 Matteo Foschi1†
Matteo Foschi1† Veria Vacchiano1†
Veria Vacchiano1† Patrizia Avoni1,2
Patrizia Avoni1,2 Alex Incensi2Stella Battaglia3
Alex Incensi2Stella Battaglia3 Vincenzo Donadio2
Vincenzo Donadio2 Elena Panzeri4
Elena Panzeri4 Maria Teresa Bassi4
Maria Teresa Bassi4 Rocco Liguori1,2
Rocco Liguori1,2 Giovanni Rizzo1,2*
Giovanni Rizzo1,2*- 1Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy
- 2UOC Clinica Neurologica, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- 3UOC Neuroradiologia, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- 4Laboratory of Molecular Biology, Scientific Institute IRCCS E. Medea, Lecco, Italy
X-linked adrenoleukodystrophy (x-ALD) is a rare genetic disorder caused by a mutation in the ABCD1 gene, which encodes for a peroxisomal very long chain fatty acid transporter. Clinically, x-ALD can present a wide spectrum of different phenotypes: asymptomatic carriers, Addison only, cerebral x-ALD, and myelopathy with/without evidence of peripheral axonopathy (Adrenomyeloneuropathy). We report on two cases of adult x-ALD, with atypical phenotypes: (Case 1) A 37-years-old male with a 2-years-long history of spastic paraparesis, urinary urgency, and subclinical adrenocortical insufficiency. As an atypical finding, the MRI showed multiple congenital brain development defects. (Case 2) A 63-years-old male with a previous diagnosis of Addison disease, with a 6-years-long history of spastic paraparesis. Two years later, he complained of severe and disabling burning pain in his feet. A nerve conduction study was normal, but a skin biopsy revealed autonomic and somatic small fiber neuropathy. In both cases, genetic testing disclosed hemizygous mutation in ABCD1 associated with x-ALD: c.1394-2A > G and p.(Thr254Met), respectively. While case 1 supports the key role of peroxisome functions in brain development, case 2 points to a possible selective and clinically relevant peripheral small fiber degeneration in x-ALD myelopathy.
Introduction
X-linked adrenoleukodystrophy (x-ALD) is a rare genetic disorder with a wide spectrum of different clinical phenotypes (1). The disease is caused by the impairment of peroxisomal beta-oxidation of very long chain fatty acids (VLCFAs), which accumulate in the central nervous system (CNS), adrenal cortex and testes (2–4). The metabolic abnormality is caused by mutations in the ABCD1 gene, which is located in the X chromosome. ABCD1 encodes for a peroxisomal membrane transporter of (adrenoleukodystrophy protein, ALDP) (5–7).
Clinically, x-ALD phenotypes include asymptomatic carriers, Addison only, childhood, adolescent, and adult cerebral x-ALD, myelopathy with or without evidence of peripheral axonal neuropathy (Adrenomyeloneuropathy, AMN) (1, 8). The pathophysiological hallmark of the neurological involvement in x-ALD, consists of a combination of slowly progressive spinal cord and peripheral nerve damage, with or without CNS inflammatory demyelination (9, 10). The latter is typical of cerebral x-ALD and it seems to have a different pathogenesis from the chronic myelopathy and peripheral axonopathy of adult x-ALD (1, 10, 11).
This paper reports on two patients with adult x-ALD showing unusual clinical features: (1) congenital brain development defects and (2) clinically relevant x-ALD related small fiber neuropathy (SFN).
Both patients explicitly agreed to their inclusion in this retrospective observational case report and gave written informed consent for publication.
