 Xue Lin1,2†
Xue Lin1,2† Yichen Xu3†
Yichen Xu3† Zhen Zhen1,4Kang Xiao5
Zhen Zhen1,4Kang Xiao5 Xu Chen6
Xu Chen6 Jigang Yang7
Jigang Yang7 Hongzhi Guan8
Hongzhi Guan8 Qi Shi5
Qi Shi5 Xiaoping Dong5
Xiaoping Dong5 Jiawei Wang1*Yanjun Guo1*
Jiawei Wang1*Yanjun Guo1*- 1Department of Neurology, Beijing Tongren Hospital, Capital Medical University, Beijing, China
- 2Department of Neurology, Beijing Puren Hospital, Beijing, China
- 3Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
- 4Department of Neurology, People's Hospital of Beijing Daxing District, Beijing, China
- 5State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
- 6Department of Neurosurgery, Beijing Friendship Hospital, Capital Medical University, Beijing, China
- 7Department of Nuclear Medicine, Beijing Friendship Hospital, Capital Medical University, Beijing, China
- 8Department of Neurology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Beijing, China
Genetic Creutzfeldt–Jakob disease (gCJD) characterized by mutations in the prion protein (PrP) gene (PRNP) contributes to approximately 10–15% of the overall human prion diseases. Here, we report a rare mutation in the PRNP gene in a Han-Chinese family. A 36-year-old man initiated with anxiety and depression followed by progressive dementia, cogwheel-like rigidity combined with tremors, and he was diagnosed with frontotemporal lobar dementia in the first 2 years. The disease progression was relatively slow, and the patient developed into akinetic mutism in 4 years. To characterize the disease, following the pedigree studies, neuropsychological examination, neuroimaging studies, real-time quaking-induced conversion (RT-QuIC) examination, and so on were conducted. We eventually identified a rare mutation of G114V combined with one octapeptide repeats deletion (1-ORPD) in the PrP in the patient by DNA sequencing. In addition, the same mutation and deletion were subsequently identified in the patient's mother without any syndromes. His maternal grandmother had a late onset of the disease in her 60s. Given that 1-OPRD has never been reported in human prion disease before, our first report that both G114V mutation and 1-OPRD appear in the family would forward our understanding of the etiological mechanisms of the gCJD.
Introduction
Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), are a group of rare, fatal neurodegenerative disorders in humans and animals, characterized by the accumulation and aggregation of prions or abnormally folded proteins (1). The abnormally folded proteins PrPSc have a high number of β-pleated sheets in their posttranslational conformation compared with the typical α-helices seen in the normal form of the protein (PrPc). The PrPSc conformation is partially resistant to proteases and acts as a template for further misfolding of the normal PrPC to abnormal PrPSc. There are three main groups of prion diseases, termed sporadic (Creutzfeldt–Jakob disease [CJD], sporadic fatal insomnia, and variably protease-sensitive prionopathy), genetic (genetic CJD, fatal familial insomnia, and Gerstmann–Straussler–Scheinker syndrome), and acquired (kuru, variant CJD, and iatrogenic CJD). For instance, approximately 85% of CJD cases are sporadic, 10–15% are inherited, and >1% of the cases are acquired (2, 3).
Based on the clinical and pathological features, inherited human prion disease, or genetic prion diseases (gPrDs), are classified as genetic CJD (gCJD) or familial CJD (fCJD), GSS disease, and fatal familial insomnia (FFI). The majority of gCJD have a common feature of rapid progression with a longer survival time; some of them also present as ataxia or Parkinson-like disorders, which have a slower decline over a few to several years (4). The clinical manifestations and neuropathological abnormalities of gPrDs may vary, and the definitive diagnosis of the gCJD requires genetic evidence except for the routine diagnostic elements such as autoimmune encephalitis (5).
The mutation in the prion protein gene (PRNP) was identified to contribute to the development of gPrDs. The PRNP gene is located at chromosome 20 and encodes prion protein (PrP) with 253 amino acids highly expressed in the central and peripheral nervous systems. The types of mutations in PRNP include missense and non-sense mutation, insertions, and deletion (6). Despite that more than 60 PRNP variants were identified to be pathogenic to date, five among them, E200K, V210I, V180I, D178N, and P102L account for around 85% of gPrD cases (7).
The G114V PRNP variant is a rare subtype in gCJD (8), and only 15 patients from 5 families were reported worldwide. The affected individuals were characterized by an early age of onset, neuropsychiatric symptoms, rapidly progressive dementia, sleep disturbance with predominant pyramidal and extrapyramidal symptoms, and long disease duration (1–5 years) (8–14). Interestingly, a PRNP gene variant with one octapeptide repeat deletion (1-OPRD) is highly related to gastric cancer but not prion diseases. In this study, for the first time, we identified a gCJD patient who carries a PRNP variant with G114V mutation and 1-OPRD.
Methods
Pedigree
The pedigree shown in Figure 1 was provided by the proband's mother (II-8).
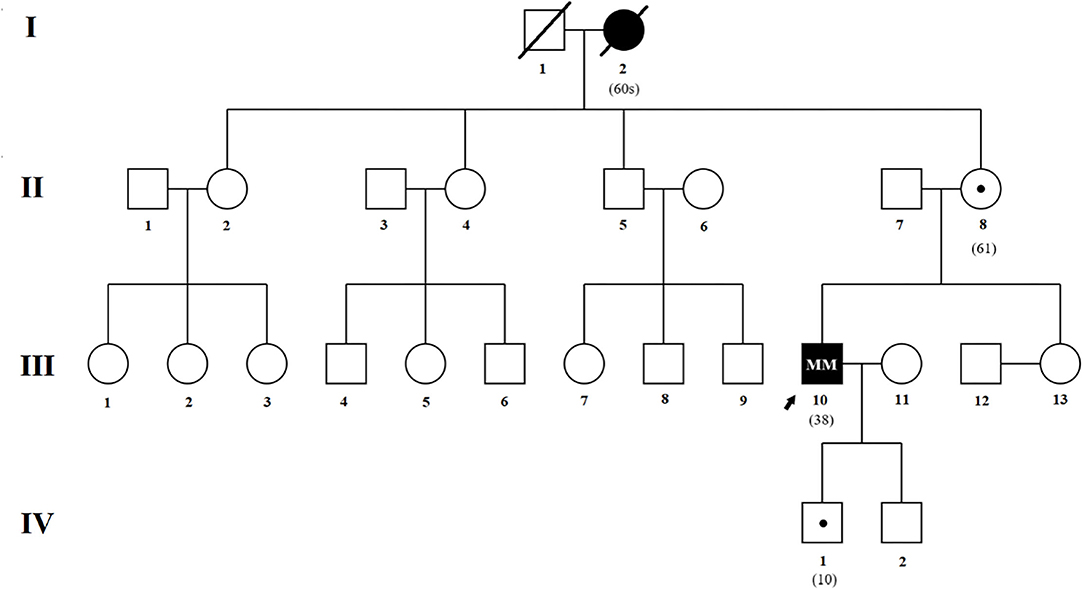
Figure 1. Genealogical tree of the family. Affected patients and asymptomatic carriers are described in the text. Squares indicate males; circles, females; slash marks, deceased; arrow, proband; solid symbols, affected individual; small solid dot with square or circle, asymptomatic carrier.
