Fast Intracortical Sensory-Motor Integration: A Window Into the Pathophysiology of Parkinson’s Disease
 Raffaele Dubbioso
Raffaele Dubbioso Fiore Manganelli
Fiore Manganelli Hartwig Roman Siebner
Hartwig Roman Siebner Vincenzo Di Lazzaro
Vincenzo Di Lazzaro- 1Department of Neurosciences, Reproductive Sciences and Odontostomatology, University Federico II of Naples, Naples, Italy
- 2Danish Research Centre for Magnetic Resonance, Centre for Functional and Diagnostic Imaging and Research, Copenhagen University Hospital Hvidovre, Hvidovre, Denmark
- 3Department of Neurology, Copenhagen University Hospital Bispebjerg, Copenhagen, Denmark
- 4Institute for Clinical Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 5Unit of Neurology, Neurophysiology, Neurobiology, Department of Medicine, University Campus Bio-Medico, Rome, Italy
Parkinson’s Disease (PD) is a prototypical basal ganglia disorder. Nigrostriatal dopaminergic denervation leads to progressive dysfunction of the cortico-basal ganglia-thalamo-cortical sensorimotor loops, causing the classical motor symptoms. Although the basal ganglia do not receive direct sensory input, they are important for sensorimotor integration. Therefore, the basal ganglia dysfunction in PD may profoundly affect sensory-motor interaction in the cortex. Cortical sensorimotor integration can be probed with transcranial magnetic stimulation (TMS) using a well-established conditioning-test paradigm, called short-latency afferent inhibition (SAI). SAI probes the fast-inhibitory effect of a conditioning peripheral electrical stimulus on the motor response evoked by a TMS test pulse given to the contralateral primary motor cortex (M1). Since SAI occurs at latencies that match the peaks of early cortical somatosensory potentials, the cortical circuitry generating SAI may play an important role in rapid online adjustments of cortical motor output to changes in somatosensory inputs. Here we review the existing studies that have used SAI to examine how PD affects fast cortical sensory-motor integration. Studies of SAI in PD have yielded variable results, showing reduced, normal or even enhanced levels of SAI. This variability may be attributed to the fact that the strength of SAI is influenced by several factors, such as differences in dopaminergic treatment or the clinical phenotype of PD. Inter-individual differences in the expression of SAI has been shown to scale with individual motor impairment as revealed by UPDRS motor score and thus, may reflect the magnitude of dopaminergic neurodegeneration. The magnitude of SAI has also been linked to cognitive dysfunction, and it has been suggested that SAI also reflects cholinergic denervation at the cortical level. Together, the results indicate that SAI is a useful marker of disease-related alterations in fast cortical sensory-motor integration driven by subcortical changes in the dopaminergic and cholinergic system. Since a multitude of neurobiological factors contribute to the magnitude of inhibition, any mechanistic interpretation of SAI changes in PD needs to consider the group characteristics in terms of phenotypical spectrum, disease stage, and medication.
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder affecting multiple neuromodulatory transmitter systems (Barone, 2010). The cardinal motor symptoms of PD are due to the progressive loss of nigrostriatal dopaminergic neurons in the midbrain. Progressive nigrostriatal dopaminergic denervation causes a dysfunction in the cortex-basal ganglia sensorimotor loops, producing slowness of movements, rigidity, tremor, and difficulties with gait and balance (Dickson, 2012). Although the basal ganglia do not receive direct somatosensory input from the periphery, several lines of evidence support the idea that the basal ganglia are important for gating sensory input for motor control through cortico-basal ganglia-thalamo-cortical re-entry loops (Haber and Calzavara, 2009). Specifically, primary and secondary somatosensory cortices in the parietal lobe send inputs to the striatum of the basal ganglia, where sensory cortical projections are topographically mapped (Künzle, 1977; Di Martino et al., 2008). The notion that the basal ganglia are relevant to sensorimotor integration is rather old. Back in 1985, Lidsky introduced the notion that the basal ganglia serve as “sensory analyzer for motor systems” which “ultimately affect movement by gating sensory inputs into other motor areas” (Lidsky et al., 1985). Lesions of the basal ganglia mostly affect automatic movements that need sensory guidance, pointing towards a role of the basal ganglia in sensory-motor control of automatic or highly trained movements (Boecker et al., 1999).
KEY CONCEPT 1. Sensorimotor integration
Sensorimotor integration is the process whereby somatosensory input is integrated by the central nervous system to shape motor program execution. Parkinson’s disease (PD) is considered a pathological model of aberrant sensory-motor integration, where movement accuracy and speed are severely affected by the altered sensory feedback.
In addition, PD patients also exhibit impairment of selecting the appropriate response while simultaneously suppressing inappropriate response tendencies (Praamstra and Plat, 2001). Interestingly, patients with PD display difficulties in suppressing automatic response activation while proactive inhibitory control appears to be intact (Praamstra and Plat, 2001; Seiss and Praamstra, 2004; Wylie et al., 2009). Using transcranial magnetic stimulation (TMS), it was also shown that impaired inhibition also manifests itself within the corticomotor output system in PD (Kleine et al., 2001). The TMS-evoked excitation of the corticomotor projections produced an increased, prolonged and less synchronized excitation of the target muscle (Kleine et al., 2001). This converging evidence shows that anatomical and functional impairment of the cortico-basal ganglia-thalamo-cortical loop in PD profoundly affects sensory-motor integration in the cortex.
