 Qilin Zhou
Qilin Zhou Yicong Lin
Yicong Lin Jing Ye
Jing Ye Liping Li
Liping Li Ningning Hu
Ningning Hu Di Wang
Di Wang Yuping Wang
Yuping Wang- 1Department of Neurology, Xuanwu Hospital, Capital Medical University, Beijing, China
- 2The Beijing Key Laboratory of Neuromodulation, Beijing, China
- 3Center of Epilepsy, Beijing Institute for Brain Disorders, Capital Medical University, Beijing, China
TBC1D24 mutation-related epileptic syndrome includes a wide spectrum of epilepsies. We describe a case with a homozygous TBC1D24 mutation inherited from consanguineous parents. The patient manifested epilepsia partialis continua (EPC) and rare secondary generalized tonic–clonic seizure without intellectual disability or developmental delay. EPC, which involved focal limbs, came with waking and went with sleep. The genetic analysis reported a novel mutation in the TBC1D24 gene, c.229_240del (p.82_84del). The homozygous mutation was inherited from her healthy parents who were heterozygous. Morphometric analysis program (MAP), an MRI post-processing technique, was used and detected a subtle abnormality of the brain. A comprehensive analysis based on semiology, electroencephalogram, somatosensory-evoked potential, and MAP suggested a potential focal structural abnormality. This case indicates a possible correlation between the TBC1D24 mutation and brain development abnormality.
Introduction
TBC1D24 mutation-related epileptic syndrome has various clinical characteristics. The correlation between phenotype and genotype is unclear. We reported a case of the homozygous TBC1D24 mutation, which was inherited from heterozygous parents. The patient’s distinct manifestation was epilepsia partialis continua (EPC). We also used the morphometric analysis program (MAP), an MRI post-processing technique, to detect a subtle lesion of the brain, aiming to describe the syndrome by genotype, phenotype, neuroimaging, and electrophysiology aspects.
Case Report
A 16-year-old female from a Chinese family with consanguineous parents, developed her first episode of jerks after a cold 11 years ago. The jerks involved right foot and leg without preceding sensation and was not relieved until she was given diazepam intravenously. The antiepileptic drugs (AEDs) were provided, but the episodes still occurred 3–4 times/year. The jerks were usually triggered by fatigue, emotions, or fever and lasted for minutes to hours. It could only be controlled by intravenous diazepam. She was diagnosed with “psychogenic attack” 6 years ago and was weaned off AEDs. The “psychogenic attacks” continued. Three years ago, the patient experienced loss of consciousness for minutes after right arm jerks. Six days before the admission, she developed right foot jerks after a long walk, but the jerks could not be relieved completely by diazepam. The jerks stopped with sleep without any treatment and reoccurred with waking. During the course of the disease, the patient had no intellectual disability or developmental delay, hearing loss, onychodystrophy, osteodystrophy, paresthesia, or cerebellar ataxia. She felt fatigue after running, long-distance walking, and climbing stairs. She had no remarkable medical history. Another individual in her family had similar paroxysmal episodes of leg jerks. Upon physical examination, no neurological deficit was found. MMSE test scored 29. The audiometry test was normal. The serum autoimmune panel, including anti-streptolysin “O,” C-reactive protein, rheumatoid factors, immunoglobulin G, immunoglobulin M, immunoglobulin A, alexin C3, alexin C4, erythrocyte sedimentation rate, anticardiolipin antibody, anti-nuclear antibody, anti-neutrophil cytoplasmic antibody, thyroid peroxidase antibody, and thyroglobulin antibody, was negative. Both amino acid and organic acid metabolism tests of serum and urine were negative.
Continuous video electroencephalogram (EEG) monitoring showed normal background without EEG change during leg jerking. One event was captured. The patient’s head and eyes deviated to the right with the right arm rising and the leg stretching straight. Then, the convulsion involved all limbs and lasted 20 s. During the event, she was alert but could not speak. The EEG showed rhythmic activity from the left frontocentral and midline regions. The activity quickly spread to the contralateral side with increased amplitude (Figure 1A). The brain MRI was reported normal. The somatosensory-evoked potential (SEP) showed remarkable reduced amplitude in the left cortex when right leg was tested. The upper limbs’ SEPs were normal. Genetic analysis through an epilepsy panel of 480 genes by Kangso Medical Inspection reported a homozygous mutation in the TBC1D24 gene, c.229_240del (p.82_84del). The homozygous mutation was inherited from her healthy parents who were heterozygous, which was consistent with autosomal recessive inheritance (Figure 1B and Supplementary Figure S1). We could not predict protein function related to this mutation through SIFT, Polyphen2, or Mutation Taster analysis software.
