara
Erick H. Vergara  María José Pérez
María José Pérez- Laboratory of Neurodegenerative Diseases, Facultad de Ciencias de la Salud, Instituto de Ciencias Biomédicas, Universidad Autónoma de Chile, Santiago, Chile
Mitochondria are highly specialized organelles essential for the synapse, and their impairment contributes to the neurodegeneration in Alzheimer’s disease (AD). Previously, we studied the role of caspase-3–cleaved tau in mitochondrial dysfunction in AD. In neurons, the presence of this AD-relevant tau form induced mitochondrial fragmentation with a concomitant reduction in the expression of Opa1, a mitochondrial fission regulator. More importantly, we showed that caspase-cleaved tau affects mitochondrial transport, decreasing the number of moving mitochondria in the neuronal processes without affecting their velocity rate. However, the molecular mechanisms involved in these events are unknown. We studied the possible role of motor proteins (kinesin 1 and dynein) and mitochondrial protein adaptors (RhoT1/T2, syntaphilin, and TRAK2) in the mitochondrial transport failure induced by caspase-cleaved tau. We expressed green fluorescent protein (GFP), GFP-full-length, and GPF-caspase-3–cleaved tau proteins in rat hippocampal neurons and immortalized cortical neurons (CN 1.4) and analyzed the expression and localization of these proteins involved in mitochondrial transport regulation. We observed that hippocampal neurons expressing caspase-cleaved tau showed a significant accumulation of a mitochondrial population in the soma. These changes were accompanied by evident mitochondrial bioenergetic deficits, including depolarization, oxidative stress, and a significant reduction in ATP production. More critically, caspase-cleaved tau significantly decreased the expression of TRAK2 in immortalized and primary hippocampal neurons without affecting RhoT1/T2 and syntaphilin levels. Also, when we analyzed the expression of motor proteins—Kinesin 1 (KIF5) and Dynein—we did not detect changes in their expression, localization, and binding to the mitochondria. Interestingly, the expression of truncated tau significantly increases the association of TRAK2 with mitochondria compared with neuronal cells expressing full-length tau. Altogether these results indicate that caspase-cleaved tau may affect mitochondrial transport through the increase of TRAK2–mitochondria binding and reduction of ATP production available for the process of movement of these organelles. These observations are novel and represent a set of exciting findings whereby tau pathology could affect mitochondrial distribution in neurons, an event that may contribute to synaptic failure observed in AD.
Introduction
Mitochondria are the mastermind organelles in charge of energy production, calcium regulation, and antioxidant defenses (reviewed in Pérez et al., 2018a). Mitochondria contribute to several synaptic processes, including neurotransmitter vesicle recycling, calcium buffering, dendritic spine formation, and neuronal plasticity (Sheng, 2017; Pérez et al., 2018a). Synaptic efficiency requires a perfect distribution and localization of the presynaptic and postsynaptic elements where mitochondria play a vital role (Macaskill and Kittler, 2010; Sheng, 2017).
Several neurodegenerative disorders, like Huntington disease (HD), Parkinson’s disease (PD), and Alzheimer’s disease (AD), showed deficiencies in axonal transport (De Vos et al., 2008). Also, accumulative studies showed several signs of mitochondrial impairment, including defects in bioenergetics, dynamics, and transport in different cells and mice models related to AD (Pérez et al., 2018b). AD is a neurodegenerative disease that is characterized by the formation of extracellular senile plaques and intraneuronal aggregates formed by abnormally modified tau protein (Mucke, 2009; Götz et al., 2019). Tau is a neuronal protein involved in the stabilization of microtubules, and its pathological modifications can also affect neuronal function, including axonal transport (Quinn et al., 2018; Tapia-Rojas et al., 2019). In this context, the genetic reduction of tau expression prevented the impairment of mitochondrial transport induced by the treatment of hippocampal neurons with Aβ peptide (Vossel et al., 2010, 2015). Also, the presence of a pathological tau modification such as hyperphosphorylation reduced mitochondrial transport through the axon (Rodríguez-Martín et al., 2013) by a mechanism that involved the impairment of different motor proteins (Shahpasand et al., 2012). Therefore, these findings indicate that tau could be a vital element involved in the regulation of mitochondrial transport in neurons.
Defects in mitochondrial health induced by pathological modifications of tau can also affect neuronal communication (Quintanilla et al., 2009, 2012, 2014; Manczak and Reddy, 2012; Pérez et al., 2018b). In this context, we studied the effects of caspase 3–cleaved tau, a relevant pathological modification in AD (de Calignon et al., 2010; Ozcelik et al., 2016), on mitochondrial function in neuronal cells. Expression of truncated tau affected mitochondrial health-inducing depolarization and mitochondrial fragmentation through the impairment of the mitochondrial fusion regulator Opa1 (Quintanilla et al., 2012, 2014; Pérez et al., 2018b). More interestingly, the expression of this cleaved tau form reduced mitochondrial movement in hippocampal neurons, suggesting that these actions could be relevant for the synaptic deficiency reported in AD (Quintanilla et al., 2012, 2014).
In neurons, mitochondrial transport is regulated for different cargo and adaptor proteins (Macaskill and Kittler, 2010; Sheng, 2017). Mitochondrial movement between axons and synaptic terminals is produced by the association of these organelles with motor proteins such as kinesin family proteins (KIFs) and dynein (Zinsmaier et al., 2009; Moore and Holzbaur, 2018). Dynein mediates the retrograde microtubular transport of several elements (from axon to soma; Zinsmaier et al., 2009; Sheng, 2017), while kinesin-1 proteins (knows as KIF5) participate in retrograde organelle transport (from neuronal soma to axon; Zinsmaier et al., 2009; Sheng, 2017). Kinesin-1 family members, also known as KIF5A, KIF5B, and KIF5C, are the main motors driving mitochondrial transport in neurons (Zinsmaier et al., 2009; Macaskill and Kittler, 2010; Ari et al., 2014; Sheng, 2017). Kinesin-1 is usually composed of two heavy kinesin chains (KHC), which contain the microtubule and nucleotide-binding domains and two kinesin light chains (KLC), which play a role in binding the cargo (Glater et al., 2006; Stokin and Goldstein, 2006). However, for mitochondrial transport, the KLC is replaced by mitochondrial accessory proteins such as Milton (also called TRAK1/2) and Miro (also called RhoT1/2 GTPases), both of which can interact directly with the KHC domain (Brickley et al., 2005). TRAK1/2 binds to KHC and also interacts with the mitochondrial membrane protein RhoT1/2 (Stowers et al., 2002; Reis et al., 2009; Zinsmaier et al., 2009). This interaction between TRAK1/2/ and RhoT1/2 ((López-Doménech et al., 2018) is essential for the axonal localization of mitochondria and for directing mitochondrial movement to the synaptic terminals according to the neuronal demands (Sheng, 2017). Also, for functional purposes, mitochondria need to remain stationary, while 20–30% of mitochondria are considered motile (Sheng, 2017). Moving mitochondria can also become stationary, and these organelles can be transported again in response to changes in bioenergetic status and synaptic activity (Misgeld and Schwarz, 2017). These actions are coordinated by syntaphilin, a protein that acts as a specific brake for axonal mitochondria (Kang et al., 2008), allowing these organelles to be anchored to microtubules (Misgeld and Schwarz, 2017).
Several reports indicate that axonal transport is impaired in AD, and this dysregulation contributes to neuronal dysfunction (Stokin and Goldstein, 2006; De Vos et al., 2008; Ari et al., 2014). For example, studies in AD brain samples showed that the expression of KIF5A, KIF1B, and KIF21B at gene and protein levels was significantly increased compared with age-matched controls (Hares et al., 2017). Also, KIF5A protein expression levels correlated inversely with the levels of APP and soluble Aβ in AD brains, indicating that these effects may be an adaptive response to impaired axonal transport in AD (Hares et al., 2017). However, it is unclear whether mitochondrial transport accessories proteins such as TRAK1/2, RhoT1/T2, and syntaphilin could be involved in mitochondrial movement deficiencies in the context of AD or any other neurodegenerative diseases.
Interestingly, several studies have shown that tau overexpression inhibits mitochondrial movement in various neuronal cell types (Stoothoff et al., 2009; Vossel et al., 2010). Also, studies by Shahpasand et al. (2012) showed that overexpression of wild-type tau or mutated forms that represent tau hyperphosphorylation in AT8 epitopes (Ser199, Ser202, and Thr205) reduced mitochondrial movement in neuronal cells. More importantly, our studies showed that the expression of caspase-cleaved tau (GFP-T4C3) in hippocampal neurons equally impairs anterograde/retrograde mitochondrial transport, reducing the number of moving mitochondrial elements compared to neurons that expressed full-length tau (Quintanilla et al., 2012, 2014). Even though the overexpression or pathological modifications of tau can affect mitochondrial transport, the mechanism behind these defects is unknown.
Therefore, we studied the events behind mitochondrial transport alterations induced by caspase-3–cleaved tau, on the principal components of the transport machinery present in hippocampal neurons (Figures 1–7). Expression of caspase-cleaved tau reduced mitochondrial localization in neuronal processes and induced different bioenergetic defects, including mitochondrial depolarization, increase of reactive oxygen species (ROS), and a decrease in ATP production. We investigated the expression levels of kinesin 1 and dynein in neuronal cells expressing full-length and truncated tau in immortalized cortical neurons with suppressed expression of tau (Bongarzone et al., 1998). Caspase-3–cleaved tau did not affect kinesin 1 and dynein expression and did not have any effects on the expression of mitochondrial accessory motor proteins such as RhoT1/T2 and syntaphilin. Interestingly, caspase-cleaved tau reduced the expression of TRAK2 in hippocampal neurons and immortalized cortical neurons. Finally, we analyzed the levels of TRAK2 and kinesin 1 in separate extracts of cytosol and mitochondria in cortical neurons transfected with green fluorescent protein (GFP) and the tau forms. These studies showed that in cells that expressed caspase-cleaved tau, TRAK2 is more present in the mitochondrial fraction compared with cells expressing full-length tau.