Sensorimotor integration at the cortical level can be probed non-invasively by pairing electrical stimulation of peripheral somatosensory afferents with focal TMS targeting the contralateral primary motor cortex (M1). In their seminal study, Tokimura et al. (2000) demonstrated that peripheral nerve stimulation at the contralateral wrist reduced the amplitude of motor evoked potentials (MEPs) when the TMS pulse was given to the primary motor hand area (M1-HAND) 2–8 ms after the arrival of the afferent volley in cortex. The term “short-latency afferent inhibition” (SAI) was coined for this conditioning-test paradigm, and SAI soon became a well-established neurophysiological technique to probe rapid intracortical sensorimotor integration in health and disease (Turco et al., 2018b). The inhibitory effect of the sensory input on the motor output provides a neurophysiological signature of fast sensory-motor integration. Three components constitute the SAI circuit that enables fast-integrative processing: the fast afferent-sensory pathway, the motor-efferent pathway and the integrative component in the sensorimotor cortex. The neuropharmacological profile of SAI is complex. The sensory input exerts its inhibitory effects on the corticospinal neurons through γ-Aminobutyric acid (GABA)-ergic intracortical circuits (Di Lazzaro et al., 2005c; Di Lazzaro and Ziemann, 2013), but its magnitude is also modulated by dopaminergic (Sailer et al., 2003) and cholinergic neuromodulatory circuits (Di Lazzaro et al., 2000). The fact that the magnitude of SAI is modulated by cholinergic drugs has provoked considerable interest in the use of SAI in patients with dementia. In patients with Alzheimer Disease (AD), a loss of SAI has been interpreted as an indicator of cortical cholinergic denervation and a normal SAI as a predictor of a positive effect of cholinergic medication on cognitive deficits (Di Lazzaro et al., 2000; Cantone et al., 2014).
KEY CONCEPT 2. Short-latency afferent inhibition (SAI)
Fast component of sensorimotor integration can be studied in vivo by examining the effects of sensory input on the motor output at the cortical level. The amplitude of a motor evoked potential (MEP) induced by transcranial magnetic simulation (TMS) over the motor cortex is reduced by a peripheral nerve stimulation few milliseconds before the TMS pulse. The magnitude of this inhibition represents the neurophysiological correlate of sensorimotor integration efficiency.
The neurophysiological and neuropharmacological properties of SAI have motivated researchers to use SAI as a tool to examine whether and how PD is associated with an impairment of fast sensory-motor integration in the pericentral sensorimotor cortex. In the same vein, researchers have examined whether the attenuation of SAI in individual patients scales with dopaminergic and cholinergic cortical neurodegeneration and is associated with particular clinical symptoms (Martin-Rodriguez and Mir, 2018). In this review, we first summarize some key features of SAI in the healthy human brain. We will then review the evidence for an alteration of SAI in PD and discuss, based on the published data, whether the individual reduction in SAI can be used as an electrophysiological biomarker of cholinergic or dopaminergic denervation of the sensorimotor cortex in PD. Finally, we will ask the question whether a reduction in SAI is associated with specific clinical manifestations of PD.
Short-Latency Afferent Inhibition in the Healthy Human Brain
Cortical Origin of SAI
Converging evidence supports the hypothesis that SAI is generated in the sensorimotor cortex, although the exact anatomic circuits generating SAI are still unknown. The most direct evidence that peripheral somatosensory input modulates the TMS-induced motor output at the cortical level comes from invasive recordings of corticospinal volleys in patients with implanted electrodes in the cervical epidural space (Tokimura et al., 2000). These studies showed that later I-waves (I2 and I3 waves) were reduced at an interval appropriate for SAI, whereas the early I-wave (I1 wave) remained unchanged. Based on these findings, it has been proposed that peripheral nerve stimulation activates glutamatergic thalamocortical projections onto intracortical GABAA-ergic interneurons which in turn, suppress the intracortical inhibitory GABAA-ergic circuits generating the late descending volleys (late I-waves) in the corticospinal tract (Di Lazzaro and Ziemann, 2013). A critical role of thalamocortical projections is substantiated by lesion studies, showing a marked reduction or loss of SAI in patients with unilateral (Oliviero et al., 2005) or bilateral (Nardone et al., 2010) paramedian thalamic stroke.
Which Factors Modulate the Expression of SAI?
The relative strength of SAI depends on the magnitude of the sensory afferent input evoked by peripheral stimulation. The greater the afferent volley evoked by peripheral stimulation, the stronger is the magnitude of SAI (Bailey et al., 2016). The expression of SAI also depends on the somatotopic relation between the sensory input and motor output. Electrical stimulation of digits close to the TMS-target muscle (i.e., homotopic stimulation) induces stronger inhibition than stimulation of digits distant to the TMS-target muscle (i.e., heterotopic stimulation; Classen et al., 2000). The somatotopic organization of SAI was studied in detail using a neuronavigated TMS mapping technique which adjusts the coil position and orientation to the individual shape of the central sulcus (Dubbioso et al., 2017c). Mapping the input-output relationship of SAI revealed a center-surround organization in the human M1-HAND. SAI was evoked by homotopic stimulation only, whereas the conditioning effect produced the opposite effect, namely short-latency afferent facilitation (SAF), in the case of heterotopic stimulation (Dubbioso et al., 2017c).
The expression of homotopic is highly state dependent. In healthy individuals, SAI is consistently expressed at rest, but attenuated during finger movements (Dubbioso et al., 2017c). In the active target muscle, SAI was reduced at movement initiation during both mixed and homotopic cutaneous nerve stimulation (Asmussen et al., 2013; Cho et al., 2016), whereas SAI was reduced during the maintenance phase of the movement (Asmussen et al., 2013) or found to be normal (Cho et al., 2016). Accordingly, SAI and SAF by homotopic or heterotopic stimulation were abolished during the tonic contraction of the target muscle (Dubbioso et al., 2017c). This state-dependent pattern of SAI modulation can be attributed to a sensorimotor gating mechanism, which attenuates the perceived intensity of stimuli generated by movements. Although SAI is closely modulated by movement, no relationship between SAI magnitude and manual dexterity has been found (Turco et al., 2018c).
The expression of SAI is not only modulated by the intrinsic sensorimotor state but also shaped by transcranial brain stimulation. Transcranial alternating current stimulation (TACS) at 20 Hz completely abolished SAI in the relaxed muscle (Guerra et al., 2016). The suppressive effect of 20 Hz TACS on SAI did not depend on the phase relationship between TACS and the timing of the TMS pulse probing SAI (Guerra et al., 2016). This finding suggests a link between SAI expression and the oscillatory state of the sensorimotor cortex, yet it remains to be shown that SAI is also suppressed by physiologically generated beta oscillations in sensorimotor cortex.