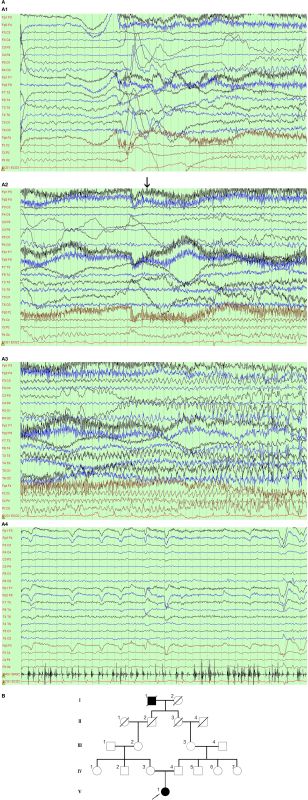
Figure 1. Ictal electroencephalogram (EEG) and pedigree of the family. (A) Ictal EEG with left frontocentral origination (arrow). A1–A3 show EEG evolution. A4 shows epilepsia partialis continua of the right lower limb without visible EEG change. (B) Pedigree of the family. The black square/circle indicates patients with the epilepsy phenotype; the double horizontal lines denote consanguineous marriage; the arrow shows the proband.
Morphometric analysis program was performed on 3-T T1-weighted images using SPM12 toolbox in MATLAB R2011b following previously established methods (1). The computed output was a junction map, which highlighted abnormal structures deviating from the average normal structures based on z-score. The normal database was provided along with the MAP program. The highlighted region might correlate with gray–white blurring on the underlying MRI. The MAP analysis identified a subtle abnormality candidate with a z-score over 4, a subtle gray–white blurring in the left medial parietal region (Figure 2).
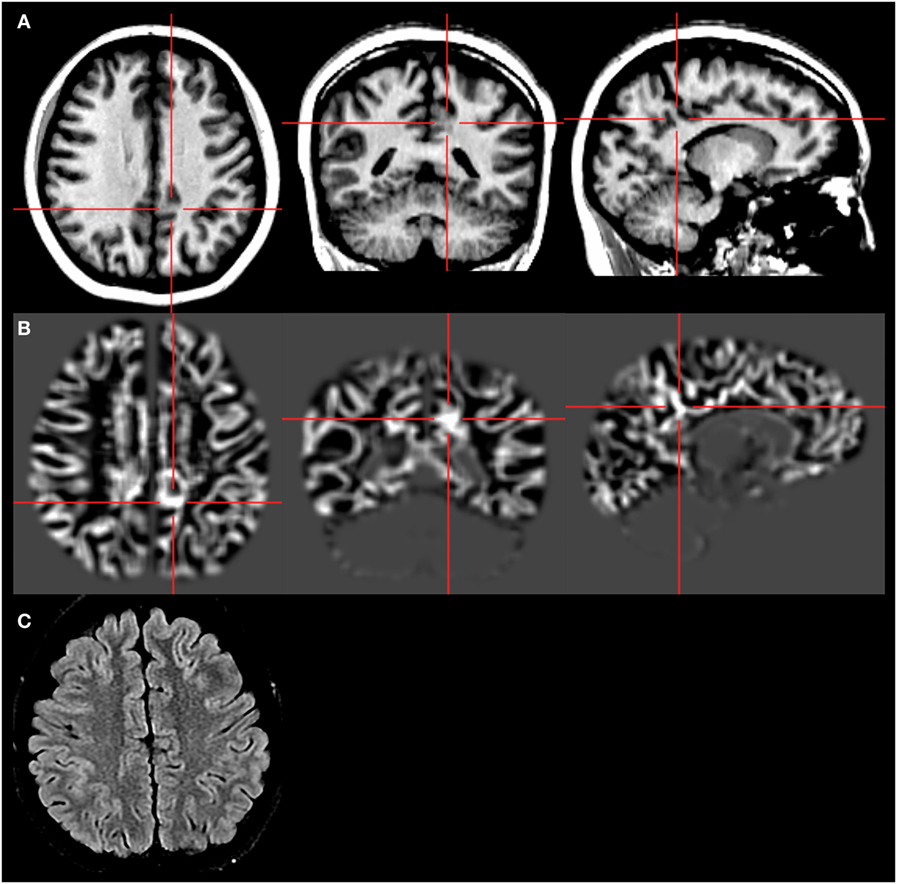
Figure 2. Morphometric analysis program (MAP) result. The crosshairs indicate the location of the MAP+ candidate. First row (A): T1-weighted magnetization prepared rapid acquisition with gradient echo images. Second row (B): gray–white matter junction z-score file, as the output of MAP processing of the T1-weighted image shown in the first row. Third row (C): T2-weighted fluid-attenuated inversion recovery images, chosen to best depict the MAP+ candidate. This was the best available image due to sub-optimal slice selection. There are no corresponding T2 changes.