Case Reports
Case 1
A 37-years-old male caught our attention due to the onset of progressive gait difficulties caused by a rigidity and weakness affecting both legs from the age of 35. At the time, he complained of urinary urgency with incontinence and erectile dysfunction. His family history was negative for neurological or endocrinological diseases. He had normal psychomotor development without learning disabilities and did not report cognitive symptoms. The neurological examination (NE) showed mild dysarthria, spastic paraparesis with a wide-based spastic gait. Deep tendon reflexes were diffusely brisk with a bilateral Achilles clonus and Babinski sign. A brain magnetic resonance imaging (MRI) scan showed multiple congenital brain development defects (Figure 1): posterior commissure agenesis, right fornix, and ipsilateral mammillary body hypoplasia, colpocephaly, right frontal parasagittal cortical thickening, two periventricular nodular heterotopic foci in the right parietal areas, and two venous drainage abnormalities in the left cerebellar hemisphere and right frontal lobe, respectively. A neuropsychological evaluation revealed no abnormalities. Electroencephalography (EEG) did not show any epileptiform discharges. A spinal MRI showed spinal cord atrophy. Electromyography (EMG) did not reveal any abnormal finding. Somatosensory evoked potentials (SEPs) showed increased central conduction time from the right arm and the left leg. No response was recorded from the right leg. Motor evoked potentials (MEPs) were absent in both legs. Visual evoked potentials (VEPs), and optic coherence tomography (OCT) were unremarkable. We used a multi-gene panel for hereditary spastic paraplegia and other motor neuron diseases (Supplementary Material). Genetic analysis revealed the presence of the hemizygous mutation c.1394-2A > G in the ABCD1 gene, leading to the diagnosis of x-ALD. Hematochemical examination disclosed normal cortisol levels with an increased adrenocorticotrophic hormone (ACTH; 352 pg/mL, n.v. 5–60 pg/mL), consistent with subclinical adrenocortical insufficiency. VLCFA plasma levels were increased. A multi-gene panel testing for cortical development defects excluded other possible genetic causes (Supplementary Material).
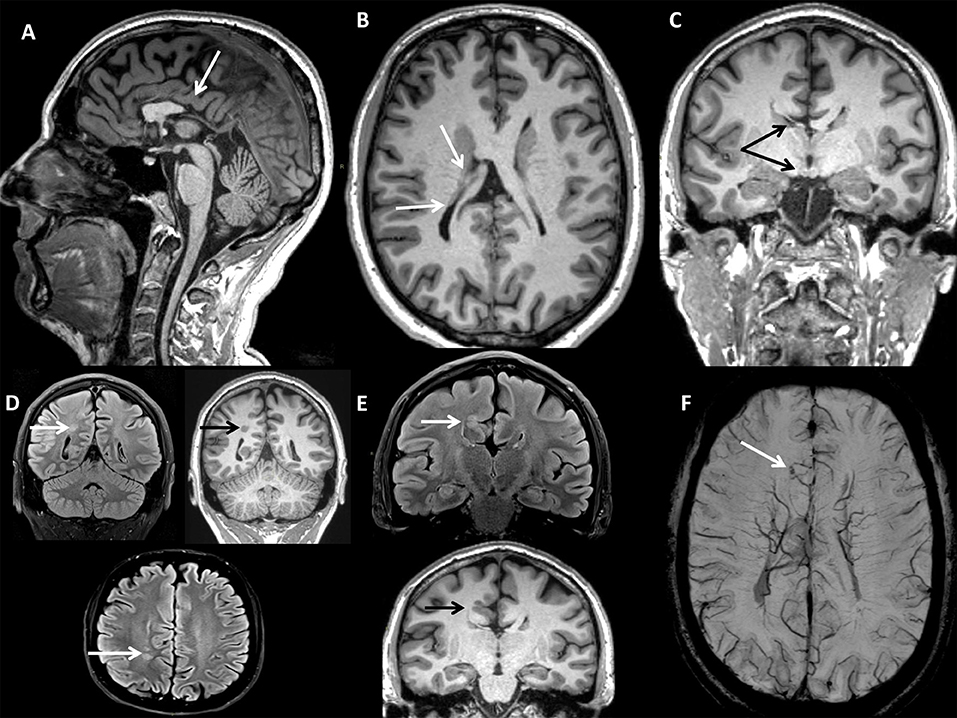
Figure 1. BRAIN MRI (CASE 1): (A) Sagittal 3D Magnetization Prepared Rapid Acquisition Gradient Echo (MPRAGE) T1-w with axial (B), and coronal (C) reconstructions showing partial corpus callosum agenesis, right fornix, and mammillary body hypoplasia, hypogenesis of bilateral lateral ventricles (colpocephaly). (D) Turbo Spin Echo Fluid-Attenuated Inversion Recovery (TSE FLAIR) T2-w axial and coronal (FLAIR T2-w & T1-w) scans showing nodular heterotopic foci. (E) TSE FLAIR T2-w & T1-w coronal scan showing right frontal parasagittal cortical thickening. (F) Susceptibility Weighted Imaging (SWI) axial scan with Maximum Intensity Projection (MIP) reconstruction showing right frontal parasagittal venous drainage abnormality with telangiectasia.