The magnitude of SAI can also be modulated by TMS interventions, for instance when electrical stimulation of the median nerve is consistently paired with TMS of the contralateral M1-HAND at an inter-stimulus interval (ISI) of 25 ms (Quartarone et al., 2006) or with TMS over the contralateral S1 at an ISI of N20-2.5 ms (Tsang et al., 2015). Sub-motor threshold 5 Hz repetitive paired associative stimulation produced a long-lasting increase in corticospinal excitability along with an attenuation of SAI (Quartarone et al., 2006; Tsang et al., 2015). Interventional TMS protocols which are thought to induce homosynaptic plasticity, such as continuous theta burst (cTBS) have also been sued to modify SAI. While cTBS over M1-HAND failed to modulate SAI, cTBS delivered over S1 reduced SAI along with an increase in cortico-spinal excitability (Tsang et al., 2014).
Cognitive processes, for instance attention and working memory, shape afferent sensory-motor integration involving distinct intracortical circuits as demonstrated by single monophasic TMS pulses that evoke different current directions in the brain (Mirdamadi et al., 2017; Suzuki and Meehan, 2018). Specifically, it has been demonstrated that SAI evoked using antero-posterior (AP), but not posterior-anterior (PA), current is reduced by a concurrent visual detection task with high attention demands. These results suggested that only AP-elicited intracortical circuits are sensitive to cross-modal attention task by altering sensory processing in premotor areas (Mirdamadi et al., 2017). Instead, a verbal working memory task modulated SAI, regardless of the TMS-induced current direction in the brain (AP or PA), reflecting a generalized effect of this cognitive task across anatomically distinct circuits upon cortico-spinal neurons in the M1-HAND (Suzuki and Meehan, 2018).
The fact that intrinsically and extrinsically induced state changes in the sensorimotor system can dynamically tune the expression of SAI needs to be born in mind when SAI is considered as “biomarker” in PD.
Influence of Neurotransmitter Systems on SAI Magnitude
Pharmacological and clinical studies provided converging evidence that the expression of SAI is modulated by several neurotransmitters such as acetylcholine, dopamine, GABA and noradrenaline (Turco et al., 2018b). SAI is significantly reduced by scopolamine, a muscarinic cholinergic antagonist, in young healthy adults (Di Lazzaro et al., 2000) and can be improved with rivastigmine, an acetylcholinesterase inhibitor, in patients with abnormal reduction of SAI, such as AD (Di Lazzaro et al., 2002). Cholinergic inhibition of pyramidal neurons has been demonstrated directly in experimental studies (Gulledge and Stuart, 2005). Interestingly, this rivastigmine effect on SAI predicted the long term response to cholinesterase inhibitor in patients with AD (Di Lazzaro et al., 2005a). The effects of scopolamine and rivastigmine suggest that SAI may be useful to probe in vivo the functional integrity of central cholinergic circuits of the human brain. These studies indicate that SAI can trace the functional impairment of central cholinergic circuits, allowing to discriminate for example cholinergic from non-cholinergic form of dementia (Di Lazzaro et al., 2006; Manganelli et al., 2014; Dubbioso et al., 2017b).
Beyond cholinergic transmission, the dopaminergic system plays a relevant role in the modulation of SAI, in accordance with a strong synaptic interaction between dopamine an acetylcholine signaling in different brain areas (Di Cara et al., 2007; Millan et al., 2007). L-dopa treatment has been shown to normalize SAI in patients with restless legs syndrome (Rizzo et al., 2010) and AD (Martorana et al., 2009; Nardone et al., 2014). Dopaminergic medication also influences SAI in patients with PD (for more details see section on PD).
KEY CONCEPT 3. Dopaminergic medication and sensory processing
Studies on PD patients consistently found reduced levels of SAI in the ON-medication state suggesting a role of dopamine replacement in driving this abnormality. Indeed, dopaminergic medication could lead to decreases in central processing or integration of sensory signals in PD patients. For instance, it has been shown that dopaminergic medication could worsen SAI and proprioception. The positive relationship between motor symptoms and SAI suppression in the ON-medication state suggests that the effect of medication may be more detrimental to SAI in patients that are less responsive to dopaminergic pharmacotherapy, for instance, patients with more prominent cholinergic involvement.
Regarding GABAergic system, in human cortical slices, it was observed that acetylcholine activated GABA neurons and triggered GABAergic postsynaptic currents (Alkondon et al., 2000). Thus, SAI may also be mediated through the interactions between cholinergic projections and specific GABAergic interneurons. This also explains the findings that the administration of positive GABAA but not GABAB receptor modulators influences SAI (Turco et al., 2018a). Zolpidem, a selective agonist of alpha1 subunit of GABAA receptor, and lorazepam, a positive allosteric modulator of GABAA receptor, significantly reduced SAI (Di Lazzaro et al., 2005b,c, 2007; Turco et al., 2018a), whereas diazepam, a non-selective agonist, induced a slight increase or no effect on SAI (Di Lazzaro et al., 2005c, 2007). This observation is presumably explained by a differential role of the different alpha subunits of GABAA receptor in the modulation of afferent inhibition with a suppression of cholinergic inhibition by alpha1 subunit activation.
Lastly, a recent study has also demonstrated that acute and chronic intake of reboxetine, a noradrenaline reuptake inhibitor, reduces SAI, likely through suppression of GABAergic neurotransmission (Kuo et al., 2017).
Fast Sensory Input and Motor Output Pathways in PD
As stated above the basal ganglia do not receive direct sensory input, yet patients with PD often report sensory symptoms (Pallis, 1971; Snider et al., 1976; Hillen and Sage, 1996). Objective somatosensory deficits are well documented in PD and have been mainly found in tasks that require the use of kinaesthetic sense such as conscious perception of limb position and motion in space proprioception and kinaesthesia (Schneider et al., 1987; Klockgether et al., 1995; Demirci et al., 1997; Jobst et al., 1997; Zia et al., 2000) or temporal or spatial discrimination (Conte et al., 2013).