Clinical Assessment
The proband (III-10) and one carrier (II-8) underwent systematic neurological and neuropsychological examinations and scoring on the Medical Research Council prion disease rating scale (MRC Scale) (15). This scoring was repeated for the proband and patient II-8 monthly over the telephone in the follow-up studies.
Genetic Analysis
Genomic DNA was extracted from peripheral blood leucocytes of 3 living members in this pedigree using the DNA Isolation Kit (Bioteke, AU1802). Qualified DNA samples were fragmented into 200~300 bp. The procedure comprises three standard steps: end-repair of fragmented DNA, A-tailing, adapter ligation, and amplification. Hybridization of pooled libraries to the capture probes and removal of non-hybridized library molecules were carried out according to the IDT and xGen Lockdown® Probes (Integrated DNA Technologies). Sample dilution, flow cell loading, and sequencing were performed according to the Illumina specifications. The DNA libraries were sequenced on the HiSeq X10 (Illumina, San Diego, USA) as paired-end 150-bp reads.
Cerebrospinal Fluid Examination
A lumbar puncture was performed on the proband. Cerebrospinal fluid (CSF) 14-3-3 protein was examined at the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention.
Real-Time Quaking-Induced Conversion
CSF and skin RT-QuIC were conducted in the proband at the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention.
The CSF assay was conducted in a black 96-well, optical-bottomed plate (Nunc, 265301) on a BMG FLUOstar plate reader (BMG LABTECH); 15 μl of each CSF sample was mixed, with 10 μg of recombinant hamster PrP90-231 in a reaction buffer containing 10 mmol/L PBS, 170 mmol/L NaCl, 10 μmol/L thioflavin T (ThT), 10 μmol/L EDTA, and 0.002% SDS in final. The final reaction volume was 100 μl. Each tested sample was quadruplicated. Each reaction contained blank (reaction buffer), negative (2 μl 10% brain homogenate from normal hamster), and positive (2 μl 10% brain homogenate from scrapie agent 263K-infected hamster) controls. The working conditions were as follows: temperature, 55 °C; shaking speed, 700 rpm; shaking/incubation time, 60/60 s; total reaction time, 60 h. ThT fluorescence (450 nm excitation and 480 nm emission) was automatically measured every 45 min as relative fluorescence units (rfu). The cutoff value was set as the average value of the negative controls plus 10 times SD. The sample was considered positive when two or more parallel wells revealed positive reactive curves.
The sites for skin biopsies were behind the right ear. After disinfection with 75% alcohol, the proband received local anesthesia with a subcutaneous injection of 2% lidocaine hydrochloride. A small piece of skin with a size of about 2 × 1 cm2 was taken with a scalpel by a neurosurgeon. The biopsy skin specimen covered the epidermis, dermis, and adipose tissues; 2% (w/v) of skin homogenate was prepared in lysis buffer (100 mM NaCl, 10 mM EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 10 mM Tris, pH 7.5). RT-QuIC reaction contained 10 μg of rHaPrP90-231, 1X PBS, 170 mM NaCl, 1 mM EDTA, 0.01 mM ThT, 0.001% SDS, together with 15 μl CSF samples or 2 μl 10−2 to 10−4 diluted skin homogenates in a final volume of 100 μl. Each sample was assayed in triplicated or quadruplicated. The assay was conducted in a black 96-well, optical-bottomed plate (Nunc, 265301) on a BMG FLUOstar plate reader (BMG LABTECH). The working conditions were optimized as follows: temperature, 55°C; vibration speed, 700 rpm; vibration/incubation time, 60/60 s; total reaction time, 60 h. ThT fluorescence (excitation wavelength, 450 nm; emission wavelength, 480 nm). Each reaction was automatically measured every 45 min and expressed as relative fluorescence units (rfu). The cutoff value was set as the mean value of the negative controls plus 10 times the standard deviation. A sample was considered to be positive when ≥2 wells revealed positive reaction curves. The positive control was 10−5 diluted the brain homogenate of the scrapie agent 263K-infected hamster, while the negative control was 10−5 diluted the brain homogenate of the normal hamster.
Ethical Approval and Consent to Participate
All patients were informed about the purpose of the study and given written consent. The study was approved by the Ethics Committee of the Beijing Tongren Hospital.
Results
Case Report
A 36-year-old Han-Chinese right-handed man (patient III-10, the proband) developed anxiety, depression, sleep disorder, and tremor in his hands after a panic attack but without known medical history. His cognitive state declined, and he was unable to perform job duties due to memory loss. At 11 months after onset, the patient became withdrawn, and more deterioration of his cognitive function was observed, indicated by the difficulty in calculating, being lost at home, and frequently forgetting the names of acquaintances. Besides, the tremors spread to bilateral limbs, which led to difficulty in cake decoration (the patient's profession). After 16 months of onset, he was diagnosed with depression and anxiety and was prescribed sertraline, fluphenazine, and piracetam. At 17 months after onset, he was scored 20 of 30 on the Chinese Mini-Mental Status Examination (MMSE), and 15 of 30 on the Montreal Cognitive Assessment (MoCA Beijing Version). The Hamilton's Depression Scale was 7. At 19 months after onset, he developed hallucinations, which made him see his sons as enemies, and occasionally, he protected himself via aggressive behaviors.
In the clinic, the diagnosis of possible behavioral variant Frontotemporal Dementia (bvFTD) was considered because of the abnormal neuropsychological profile, such as early apathy and executive/gene deficits with relative sparing of memory and visuospatial functions.
A thorough neurological examination 20 months after the onset revealed a total deterioration of the cognitive state, dysarthria, slight hypermyotonia, and deep tendon hyperreflexias in the bilateral limbs, and the patient presented cogwheel-like rigidity. He had difficulty finishing the finger-to-nose test and heel-knee-tibia test due to tremors in his limbs. The patient was unsteady when walking on a straight line. No involuntary movement was observed. He scored 15 of 30 on the Chinese MMSE, and 9 of 30 on the MoCA Beijing Version. The Hamilton's Depression Scale was 7.
To our surprise, the brain MRI demonstrated abnormal intensities in the bilateral caudate nucleus, putamen, and cerebral cortex. A series of laboratory examinations were performed for the rapidly progressive early-onset dementia. The autoimmune screening and tumor marker identification were shown to be unremarkable or negative. An extensive panel for paraneoplastic antibodies including Amphiphysin, CV2, PNMA2 (Ma2/Ta), Ri, Yo, Hu, titin, SOX1, recoverin, zic4, GAD65, and Tr (DNER) were tested, and all were negative. Serology and cerebrospinal fluid (CSF) tests for HIV, cryptococcus, syphilis, tuberculosis, bacteria, fungus, and virus showed no evidence of inflammation. In addition, CSF 14-3-3 protein was tested to be negative. RT-QuIC tests of skin and CSF were also negative (Figure 2).