Case 2
A 63-years-old male with a 6-years-long history of progressive gait impairment, received a diagnosis of Addison's disease at the age of 13, and had been chronically treated with cortone acetate from the age of 53. From the age of 57, he noticed a progressive tendency to drag both his feet, which is associated with orthostatic imbalance. His family history was negative for neurological or endocrinological diseases. Psychomotor development was normal and the patient did not report cognitive symptoms. On admission, NE showed diffuse skin pigmentation, and spastic paraparesis (right > left). The deep tendon reflexes were brisk with bilateral Achilles clonus, Babinski, and a right-hand Hoffman sign. Hematochemical investigations, including liver and renal functions, vitamin B12, folic acid, creatinine phosphokinase (CPK), thyroid hormone levels, and a complete screening for autoimmune disease, were all unremarkable. EMG excluded a peripheral neuropathy. SEP showed an increased latency in the central responses from the upper and lower limbs. No motor responses were recorded from the lower limbs. A brain MRI showed T2-hyperintensity of the corticospinal tracts (left > right) with a bilateral hypointensity of the pre-central gyrus in susceptibility weighted imaging (SWI) sequences. A spinal MRI showed atrophy of the spinal cord. Neuropsychological evaluation uncovered no abnormalities. Plasma levels of VLCFA were increased. Genetic analysis of the ABCD1 gene, disclosed the presence of the hemizygous base change c.761C > T, leading to the amino acid substitution p.(Thr254Met). This change is known in ClinVar, as likely pathogenic and classified pathogenic, according to the ACMG guidelines for variant classification, confirming the diagnosis of x-ALD. From the age of 65, the patient started to complain of a severe burning pain and painful dysesthesia affecting the lower limbs and feet. Within a few months, the pain rapidly became his main complaint impairing his quality of life. The patient was treated with common analgesics, gabapentin, amitriptyline, duloxetine, and cannabis without substantial improvement or side effects. High doses of pregabalin mildly attenuated the symptoms. EMG was repeated with negative results. Therefore, the patient underwent a skin biopsy. The immunofluorescence (IF) analysis (12) revealed a prevalently somatic SFN (Figure 2). We repeated blood tests including hepatic and renal function, thyroid hormones levels, serological screening for infectious diseases, and a glucose challenge test. All tests turned out to be in range, excluding the presence of risk factors potentially associated with SFN (13). His family history was negative for symptoms possibly related with SNF. We performed whole exome sequencing to search for the possible presence of concomitant mutations/variants in other genes that could explain the complex clinical phenotype. Among genes causing hereditary neuropathies, whole exome analysis identified only two heterozygous variants in SBF1 (c.3044G>A, p.Arg1015Gln-rs372268920) and WNK1 (c.2228C > T, p.Pro743Leu -rs528772088), genes with a very low allele frequency in the ExAC database (0.0003 and 0.0008, respectively). In both cases bioinformatics analysis predict likely deleterious effects on protein function. None of them are known in the ClinVar database. However, both genes are associated with recessive diseases: SBF1 with Charcot Marie Tooth disease type 4B3, (14) and WNK1 with a hereditary sensory and autonomic neuropathy, type 2 (15). Therefore, the sole presence of these variants may somewhat contribute to the SFN phenotype, but cannot be considered pathogenic mutations. Furthermore, given the known relationship between WNK1 mutations and pseudohypoaldosteronism type 2 (16), the endocrinological history was deepened, by searching for specific features (hypertension, hyperkalemia, or hyperchloremic metabolic acidosis). The patient had no antecedents of suggestive symptoms and repeated blood, and urine analyses never showed electrolytes or pH alterations.