The patient was given levetiracetam with a gradually increasing dosage to 1.25 g twice/day and sodium valproate with a dosage of 0.5 g twice/day and was completely relieved several days later. Upon follow-up, the patient was weaned off the sodium valproate and did not report any symptoms in the 10-month follow-up.
Both patient and parents provided written informed consent for the study and publication.
Discussion
We report here a novel homozygous mutation in TBC1D24: the c.229_240del (p.82_84del). The variants were inherited from the patient’s consanguineous parents who were healthy. Another family member, who probably carried the homozygous gene mutation, had similar symptoms. The phenotypes were characterized as EPC and rare secondary tonic–clonic seizure, without intellectual disability or hearing loss. EPC resolved with sleep and reappeared with waking and was not associated with EEG changes. The tonic–clonic seizure probably originated from the left central region. The clinical manifestation was consistent with reported cases of the TBC1D24 mutation. Balestrini et al. (2) summarized the clinical phenotypes and genotypes of 48 patients with TBC1D24 mutation-related epileptic syndrome. The authors found that this syndrome comprised a group of epilepsies with clinical heterogeneity. Most patients suffered the first seizure between the ages of 7 months and 8 years. The most common seizure types were myoclonus or clonic seizures. Myoclonus could be focal or generalized, always appeared in clusters and sustained for many days. In 2017, two articles expanded the phenotypic spectrum. Ragona et al. (3) reported a TBC1D24 mutation case that was related to alternating hemiplegia. Banuelos et al. (4) described a TBC1D24 mutation that was linked to tonic–clonic and myoclonic epilepsy in autosomal dominant inheritance, psychosis, parkinsonism, and intelligent obstacle with autosomal recessive inheritance. Trivisano et al. (5) reported two TBC1D24 mutation patients died from sudden unexpected death (SUDEP). The mechanism of SUDEP is still unclear. Some risk factors such as generalized tonic–clonic seizures, nocturnal seizures, early onset epilepsies, and polytherapy may cause SUDEP in patients (5, 6).
The TBC1D24 gene encodes a protein with an N-terminal Tre2/Bub2/Cdc16(TBC) domain, which interacts with GTPases. TBC1D24 could affect the migration and maturation of neurons by regulating the activity of ADP-ribosylation factor (ARF-6) (7). ARF-6 is a type of GTPase associated with vesicle membrane transport that plays a pivotal role in brain development (8). Therefore, the TBC1D24 mutation may potentially affect brain development, although the specific mechanism has not yet been clarified. Recently, Fischer et al. (9) found an unanticipated cationic pocket that bound phosphoinositides in a study of the crystal structure of the TBC domain of the Drosophila ortholog Skywalker, which had the same domain organization as humans. The most common mutations affect the phosphoinositide-binding pocket and inhibit lipid binding, consequently causing impaired synaptic-vesicle trafficking, seizures, and many other neurological defects in flies via restricting Skywalker diffusion in presynaptic terminals. The TBC1D24 gene encoded protein also contains a TLDc domain, which participates in oxidative stress (OS) resistance. The OS response is essential for the dynamic balance of reactive oxygen species (ROS), the important intracellular messengers. The imbalance in the numbers of ROS can do harm to DNA, proteins, and lipids and results in neuronal hyperactivity, synaptic dysfunction, and neuronal loss (10).
In Balestrini’s review (2), 16 out of 48 patients with the TBC1D24 mutation had visible structural abnormalities on MRI, including delayed myelination, hippocampal sclerosis, vermian hypoplasia, cerebellar atrophy, and cerebellar signal hyperintensity. In our case, the MRI was reported normal. We further performed an MRI post-processing procedure and found a subtle gray–white blurring in the left medial parietal region. The focal feature, based on MAP, SEP, EEG, and semiology, most likely indicated a restricted brain structural abnormality in the left medial centro-parietal region. We hypothesize that the TBC1D24 mutation might lead to a structural abnormality during brain development. Re-reviewing the MRI, MRI post-processing, or a combination of multi-modality neuroimaging may help detect abnormalities in patients with the TBC1D24 mutation and probably has implications for elucidating the mechanism of epileptogenesis.