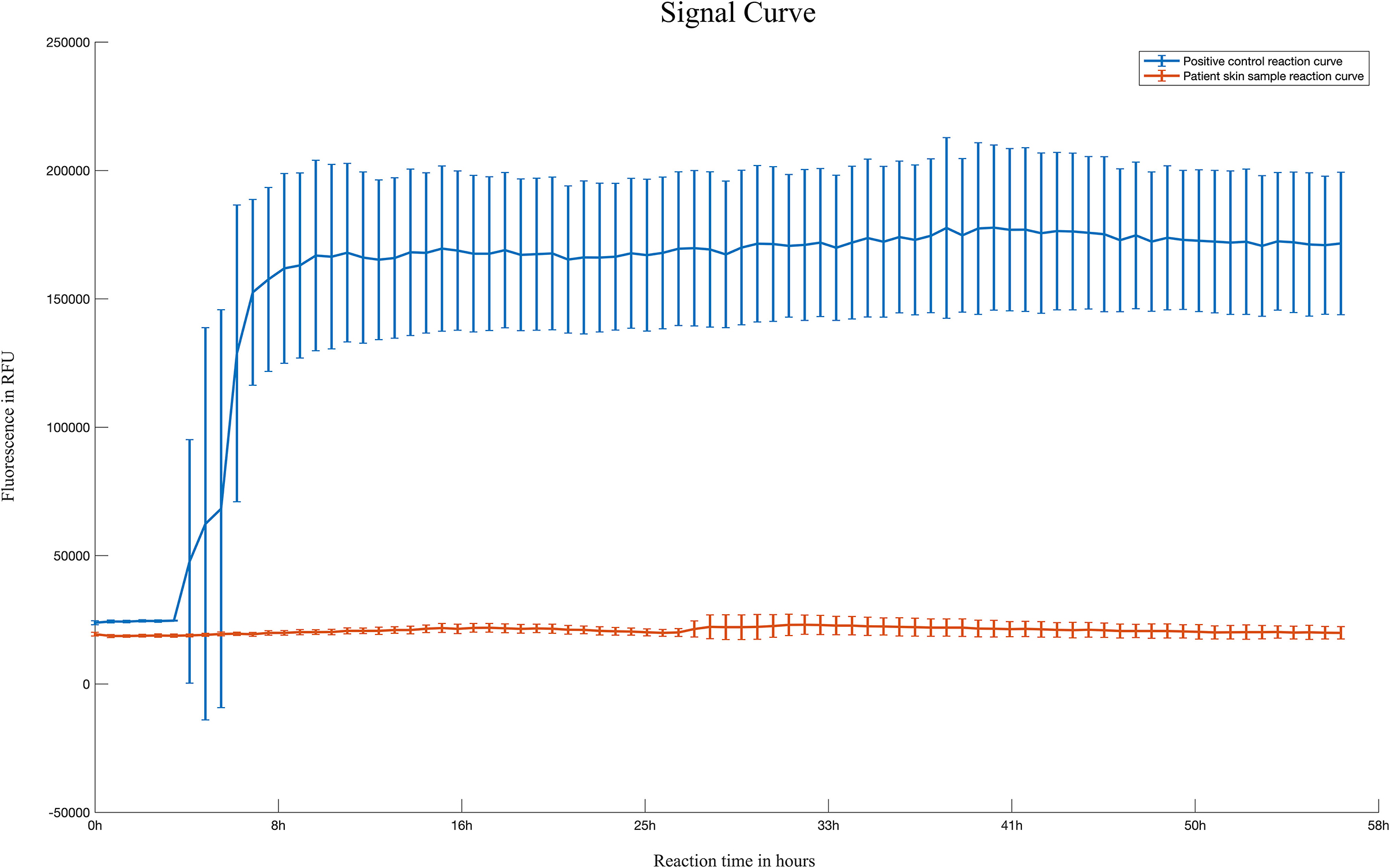
Figure 2. Reaction curve of skin RT-QuIC of the patient. Reaction curves of skin RT-QuIC of the patient. Four replicate reactions of patient skin samples and four replicate reactions of the positive control (hamster scrapie 263K strain brain smear with a 10−7 dilution) were used in the RT-QuIC. Each line represents the reaction curve of positive control and patient sample, respectively. The means with standard deviations of those averages are shown as a function of RT-QuIC reaction time. The reaction curve did not show the peak of fluorescence in the patient sample (bottom), indicating a negative result of RT-QuIC. X-axis, hours post-reaction; Y-axis, fluorescence values.
Diffusion-weighted imaging (DWI) sequences displayed restricted diffusion in the bilateral frontal and parietal cortex (Figures 3A–D). DWI hyperintensities were revealed in the bilateral basal ganglia, bilateral pulvinar, and dorsomedial thalamus (Figures 3E–J). Fluorodeoxyglucose PET (FDG-PET) exhibited hypometabolism in the bilateral cerebral cortex and right basal ganglia (Figure 4).
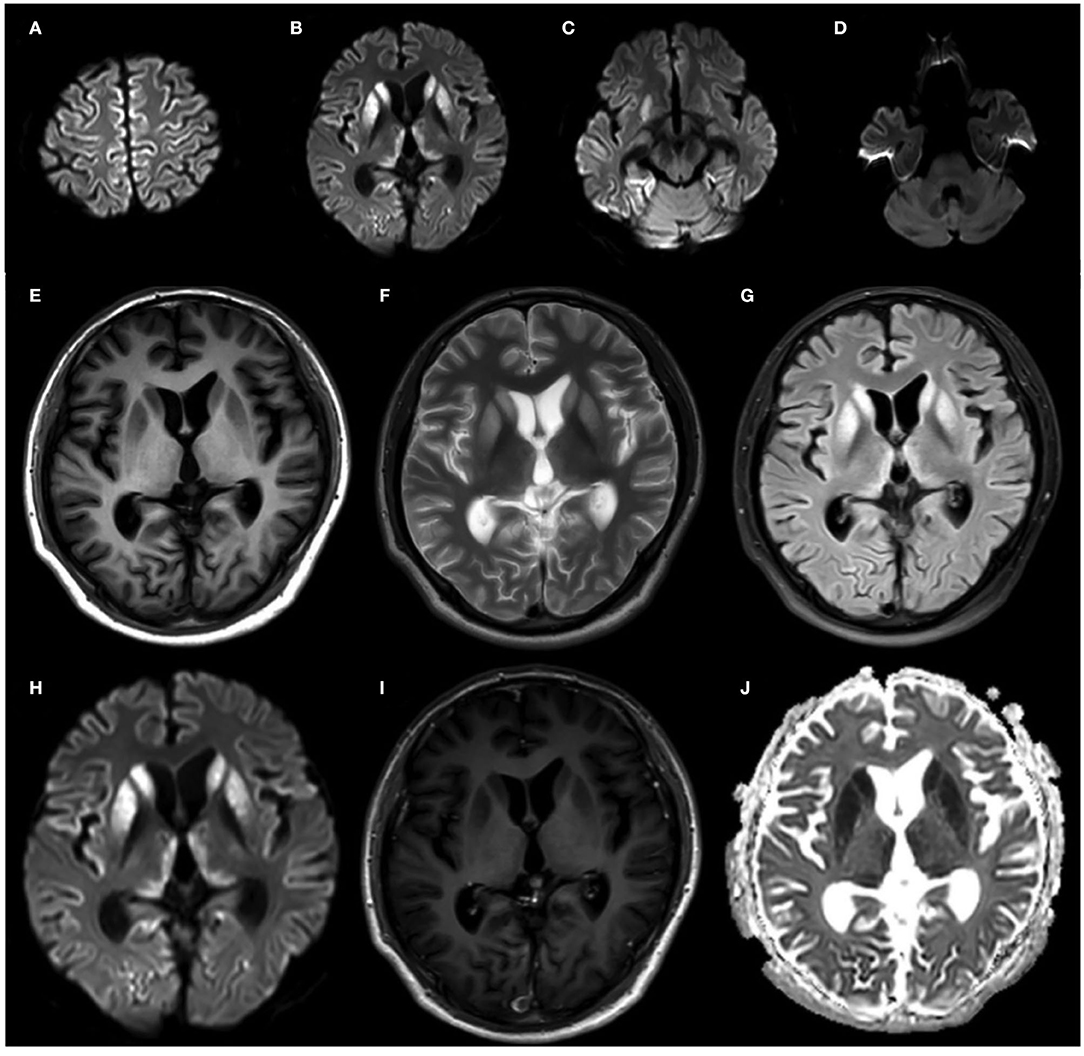
Figure 3. Brain MRI of the proband at 20 months after onset. (A–D) DWI sequences displayed restricted diffusion in the bilateral frontal and parietal cortex. (E–J) Bilateral basal ganglia hyperintensities, with slight bilateral pulvinar and dorsomedial thalamus hyperintensities on FLAIR (I) and DWI (J) sequences.