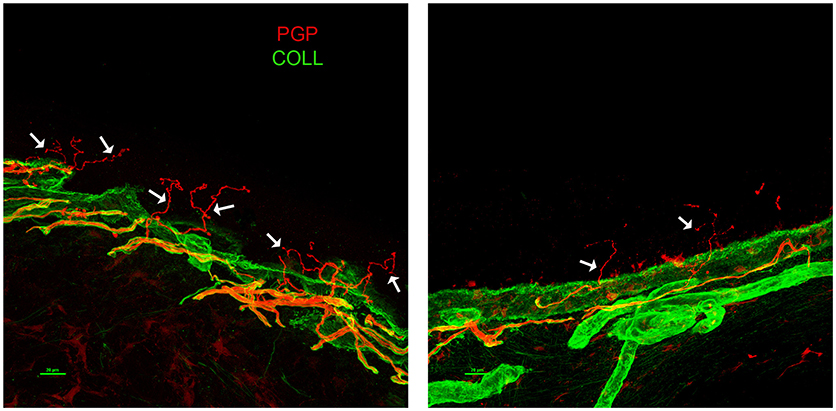
Figure 2. Skin Biopsy Immunoflorescence (Case 2) showing prevalently somatic small fiber neuropathy (SFN). A confocal study of epidermal innervation in the patient and a control subject: leg epidermal innervation disclosed by confocal microscope (×40) in an age-matched control subject (left panel), and the x-ALD patient case 2 (right panel). Nerve fibers are marked in red by a pan-neuronal marker PGP 9.5 whereas, the collagen staining is shown in green. The basement membrane separating epidermis from dermis is marked by collagen staining. Free-ending PGP immunoreactive fibers are evident in the epidermis of the control (arrows) whereas, the patient showed a marked decrease of such fibers, often fragmented (arrows). PGP, protein gene product; COLL, collagen.
The clinical and demographic characteristics of case 1 and 2 are summarized in Table 1, along with the results of the principal diagnostic investigations.
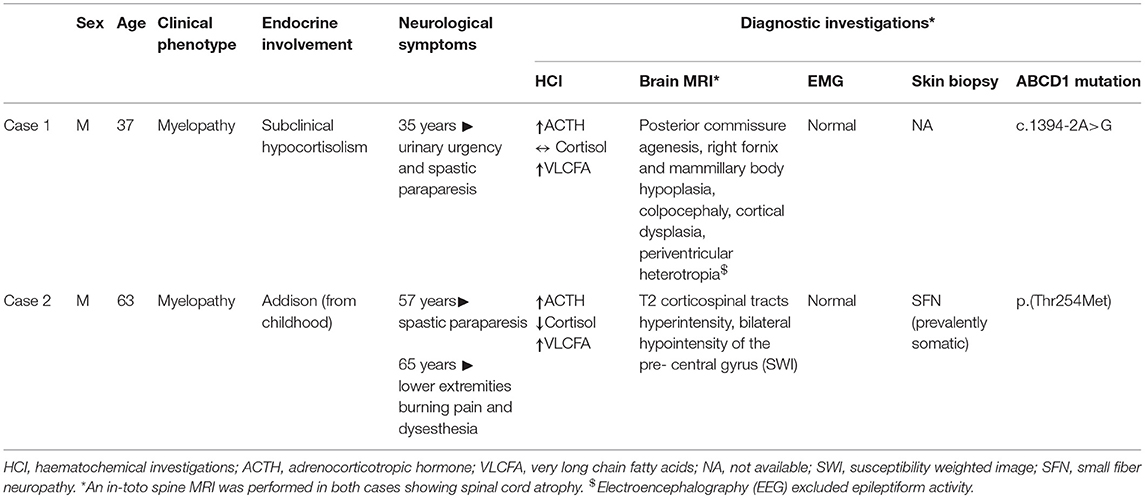
Table 1. Clinical features and diagnostic investigations of case 1 and case 2.
Discussion and Conclusions
We have described two cases of adult x-ALD and their atypical clinical features. Case 1 clinically corresponds to a pure adrenomyelopathy without cerebral demyelination. The patient carried the c.1394-2A > G mutation of the ABCD1 gene. This mutation has previously been reported only in one Indian patient affected by childhood cerebral x-ALD and without evidence of neuronal migrations defects (17).