Conclusion
Our report reveals a novel mutation and expands the genotypic spectrum of TBC1D24 mutation-related epileptic syndrome. A potential regional epileptogenic brain structural abnormality may develop in cases of TBC1D24 mutation.
Ethics Statement
All clinical data in this case report were either provided by the patient and her parents or collected by our team’s members with the consent of them. There was no additional invasive test or experimental drugs used out of order for the patient. A written informed consent was obtained from the patient and her parents for the participation in the study and the publication of this report. The case report is exempt from institutional review board approval.
Author Contributions
QZ acquired the clinical data, reviewed the literature, and drafted the manuscript. YL designed the study, oversaw data acquisition, supervised the initial drafting, performed post-MRI processing, and critically revised the manuscript. JY and YW analyzed the clinical data and critically revised the manuscript. LL and NH analyzed and interpreted the electrophysiological data and critically revised the manuscript. DW reviewed the literature and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the National High Technology Research and Development Program of China (2015AA020514) and Beijing Municipal Science and Technology Subject (Z151100003915117).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fneur.2017.00750/full#supplementary-material.
References
1. Huppertz HJ, Grimm C, Fauser S, Kassubek J, Mader I, Hochmuth A, et al. Enhanced visualization of blurred gray-white matter junctions in focal cortical dysplasia by voxel-based 3D MRI analysis. Epilepsy Res (2005) 67(1–2):35–50. doi:10.1016/j.eplepsyres.2005.07.009
2. Balestrini S, Milh M, Castiglioni C, Luthy K, Finelli MJ, Verstreken P, et al. TBC1D24 genotype-phenotype correlation: epilepsies and other neurologic features. Neurology (2016) 87(1):77–85. doi:10.1212/WNL.0000000000002807
3. Ragona F, Castellotti B, Salis B, Magri S, DiFrancesco JC, Nardocci N, et al. Alternating hemiplegia and epilepsia partialis continua: a new phenotype for a novel compound TBC1D24 mutation. Seizure (2017) 47:71–3. doi:10.1016/j.seizure.2017.03.003
4. Banuelos E, Ramsey K, Belnap N, Krishnan M, Balak C, Szelinger S, et al. Case report: novel mutations in TBC1D24 are associated with autosomal dominant tonic-clonic and myoclonic epilepsy and recessive parkinsonism, psychosis, and intellectual disability. F1000Res (2017) 6:553. doi:10.12688/f1000research.10588.1
5. Trivisano M, Bellusci M, Terracciano A, De Palma L, Pietrafusa N, Valeriani M, et al. TBC1D24 gene mutations are associated with high risk of sudden unexpected death. Epilepsy Behav (2017) 72(2017):208–9. doi:10.1016/j.yebeh.2017.04.010
6. Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol (2016) 15:1075–88. doi:10.1016/S1474-4422(16)30158-2
7. Falace A, Filipello F, La Padula V, Vanni N, Madia F, De Pietri Tonelli D, et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am J Hum Genet (2010) 87(3):365–70. doi:10.1016/j.ajhg.2010.07.020
8. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Bio (2009) 10(8):513–25. doi:10.1038/nrm2728
9. Fischer B, Lüthy K, Paesmans J, Koninck CD, Maes I, Swerts J, et al. Skywalker-TBC1D24 has a lipid-binding pocket mutated in epilepsy and required for synaptic function. Nat Struct Mol Biol (2016) 23(11):965–73. doi:10.1038/nsmb.3297
Keywords: TBC1D24 gene, epilepsia partialis continua, homozygous mutation, morphometric analysis program, MRI post-processing technique
Citation: Zhou Q, Lin Y, Ye J, Li L, Hu N, Wang D and Wang Y (2018) Homozygous TBC1D24 Mutation in a Case of Epilepsia Partialis Continua. Front. Neurol. 8:750. doi: 10.3389/fneur.2017.00750
Received: 19 October 2017; Accepted: 28 December 2017;
Published: 24 January 2018
Edited by:
John R. Mytinger, The Research Institute at Nationwide Children’s Hospital, United StatesReviewed by:
Brahim Tabarki, University of Sousse, TunisiaGouri Rao Passi, Choithram Hospital and Research Centre, India
Copyright: © 2018 Zhou, Lin, Ye, Li, Hu, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yicong Lin, linyc_1@163.com