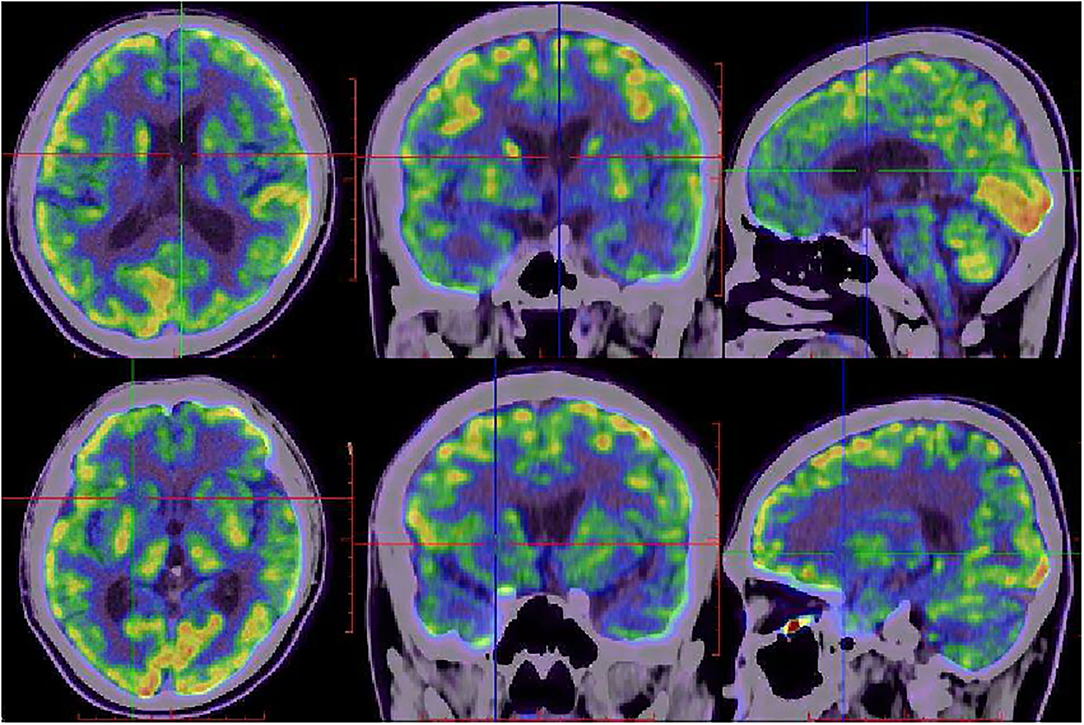
Figure 4. FDG-PET images of the proband at 20 months after onset. FDG-PET showed hypometabolism in bilateral frontal, parietal, temporal lobes, bilateral caudate nucleus, putamen, and thalamus, with right lateralized basal ganglia hypometabolism.
An empiric course of pulse IV gamma globulin was tried without notable improvement.
At 22 months after onset, the hypermyotonia of the patient in the bilateral limbs became more obvious. He had occasional urinary incontinence, and his ability to study was relatively reserved when he scored 21 of 30 on the Chinese MMSE. The MRC Scale score was 13 of 20. The Hamilton's Depression Scale was 6. EEG showed diffuse slow waves. MRI scanning indicated no obvious change compared to images 2 months earlier.
A follow-up study with the MRC Scale revealed gradually developed aphasia, gait disorder, and fecal incontinence 30 months after onset. The patient is still alive, 4 years after the onset, while the MRC Scale score was 2 of 20, and his swallowing function and mobility were still preserved (Table 1).
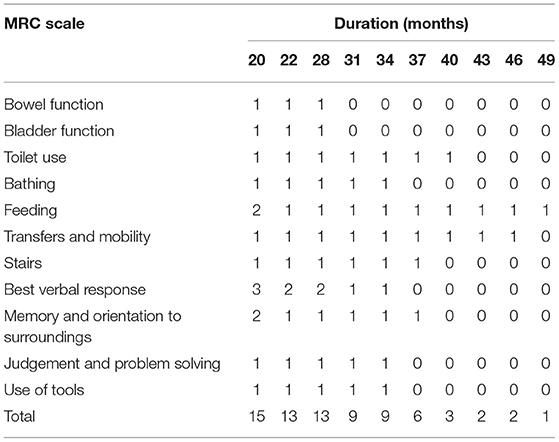
Table 1. The MRC Scale of the proband.
The clinical features indicated that the patient might have prion disease. To determine the etiology of the disease, we extracted genome DNA from peripheral blood leucocytes of the patient and performed a direct DNA sequencing of the PRNP coding sequence. Unexpectedly, a rare mutation of G114V and 1-OPRD of the PrP in the patient was identified (Figure 5). Given that only a few patients with gCJD were reported to carry the G114V PRNP variant, we suspected that the mutation in the patient might be inherited from his parents, and we, thus, enrolled his immediate family members in this study for a genetic investigation.