Although not experimentally proven, at the transcript level, the base change alters the wild type (WT) acceptor site, most probably affecting ABCD1 RNA splicing. With these limits, our cases present two peculiarities: (1) the presence of brain development defects on the MRI, not previously observed in the adult x-ALD spectrum; and (2) the late onset with a typical adrenomyelopathy phenotype. Brain development alterations can be related to specific metabolic dysfunctions. In particular, they may result from a complete loss of all peroxisomal functions, or from defects in individual enzymatic reactions (18). Indeed, pathological aberrations of the central nervous system are prominent features in most peroxisomal disorders. Specifically, peroxisome biogenesis disorders (PBD) include: (1) Zellweger spectrum disorders (Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum), related to mutations in 13 different peroxin (PEX) genes (PEX1, PEX2, PEX3, PEX5, PEX6, PEX10, PEX11b, PEX12, PEX13, PEX14, PEX16, PEX19, PEX26) and (2) rhizomelic chondrodysplasia punctata (RCDP) type 1, caused by mutations in the PEX 7 gene (18). In Zellweger syndrome, typical brain pathological features are abnormalities in the cytoarchitecture of the cerebral cortex with microgyric and pachygyric areas. The most frequent finding is a local thickening of small convolutions around the central sulcus (centrosylvian pachygyria), sometimes with an excess of local convolutions in the same area (polymicrogyria) (19). The heterotopic localization of Purkinje cells within the cerebellar white matter is often observed (18). Several metabolic diseases related to a single enzyme deficiency (SED) can cause neuronal migration defects (20, 21). For instance, neocortical dysplasia has been reported in some cases of acyl-CoA oxidase deficiency, in association with typical cerebral and/or cerebellar white matter abnormalities on the MRI (20). Another example is peroxisomal D-bifunctional protein (DBP) deficiency (21). These two conditions share a disruption in the peroxisomal VLCFA degradation pathway, with x-ALD. Less severe cerebral migratory abnormalities have occasionally been reported in neonatal x-ALD (22). These observations suggest that multiple peroxisomal functions are required for proper brain development.
The congruence of the biochemical patterns of VLCFAs in x-ALD, Zellweger spectrum disorder and SED syndromes (19–21) suggests that their accumulation could cause specific morphogenesis abnormalities. Pathologically increased VLCFAs have been related to direct and indirect biochemical disruptions (22). Specifically, VLCFAs inhibit intracellular biochemical processes and interfere with the metabolism of other unsaturated fatty acids (such as arachidonic acid–C20:4) that play an important role in histogenesis (22). We can only speculate that the neurogenesis aberrations rarely reported in x-ALD, can be explained by the same mechanisms.
Case 2 presented adrenocortical deficiency, myelopathy, and a painful syndrome due to SFN. SFN has recently been observed as a frequent finding in x-ALD, although only in a small case series (23). It appears to be more prevalent in older subjects with an AMN phenotype (23). Similar to large fiber degeneration in AMN, SFN probably starts in an early asymptomatic stage and progresses with age. The pathogenesis of x-ALD-related SFN is still unclear. The frequent association with peripheral axonopathy suggests a myelopathy-related dying back process (23) but small fibers are rarely affected selectively, as in our case. Our report represents the first histologic characterization of x-ALD related SFN through a skin biopsy analysis with IF (12). The negativity of all tests performed to exclude other possible secondary causes (diabetes, infectious diseases, hepatic, or thyroid alterations, autoimmunity) (13) support a probable relationship with x-ALD. The whole exome sequencing excluded concomitant mutations in other genes causing hereditary neuropathies, except for two heterozygous variants in the SBF1 and WNK1 genes, which are both involved in different forms of recessive neuropathies (CMT4B3 and HSN2, respectively) (14, 15). Although not causative, we cannot exclude the possibility that these variants contribute to the phenotypic component of SFN observed in our patient. Indeed, carriers of WNK1 recessive mutations show greater sensitivity to thermal and cold pain stimuli (24). However, although WNK1 mutations are associated with autosomal dominant pseudohypoaldosteronism (16), our patient had no clinical or laboratoristic features of pseudohypoaldosteronism. Unfortunately, we did not have access to parental DNA to assess the segregation of all these variants.