The fast-afferent sensory volley eliciting SAI can be studied by recording the somatosensory evoked potentials (SSEPs) evoked by stimulation of the peripheral nerve. Most SSEP studies in PD have employed electrical stimulation of a mixed nerve that reflects activation of proprioceptive as well as cutaneous inputs. SSEP studies in patients with PD found a reduced amplitude of the late N30 component of the SSEP, while the early N20-P25 components were found to be normal (Rossini et al., 1989; Cheron et al., 1994; Ulivelli et al., 1999). SSEP studies following proprioceptive stimulation during passive flexion (Mima et al., 1996) or electric stimulation (Restuccia et al., 1999) of the proximal interphalangeal joint of the finger demonstrated that the origin of the N30 waveform is more complex than the early components, containing information from cutaneous afferents as well as from joint and tendinous inputs. Therefore, it has been hypothesized that the defective proprioception described in PD might be related to the depression of the N30 component.
The early cortical components of the SSEP, namely the dipole N20/P20 and P25 component, reflect early sensory processing in the pericentral cortex and are thought to give rise to fast sensory afferent inhibition. Since these early components are intact in PD patients, alterations of SAI in PD patients cannot be attributed to a dysfunction of the afferent sensory pathway. The same consideration applies to the fast cortico-motor output pathway which is unaffected in PD. Indeed, a TMS study, which recorded MEPs at increasing stimulus intensities, demonstrated a normal gain function of corticospinal excitability in PD patients (Kojovic et al., 2012).
However, context-dependent modulation of early cortical sensory processing is impaired in PD. Normal movement-related attenuation of perceived stimuli, referred to as sensorimotor gating, is deficient in patients with PD while they are off dopaminergic treatment and can be restored by dopamine replacement therapy (Macerollo et al., 2016). In contrast to healthy controls, the early N20-P25 SSEP components were not modulated at all by movement in patients with PD. The authors speculated that abnormalities in sensory gating may contribute to the difficulties in movement initiation observed in PD (Macerollo et al., 2016). Another study on PD patients treated with deep brain stimulation (DBS) of the subthalamic nucleus or globus pallidum found that movement of the hand ipsilateral to median nerve stimulation gated the subcortical triphasic negative–positive–negative potentials—at latencies of 14–18–22.5 ms, similar to cortical gating observed with SSEP at N20, P20, and N30 (Insola et al., 2004). Converging evidence suggests that sensory gating preceding the onset of movement seems to be mediated by motor cortical areas that contribute to preparation and execution of movement (Cohen and Starr, 1987; Seki and Fetz, 2012; Macerollo et al., 2018).
Cortical Sensorimotor Integration in PD
Based on the work summarized in the previous sections, it can be concluded that SAI is a cortical process, albeit the exact circuit underlying this fast sensory-motor integration is still unknown. In addition, the lack of major impairment in the fast afferent sensory-to-cortical and efferent cortical-to-motor pathways in PD suggest that abnormalities of SAI in PD are caused by a dysfunction in fast intracortical sensorimotor integration.
Patients with electrodes implanted for DBS provide a unique opportunity to study the interplay between the subcortical target site and the cortex. Two electrophysiological studies showed that continuous high-frequency DBS of the STN modifies SAI in PD, confirming a close relationship between fast sensory-motor cortical integration and the basal ganglia. A first study examined medicated patients with the STN-DBS switched on or off (Sailer et al., 2007). SAI was reduced in the off-stimulation and was acutely restored after STN stimulation was resumed, suggesting that STN stimulation might normalize pathways that are adversely affected by dopaminergic medications (Sailer et al., 2007).
A second study focused on the long-term effect of STN-DBS on SAI and spatial proprioception (Wagle Shukla et al., 2013). SAI and proprioception were first normalized after 6 months, but not after 1 month of DBS. This study underscores the importance of chronic stimulation in the modulation of sensorimotor integration and proprioception.
The authors considered two possible mechanisms underlying SAI modulation by STN DBS. High-frequency DBS of the STN might normalize synchronization between basal ganglia structures, which might restore the ability of thalamocortical relay cells to respond to depolarizing inputs involved in sensorimotor integration (Brown et al., 2001; Rubin and Terman, 2004). Alternatively, STN DBS might have a direct effect on cortical structures through antidromic stimulation of the cortico-subthalamic pathway. In addition, the delayed effect of STN DBS on SAI may reflect long-term plastic changes in the sensorimotor cortex (Udupa et al., 2016). Whatever the underlying mechanisms may be, the modulatory effects of STN-DBS on SAI corroborate a sensorimotor integrative function of the STN as suggested by animal studies. Many STN neurons in the monkey (Wichmann et al., 1994) and patients with PD (Hutchison et al., 1998; Rodriguez-Oroz et al., 2001; Theodosopoulos et al., 2003) respond to cutaneous stimuli and passive movements. Alteration of sensory properties of the STN has been observed in animal models of PD. Peripheral sensory stimulation by hind paw pinch led to a greater increase in STN activity in dopamine-depleted rats than controls, suggesting altered STN sensitivity to afferent sensory inputs in the parkinsonian state (Magill et al., 2001).
Is SAI Abnormal in PD?
To answer this question, we conducted a literature search on Pubmed1 using the following search strings: “Short afferent inhibition” OR “SAI” AND “PD.”
Exclusion criteria were as follows:
i. review articles or letter to the editors reporting no original data.
ii. studies about atypical parkinsonism [i.e., Progressive Supranuclear Palsy (PSP), Multisystemic Atrophy or Cortical Basal Syndrome], or dystonia not including PD population as control group.
This search resulted in 22 studies on the final search on November 2, 2018 (Tables 1, 2). Fourteen studies reported a reduction of SAI in patients with PD. The average disease duration was 6.14 ± 4.39 years, mean ON UPDRS-III score was 24.03 ± 13.11, and the mean L-dopa equivalent dose 667.02 ± 314.51 mg across all positive studies (Table 1). In the remaining eight studies, six reported normal (Degardin et al., 2012; Zamir et al., 2012; Picillo et al., 2015; Dubbioso et al., 2017a; Ponzo et al., 2017; Nelson et al., 2018) and two found an enhanced SAI in PD patients (Di Lazzaro et al., 2004; Nardone et al., 2005). Mean disease duration was 5.63 ± 2.69 years, mean ON UPDRS-III score was 18.32 ± 8.52 and L-dopa equivalent dose was 633.94 ± 206.30 mg across all negative studies (Table 2).