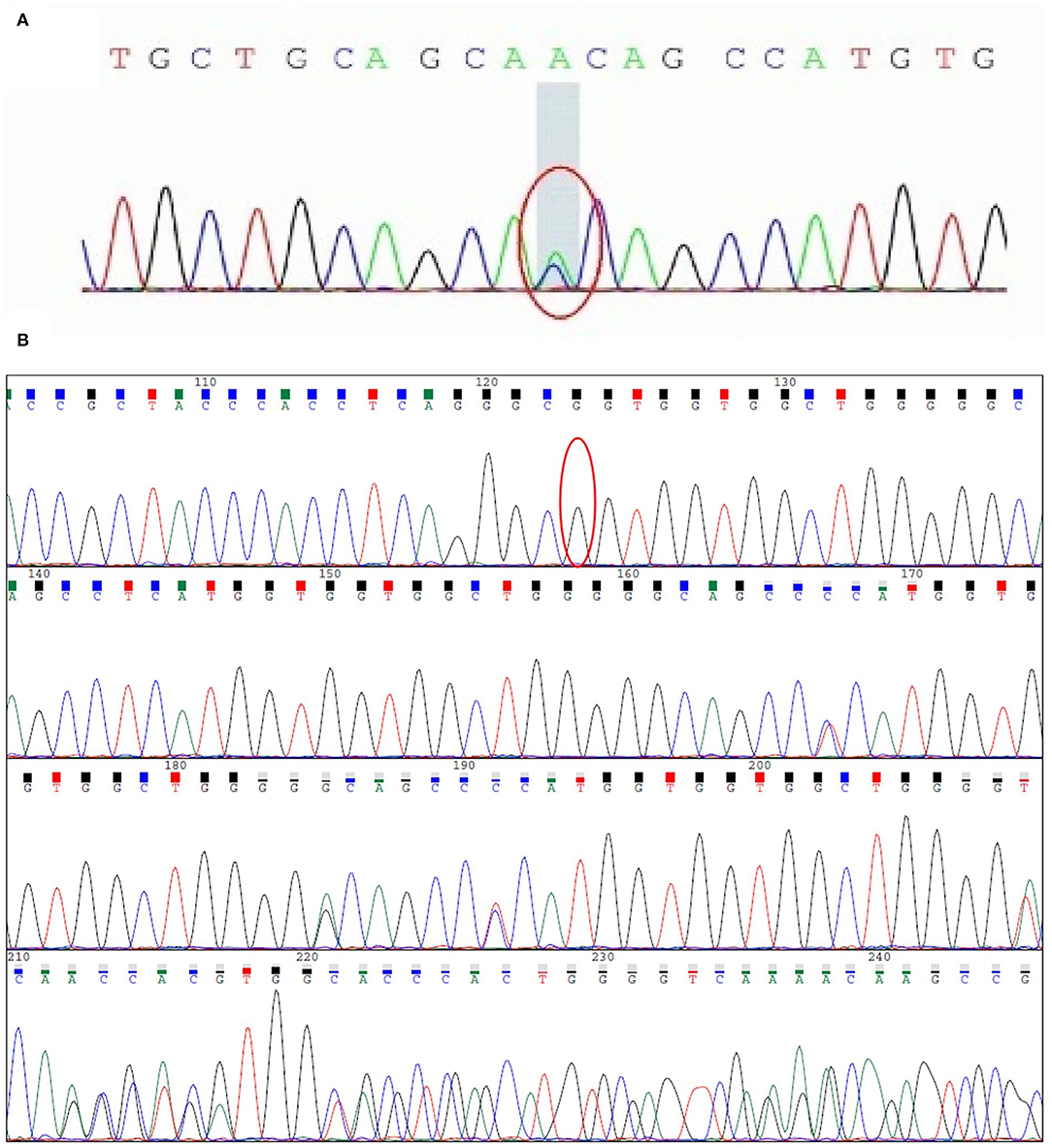
Figure 5. Sequencing analyses in the PRNP gene from patient III-10. (A) c.341G>T (p.G114V) variant of the PRNP gene was detected. The mutation leads to the alteration of amino acid from glycine-to-valine. This alteration will lead to prion disease. (B) c.204_227 delTCATGGTGGTGGCTGGGGGCAGCC (1-OPRD) variant was detected. PRNP gene comprises two exons with the entire open reading frame (ORF) of 762 bp which is contained with exon 2. Nuclides between PRNP codon 51-91 into N-terminal are 5 groups of 24 base pair (bp) repeats, a deletion of the repeat TCATGGTGGTGGCTGGGGGCAGCC was found at the end of the PRNP gene (bottom). This mutation can be found in a minority of people and its pathogenicity has not yet been determined.
Although the medical record was not available, the maternal grandmother (patient I-2) of the proband was found to have progressive dementia in her 60s, which was within 1 year before her death, and subsequently developed a tremor in her last few months. The proband's mother (carrier II-8) received examination when she was 61 years old, but no neuropsychiatric symptoms were observed. She scored 29 of 30 on the Chinese MMSE and 21 of 30 on the MoCA (Beijing Version). Although the EEG showed slow waves in the left temporal lobe, both cranial MRI (including DWI) and FDG-PET were unremarkable. A follow-up study revealed that she suffered an acute cerebral infarction in the callosum 18 months after the first examination. No cortical ribbon was found by DWI to date (Table 2). Whereas DNA sequencing revealed a G114V mutation and 1-OPRD of PrP in this individual. The elder son of the proband (carrier IV-1) received DNA sequencing at age 10, and G114V mutation and 1-OPRD were also found, although he did not show any clinical symptoms. No further examination was performed due to his age. No autopsy or biopsy were performed on any of the patients.

Table 2. Summary of clinical features in affected family members.
Genetic Analysis
Direct sequencing of the PRNP coding sequence disclosed a heterozygous missense mutation (Figure 5A) at the second position of codon 114, leading to a GGT-to-GTT substitution and a glycine-to-valine change in the PrP (G114V). 1-OPRD was also identified by the sequencing (Figure 5B). Both the mutation and deletion were found in 3 family members (Figure 1), including patients III-10, and asymptomatic carriers II-8 and IV-1.
Neuropsychological Assessment
At 20 months and 22 months after onset, a series of cognitive investigations were carried out to evaluate the proband's cognitive state (Table 3). At 20 months after onset, he scored 15 of 30 on the Chinese MMSE, 9 of 30 on the MoCA Beijing Version, 0 of 3 on the Clock Drawing Test (CDT), 38 of 40 on the Boston Naming Test (BNT), and 3 of 10 on the Copying test. The Hamilton's Depression Scale was 7. The Digit span test (DST) showed 7 in order and 3 in inverted order. The symbol digit modalities test (SDMT) score was 0 of 110. The score of the Rey auditory verbal learning test (RAVLT) exhibited that the ability to learn and recall was significantly reduced. The proband could not finish both the Trail making test (TMT)-A and B. The activity of daily living (ADL) scale and instrumental activities of daily living (IADL) scale displayed 15 of 40 and 32 of 40, respectively. Besides, he got 6 on the clinical dementia rating scale sum of boxes (CDR-SB). The MRC Prion Disease Rating Scale (MRC Scale) was scored 15 of 20.
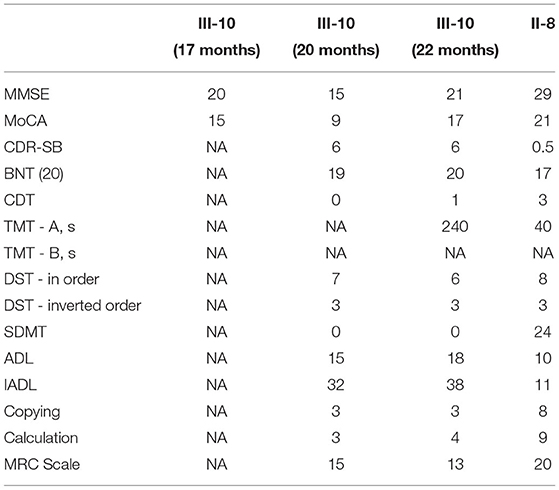
Table 3. Summary of cognitive abilities test in examined family members.
At 22 months after onset, he scored 21 of 30 on the Chinese MMSE, 17 of 30 on the MoCA Beijing Version, 20 of 20 on BNT, 1 of 3 on CDT, and 3 of 10 on the Copying test. DST showed 6 in order and 3 in inverted order. The Hamilton's Depression Scale was 7. SDMT score was 0 of 110. He still got 6 on CDR-SB. The score of RAVLT showed that the patient's ability of learning and recall was increasingly damaged. The TMT-A was finished correctly in 240 s while the TMT-B was not finished. ADL displayed 18 of 40 in PSMS and 38 of 40 in IADL. The score of the MRC Scale was 13 of 20.