Our skin specimen revealed a prevalently somatic small fiber loss. The somatic subtype accounts for about 30% of all peripheral small fibers (25). In contrast to autonomic fibers, somatic fibers are thicker and sparsely myelinated (25, 26). Therefore, the most plausible hypothesis is that somatic fibers undergo the same degenerative process, affecting the large myelinated nerves in the AMN, though this occurs at an earlier stage of disease progression. Another peculiarity of our report is the clinical presentation with predominant SFN-related symptoms (pain and dysesthesia unresponsive to commonly used therapies), in contrast to previously reported samples (23) in which SFN was asymptomatic or paucisymptomatic. The patient did not report any signs or symptoms that might be linked to dysautonomia (such as gastrointestinal motility disturbances, orthostatic hypotension, pupillary abnormalities, or sweating alterations). Further studies, including routinely performed skin biopsy analyses, are needed to clarify the actual prevalence of SFN in adult x-ALD with/without AMN. These data (along with histopathology) should contribute to a more complete characterization of x-ALD-related SFN, to improve the understanding of the pathogenic mechanisms underlying small fiber involvement.
In conclusion, while noting that extensive genetic analyses should be interpreted with caution in patients whose symptoms are related to different coexisting causes, our two cases expand the observed clinical heterogeneity of adult x-ALD, including the possible presence of brain development defects and clinically relevant SFN.
Author Contributions
MF, VV, and GR collected clinical data and drafted the manuscript. RL and PA contributed to the clinical data collection and clinical management of the patients, and to the manuscript revision. MB and EP performed and interpreted genetic analyses, and revised the manuscript. VD and AI performed skin biopsy analysis, created Figure 2, and contributed to the revision. SB performed MRI, created Figure 1, and revised the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
Italian Ministry of Health Funding Grant n. RC 2017-2018-2019 and 5XMILLE.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank David Neil Manners for the English editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00070/full#supplementary-material
References
1. Engelen M, Kemp S, Poll-The BT. X-linked adrenoleukodystrophy: pathogenesis and treatment. Curr Neurol Neurosci Rep. (2014) 14:486. doi: 10.1007/s11910-014-0486-0
2. Igarashi M, Schaumburg HH, Powers J, Kishmoto Y, Kolodny E, Suzuki K. Fatty acid abnormality in adrenoleukodystrophy. J Neurochem. (1976) 26:851–60. doi: 10.1111/j.1471-4159.1976.tb04462.x
3. Powers JM, Schaumburg HH. Adreno-leukodystrophy (sex-linked Schilder's disease). A pathogenetic hypothesis based on ultrastructural lesions in adrenalcortex, peripheral nerve and testis. Am J Pathol. (1974) 76:481–91.
4. Dubey P, Raymond GV, Moser AB, Kharkar S, BezmanL, Moser HW. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J Pediatr. (2005) 146:528–32. doi: 10.1016/j.jpeds.2004.10.067
5. Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H, et al. Putative X- linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature (1993) 361:726–30. doi: 10.1038/361726a0
6. Migeon BR, Moser HW, Moser AB, Axelman J, Sillence D, Norum RA. Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci USA. (1981) 78:5066–70. doi: 10.1073/pnas.78.8.5066
7. Mosser J, Lutz Y, Stoeckel ME, Sarde CO, Kretz C, Douar AM, et al. The gene responsible for adrenoleukodystrophy encodes a peroxisomal membrane protein. Hum Mol Genet. (1994) 3:265–71. doi: 10.1093/hmg/3.2.265
8. Kemp S, Berger J, Aubourg P. X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta (2012) 1822:1465–74. doi: 10.1016/j.bbadis.2012.03.012
9. van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol. (2001) 49:186–94. doi: 10.1002/1531-8249(20010201)49:2<186::AID-ANA38>3.0.CO;2-R
10. Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, et al. X- linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. (2012) 7:51. doi: 10.1186/1750-1172-7-51
11. Moser HW, Smith KD, Watkins PA, Powers J, Moser AB. X-linked adrenoleukodystrophy. In: Scriver CR, Sly WS, Childs B, Beaudet AL, Valle D, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw Hill (2001). p. 3257–301.