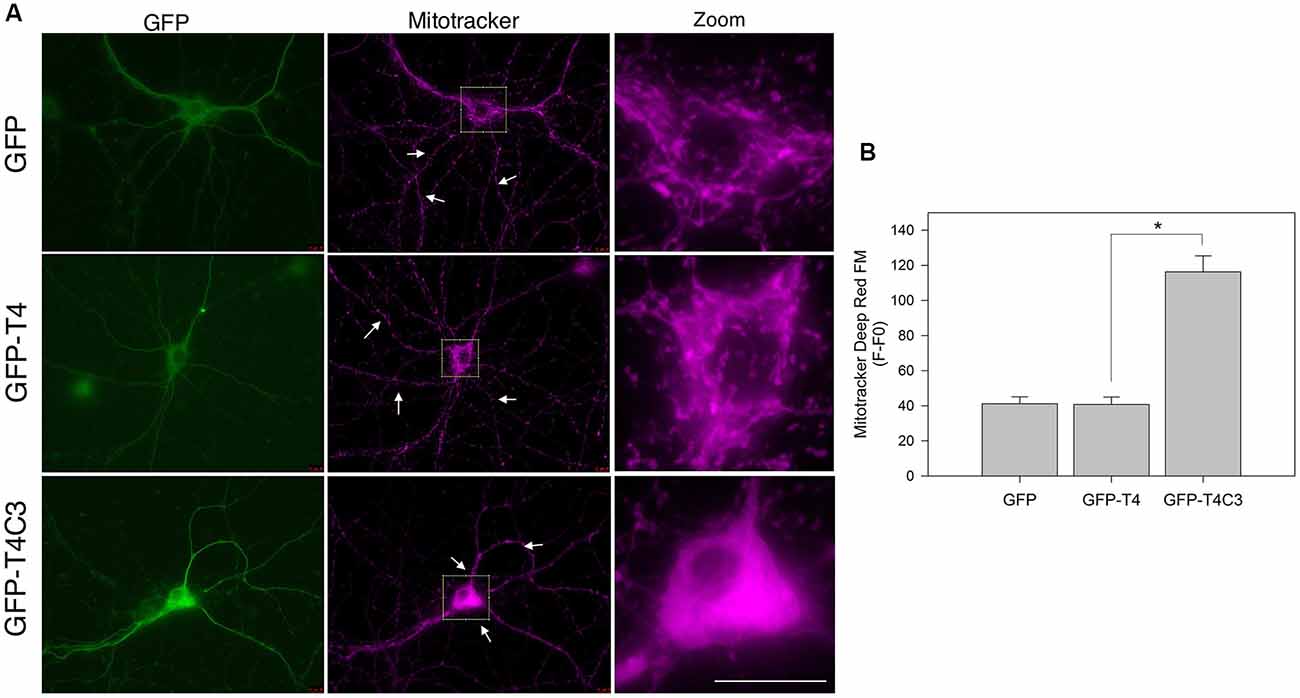
Figure 1. Expression of caspase-3–cleaved tau reduces mitochondrial localization in the neuronal processes of hippocampal neurons. (A) Representative images of rat hippocampal neurons transfected with green fluorescent protein (GFP), GFP-T4 (full-length tau), and GFP-T4C3 (caspase-cleaved tau) were loaded with MitoTracker Deep Red FM to determine mitochondrial localization. Arrows indicate isolated mitochondria in neurons. Zoom images shows a magnified were region of neuronal soma showing mitochondrial localization in this area. (B) Quantification of fluorescence intensity of MitoTracker Deep Red FM (F) per area minus background fluorescence (F0), which represents mitochondrial localization in the somatic region of hippocampal neurons. Caspase-cleaved tau increases mitochondrial localization in the soma compared with neurons expressing vector (GFP) or full-length tau (GFP-T4). Data are mean ± standard error (SE), n = 3. *p < 0.001 indicates differences between groups calculated by ANOVA test. Bar = 20 μm.
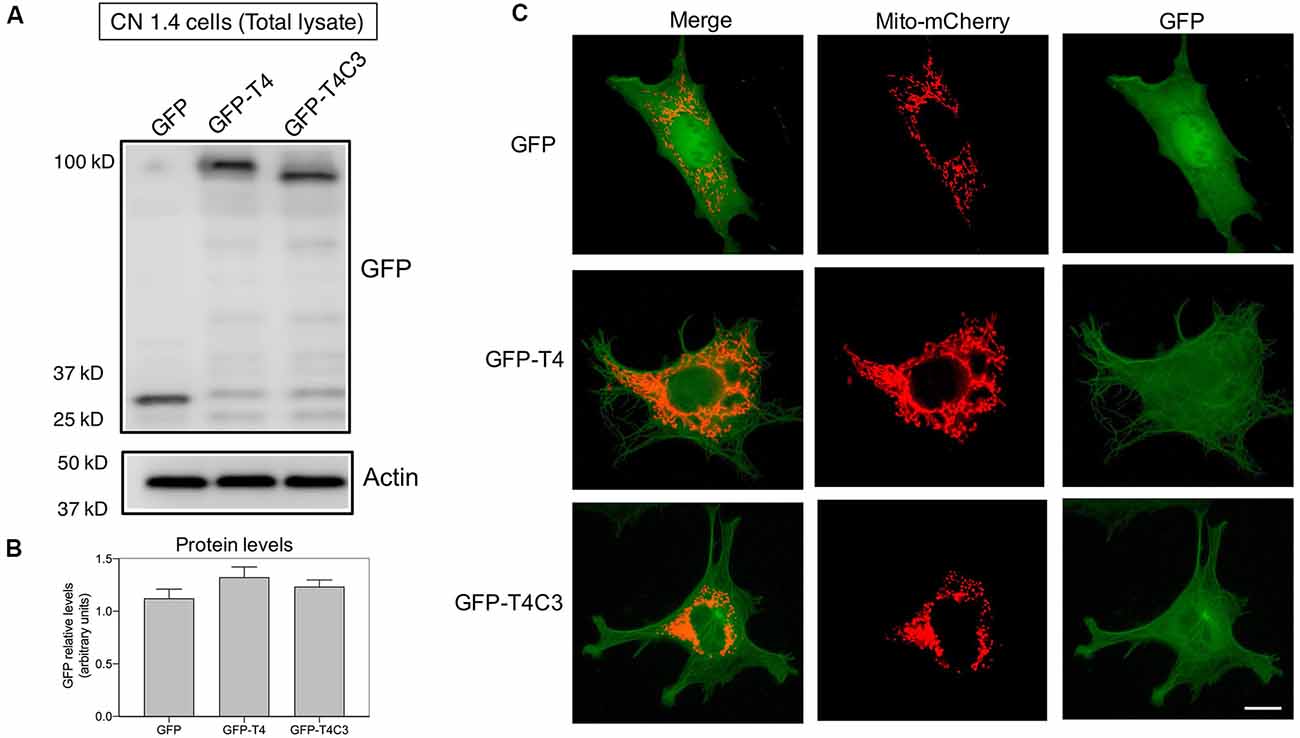
Figure 2. Expression of pathological forms of tau in immortalized cortical neurons. (A) CN1.4 cells were transfected with GFP and tau constructs (GFP-T4, GFP-T4C3) to determine the expression of the different forms of tau using GFP by western blot. Whole-cell extracts were collected, and GFP levels were evaluated 48 h after transfection. Representative images show that GFP expression levels are similar, indicating equal expression levels of the different forms of tau. Images also show the levels of β-actin as a loading control. (B) Data represent the mean ± SE, n = 4. GFP levels were normalized against β-actin levels. (C) CN1.4 cells were double transfected with Mito-mCherry (a mitochondrial construct) and the GFP-tau constructs (GFP-T4, GFP-T4C3). Representative fluorescent images were taken in live cells. Bar = 10 μm.
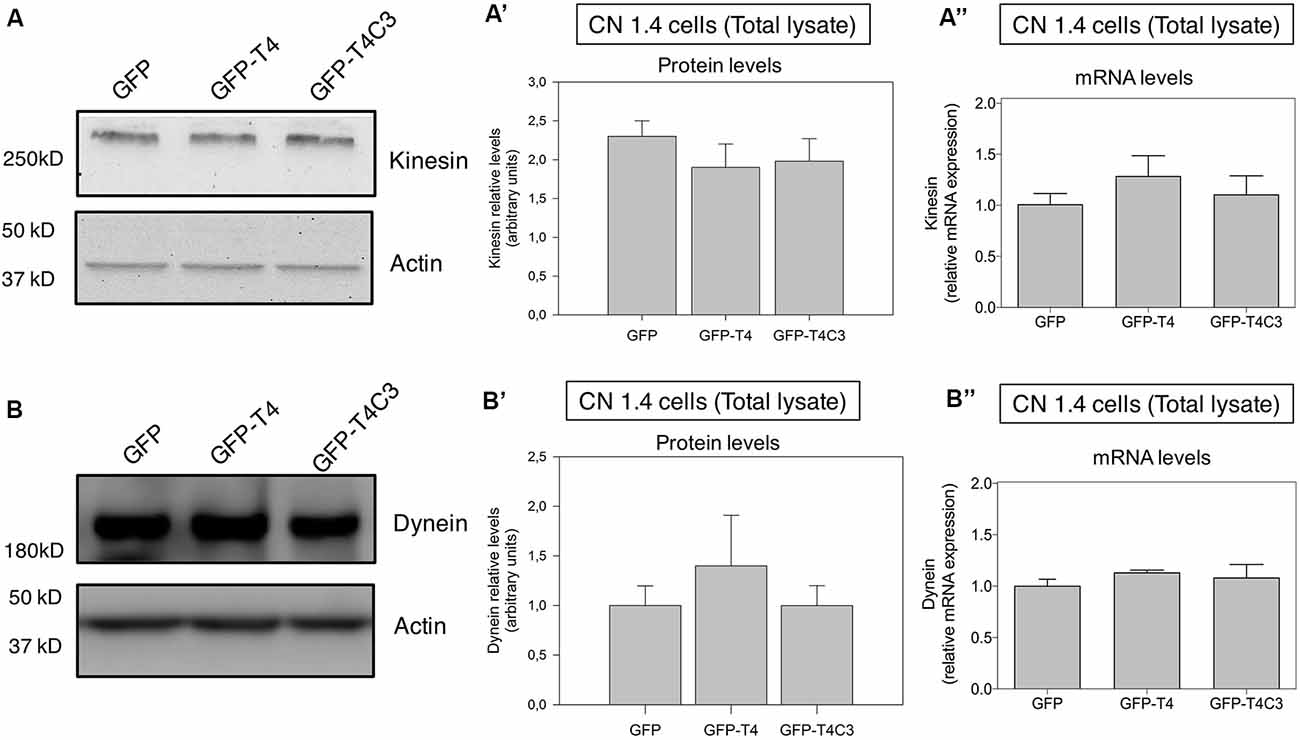
Figure 3. Effects of caspase-cleaved tau on kinesin 1 and dynein expression in immortalized cortical neurons. Panels (A,B) show representative western blot images of CN 1.4 cells transfected with GFP, GFP-T4 (normal tau), and GFP-T4C3 (truncated tau) showing the relative kinesin 1 (KIF5) or dynein expression levels. Also, Panels (A′,B′) show a quantitative analysis of the relative expression levels of kinesin 1 (A′) and dynein (B′) in immortalized cells transfected with GFP and GFP-tau (s) forms. Data are mean ± SE, n = 3. Total RNA was extracted from immortalized cortical neurons transfected with GFP, GFP-T4 (full-length tau), and GFP-T4C3 (truncated tau), and mRNA expression levels of kinesin (A″) and dynein (B″) were measured by RT-PCR (Jara et al., 2018). Data represent mean ± SE, n = 4.