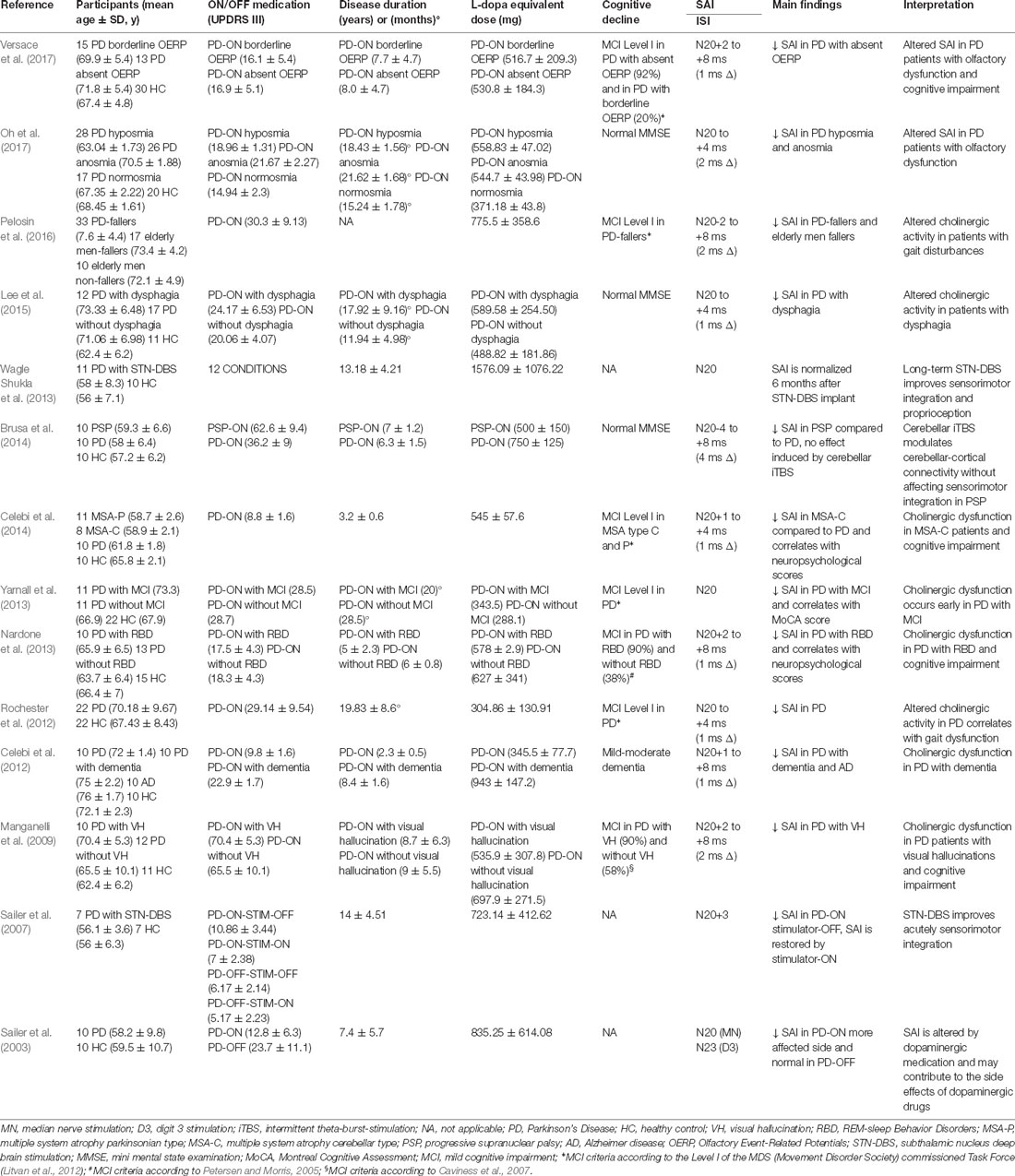
Table 1. Studies showing short-latency afferent inhibition (SAI) alteration in Parkinson’s Disease (PD) patients.
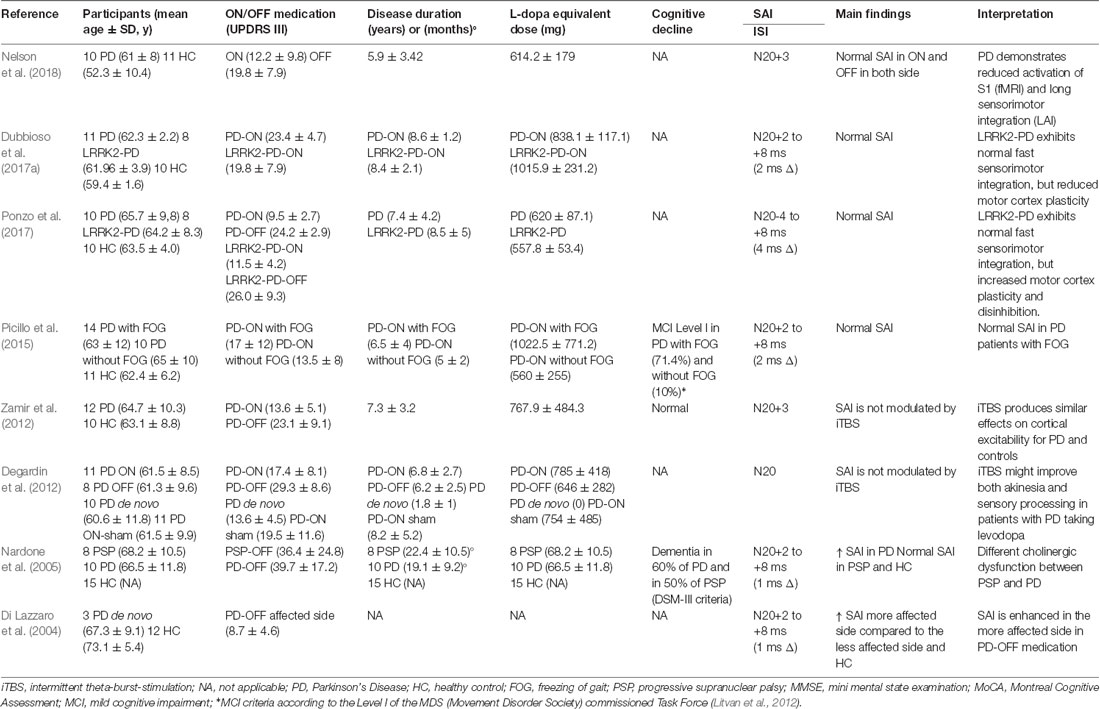
Table 2. Studies showing normal or enhanced SAI in PD patients.