12. Donadio V, Nolano M, Provitera V, Stancanelli A, Lullo F, Liguori R, et al. Skin sympathetic adrenergic innervation: an immunofluorescence confocal study. Ann Neurol. (2006) 59:376–81. doi: 10.1002/ana.20769
13. Gibbons CH. Small fiber neuropathies. Continuum (2014) 20:1398–412. doi: 10.1212/01.CON.0000455874.68556.02
14. Nakhro K, Park J-M, Hong YB, Park JH, Nam SH, Yoon BR, et al. SET binding factor 1 (SBF1) mutation causes charcot-marie-tooth disease type 4B3. Neurology (2013) 81:165–73. doi: 10.1212/WNL.0b013e31829a3421
15. Shekarabi M, Girard N, Riviere J-B, Dion P, Houle M, Toulouse A, et al. Mutations in the nervous system-specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest. (2008) 118:2496–505. doi: 10.1172/JCI34088
16. Bergaya S, Vidal-Petiot E, Jeunemaitre X, Hadchouel J. Pathogenesis of pseudohypoaldosteronism type 2 by WNK1 mutations. Curr Opin Nephrol Hypertens. (2012) 21:39–45. doi: 10.1097/MNH.0b013e32834d2fde
17. Kumar N, Shukla P, Taneja KK, Kalra V, Bansal SK. De novo ABCD1 gene mutation in an Indian patient with adrenoleukodystrophy. Pediatr Neurol. (2008) 39:289–92. doi: 10.1016/j.pediatrneurol.2008.07.006
18. Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta (2006) 1763:1733–48. doi: 10.1016/j.bbamcr.2006.09.010
19. Evrard P, Caviness VS Jr, Prats-Vinas J, Lyon G. The mechanism of arrest of neuronal migration in the Zellweger malformation: a hypothesis based upon cytoarchitectonic analysis. Acta Neuropathol. (1978) 41:109–17. doi: 10.1007/BF00689761
20. Ferdinandusse S, Denis S, Hogenhout EM, Koster J, van Roermund CW, Moser AB, et al. Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency. Hum Mutat. (2007) 28:904–12. doi: 10.1002/humu.20535
21. Ferdinandusse S, Ylianttila MS, Gloerich J, Koski MK, Oostheim W, Waterham HR, et al. Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype–phenotype analysis. Am J Hum Genet. (2006) 78:112–24. doi: 10.1086/498880
22. Kelley RI, Datta NS, Dobyns WB, Hajra AK, Moser AB, Noetzel MJ, et al. Neonatal adrenoleukodystrophy: new cases, biochemical studies, and differentiation from Zellweger and related peroxisomal polydystrophy syndromes. Am J Med Genet. (1986) 23:869–901. doi: 10.1002/ajmg.1320230404
23. Horn MA, Nilsen KB, Jørum E, Mellgren SI, Tallaksen CM. Small nerve fiber involvement is frequent in X-linked adrenoleukodystrophy. Neurology (2014) 82:1678–83. doi: 10.1212/WNL.0000000000000415
24. Loggia ML, Bushnell MC, Tétreault M, Thiffault I, Bhérer C, Mohammed NK, et al. Carriers of recessive WNK1/HSN2 mutations for hereditary sensory and autonomic neuropathy type 2 (HSAN2) are more sensitive to thermal stimuli. J Neurosci. (2009) 29:2162–6. doi: 10.1523/JNEUROSCI.4633-08.2009
25. Lauria G, Cornblath DR, Johansson O, McArthur JC, Mellgren SI, Nolano M, et al. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur J Neurol. (2005) 12:747–58. doi: 10.1111/j.1468-1331.2005.01260.x
Keywords: adrenoleukodystrophy, small fiber neuropathy, skin biopsy, MRI, adrenomyelopathy, brain development defects, ABCD1, very long chain fatty acids
Citation: Foschi M, Vacchiano V, Avoni P, Incensi A, Battaglia S, Donadio V, Panzeri E, Bassi MT, Liguori R and Rizzo G (2019) Broadening the Spectrum of Adulthood X-Linked Adrenoleukodystrophy: A Report of Two Atypical Cases. Front. Neurol. 10:70. doi: 10.3389/fneur.2019.00070
Received: 14 October 2018; Accepted: 18 January 2019;
Published: 06 February 2019.
Edited by:
Ignacio Mata, Cleveland Clinic, United StatesReviewed by:
Oswaldo Lorenzo-Betancor, University of Washington, United StatesMario Reynaldo Cornejo-Olivas, Instituto Nacional de Ciencias Neurológicas, Peru
Copyright © 2019 Foschi, Vacchiano, Avoni, Incensi, Battaglia, Donadio, Panzeri, Bassi, Liguori and Rizzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Rizzo, g.rizzo@unibo.it
†These authors have contributed equally to this work