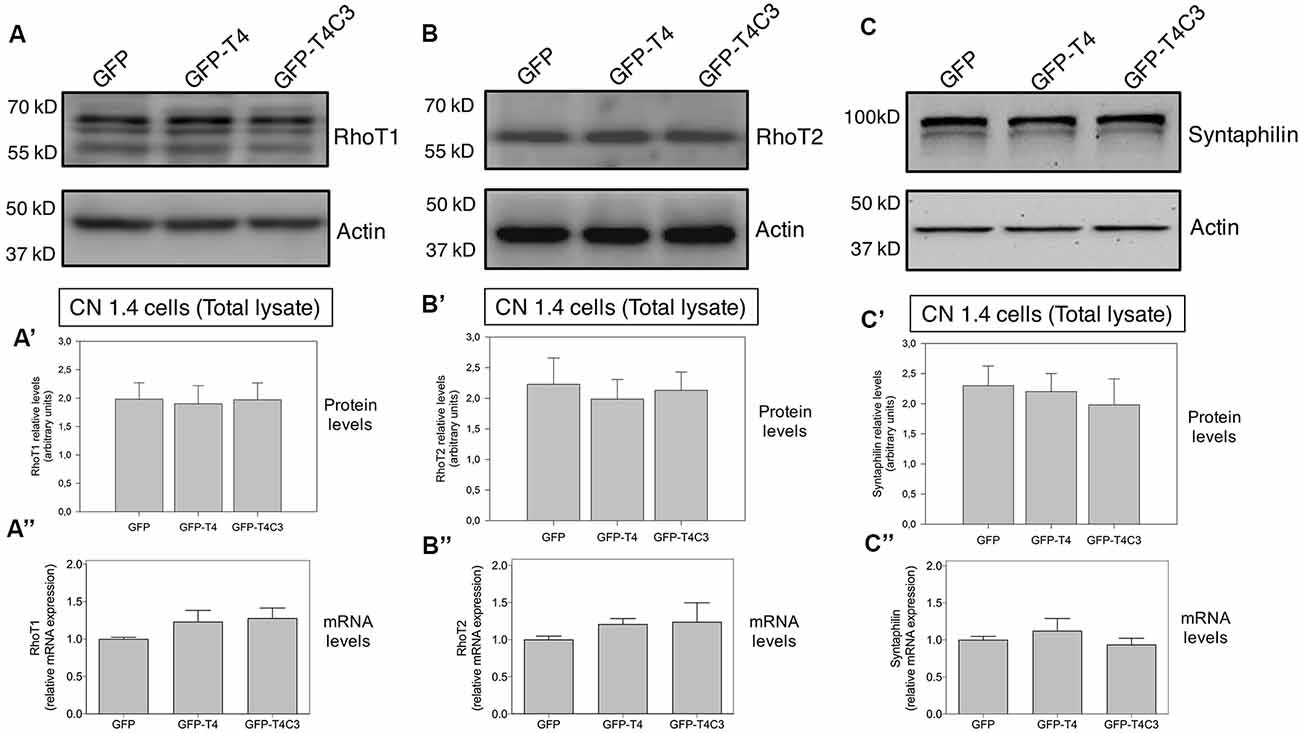
Figure 4. Effects of caspase-cleaved tau on the expression of mitochondrial accessory proteins—RhoT1/T2 and syntaphilin—in immortalized cortical neurons. Panels (A–C) show representative western blot images of CN 1.4 cells transfected with GFP, GFP-T4 (normal tau), and GFP-T4C3 (truncated tau), indicating the relative RhoT1 (A′), RhoT2 (B′), and (C′) syntaphilin expression levels. Also, Panels (A′–C′) show quantitative analysis of the relative expression levels of RhoT1 (A′), RhoT2 (B′), and syntaphilin (C′) in immortalized cells transfected with GFP and GFP-tau (s) forms. Data are mean ± SE, n = 3. Total RNA was extracted from immortalized cortical neurons transfected with GFP, GFP-T4 (full-length tau), and GFP-T4C3 (truncated tau) and mRNA expression levels of (A″) RhoT1, (B″) RhoT2, and (C″) syntaphilin were measured by RT-PCR (Jara et al., 2018). Data represent mean ± SE, n = 4.
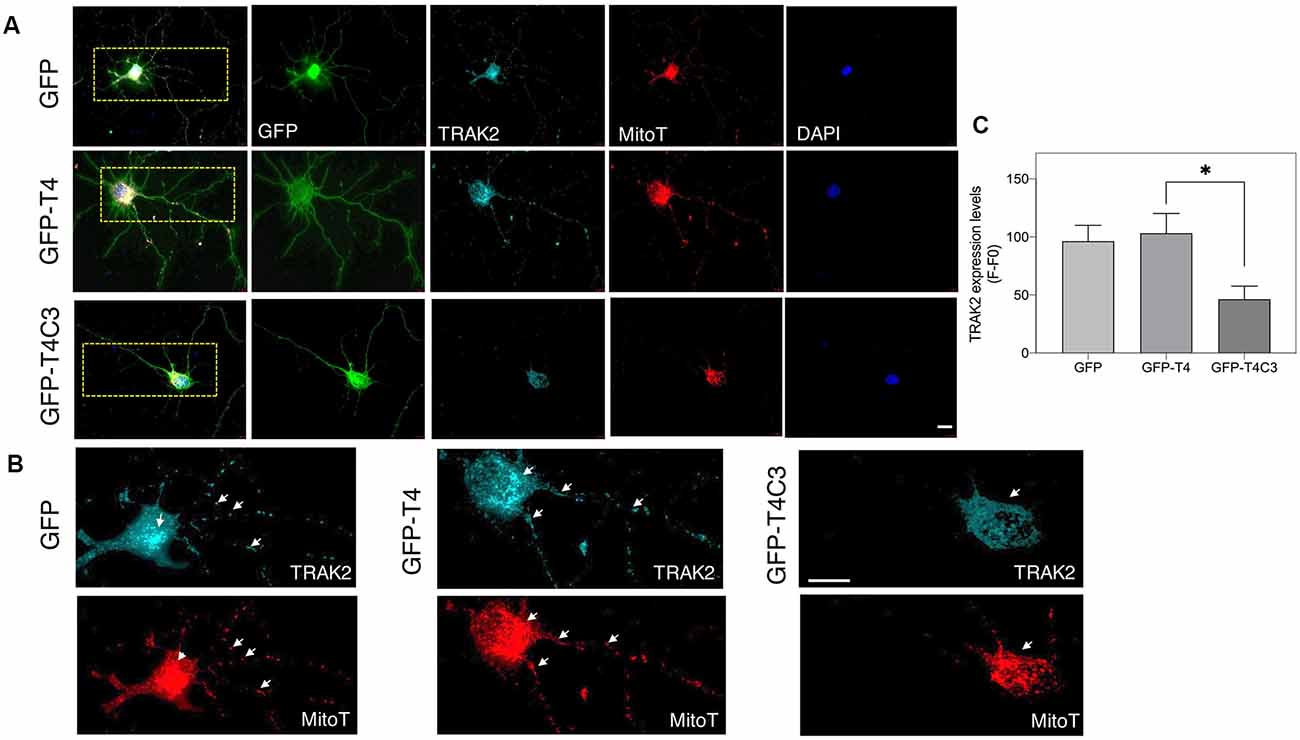
Figure 5. Truncated tau reduces TRAK2 expression and increases its localization in the neuronal soma of hippocampal neurons. (A) Representative immunofluorescence images of rat hippocampal neurons transfected with GFP, GFP-T4 (normal tau), and GFP-T4C3 (truncated tau) showing TRAK2 expression and mitochondrial staining (MitoTracker Red CMXRos, a fixable mitochondrial dye). Expression of GFP-T4C3 reduces TRAK2 levels compared with neurons expressing GFP-T4 (normal tau). (B) Higher magnification images from (A) showing a detail of the TRAK2 localization and expression pattern in hippocampal neurons transfected with the indicated conditions. Cells transfected with caspase-cleaved tau show an apparent reduction in TRAK2 expression and an apparent increase in the localization of TRAK2 with MitoTracker Red CMXRos, suggesting an increase in the association of TRAK2 with mitochondria. White arrows indicate presence of mitochondria in neuronal processes, which is reduced in neurons that expressed caspase-cleaved tau. (C) Quantitative analysis of TRAK2 fluorescence levels obtained from hippocampal neurons transfected with GFP, GFP-T4, and GFP-T4C3. Truncated tau reduced TRAK2 expression compared to neurons that expressed full-length tau (GFP-T4). Data are mean ± SE, n = 4. *p < 0.05 indicates differences between groups calculated by ANOVA test. Immunofluorescence studies are representative of four independent experiments. Bar = 20 μm.
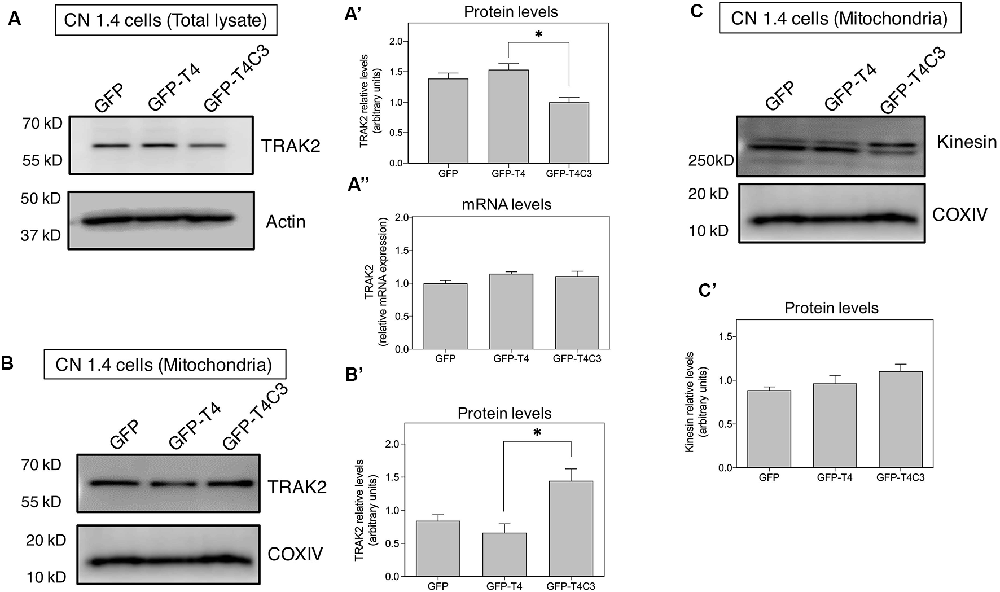
Figure 6. Truncated tau reduces TRAK2 expression and increases its association with mitochondrial fraction in immortalized cortical neurons. (A) Representative western blot images of CN 1.4 cells transfected with GFP, GFP-T4 (normal tau), and GFP-T4C3 (truncated tau) indicating the relative TRAK2 expression levels. Caspase-cleaved tau decreases TRAK2 expression compared with cells that were expressing GFP or GFP-T4 (normal tau). (A′) Quantitative analysis of the relative expression levels of TRAK2 in immortalized neurons transfected with indicated conditions. Data are mean ± SE, n = 5. *p < 0.05 indicates differences between groups calculated by ANOVA test. (A″) Total RNA was extracted from immortalized cortical neurons transfected with GFP, GFP-T4 (full-length tau), and GFP-T4C3 (truncated tau) and mRNA expression levels of TRAK2 were measured by RT-PCR. Data represent mean ± SE, n = 3. (B) Representative western blot images of CN 1.4 cells for mitochondrial extracts transfected with the conditions indicated. (B′) Quantitative graph showing that caspase-cleaved tau expression increases TRAK2 presence in mitochondrial extracts compared with cells transfected with the full-length tau form (GFP-T4). Data are mean ± SE, n = 4. *p < 0.05 indicates differences between groups calculated by ANOVA test. (C) Representative western blot images and quantitative analysis (C′) of kinesin 1 protein levels in mitochondrial extracts of CN 1.4 cells transfected with the conditions indicated (n = 4).