Importantly, these studies found consistent reduction of SAI mainly in medicated PD patients, whereas the off state was not associated with SAI alterations. The idea that nigrostriatal dopaminergic denervation does not reduce SAI or might even enhance cortical inhibition is supported by two studies. The first one, a small study on three drug-free patients with pure hemiparkinsonism (Di Lazzaro et al., 2004) showed enhanced SAI on the affected side. The second one, performed on 10 PD patients in off-state confirmed the increased cortical inhibition respect to patients with PSP and healthy controls (Nardone et al., 2005). The enhancement of SAI in the affected side might be related to an increase of cholinergic muscarinic activity in the contralateral cerebral cortex. Altered muscarinic cortical activity in PD is also supported by several post-mortem studies that have shown an increase in the total number of muscarinic cholinergic receptors in the cerebral cortex (Ruberg et al., 1982; Sirviö et al., 1989; Lange et al., 1993).
An intriguing and alternative hypothesis might be that a reduced thalamo-cortical drive caused by nigrostriatal dopaminergic denervation may increase SAI, an effect which might be obscured by chronic dopamine replacing therapy.
In 2003, Sailer et al. (2003) systematically examined the effect of dopaminergic therapy in 10 PD patients on and off medication. Patients only showed a reduction in SAI when they were on medication, and the medication-induced reduction in SAI only emerged on the more affected side. The medication-related SAI reduction can be restored acutely with STN-DBS (Sailer et al., 2007). This finding was largely confirmed by a recent meta-analysis which only found a consistent reduction in SAI across studies for PD patients on medication, but the attenuating effect of medication on SAI in PD was retrieved in the meta-analysis regardless of the affected side (Martin-Rodriguez and Mir, 2018). Moreover, the meta-analysis revealed an association between SAI changes and disease severity as well as cognitive deficits. Specifically, SAI impairment scaled with cognitive deficits in the four major cognitive domains, although the strongest association was found for visuospatial and executive deficits.
Prompted by these findings, we pooled SAI data from our database and three previous studies (Manganelli et al., 2009; Picillo et al., 2015; Dubbioso et al., 2017a). The pooled data set included measurements from 81 PD patients (57 men) with a mean age of 64.37 ± 7.57 years, average disease duration of 8.41 ± 4.61 years, mean ON UPDRS-III score of 15.48 ± 10.89, and a daily L-dopa equivalent dose of 833.55 ± 470.70 mg. SAI was tested in all patient on the more affected side while they were taking their normal medication. SAI measurements covered five interstimulus intervals adjusted to the individual N20 wave latency (N20+0 ms, N20+2 ms, N20+4 ms, N20+6 ms, N20+8 ms). Patient’s UPDRS III motor score in the ON medication state was the only variable that showed a positive linear correlation with SAI at an ISI of N20+4 ms (ρ = 0.405; p < 0.01) and with the mean SAI across all five interstimulus intervals (ρ = 0.401; p < 0.01, Figures 1A,B). Indeed, neither disease duration nor daily dopaminergic medication showed a significant relationship with SAI (all p ≥ 0.105). Overall, the results suggest that the attenuating effect of medication state on SAI is more pronounced in patients in whom dopamine replacement therapy shows limited efficacy to normalize parkinsonian motor symptoms as indicated by high UPDRS scores in the on-medication state. It is conceivable that patients who show a less favorable response to dopamine replacement therapy may also have more cholinergic deficits and hence the reduction in SAI may, at least in part, resulting from a co-existing cholinergic deficit at the cortical level. Interestingly, patients with atypical parkinsonism (i.e., Progressive Supranuclear Palsy or Multisystemic Atrophy) that usually respond insufficiently to dopaminergic medication, might exhibit reduced levels of SAI (Brusa et al., 2014; Celebi et al., 2014).
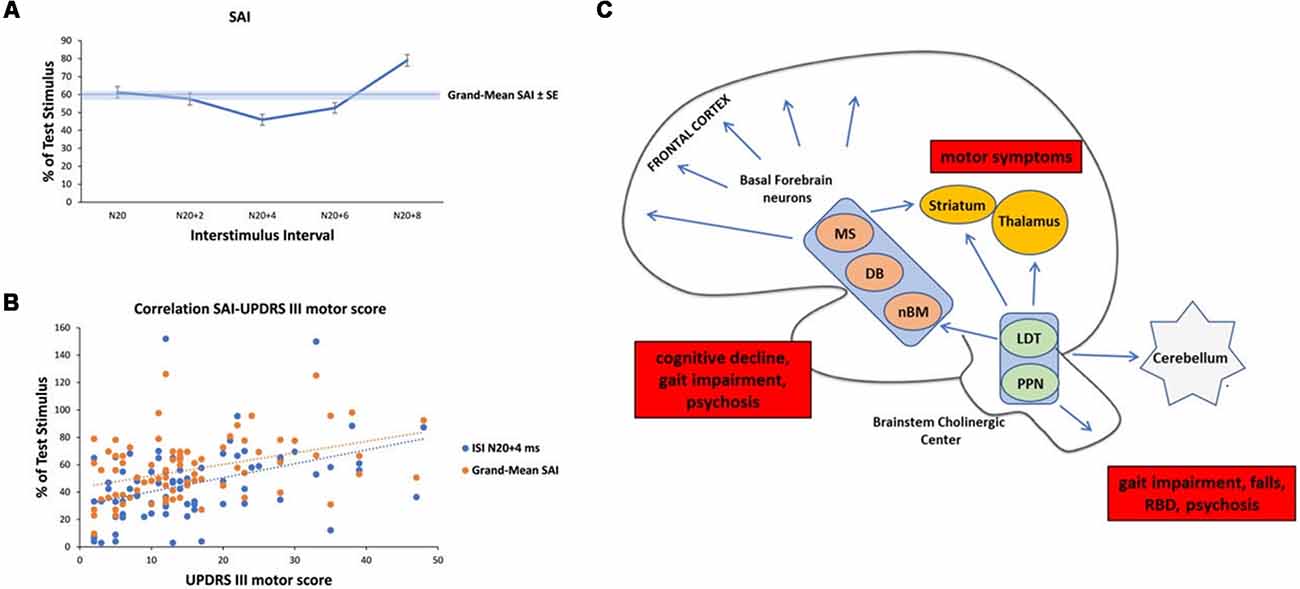
Figure 1. Analysis of short-latency afferent inhibition (SAI) in our Parkinson’s Disease (PD) cohort and schematic representation of cholinergic sources in the human brain with their clinical correlates in PD. (A) Temporal evolution of SAI in our cohort of PD patients. The horizontal axis shows inter-stimulus interval (ISI) values (the time between the peripheral stimulation and cortical stimulation). ISIs were determined by adding 0, 2, 4, 6, and 8 ms to the latency of the N20 component. The vertical axis shows the percentage of test motor evoked potential (MEP) at each ISI. (B) Linear positive correlation between SAI at ISI N20+4 ms, Grand-Mean SAI and UPDRS III motor score in medicated patients. (C) Schematic representation of the three major sources of cholinergic projections in the brain and main clinical correlates in PD (red boxes). Basal forebrain neurons, including the nucleus basalis of Meynert (nbM), medial septal nucleus (MS) and diagonal band of Broca (DB) provide the cholinergic projections