Discussion
In this report, we have described a rare G114V mutation and 1-OPRD in PrP in a Chinese family comprising two affected individuals (III-10 and I-2) and two carriers (II-8 and IV-1) (Figure 1). II-8 and III-10 were alive at examinations. To the best of our knowledge, our report is the first description of a CJD case with both G114V mutation and 1-OPRD PRNP variant.
The proband developed anxiety, depression and progressive dementia, myoclonus, pyramidal, and extrapyramidal syndrome in his late 30s. The DWI sequences in cranial MRI of the patient displayed restricted diffusion in the cortical ribbon, bilateral basal ganglia, bilateral pulvinar, and dorsomedial thalamus hyperintensities. FDG-PET exhibited hypometabolism in the cerebral cortex and the right basal ganglia. The patient was suspected of having CJD while the 14-3-3 protein in CSF was not detected and the RT-QuIC for PrPSc was negative. Finally, the genetic analysis showed the G114V PRNP variant with 1-OPRD.
G114V is a rare mutation linked to the early age of onset, ranging from 18 to 45 years, and long disease duration (1 to 5 years). Since 2005, only 15 patients with G114V-associated genetic prion diseases were identified (the major clinical characteristics are summarized in Table 4), and the appearance of G114V mutation seems to have no obvious racial difference. Among the 15 cases, progressive dementia appeared in all cases (100%), extrapyramidal syndromes were reported in 12 cases (80%), and neuropsychiatric symptoms were described in 10 cases (66%). Myoclonus was mentioned in 9 cases (60%) and pyramidal syndromes were noted in 8 cases (53%). Additionally, mild cerebellar signs were reported in a Uruguayan family, and aphasia was described in an American-born Polish descent family (9, 12). Almost all of the patients had neuropsychiatric symptoms and progressive dementia at the onset. In all 15 cases, progressive dementia gradually appeared and lasted the entire clinical course.
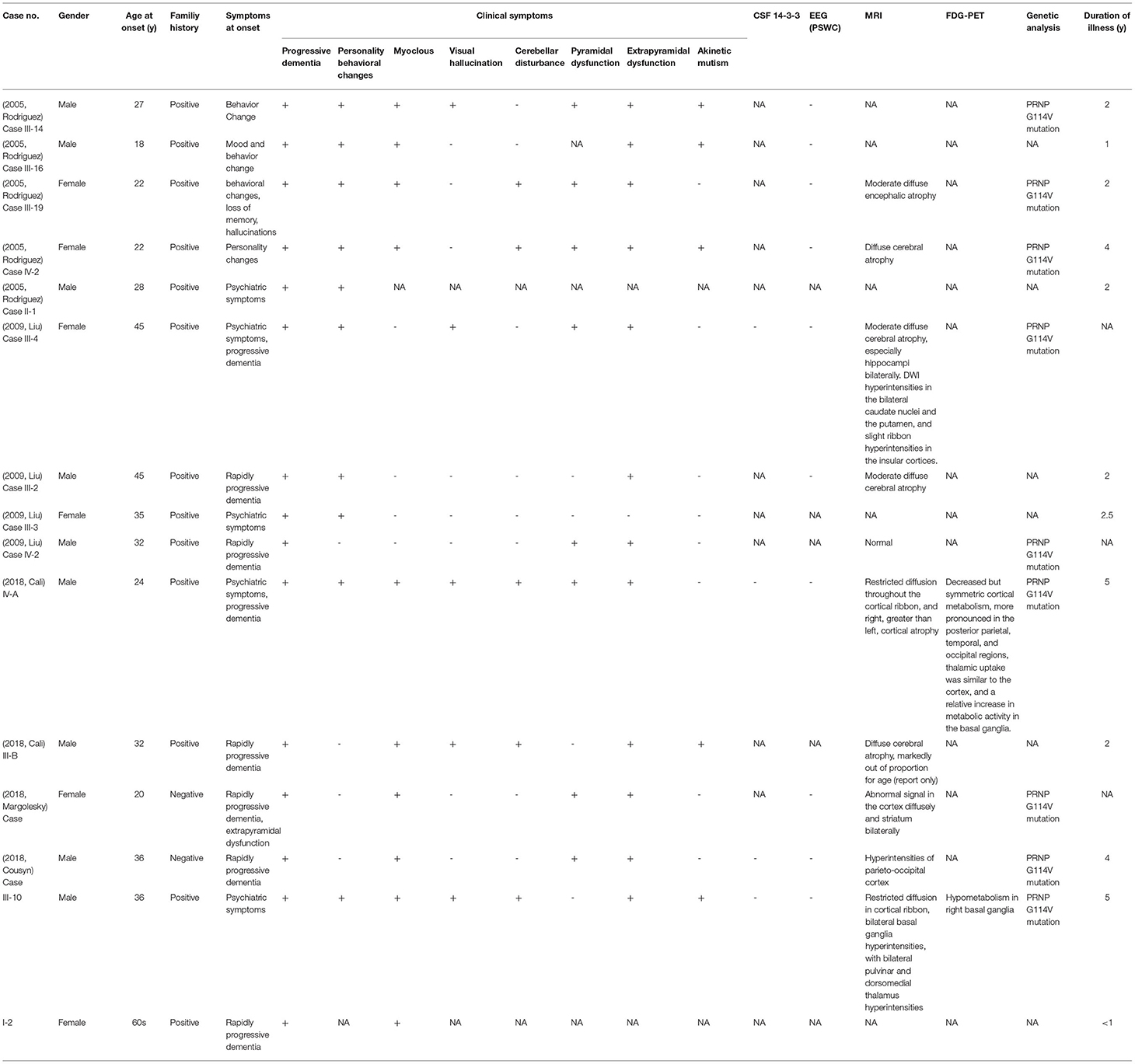
Table 4. Features of the 15 reported cases with PrP G114V mutation.
The 14-3-3 protein is one of the most common laboratory markers in the diagnosis of CJD for its high sensitivity and specificity. But Muayqil et al. (16) indicate that it can be positive in different neurological diseases, including encephalitis and acute stroke. The 14-3-3 protein was shown to be negative in the proband CSF. Interestingly, CSF 14-3-3 is negative in all reported G114V patients (8–14), which suggests that 14-3-3 protein is not a specific index for this phenotype.
RT-QuIC for PrPSc in skin and CSF were also negative in the proband, which was not available in other cohorts. These results were first reported in G114V-gCJD. The diagnostic value of RT-QuIC in the detection of sCJD from CSF samples has been demonstrated previously (17). Orrù et al. also reported that the RT-QuIC-based method detects CJD patients with an overall sensitivity and specificity of 100% (18). Besides, according to Xiao et al. skin specimen was ideal for the RT-QuIC test in Chinese patients (19). The RT-QuIC assay was negative in many other gCJDs, such as V180I, V210I mutation (20, 21).
A detailed neuropsychological investigation revealed that the proband was impaired in language and recall and developed time disorientation, attention deficit disorder, dysexecutive syndrome, and visuospatial dysfunction, which is consistent with previous reports. For example, Cousyn et al. reported dysexecutive syndrome and impaired episodic memory with spatial disorientation in their cases (14). A major axis of the frontoparietal dysfunction was also reported in a study by Caine et al. which includes patients with inherited, acquired, and sporadic CJD, suggesting characteristic cognitive features in prominent executive impairment, parietal dysfunction, a largely expressive dysphasia with reduced motor speed, and it strongly correlated with volume reduction in the frontal and parietal gray matter revealed by MRI (22).