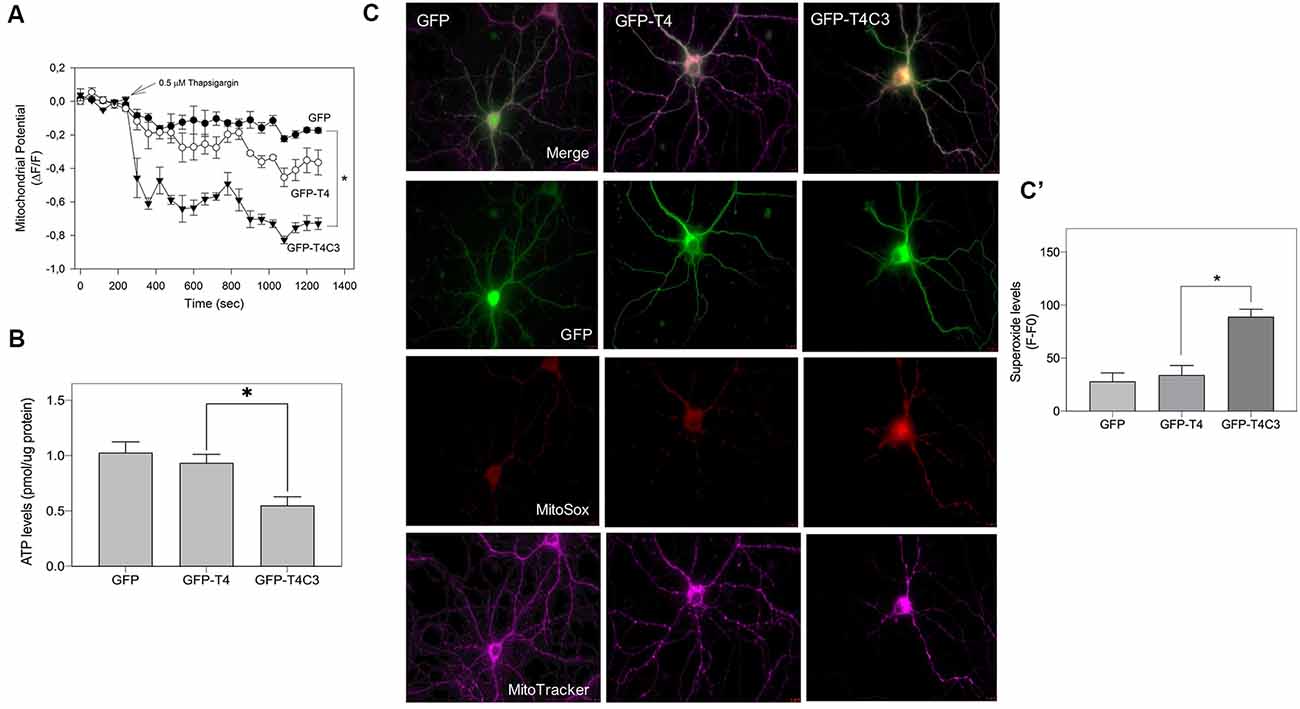
Figure 7. Caspase-3–cleaved tau expression reduces ATP production and mitochondrial membrane potential in immortalized cortical neurons. (A) CN 1.4 cells were transfected with the indicated conditions and loaded with MitoTracker red CM-H2XRos to evaluate mitochondrial membrane potential levels (Quintanilla et al., 2009, 2012, 2013, 2014; Pérez et al., 2018b). Cells were challenged with thapsigargin (0.5 μM, 30 min) to mobilize a small calcium concentration, and changes in mitochondrial potential were determined by live-cell imaging. Treatment with thapsigargin significantly decreases mitochondrial potential levels in cells transfected with caspase-cleaved tau (A). The graph shows representative plots of mitochondrial potential changes in cells transfected with the indicated conditions. Data represent the mean ± SE, n = 4. Statistical differences, *p < 0.05, were estimated using one-way ANOVA. (B) Immortalized cortical neurons were transfected with GFP, GFP-T4, and GFP-T4C3, and ATP levels were evaluated using a bioluminescence assay kit (see “Materials and Methods” section). Data represent the mean ± SE, n = 3. Statistical differences, *p < 0.05, were estimated using one-way ANOVA. (C) Representative fluorescence images of hippocampal neurons transfected with GFP, GFP-T4 (normal tau), and GFP-T4C3 (caspase-cleaved tau) loaded with MitoSox (superoxide indicator) and MitoTracker Deep Red (mitochondrial marker). Caspase-cleaved tau induces a significant increase in superoxide levels compared with cells positive for GFP-T4 (normal tau). (C′) Graph bars show a quantitative analysis of MitoSox fluorescence levels obtained from hippocampal neurons transfected with GFP, GFP-T4, and GFP-T4C3. Truncated tau increased superoxide production compared to neurons that expressed full-length tau (GFP-T4). Data are mean ± SE, n = 3. *p < 0.05 indicates differences between groups calculated by ANOVA test. Bar = 20 μm.
Materials and Methods
Cell Culture
Conditionally immortalized neuronal cell lines (CN1.4 cells; Bongarzone et al., 1998; Quintanilla et al., 2009; Pérez et al., 2018b) were cultured in DMEM media (Mediatech CellGro, Corning, NY, USA) supplemented with 5% inactivated fetal bovine serum (Mediatech Inc., Manassas, VA, USA) and 1% penicillin/streptomycin (Mediatech Inc., CellGro, Corning, NY, USA) and incubated at 33°C and 5% CO2. These cells were immortalized by retroviral infection of enriched cortical neuronal cultures that adopt a morphology similar to that of cortical neurons and express low endogenous levels of tau (Bongarzone et al., 1998; Krishnamurthy and Johnson, 2004). Hippocampal neuronal cultures were prepared from Sprague–Dawley dam pregnant rats at embryonic day 18, according to Quintanilla et al. (2012, 2014). Pregnant dams were obtained from Bioterio Central, P. Universidad Catolica de Chile, Santiago, Chile. Cultures were maintained in neurobasal growth medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with B27 (Thermo Fisher, Scientific), L-glutamine, penicillin, and streptomycin. On day 3, the cultured neurons were treated with cytosine arabinose (Sigma Aldrich, St. Louis, MO, USA) for 72 h to obtain cultures highly enriched in neurons (~5% glia), and on day 6, the cultured neurons were transfected (Quintanilla et al., 2014; Pérez et al., 2018b). All procedures were performed in agreement with the Institutional Bioethics Committee of Universidad Autonoma de Chile (Resolution 0049-17, May 2017).
Tau Constructs
Tau constructs tagged with GFP, GFP-full-length tau (GFP-T4 in the figures), and GFP-cleaved tau [GFP-T4C3 in the figures; GFP-tau (s)] were generated as previously described (Quintanilla et al., 2009, 2012, 2014). CN1.4 cells and hippocampal neurons were transiently transfected with plasmids containing tau constructs using Lipofectamine 2000 (Thermo Fisher Scientific) diluted in OptiMEM (Thermo Fisher Scientific; Quintanilla et al., 2012, 2014; Pérez et al., 2018b). Complementary CN 1.4 cells transfected with GFP-related constructs were transfected with Mito-mCherry (Pérez et al., 2018b) to evaluate mitochondrial morphology (Figure 2C). Media were changed at 24 h posttransfection, and analyses were conducted 48 h posttransfection in CN 1.4 cells and 15 days in vitro for studies in primary hippocampal neurons. The GFP plasmid expression was verified with live-cell imaging, observing a 40% and 8% of transfection efficiency in CN 1.4 and primary neurons, respectively (Pérez et al., 2018b). Also, GFP, GFP-full-length tau (GFP-T4), and GFP-truncated tau (GFP-T4C3) expression levels were estimated in CN 1.4 cells by detecting GFP expression using western blot (Figure 2).
Western Blot Analysis
CN1.4 cells were lysed in Triton lysis buffer, including a protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany) and a phosphatase inhibitor (Jara et al., 2018; Pérez et al., 2018b). Thirty micrograms of total protein extracts were resolved by electrophoresis on a sodium dodecyl sulfate (SDS)-polyacrylamide gel and transferred to PVDF membranes (Jara et al., 2018; Pérez et al., 2018b). Membranes were incubated with primary antibodies: polyclonal anti-KIF5A (1:500, Cat. No. PA1-642, Thermo Fisher Scientific), polyclonal anti-dynein (1:500, Cat. No. SC-514579, Santa Cruz Biotechnology, Dallas, TX, USA), polyclonal anti-RhoT1 (1:500, Cat. No. SC-398520, Santa Cruz Biotechnology), polyclonal anti-RhoT2 (1:500, Cat. No. SC-398521, Santa Cruz Biotechnology), polyclonal anti-syntaphilin (1:1,000, Cat. No. PA5-88474, Thermo Fisher Scientific), polyclonal anti-TRAK2 (1:1,000, Cat. No. PA5-21858, Thermo Fisher Scientific), monoclonal anti-COX4 (1:500, Cat. No. SC-376731, Santa Cruz Biotechnology), and monoclonal anti-VDAC (1:500, Cat. No. SC-390996, Santa Cruz Biotechnology) overnight. The equal loading and transfer of protein to the membranes were determined by reprobing with a monoclonal anti-β-actin antibody (1:2,000, Cat. No. SC-8432, Santa Cruz Biotechnology). Protein signal was detected using a horseradish peroxide (HRP)-linked goat anti-mouse or anti-rabbit secondary antibody (1:2,000, Thermo Fisher Scientific), as indicated. Finally, the immunoreactivity of the protein signal was detected using enhanced chemiluminescence reagent (ECL, Thermo Fisher Scientific; Jara et al., 2018; Pérez et al., 2018b). Expression levels of the indicated proteins were estimated related to the intensity of housekeeping protein signals using ImageJ software (NIH, Bethesda, MD, USA).