to the cerebral cortex and are responsible for cognitive impairment, gait impairment and psychosis. The pedunculopontine nucleus-laterodorsal tegmental complex [referred to as the pedunculopontine tegmental nucleus (PPN) and LDT], a brainstem center, provides cholinergic inputs primarily to the thalamus, but also has connections to the cerebellum, several brainstem nuclei, some striatal fibers, and the spinal cord. This system is mainly involved in walking disturbances, rem-sleep behavior disorders (RBDs) and psychosis. In addition, small populations of intrinsic cholinergic neurons are present in the hippocampus, striatum (cholinergic interneurons), parts of the reticular formation, and cerebellum. The cholinergic interneurons might be the main cause of motor symptoms in PD.
Does SAI in PD Scale With Gait Problems and Non-motor Symptoms?
PD causes a wide range of non-motor symptoms which may even precede the manifestation of the classic motor symptoms (Riedel et al., 2010). Non-motor symptoms include cognitive dysfunctions and decline, apathy, psychiatric disturbances (depression, psychosis, impulse control), autonomic failure (gastrointestinal, cardiovascular, urinary, sexual ability, thermoregulation), sleep disorders, and pain syndrome (Chaudhuri and Schapira, 2009). In recent years, SAI has been increasingly used in PD to identify whether specific motor and non-motor symptoms scale with abnormalities in SAI, presumably due to cholinergic and dopaminergic cortical dysfunction.
KEY CONCEPT 4. Motor and non-motor symptoms
Sources of considerable burden in people with PD are the typical motor symptoms, such as resting tremor, rigidity, bradykinesia, postural instability and non-motor symptoms, namely cognitive declines, psychiatric disturbances, autonomic failures, sleep difficulties, and pain syndrome. These symptoms are variously associated with dopaminergic/cholinergic neurodegeneration and SAI alteration at the cortical level.
Among motor disturbances, gait abnormalities and falls have attracted attention for their association with cognitive decline and cholinergic dysfunction in PD (Newman et al., 2012; Perez-Lloret and Barrantes, 2016). Indeed, reduced SAI has been proven to be an independent predictor of slower gait speed (Rochester et al., 2012) and associated with a higher falls risk in PD (Pelosin et al., 2016). However, in a recent study performed on PD with freezing of gait (FOG; Picillo et al., 2015) the authors failed to prove alteration of SAI in this subtype of patients. Since gait disturbances in PD are heterogeneous and may be underpinned by different neurotransmitters and circuits, it is to be expected that the relation between SAI and gait deficits may be complex and the modulatory role of medication state also needs to be factored in when addressing this issue.
Regarding non-motor symptoms, SAI abnormalities have been found associated with dementia and Mild Cognitive Impairment (MCI; Celebi et al., 2012; Yarnall et al., 2013), confirming the role of cholinergic dysfunction in the development of cognitive impairment in PD. Indeed, SAI has been found to be reduced in PD patients with those symptoms associated with a higher risk of cognitive decline, such as visual hallucinations (VH; Manganelli et al., 2009), dysphagia (Lee et al., 2015), olfactory dysfunction (Oh et al., 2017; Versace et al., 2017) and REM-sleep Behavior Disorders (RBDs; Nardone et al., 2013). These studies are in agreement with the idea that the cholinergic dysfunction makes a major contribution to non-motor symptoms and associated cognitive deficits in PD (Marra et al., 2012; Newman et al., 2012).
A recent review summarized central cholinergic sources of the healthy human brain in two main tracks (Newman et al., 2012). On the one hand, brainstem nuclei, including the pedunculopontine tegmental nucleus (PPN) and the laterodorsal pontine tegmentum, send cholinergic projections to the thalamus, basal ganglia, basal forebrain and to a much lesser extent, the cerebral cortex. On the other hand, the magnocellular basal forebrain-cholinergic systems, including the nucleus basalis magnocellularis and nucleus basalis of Meinert (NBM) send major projections to neocortex, entorhinal cortex, limbic cortices, cingulate cortex, and hippocampus. In addition, small populations of intrinsic cholinergic neurons are present in the hippocampus, striatum (cholinergic interneurons), parts of the reticular formation, and cerebellum (Bohnen and Albin, 2011; Manganelli et al., 2013; Dubbioso et al., 2015). These cholinergic nuclei and their projections have selectively degenerated in PD (Bohnen and Albin, 2011; Perez-Lloret and Barrantes, 2016). Thus, we speculate that SAI in PD may mainly reflect a cortical cholinergic deficit due to cholinergic neurodegeneration. The cortical cholinergic imbalance may derive from many sources that are variably impaired according to disease severity and symptoms. For example, degeneration of cholinergic striatal tone is responsible for motor symptoms, alteration of the NBM and/or PPN nuclei for gait impairment and falls, cognitive decline, RBD, psychosis (Perez-Lloret and Barrantes, 2016), see Figure 1C.