The MRC Scale examination assesses domains of cognitive function, speech, mobility, personal care/feeding, and continence, according to their relative importance documented by the carer's interviews, which can describe the disease progression of a patient. The 20-point functionally oriented scale used in this study is advantageous over single scales for its simplicity of administration and the ability to capture the rapid changes that can characterize the disease. The scores of the MRC Scale for the patient from 20 months to 49 months after disease onset are summarized in Table 1, which showed that memory and orientation to surroundings were relatively reserved through long disease duration.
The DWI imaging revealed hyperintensities in the bilateral basal ganglia, pulvinar, and dorsomedial thalamus of the patient. However, hyperintensities in the cortex were restricted to the bilateral frontal and parietal cortex. A similar phenomenon was noted in previously reported one Chinese and two American families carrying the G114V mutation. Compared to another type of gCJD with E200K mutant or sCJD, the ribbon sign is less evident in the cortex in all the G114V patients.
Recently, FDG-PET has been suggested to be used in the diagnosis of CJD, but literature about utilizing FDG-PET in gCJDs is limited (23). In our report, FDG-PET exhibited hypometabolism in the right basal ganglia and cortex of the proband, which is consistent with a previous report that a relative increase in metabolic activity in the basal ganglia as revealed by FDG-PET in a G114V case (12). Interestingly, the basal ganglia and thalamus were unaffected in the context of metabolism of sCJD, as reported previously (24). Thus, measuring the metabolism activity in basal ganglia may help diagnose gCJD.
The EEG of the proband showed diffuse slow waves without typical periodic sharp wave complexes, which are similar to previous reports.
Amino acid 114 is located within a potential membrane-spanning domain of PrPC, which not only belongs to a highly conserved palindromic sequence but also to an amyloidogenic region that is essential for the conversion of PrP (25). G114V has been demonstrated to associate with some pathways, including oxidative phosphorylation, regulation of actin cytoskeleton, MAPK signaling and proteasome, axon guidance, gap junction, and purine metabolism (26). Cali et al. indicated that the conformation of PrPSc linked to the PRNP-G114V variant might be the principal barrier to transmission (12).
In addition to the G114V mutation, we also detected 1-OPRD in the PrP in the gCJD patient, which has never been reported in the prion diseases. The PRNP gene comprises two exons with the entire open reading frame (ORF) of 762 bp which is contained with exon 2. Nuclides between PRNP codon 51-91 into N-terminal are 5 groups of 24 base pair (bp) repeats, which are also called octapeptide repeats region (OPRP). RNP gene are transmitted in an autosomal dominant manner and include point mutations (such as G114V, E200K, and T188K), insertion of one-nine 24 bp extra repeats or two 24 bp repeats deletion between PRNP codon 51-91 (27, 28). Insertional mutations of one or more extra octapeptide (from 2 to 12 OR insertions) are associated with CJD (4, 29). Two-octapeptide repeat region (2-OPRD) deletions were reported in two unrelated patients and were considered to be pathogenic (6, 30). The two 2-OPRD cases were characterized by late age of onset (86 and 62 years old) and rapidly progressive dementia. 1-OPRD PRNP variant has also been found in healthy individuals with a frequency of less than 1% (31). Although the biological functions of 1-OPRD largely remain unclear, one report suggested that single octapeptide deletion selectively reduces the expression level of pathogenic N-terminal mutants. Besides, 1-OPRD also induces the release of a pathogenic PrP mutant that the mutation occurred within the internal hydrophobic domain from the cell surface (32). 1-OPRD also exists in HeLa cells and several gastric cancer cell lines. Around 66.7% variant frequency in gastric cancer cell lines and 8.9% in gastric cancer tissues were reported, which was significantly higher than that in the normal population (less than 1%) (33, 34). Our report first described 1-OPRD in a familial prion disease, which suggests that 1-OPRD plays a role in the pathogenesis of prion disease.
In summary, we reported a rare pedigree with PrP G114V mutation combined with 1-OPRD, the first report in human gCJD. The proband and his mother and his elder son carry the same mutations. The proband displayed symptoms at the age of 36, but his mother did not develop prion-like disorders in her 60s. The phenomenon would help to forward our understanding of the etiology of gCJD and warrants future study on its pathologic mechanisms.
Data Availability Statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Beijing Tongren Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
XL and YX drafted the initial manuscript and reviewed the literature. XL, YX, and ZZ were in charge of the patients' follow up and cognitive assessments. KX, XC, QS, and XD performed the Skin RT-Quic. JY and HG provide part of imaging. JW and YG revised the manuscript and in charge of all the clinical data analysis, gene analysis, and imaging. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China grant (81771313 and 81301032), Open Project of State Key Laboratory of Infectious Disease Prevention and Control (2020SKLID311).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor LW declared a shared parent affiliation with the author(s) XL, YX, ZZ, XC, JY, and YG at the time of review.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. (2012) 11:618–28. doi: 10.1016/S1474-4422(12)70063-7
2. Prusiner SB. The prion diseases. Brain Pathol. (1998) 8:499–513. doi: 10.1111/j.1750-3639.1998.tb00171.x
3. Ladogana A, Puopolo M, Poleggi A, Almonti S, Mellina V, Equestre M, et al. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology. (2005) 64:1592–7. doi: 10.1212/01.WNL.0000160118.26865.11
4. Takada LT, Kim MO, Cleveland RW, Wong K, Forner SA, Gala II, et al. Genetic prion disease: Experience of a rapidly progressive dementia center in the United States and a review of the literature. Am J Med Genet B Neuropsychiatr Genet. (2017) 174:36–69. doi: 10.1002/ajmg.b.32505
5. Zerr I, Poser S. Clinical diagnosis and differential diagnosis of CJD and vCJD. With special emphasis on laboratory tests. APMIS. (2002) 110:88–98. doi: 10.1034/j.1600-0463.2002.100111.x
6. Beck JA, Mead S, Campbell TA, Dickinson A, Wientjens DP, Croes EA, et al. Two-octapeptide repeat deletion of prion protein associated with rapidly progressive dementia. Neurology. (2001) 57:354–6. doi: 10.1212/WNL.57.2.354
7. Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. (2016) 8:322ra9. doi: 10.1126/scitranslmed.aad5169