Preparation of Mitochondrial Extracts
Transfected CN 1.4 cells with GFP or GFP-tau forms (GFP-full-length tau and GFP-caspase-cleaved tau) were suspended in MSH buffer (230 mM mannitol, 70 mM sucrose, 5 mM HEPES, pH 7.4) supplemented with 1 mM EDTA and protease inhibitor cocktail. Homogenates were centrifuged at 600 g for 10 min at 4°C to discard nuclei and cell debris (Jara et al., 2018). The supernatant was centrifuged at 8,000 g for 10 min; the new mitochondrial pellet was washed twice in MSH without EDTA. Protein concentration was determined using a standard BCA kit (Thermo Fisher Scientific), and western blot was made accordingly (Jara et al., 2018).
Mitochondrial Localization and Membrane Potential Determinations
Mitochondrial localization was evaluated in hippocampal neurons expressing GFP, GFP-full-length tau (GFP-T4), and GFP-caspase-cleaved tau (GFP-T4C3) using MitoTracker Deep Red FM (Molecular Probes, Thermo Fisher Scientific) dye according to Quintanilla et al. (2013, 2014), with modifications. Hippocampal neurons were incubated with this dye for 35 min in Krebs–Ringer–HEPES (KRH)–glucose buffer and then were photographed in a high-resolution fluorescence microscope (LX6000 X, Leica; Quintanilla et al., 2012, 2013, 2014). Mitochondrial staining was expressed as the average of the fluorescence signal (F) per area in every image (30 images per condition and experiment) minus the intensity of background fluorescence (F0). The mitochondrial potential was determined using the mitochondrial dye MitoTracker Red-H2XRos (Molecular Probes, Thermo Fisher Scientific), as previously described (Quintanilla et al., 2009, 2012, 2013, 2014; Pérez et al., 2018b). Mitochondrial potential levels were expressed as MitoTracker Red-H2XRos fluorescence and are presented as the pseudo ratio (ΔF/F0), where F is the average of the fluorescence signal (F) per area in every image and (F0) represents the intensity of background fluorescence. Cells were incubated with the dye (100 nM) in KRH–glucose buffer at 37°C for 45 min, and then cells were treated with thapsigargin (0.5 μM, Tocris Bioscience, Minneapolis, MN, USA) for 30 min. The intensity of the signal was analyzed in 10 cells per experiment, in three different studies using ImageJ software (Quintanilla et al., 2009, 2012, 2013, 2014; Pérez et al., 2018b).
Determination of ROS (Superoxide) Levels
Superoxide levels were evaluated with MitoSox™ (Molecular Probes, Thermo Fisher Scientific) and the mitochondrial dye MitoTracker Deep Red FM (Pérez et al., 2020). Transfected cultured neurons were incubated with 0.5 μM MitoSox and 0.3 μM MitoTracker Deep Red in KRH–glucose buffer at 37°C for 30 min (Quintanilla et al., 2013; Pérez et al., 2020). For superoxide determinations with cell imaging, fluorescence images were acquired using the same exposure time and gain to minimize dye photo-bleaching (Quintanilla et al., 2013; Pérez et al., 2020). MitoSox fluorescence (F) was determined in each cell, and background (F0) of each image was subtracted before normalization per cell area. Normalization was made to ameliorate differences of the dye distribution inside of the cells that present differences in neuronal morphology. Fluorescence images were acquired by live epifluorescence microscopy (Leica LX6000, Wetzlar, Germany) using a 63× oil-immersion objective.
Immunofluorescence Studies
Hippocampal neurons transfected with GFP or GFP-tau (s) forms were incubated with MitoTracker Red CMXRos (fixable mitochondrial dye, Molecular Probes, Thermo Fisher Scientific) for 35 min in KRH–glucose to evaluate mitochondrial localization. Later, neurons were fixed in 4% paraformaldehyde-KRH for 15 min at 37°C (Quintanilla et al., 2013; Pérez et al., 2018b). Hippocampal cultured neurons were blocked with 10% bovine serum KRH for 1 h and incubated with a primary polyclonal anti-TRAK2 (1:200, Cat. No. PA5-21858, Thermo Fisher Scientific) antibody overnight. After that, the cells were washed in KRH–glucose, probed with the goat secondary polyclonal antibody Alexa Fluor 647 (1:1,000, Cat. No. A32733, Molecular Probes, Thermo Fisher Scientific) for 3 h, and then stained with DAPI (1:5,000, Sigma Aldrich) for 15 min. Estimation of TRAK2 expression in hippocampal neurons was calculated in the neuronal soma, obtaining fluorescence intensity (F) and background levels (F0) from 25 images (1 or 2 neurons per image) per experiment (total, n = 3) using ImageJ software (NIH).
Determination of ATP Levels
Total ATP levels were measured in immortalized cortical neurons transfected with GFP, GFP-T4 (full-length tau), and GFP-T4C3 (truncated tau) lysates using a luciferin/luciferase bioluminescence assay kit (ATP Determination Kit No. A22066, Molecular Probes, Invitrogen; Jara et al., 2018). The amount of ATP in each sample was calculated from standard curves and normalized to total protein concentration (Jara et al., 2018).
Real-Time Polymerase Chain Reaction
Dynein, RhoT1/T2, syntaphilin, kinesin, and TRAK2 mRNA levels were analyzed by real-time polymerase chain reaction (RT-PCR). Total RNA was isolated from 100 mg of CN 1.4 lysates transfected with GFP, GFP-T4, and GFP-T4C3 using the TRIzol reagent (Life Technologies, Thermo Fisher Scientific) following the manufacturer’s instructions (Jara et al., 2018). Residual genomic DNA was removed with amplification grade RNase free DNase I (Invitrogen, Thermo Fisher Scientific). RNA yield and purity were measured in a microplate reader (TECAN, Infinite 200 PRO series). One microgram of total RNA was reverse transcribed using the ImProm-II Reverse Transcription System (Promega, Madison, WI, USA) to obtain cDNA, which was stored at –20°C for future use. For quantitative PCR (qPCR) analysis, each cDNA sample was diluted 10× in nuclease-free water. RT-PCR was performed in triplicate in a LightCycler 96 System (Roche Diagnostics GmbH, Roche Applied Science, Mannheim, Germany) using KAPA SYBR FAST qPCR Master Mix (2×) in a final reaction volume of 10 μl (Jara et al., 2018). Amplification conditions consisted of an initial hot start at 95°C for 10 min followed by amplification for 40 cycles (95°C for 15 s, Tm°C for 20 s, and 72°C for 20 s). Melting curves were analyzed immediately after amplification from 55 to 95°C. Expression values were normalized to 18S rRNA levels using the ΔCt method. The following primers were used in RT-PCR:

Statistical Analysis
For statistical analysis, one-way ANOVA and Dunn’s a posteriori test were calculated with SigmaPlot software. Differences were considered significant if p < 0.05 or p < 0.001, as indicated.
Results
Caspase-Cleaved Tau Expression Reduces the Mitochondrial Population in Neuronal Processes
Previously our group studied the role of pathological forms of tau on mitochondrial function and its consequences against neuronal viability (Quintanilla et al., 2009, 2012, 2014; Pérez et al., 2018b). We showed that the expression of caspase-3–cleaved tau at D421 (truncated tau) and hyperphosphorylated tau at PHF1 epitopes (S396, S404) affects mitochondrial membrane potential and increases ROS production (Quintanilla et al., 2009, 2012, 2014). More importantly, neurons expressing caspase-cleaved tau showed a significant defect in mitochondrial transport (Quintanilla et al., 2012, 2014). Interestingly, truncated tau decreases the number of moving mitochondria in neuronal processes without affecting the velocity rate of individual mitochondria (Quintanilla et al., 2012, 2014). However, the mechanism involved in these alterations remains unexplored. To address this question, we first analyzed the distribution of a mitochondrial population in hippocampal rat neurons transfected with GFP, GFP-full-length (GFP-T4), and GFP-truncated tau at D421 (GFP-T4C3) using MitoTracker Deep Red FM dye (Figure 1). We observed that expression of truncated tau reduced the number of mitochondria present in neuronal processes compared with neurons transfected with full-length tau (GFP-T4) and GFP (vector; Figure 1, white arrows). We observed that caspase-cleaved tau significantly increases mitochondrial accumulation in the soma of hippocampal neurons compared to cells transfected with full-length tau (Figure 1A, Zoom). Quantification of MitoTracker Deep Red FM fluorescence indicated that truncated tau increases somatic mitochondrial localization compared with the presence of these organelles in neuronal processes shown in neurons transfected with full-length tau and GFP (Figure 1B). These studies indicate that caspase-cleaved tau affects mitochondrial localization in hippocampal neurons, which is in agreement with our previous findings that showed significant alterations in the number of moving mitochondrial elements in neurons expressing truncated tau (Quintanilla et al., 2012, 2014).
Caspase-Cleaved Tau Expression Does Not Modify the Expression of Kinesin 1 and Dynein Motor Proteins in Immortalized Cortical Neurons
Neurons are polarized cells with axons and neuronal processes that extend a great distance to establish neuronal synapses (Stokin and Goldstein, 2006). This process requires a constant supply of energy, and for that, mitochondrial transport and localization are essential (Zinsmaier et al., 2009). The constant process of anterograde/retrograde mitochondrial transport between axon and dendritic synaptic terminals is produced by association with motor proteins: kinesin family proteins (KIFs) and dynein with microtubular structures (Moore and Holzbaur, 2018). Deficiencies in the expression or mutations in both motor protein components have shown defects in mitochondrial transport, reducing mitochondrial localization in axons (Ari et al., 2014; Hares et al., 2017; Chaudhary et al., 2018). More importantly, we showed that the expression of caspase-cleaved tau equally decreases anterograde/retrograde mitochondrial movement in hippocampal neurons compared with cells expressing full-length tau (Quintanilla et al., 2014). Therefore, we expressed GFP, full-length (GFP-T4), and caspase-3–cleaved tau (GFP-T4C3; Figures 2A,B) in CN 1.4 cells (Bongarzone et al., 1998; Ding et al., 2006), an immortalized cortical neuronal model with a minor endogenous expression of tau (Figure 2). Transfection of GFP-related constructs induced a similar expression of GFP and GFP-full-length and GFP-truncated tau levels in immortalized cortical neurons (Figures 2A,B). Also, we expressed GFP and GFP-tau (s) forms and Mito-mCherry in CN 1.4 cells (Figure 2C). The expressions of GFP and GFP-tau (s) forms were very similar and cells expressing truncated tau showed a significant degree of mitochondrial fragmentation, corroborating our previous findings (Quintanilla et al., 2009; Pérez et al., 2018b). Further, we studied kinesin 1 and dynein expression levels in immortalized cortical neurons transfected with GFP, GFP-full-length tau (GFP-T4), and GFP-truncated tau (GFP-T4C3) forms by western blot (Figure 3). We observed that expression of caspase-cleaved tau did not affect the protein levels of kinesin 1 (Figures 3A,A′) or dynein (Figures 3B,B′) compared with cells that expressed GFP and GFP full-length tau (Figures 3A′,B′). Also, truncated tau expression did not affect mRNA expression of kinesin 1 (Figure 3A″) and dynein (Figure 3B″) compared with immortalized cortical neurons transfected with full-length tau.
Caspase-Cleaved Tau Expression Does Not Modify RhoT1/T2 (Miro1/2) Protein Levels in Immortalized Cortical Neurons
Mitochondrial motor proteins kinesin (KIF5) and dynein are regularly associated with motor adaptor proteins and mitochondrial membrane receptors (Reis et al., 2009; Nemani et al., 2018). These complexes contribute to mitochondrial trafficking and the precise regulation of the distribution of mitochondria in response to different changes in neuronal function (Sheng, 2017). In this context, Miro (know as RhoT1/T2) is a member of the mitochondrial outer membrane Rho GTPase family that acts as a mitochondrial receptor and binds with the motor adaptor protein Milton (or TRAK1/2; Reis et al., 2009; Nemani et al., 2018). In the same context, some studies have shown that dMiro mutant impairs anterograde mitochondrial transport depleting axonal and synaptic mitochondria (López-Doménech et al., 2016). Therefore, in our model, we studied the levels of RhoT1/T2 in immortalized cortical neurons transfected with GFP, full-length, and caspase-cleaved tau (Figures 4A,B). These studies showed that caspase-cleaved tau did not affect RhoT1/T2 protein expression (Figures 4A,A′,B,B′) and mRNA levels (Figures 4A″,B″), indicating that mitochondrial transport impairment is likely not caused by dysfunction of these proteins.
Caspase-Cleaved Tau Does Not Affect the Mitochondrial Adaptor Syntaphilin in Neuronal Cells
To investigate how truncated tau could affect mitochondrial transport in hippocampal neurons, we studied the expression of syntaphilin, which is a mitochondrial docking protein that contributes to specifically immobilizing mitochondria in axons (Kang et al., 2008; Chen et al., 2009). Syntaphilin is a mitochondrial outer membrane protein whose function is binding mitochondria to microtubules to reduce mitochondrial movement in axons (Kang et al., 2008; Chen et al., 2009). Interestingly, several studies have shown that reducing syntaphilin expression increases axonal mitochondria movement, therefore reducing mitochondrial density within the axons (Lin et al., 2017). We analyzed the expression of syntaphilin in immortalized cortical neuronal cells in response to GFP, full-length, and caspase-cleaved tau expression (Figure 4C). Complementary western blot analysis (Figures 4C,C′) and qPCR analyses (Figure 4C″) did not show any significant changes in syntaphilin protein (Figure 4C″) and mRNA levels (Figure 4C″) in any condition suggesting that accumulation of mitochondria in neuronal soma is not influenced by syntaphilin.
Truncated Tau Affects the Expression and Localization of the Mitochondrial Adaptor Protein TRAK2 in Neuronal Cells
Milton (as is known in Drosophila) is an adaptor protein that binds the mitochondria receptor RhoT1/T2 (Miro1/2; López-Doménech et al., 2018) to kinesin (KIF5; van Spronsen et al., 2013; Loss and Stephenson, 2017). It is known that Milton has two mammalian orthologues, TRAK1 and TRAK2, whose function is vital for mitochondrial localization at synaptic terminals (van Spronsen et al., 2013; Loss and Stephenson, 2017). In the same context, increased expression of TRAK2 increases mitochondrial movement, and its genetic ablation reduces mitochondrial localization in neuronal processes (van Spronsen et al., 2013). Therefore, we studied TRAK2 expression in hippocampal neurons transfected with full-length (GFP-T4) and caspase-cleaved tau (GFP-T4C3) using immunofluorescence methods (Figure 5). We observed a significant reduction in TRAK2 expression in neurons expressing caspase-cleaved tau compared with neuronal cells transfected with normal tau (Figure 5A, white arrows, Figures 5B,C). Quantification of immunofluorescence staining of TRAK2 in hippocampal neurons showed a significant decrease in TRAK2 expression compared to neurons with GFP-full-length tau or GFP (vector) expression (Figure 5C). Also, we complemented immunofluorescence studies using a fixable mitochondrial marker, MitoTracker Red CMXRos, to observe mitochondrial localization (Quintanilla et al., 2013). Interestingly, we observed that truncated tau (GFP-T4C3) expression induced a change in TRAK2 distribution, increasing its accumulation in neuronal soma compared to neurons transfected with GFP or GFP-full-length tau (Figure 5B, white arrows). Complementary TRAK2 expression was evaluated by western blot (Figures 6A,A′) and qPCR analyses (Figure 6A″) in immortalized cortical neurons transfected with GFP, full-length, or caspase-cleaved tau. Truncated tau significantly reduced TRAK2 protein expression levels compared to cells expressing GFP or GFP-full-length tau (Figures 6A,A′). Interestingly, truncated tau expression did not affect mRNA levels of TRAK2 in immortalized cortical neurons (Figure 6A″), indicating that this tau form could be affecting TRAK2 through the modification of a posttranslational mechanism. These studies showed that truncated tau reduced TRAK2 expression levels in immortalized cortical neurons, corroborating our observations in hippocampal neurons (Figures 5A,C).
Caspase-Cleaved Tau Increases Mitochondrial Localization of TRAK2 in Immortalized Cortical Neurons
Previously, we showed that caspase-cleaved tau reduced TRAK2 expression (Figures 5, 6A,A′) and increased its localization in the neuronal soma (Figure 5B). To study if truncated tau expression affects TRAK2 distribution in neuronal cells, we transfected GFP, full-length (GFP-T4), and caspase-cleaved tau (GFP-T4C3) in immortalized cortical neurons and studied TRAK2 levels in cytosol and mitochondrial fractions (Figures 6A,B). Total and mitochondrial extracts were separated, and we determined TRAK2 expression levels by western blot (Figures 6A,B). These studies showed that the expression of caspase-cleaved tau increased TRAK2 localization with mitochondria, an effect that is evident in western blot assay from mitochondrial extracts (Figures 6B,B′). As a motor protein, kinesin 1 is responsible for mitochondrial transport, and it must be correctly associated with TRAK2 and then with mitochondria to generate movement of mitochondria through microtubules (Sheng, 2017). In this context, we evaluated kinesin 1 localization in the mitochondrial extracts in immortalized cortical neurons transfected with GFP and GFP-tau (s) forms (Figures 6C,C′). Interestingly, we observed that neuronal cells expressing caspase-cleaved tau showed no differences in kinesin 1 levels (KIF 5) in mitochondria preparations compared with neuronal cells transfected with full-length tau (Figure 6C′). These results indicate that the presence of caspase-cleaved tau modifies the TRAK2 interaction with mitochondria, and this effect could be responsible for the accumulation of mitochondria in neuronal soma.
Expression of Truncated Tau Induces Mitochondrial Depolarization, ATP Reduction, and ROS Increase in Neuronal Cells
Mitochondrial bioenergetics, which includes ROS production, membrane potential, and ATP production, are an essential element for the movement of mitochondria (Pérez et al., 2018a). Therefore, we studied mitochondrial membrane potential levels, ATP production, and ROS levels in immortalized cortical cells and hippocampal neurons transfected with truncated and full-length tau (Figure 7). The mitochondrial potential was estimated using MitoTracker Red in situ in transfected cells treated with thapsigargin (0.5 μ, 30 min; Figure 7A). Thapsigargin treatment mildly reduced mitochondrial potential levels in cells transfected with GFP and GFP-full-length tau (Figure 7A). However, truncated tau expression strongly decreased mitochondrial potential in CN 1.4 cells exposed to thapsigargin, indicating that this tau form significantly affects mitochondrial health (Figure 7A). In the same context, we evaluated ATP production in immortalized cortical neurons expressing GFP, GFP-full-length, and GFP-truncated tau (Figure 7B). Interestingly, we observed that truncated tau significantly reduced ATP production compared with cells that expressed full-length tau (Figure 7B). Complementarily, we evaluated superoxide levels in hippocampal neurons transfected with GFP, normal (GFP-T4), and caspase-cleaved tau (GFP-T4C3) using MitoSox dye in live-cell imaging (Figure 7C). Expression of caspase-cleaved tau (GFP-T4C3) increased superoxide levels (Figure 7C) compared to hippocampal neurons expressing full-length tau (GFP-T4; Figure 7C′). Therefore, these results indicate that the presence of truncated tau affects mitochondrial bioenergetics, events that could contribute to the reduction in mitochondrial movement in neurons (Shidara and Hollenbeck, 2010; Watters et al., 2020).
Discussion
Despite the recognized importance of mitochondria in the synapse, there are still unknown aspects regarding the mechanism of how mitochondrial health affected by the presence of pathological forms of tau contributes to synaptic and cognitive alterations in AD (Pérez et al., 2018a). In this context, the study of mitochondrial transport alterations produced by pathological forms of tau is a pivotal aspect. Tau is a microtubule-associated protein (MAP) that interacts with microtubules, and nonphysiological modifications of tau could cause a destabilization of microtubular elements contributing to neurodegeneration in AD (Tapia-Rojas et al., 2019). Appropriate localization of mitochondria to the synapse is required due to the high demand for ATP production and Ca2+ buffering (Macaskill and Kittler, 2010; Sheng, 2017). Previously, we showed that expression of caspase-cleaved tau at D421 by caspase 3 affects mitochondrial transport in hippocampal and cortical cultured neurons (Quintanilla et al., 2012, 2014). Truncated tau reduced the number of moving mitochondria at retrograde and anterograde directions compared with neurons expressing full-length tau (Quintanilla et al., 2014). Here, we complement these observations with our novel findings suggesting an essential role of mitochondria-associated transport accessory protein, TRAK2, in the impairment of movement of mitochondria produced by truncated tau. The expression of this tau form increased the accumulation of mitochondria in the soma, reducing the mitochondrial population in the neuronal processes, including the axon (Figure 1). Using an immortalized cortical neuronal model that does not express tau (Bongarzone et al., 1998; Quintanilla et al., 2009), we studied the effects that full-length and truncated tau could have on the expression of the motor proteins kinesin 1 and dynein, and mitochondria motor accessory proteins RhoT1/T2, synthaphilin, and TRAK2. Truncated tau did not affect the expression of major motor proteins (kinesin 1 and dynein) and also RhoT1/T2 and syntaphilin (Figures 3, 4).