8. Shi Q, Zhang BY, Gao C, Han J, Wang GR, Chen C, et al. The diversities of PrP(Sc) distributions and pathologic changes in various brain regions from a Chinese patient with G114V genetic CJD. Neuropathology. (2012) 32:51–9. doi: 10.1111/j.1440-1789.2011.01237.x
9. Rodriguez MM, Peoc'h K, Haik S, Bouchet C, Vernengo L, Mañana G, et al. A novel mutation (G114V) in the prion protein gene in a family with inherited prion disease. J Neurology. (2005) 64:1455–7. doi: 10.1212/01.WNL.0000158618.39527.93
10. Ye J, Han J, Shi Q, Zhang BY, Wang GR, Tian C, et al. Human prion disease with a G114V mutation and epidemiological studies in a Chinese family: a case series. J Med Case Reports. (2008) 2:331. doi: 10.1186/1752-1947-2-331
11. Liu Z, Jia L, Piao Y, Lu D, Wang F, Lv H, et al. Creutzfeldt-Jakob disease with PRNP G114V mutation in a Chinese family. Acta Neurol Scand. (2010) 121:377–83. doi: 10.1111/j.1600-0404.2009.01236.x
12. Cali I, Mikhail F, Qin K, Gregory C, Solanki A, Martinez MC, et al. Impaired transmissibility of atypical prions from genetic CJDG114V. Neurol Genet. (2018) 4:e253. doi: 10.1212/NXG.0000000000000253
13. Margolesky J, Mario S. Twenty-year-old African American woman with prion disease associated with the G114V PRNP variant. Neurol Genet. (2018) 4:e229. doi: 10.1212/NXG.0000000000000229
14. Cousyn L, Grabli D, Seilhean D, Azuar C, Huiban C, Epelbaum S, et al. First European case of Creutzfeldt-Jakob disease with a PRNP G114V mutation. Cortex. (2019) 117:407–413. doi: 10.1016/j.cortex.2018.08.014
15. Thompson AG, Lowe J, Fox Z, Lukic A, Porter MC, Ford L, et al. The Medical Research Council prion disease rating scale: a new outcome measure for prion disease therapeutic trials developed and validated using systematic observational studies. Brain. (2013) 136:1116–27. doi: 10.1093/brain/awt048
16. Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. (2012) 79:1499–506. doi: 10.1212/WNL.0b013e31826d5fc3
17. Cramm M, Schmitz M, Karch A, Mitrova E, Kuhn F, Schroeder B, et al. Stability and Reproducibility Underscore Utility of RT-QuIC for Diagnosis of Creutzfeldt-Jakob Disease. Mol Neurobiol. (2016) 53:1896–904. doi: 10.1007/s12035-015-9133-2
18. Orrù CD, Groveman BR, Hughson AG, Manca M, Raymond LD, Raymond GJ, et al. RT-QuIC assays for prion disease detection and diagnostics. Methods Mol Biol. (2017) 1658:185–203. doi: 10.1007/978-1-4939-7244-9_14
19. Xiao K, Yang X, Zhou W, Chen C, Shi Q, Dong X. Validation and application of skin RT-QuIC to patients in China with probable CJD. Pathogens. (2021) 10:1642. doi: 10.3390/pathogens10121642
20. Xiao K, Zhou W, Gao LP, Wu YZ, Wang Y, Chen C, et al. Clinical and laboratory features of three rare Chinese V210I gCJD patients. Pathogens. (2020) 9:800. doi: 10.3390/pathogens9100800
21. Nomura T, Iwata I, Naganuma R, Matsushima M, Satoh K, Kitamoto T, et al. A patient with spastic paralysis finally diagnosed as V180I genetic Creutzfeldt-Jakob disease 9 years after onset. Prion. (2020) 14:226–31. doi: 10.1080/19336896.2020.1823179
22. Caine D, Tinelli RJ, Hyare H, De Vita E, Lowe J, Lukic A, et al. The cognitive profile of prion disease: a prospective clinical and imaging study. Ann Clin Transl Neurol. (2015) 2:548–58. doi: 10.1002/acn3.195
23. Renard D, Vandenberghe R, Collombier L, Kotzki PO, Pouget JP, Boudousq V. Glucose metabolism in nine patients with probable sporadic Creutzfeldt-Jakob disease: FDG-PET study using SPM and individual patient analysis. J Neurol. (2013) 260:3055–64. doi: 10.1007/s00415-013-7117-6
24. Kim EJ, Cho SS, Jeong BH, Kim YS, Seo SW, Na DL, et al. Glucose metabolism in sporadic Creutzfeldt-Jakob disease: a statistical parametric mapping analysis of (18) F-FDG PET. Eur J Neurol. (2012) 19:488–93. doi: 10.1111/j.1468-1331.2011.03570.x
25. Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, et al. A transmembrane form of the prion protein in neurodegenerative disease. Science. (1998) 279:827–34. doi: 10.1126/science.279.5352.827
26. Tian C, Liu D, Chen C, Xu Y, Gong HS, Chen C, et al. Global transcriptional profiling of the postmortem brain of a patient with G114V genetic Creutzfeldt-Jakob disease. Int J Mol Med. (2013) 31:676–88. doi: 10.3892/ijmm.2013.1239
27. Das NR, Miyata H, Hara H, Chida J, Uchiyama K, Masujin K, et al. The N-terminal polybasic region of prion protein is crucial in prion pathogenesis independently of the octapeptide repeat region. Mol Neurobiol. (2020) 57:1203–16. doi: 10.1007/s12035-019-01804-5
28. Wang XF, Guo YJ, Zhang BY, Zhao WQ, Gao JM, Wan YZ Li F, et al. Creutzfeldt-Jakob disease in a Chinese patient with a novel seven extra-repeat insertion in PRNP. J Neurol Neurosurg Psychiatry. (2007) 78:201–3. doi: 10.1136/jnnp.2006.09433
29. Hara H, Miyata H, Das NR, Chida J, Yoshimochi T, Uchiyama K, et al. Prion protein devoid of the octapeptide repeat region delays bovine spongiform encephalopathy pathogenesis in mice. J Virol. (2017) 92:e01368–17. doi: 10.1128/JVI.01368-17
30. Capellari S, Parchi P, Wolff BD, Campbell J, Atkinson R, Posey DM, et al. Creutzfeldt–Jakob disease associated with a deletion of two repeats in the prion protein gene. Neurology. (2002) 59:1628–30. doi: 10.1212/01.WNL.0000035533.86833.28
31. Yu SL, Jin L, Sy MS, Mei FH, Kang SL, Sun GH, et al. Polymorphisms of the PRNP gene in Chinese populations and the identification of a novel insertion mutation. Eur J Hum Genet. (2004) 12:867–70. doi: 10.1038/sj.ejhg.5201245
32. Lee Y, Lee D, Choi I, Song Y, Kang MJ, Kang SW. Single octapeptide deletion selectively processes a pathogenic prion protein mutant on the cell surface. Biochem Biophys Res Commun. (2016) 470:263–8. doi: 10.1016/j.bbrc.2016.01.074
33. Liang J, Wang JB, Pan YL, Wang J, Liu LL, Guo XY, et al. High frequency occurrence of 1-OPRD variant of PRNP gene in gastric cancer cell lines and Chinese population with gastric cancer. Cell Biol Int. (2006) 30:920–3. doi: 10.1016/j.cellbi.2006.05.015
Keywords: genetic Creutzfeldt-Jakob disease, prion, PRNP, G114V mutation, one octapeptide repeat deletions
Citation: Lin X, Xu Y, Zhen Z, Xiao K, Chen X, Yang J, Guan H, Shi Q, Dong X, Wang J and Guo Y (2022) Case Report: Genetic Creutzfeldt–Jakob Disease With a G114V Mutation and One Octapeptide Repeat Deletion as a Mimic of Frontotemporal Dementia. Front. Neurol. 13:888309. doi: 10.3389/fneur.2022.888309
Received: 02 March 2022; Accepted: 04 May 2022;
Published: 24 June 2022.
Edited by:
Liyong Wu, Xuanwu Hospital, Capital Medical University, ChinaReviewed by:
Jesus R. Requena, University of Santiago de Compostela, SpainLi Cui, First Affiliated Hospital of Jilin University, China
Copyright © 2022 Lin, Xu, Zhen, Xiao, Chen, Yang, Guan, Shi, Dong, Wang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanjun Guo, yguo12@ccmu.edu.cn; Jiawei Wang, wangjwcq@163.com
†These authors have contributed equally to this work