Interestingly, truncated tau significantly reduced TRAK2 expression (Figures 5, 6A) and increased its association with the mitochondria (Figure 6B) compared with cells expressing full-length tau (Figure 6B′). Also, truncated tau expression did not significantly affect kinesin 1 association with mitochondria compared with cells expressing full-length tau and GFP alone (Figures 6C,C′). These observations are related to our previous results that showed that truncated tau expression affects mitochondrial movement equally in the retrograde and anterograde directions, suggesting that defects in kinesin 1 or dynein function may not be involved (Quintanilla et al., 2014). Therefore, it is possible that the defects in the movement of mitochondria could be caused by an apparent malfunction of the mitochondrial adaptor protein TRAK2 induced by truncated tau (van Spronsen et al., 2013).
In different neuronal cell cultures where tau is highly overexpressed (Dixit et al., 2008; Stoothoff et al., 2009; Stamer et al., 2002) or in entirely in vitro systems (Morfini et al., 2007), tau can inhibit axonal transport by directly binding to microtubules and preventing the movement of the anterograde motor proteins (Morfini et al., 2007). However, other studies showed clear evidence to the contrary (Yuan et al., 2008; LaPointe et al., 2009). For example, in the squid axoplasm model, the addition of tau at concentrations approximately 20-fold higher than physiological levels did not affect fast axonal transport (Yuan et al., 2008). However, in the same model, a tau construct of only the amino-terminal portion of tau (without the microtubule-binding domain) inhibited transport, as did tau in a conformation that exposed the amino terminus (Götz et al., 2006). These data suggest that anomalous modification of tau may selectively impair axonal transport of mitochondria (Götz et al., 2006) rather than creating a “general roadblock” effect at the level of the microtubules (Miller and Sheetz, 2004; Götz et al., 2006; Ittner et al., 2008). Also, mitochondria with lower mitochondrial membrane potential and bioenergetic impairment are not transported efficiently in the anterograde direction (kinesin-dependent transport; Miller and Sheetz, 2004). Interestingly, we found that caspase-cleaved tau expression affects mitochondrial bioenergetics inducing depolarization, ROS increase, and ATP reduction (Figure 7). Therefore, these mitochondrial defects could contribute to the deficits in mitochondrial transport produced by truncated tau expression.
Mitochondrial transport is actively regulated by local increases in calcium concentration (Yi et al., 2004). Mitochondrial arrest at the synaptic terminal occurs in order for mitochondria to be recruited near the synaptic vesicles, which require energy for release of neurotransmitters (Zinsmaier et al., 2009). Reduction in syntaphilin expression significantly increases the number of mitochondria moving through the axon, improving synaptic activity (Kang et al., 2008). Previously, we showed that truncated tau expression affects mitochondrial bioenergetics (Figure 7) and mitochondrial calcium capacity (Quintanilla et al., 2009). However, in this work, we did not detect significant changes in syntaphilin expression (protein and mRNA levels) in immortalized cortical neurons transfected with GFP and GFP-tau forms (Figures 4C″).
An interesting observation is that the expression of truncated tau increases the association of TRAK2 with mitochondria compared with hippocampal neurons and immortalized cortical neurons transfected with full-length tau (Figures 6B,B′). Also, in mitochondrial extracts, the presence of kinesin 1 or dynein (data not shown) was not affected in neuronal cells expressing truncated tau (Figures 6C,C′). Therefore, the fibrogenic and destabilizing effect of truncated tau on microtubule structures (Ding et al., 2006) could increase TRAK2 attachment to the mitochondria, producing adverse effects against mitochondrial transport. Also, this increased interaction between TRAK2 and mitochondria induced by caspase-cleaved tau could be facilitated by the reduction of ATP production, and mitochondrial depolarization was observed in cells that express this tau form (Figure 7B).
Tau family MAPs also reportedly inhibit kinesin-dependent transport along microtubules (Vershinin et al., 2007; Shigematsu et al., 2018). This inhibition is primarily caused by direct competition between MAPs and kinesin for microtubule binding, reducing the attachment frequency of kinesin (Trinczek et al., 1999). Interestingly, recent work from Shigematsu et al. (2018) studied the ability of tau family MAPs 4R- and 5R-MAP4 [tau isoforms containing (3R) or five (5R) tubulin-binding domains] to form a complex with kinesin-1 and microtubules using cryo-EM studies. These studies showed that both modifications coexist with kinesin-1 on the microtubules and produce an inhibitory effect of MAP4 on kinesin-dependent transport along the microtubules (Shigematsu et al., 2018). However, our present observations indicate that the association between kinesin-1 and mitochondria is not affected by caspase-cleaved tau, which correlates with our previous observations that the direction of mitochondrial movement (anterograde/retrograde) was equally affected by truncated tau expression in hippocampal neurons (Quintanilla et al., 2014). Importantly, these actions could be augmented by the adverse effects that caspase-cleaved tau induces against mitochondria health (Quintanilla et al., 2009, 2012, 2014; Pérez et al., 2018b) and ATP production (Figure 7B). These combinatory actions may reduce mitochondrial localization in neuronal processes, compromising energy supply and calcium regulation for the synaptic process (Macaskill and Kittler, 2010).
Finally, our findings show for the first time that pathological forms of tau can affect mitochondrial transport, enhancing the association of TRAK2 with mitochondria (Figures 6B,B′) and reducing ATP production (Figure 7B). Recent studies have shown that TRAK2 contributes to both axonal and dendritic mitochondrial transport, since TRAK2-shRNA inhibited mobility in these processes in cortical and hippocampal neurons (Brickley and Stephenson, 2011; Loss and Stephenson, 2017). Therefore, it is possible that in our model, a decrease in TRAK2 expression induced by caspase-cleaved tau could be an essential contributor to the reduction in mitochondrial movement in hippocampal neurons. TRAK2 cooperatively binds to kinesin 1 (KIF5; Brickley et al., 2005) and RhoT1/T2 (Loss and Stephenson, 2017) in order to facilitate mitochondrial transport (Reis et al., 2009); however, our evidence indicates that kinesin 1 is likely not involved in the (cannot rule out changes in functional properties by posttransational modifications) impairment of mitochondrial movement (Quintanilla et al., 2014). In the same context, it is possible that caspase-cleaved tau could also affect the binding or interaction of RhoT1/T2 with TRAK2 or with mitochondria to facilitate their movement. While we did not explore this possibility in this work, it could certainly be interesting to test it. Nevertheless, this possibility does not reduce the importance of our finding that pathological modifications of tau could affect mitochondrial movement by modifying the function of TRAK2 and the bioenergetic status of mitochondria.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Bioethics Committee of the Universidad Autónoma de Chile, Santiago, Chile (Resolution 0049-17, May 2017).
Author Contributions
RQ conceived the study, performed some experiments, analyzed the data, and wrote the manuscript. CT-M performed some experiments and analyzed data. EV, MP, and AA performed the experiments. All authors read and approved the final version of the manuscript.
Funding
This work was supported by Fondo de Ciencia y Tecnología (FONDECYT), Chile (Grants: #1170441, 1200178; to RQ), and Comisión Nacional de Investigación Científica y Tecnológica (CONICYT)-PIA Anillo ACT1411 (to RQ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ari, C., Borysov, S. I., Wu, J., Padmanabhan, J., and Potter, H. (2014). Alzheimer amyloid beta inhibition of Eg5/kinesin 5 reduces neurotrophin and/or transmitter receptor function. Neurobiol. Aging 35, 1839–1849. doi: 10.1016/j.neurobiolaging.2014.02.006
Bongarzone, E. R., Foster, L., Byravan, S., Casaccia-Bonnefil, P., Schonmann, V., and Campagnoni, A. T. (1998). Two neuronal cell lines expressing the myelin basic protein gene display differences in their in vitro survival and in their response to glia. J. Neurosci. Res. 54, 309–319. doi: 10.1002/(sici)1097-4547(19981101)54:3<309::aid-jnr2>3.0.co;2-5
Brickley, K., Smith, M. J., Beck, M., and Stephenson, F. A. (2005). GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J. Biol. Chem. 280, 14723–14732. doi: 10.1074/jbc.m409095200
Brickley, K., and Stephenson, F. A. (2011). Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J. Biol. Chem. 286, 18079–18092. doi: 10.1074/jbc.m111.236018
Chaudhary, A. R., Berger, F., Berger, C. L., and Hendricks, A. G. (2018). Tau directs intracellular trafficking by regulating the forces exerted by kinesin and dynein teams. Traffic 19, 111–121. doi: 10.1111/tra.12537
Chen, Y. M., Gerwin, C., and Sheng, Z. H. (2009). Dynein light chain LC8 regulates syntaphilin-mediated mitochondrial docking in axons. J. Neurosci. 29, 9429–9438. doi: 10.1523/jneurosci.1472-09.2009
de Calignon, A., Fox, L. M., Pitstick, R., Carlson, G. A., Bacskai, B. J., Spires-Jones, T. L., et al. (2010). Caspase activation precedes and leads to tangles. Nature 464, 1201–1204. doi: 10.1038/nature08890
De Vos, K. J., Grierson, A. J., Ackerley, S., and Miller, C. C. J. (2008). Role of axonal transport in neurodegenerative diseases. Ann. Rev. Neurosci. 31, 151–173. doi: 10.1146/annurev.neuro.31.061307.090711
Ding, H., Matthews, T. A., and Johnson, G. V. (2006). Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J. Biol. Chem. 281, 19107–19114. doi: 10.1074/jbc.m511697200
Dixit, R., Ross, J. L., Goldman, Y. E., and Holzbaur, E. L. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. doi: 10.1126/science.1152993
Glater, E. E., Megeath, L. J., Stowers, R. S., and Schwarz, T. L. (2006). Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 173, 545–557. doi: 10.1083/jcb.200601067
Götz, J., Halliday, G., and Nisbet, R. M. (2019). Molecular pathogenesis of the tauopathies. Annu. Rev. Pathol. 14, 239–261. doi: 10.1146/annurev-pathmechdis-012418-012936
Götz, J., Ittner, L. M., and Kins, S. (2006). Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer’s disease? J. Neurochem. 98, 993–1006. doi: 10.1111/j.1471-4159.2006.03955.x
Hares, K., Miners, J. S., Cook, A. J., Rice, C., Scolding, N., Love, S., et al. (2017). Overexpression of kinesin superfamily motor proteins in Alzheimer’s disease. J. Alzheimers Dis. 60, 1511–1524. doi: 10.3233/JAD-170094
Ittner, L. M., Fath, T., Ke, Y. D., Bi, M., van Eersel, J., Li, M. K., et al. (2008). Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc. Natl. Acad. Sci. U S A 105, 15997–16002. doi: 10.1073/pnas.0808084105
Jara, C., Aránguiz, A., Cerpa, W., Tapia-Rojas, C., and Quintanilla, R. A. (2018). Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 18, 279–294. doi: 10.1016/j.redox.2018.07.010
Kang, J.-S., Tian, J.-H., Pan, P.-Y., Zald, P., Li, C., Deng, C., et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. doi: 10.1016/j.cell.2007.11.024
Krishnamurthy, P. K., and Johnson, G. V. W. (2004). Mutant (R406W) human tau is hyperphosphorylated and does not efficiently bind microtubules in a neuronal cortical cell model. J. Biol. Chem. 279, 7893–7900. doi: 10.1074/jbc.m311203200
LaPointe, N. E., Morfini, G., Pigino, G., Gaisina, I. N., Koziwoski, A. P., Binder, L. I., et al. (2009). The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res. 87, 440–451. doi: 10.1002/jnr.21850
Lin, M.-Y., Cheng, X.-T., Tammineni, P., Xie, Y., Zhou, B., Cai, Q., et al. (2017). Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94, 595.e6–610.e6. doi: 10.1016/j.neuron.2017.04.004
López-Doménech, G., Covill Cooke, C., Ivankovic, D., Halff, E. F., Sheehan, D. F., Norkett, R., et al. (2018). Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 37, 321–336. doi: 10.15252/embj.201696380
López-Doménech, G., Higgs, N. F., Vaccaro, V., Ros, H., Arancibia-Carcamo, I. L., Macaskill, A. F., et al. (2016). Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep. 17, 317–327. doi: 10.1016/j.celrep.2016.09.004
Loss, O., and Stephenson, F. A. (2017). Developmental changes in trak-mediated mitochondrial transport in neurons. Mol. Cell. Neurosci. 80, 134–147. doi: 10.1016/j.mcn.2017.03.006
Macaskill, A. F., and Kittler, J. T. (2010). Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 20, 102–112. doi: 10.1016/j.tcb.2009.11.002
Manczak, M., and Reddy, P. H. (2012). Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 21, 5131–5146. doi: 10.1093/hmg/dds360
Miller, K. E., and Sheetz, M. P. (2004). Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 117, 2791–2804. doi: 10.1242/jcs.01130
Misgeld, T., and Schwarz, T. L. (2017). Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666. doi: 10.1016/j.neuron.2017.09.055
Moore, A. S., and Holzbaur, E. L. F. (2018). Mitochondrial-cytoskeletal interactions: dynamic associations that facilitate network function and remodeling. Curr. Opin. Psychol. 3, 94–100. doi: 10.1016/j.cophys.2018.03.003
Morfini, G., Pigino, G., Mizuno, N., Kikkawa, M., and Brady, S. T. (2007). Tau binding to microtubules does not directly affect microtubule-based vesicle motility. J. Neurosci. Res. 85, 2620–2630. doi: 10.1002/jnr.21154
Nemani, N., Carvalho, E., Tomar, D., Dong, Z., Ketschek, A., Breves, S. L., et al. (2018). MIRO-1 determines mitochondrial shape transition upon GPCR activation and Ca2+ stress. Cell Rep. 23, 1005–1019. doi: 10.1016/j.celrep.2018.03.098
Ozcelik, S., Sprenger, F., Skachokova, Z., Fraser, G., Abramowski, D., Clavaguera, F., et al. (2016). Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol. Psychiatry 21, 1790–1798. doi: 10.1038/mp.2015.228
Pérez, M. J., Jara, C., and Quintanilla, R. A. (2018a). Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front. Neurosci. 12:441. doi: 10.3389/fnins.2018.00441
Pérez, M. J., Vergara-Pulgar, K., Jara, C., Cabezas-Opazo, F., and Quintanilla, R. A. (2018b). Caspase-cleaved tau impairs mitochondrial dynamics in Alzheimer’s disease. Mol. Neurobiol. 55, 1004–1018. doi: 10.1007/s12035-017-0385-x
Pérez, M. J., Loyola, R., Canelo, F., Aránguiz, A., Tapia-Monsalves, C., Osorio-Fuentealba, C., et al. (2020). NADPH oxidase contributes to oxidative damage and mitochondrial impairment induced by acute ethanol treatment in rat hippocampal neurons. Neuropharmacology 171:108100. doi: 10.1016/j.neuropharm.2020.108100
Quinn, J. P., Corbett, N. J., Kellett, K. A. B., and Hooper, N. M. (2018). Tau proteolysis in the pathogenesis of tauopathies: neurotoxic fragments and novel biomarkers. J. Alzheimers Dis. 63, 13–33. doi: 10.3233/jad-170959
Quintanilla, R. A., von Bernhardi, R., Godoy, J. A., Inestrosa, N. C., and Johnson, G. V. W. (2014). Phosphorylated tau potentiates Aβ-induced mitochondrial damage in mature neurons. Neurobiol. Dis. 71, 260–269. doi: 10.1016/j.nbd.2014.08.016
Quintanilla, R. A., Dolan, P. J., Jin, Y. N., and Johnson, G. V. W. (2012). Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol. Aging 33, 619.e25–635.e25. doi: 10.1016/j.neurobiolaging.2011.02.007
Quintanilla, R. A., Jin, Y. N., von Bernhardi, R., and Johnson, G. V. W. (2013). Mitochondrial permeability transition pore induces mitochondria injury in Huntington disease. Mol. Neurodegener. 8:45. doi: 10.1186/1750-1326-8-45
Quintanilla, R. A., Matthews-Roberson, T. A., Dolan, P. J., and Johnson, G. V. W. (2009). Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J. Biol. Chem. 284, 18754–18766. doi: 10.1074/jbc.m808908200
Reis, K., Fransson, A., and Aspenstrom, P. (2009). The Miro GTPases: at the heart of the mitochondrial transport machinery. FEBS Lett. 583, 1391–1398. doi: 10.1016/j.febslet.2009.04.015
Rodríguez-Martín, T., Cuchillo-Ibáñez, I., Noble, W., Nyenya, F., Anderton, B. H., and Hanger, D. P. (2013). Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 34, 2146–2157. doi: 10.1016/j.neurobiolaging.2013.03.015
Shahpasand, K., Uemura, I., Saito, T., Asano, T., Hata, K., Shibata, K., et al. (2012). Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J. Neurosci. 32, 2430–2441. doi: 10.1523/jneurosci.5927-11.2012
Sheng, Z.-H. (2017). The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 27, 403–416. doi: 10.1016/j.tcb.2017.01.005
Shidara, Y., and Hollenbeck, P. J. (2010). Defects in mitochondrial axonal transport and membrane potential without increased reactive oxygen species production in a Drosophila model of friedreich ataxia. J. Neurosci. 30, 11369–11378. doi: 10.1523/jneurosci.0529-10.2010
Shigematsu, H., Imasaki, T., Doki, C., Sumi, T., Aoki, M., Uchikubo-Kamo, T., et al. (2018). Structural insight into microtubule stabilization and kinesin inhibition by Tau family MAPs. J. Cell Biol. 217, 4155–4163. doi: 10.1083/jcb.201711182
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063. doi: 10.1083/jcb.200108057
Stokin, G. B., and Goldstein, L. S. (2006). Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 75, 607–627. doi: 10.1146/annurev.biochem.75.103004.142637
Stoothoff, W., Jones, P. B., Spires-Jones, T. L., Joyner, D., Chhabra, E., Bercury, K., et al. (2009). Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J. Neurochem. 111, 417–427. doi: 10.1111/j.1471-4159.2009.06316.x
Stowers, R. S., Megeath, L. J., Gorska-Andrzejak, J., Meinertzhangen, I. A., and Schwarz, T. L. (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. doi: 10.1016/s0896-6273(02)01094-2
Tapia-Rojas, C., Cabezas-Opazo, F., Deaton, C. A., Vergara, E. H., Johnson, G. V. W., and Quintanilla, R. A. (2019). Its all about tau. Prog. Neurobiol. 175, 54–76. doi: 10.1016/j.pneurobio.2018.12.005
Trinczek, B., Ebneth, A., Mandelkow, E. M., and Mandelkow, E. (1999). Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 112, 2355–2367.
van Spronsen, M., Mikhaylova, M., Lipka, J., Schlager, M. A., van den Heuvel, D. J., Kuijpers, M., et al. (2013). TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485–502. doi: 10.1016/j.neuron.2012.11.027
Vershinin, M., Carter, B. C., Razafsky, D. S., King, S. J., and Gross, S. P. (2007). Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. U S A 104, 87–92. doi: 10.1073/pnas.0607919104
Vossel, K. A., Xu, J. C., Fomenko, V., Miyamoto, T., Suberbielle, E., Knox, J. A., et al. (2015). Tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. J. Cell Biol. 209, 419–433. doi: 10.1083/jcb.201407065
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2010). Tau reduction prevents Aβ-induced defects in axonal transport. Science 330:198. doi: 10.1126/science.1194653
Watters, O., Connolly, N. M. C., König, H.-G., Düssmann, H., and Prehn, J. H. M. (2020). AMPK preferentially depresses retrograde transport of axonal mitochondria during localised nutrient deprivation. J. Neurosci. doi: 10.1523/jneurosci.2067-19.2020 [Epub ahead of print].
Yi, M., Weaver, D., and Hajnóczky, G. (2004). Control of mitochondrial motility and distribution by the calcium signal. J. Cell Biol. 167, 661–672. doi: 10.1083/jcb.200406038
Yuan, A., Kumar, A., Peterhoff, C., and Duff, K. (2008). Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci. 28, 1682–1687. doi: 10.1523/jneurosci.5242-07.2008
Keywords: kinesin, mitochondria, tau, TRAK2/Milton, transport, truncated tau
Citation: Quintanilla RA, Tapia-Monsalves C, Vergara EH, Pérez MJ and Aranguiz A (2020) Truncated Tau Induces Mitochondrial Transport Failure Through the Impairment of TRAK2 Protein and Bioenergetics Decline in Neuronal Cells. Front. Cell. Neurosci. 14:175. doi: 10.3389/fncel.2020.00175
Received: 05 January 2020; Accepted: 22 May 2020;
Published: 30 July 2020.
Edited by:
Tomas Luis Falzone, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Heather M. Wilkins, University of Kansas Medical Center Research Institute, United StatesFabienne E. Poulain, University of South Carolina, United States
Copyright © 2020 Quintanilla, Tapia-Monsalves, Vergara, Pérez and Aranguiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodrigo A. Quintanilla, rodrigo.quintanilla@uautonoma.cl