Conclusion and Outlook
In this review, we have discussed the possible contributions of the fast-afferent somatosensory pathway, the intracortical integrative component and the fast-efferent corticomotor pathway to alterations of SAI in PD. We concluded that PD-related changes in SAI are most likely caused at the cortical level, where sensory input is rapidly integrated into a motor output. This makes SAI a useful tool to probe how PD impacts on the sensorimotor integration processing at the cortical level.
Studies performed on PD patients have shown variable results, ranging from reduced to normal or even enhanced SAI findings. Several factors may be responsible for these heterogenous results such as between-group differences in disease severity, disease duration, dopamine replacement therapy and cognitive status. While patients with PD show normal levels of SAI in the off-medication state, SAI is reduced in the on-medication state, suggesting a role of dopamine replacement in driving this abnormality. Interestingly, previous research has suggested that dopaminergic medication could lead to decreases in central processing or integration of sensory signals in PD patients. For instance, it has been shown that dopaminergic medication could worsen SAI and proprioception (distal and spatial errors) and were normalized by chronic STN-DBS, likely through long-term plastic changes in the basal ganglia thalamocortical circuit (Wagle Shukla et al., 2013). Yet, pharmacological studies which systematically study dose-dependent effects of dopamine replacement therapy on SAI magnitude in PD are still lacking. The positive relationship between residual parkinsonian motor symptoms and SAI suppression in the on-medication state suggests that the effect of dopamine replacement may be more detrimental to SAI in patients that are less responsive to dopaminergic pharmacotherapy, for instance, patients with more prominent cholinergic involvement. This would also explain why non-motor symptoms have been associated with a reduction of SAI in PD.
Some important aspects of SAI still remain to be explored in PD. For instance, by systematically varying the intensity of peripheral stimulation one may derive a stimulus-response curve of SAI that may be more sensitive to intrinsic disease-related but also therapy-related changes in SAI. Furthermore, the application of homotopic or heterotopic somatosensory stimulation may reveal interesting insights into the altered center-surround organization of fast sensorimotor integration at the cortical level (Dubbioso et al., 2017c).
Future research on SAI in PD should focus on validating SAI as a biomarker of central cholinergic activity through a multimodal approach by combining neurophysiological results with neuroimaging. For example, correlation analysis with structural data (i.e., analysis of gray matter volume, diffusion tensor imaging) of the main cholinergic system nuclei, would reveal a structure-function relationship between SAI changes and structural cholinergic denervation. The introduction of new PET radioligands, such as (18 F) fluoroethoxybenzoyesamicol [(18 F) FEOBV], a ligand which shows a high affinity for the vesicular acetylcholine transporter, will enable the researcher to simultaneously examine functional changes of the cholinergic system in vivo. This line of research will help to clarify the role of impaired cholinergic neurotransmission in the development of motor and non-motor symptoms in PD.
Author Contributions
All authors prepared the manuscript draft and approved the final manuscript.
Funding
HS received support from Lundbeck Fonden (Grant of Excellence: Mapping, Modulation and Modelling the Control of Actions; Grant no. 59-A5399) and Novo Nordisk Fonden (Synergy grant “Biophysically Adjusted State-Informed Cortex Stimulation,” NNF14OC0011413). HS holds a 5-year professorship in precision medicine at the Faculty of Health Sciences and Medicine, University of Copenhagen which is sponsored by the Lundbeck Foundation (R186-2015-2138). HS has received honoraria as speaker from Sanofi Genzyme, Denmark and Novartis, Denmark, as consultant from Sanofi Genzyme, Denmark and as senior editor (NeuroImage) from Elsevier Publishers, Amsterdam, Netherlands. He has received royalties as book editor from Springer Publishers, Stuttgart, Germany. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
HS has received honoraria as speaker from Sanofi Genzyme, Denmark and Novartis, Denmark, as consultant from Sanofi Genzyme, Denmark and as senior editor (NeuroImage) from Elsevier Publishers, Amsterdam, Netherlands. He has received royalties as book editor from Springer Publishers, Stuttgart, Germany.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Alkondon, M., Pereira, E. F., Eisenberg, H. M., and Albuquerque, E. X. (2000). Nicotinic receptor activation in human cerebral cortical interneurons: a mechanism for inhibition and disinhibition of neuronal networks. J. Neurosci. 20, 66–75. doi: 10.1523/jneurosci.20-01-00066.2000
Asmussen, M. J., Jacobs, M. F., Lee, K. G. H., Zapallow, C. M., and Nelson, A. J. (2013). Short-latency afferent inhibition modulation during finger movement. PLoS One 8:e60496. doi: 10.1371/journal.pone.0060496
Bailey, A. Z., Asmussen, M. J., and Nelson, A. J. (2016). Short-latency afferent inhibition determined by the sensory afferent volley. J. Neurophysiol. 116, 637–644. doi: 10.1152/jn.00276.2016
Barone, P. (2010). Neurotransmission in Parkinson’s disease: beyond dopamine. Eur. J. Neurol. 17, 364–376. doi: 10.1111/j.1468-1331.2009.02900.x
Boecker, H., Ceballos-Baumann, A., Bartenstein, P., Weindl, A., Siebner, H. R., Fassbender, T., et al. (1999). Sensory processing in Parkinson’s and Huntington’s disease: investigations with 3D H215O-PET. Brain 122, 1651–1665. doi: 10.1093/brain/122.9.1651
Bohnen, N. I., and Albin, R. L. (2011). The cholinergic system and Parkinson’s Disease. Behav. Brain Res. 221, 564–573. doi: 10.1016/j.bbr.2009.12.048
Brown, P., Oliviero, A., Mazzone, P., Insola, A., Tonali, P., and Di Lazzaro, V. (2001). Dopamine dependency of oscillations between subthalamic nucleus and pallidum in Parkinson’s disease. J. Neurosci. 21, 1033–1038. doi: 10.1523/jneurosci.21-03-01033.2001
Brusa, L., Ponzo, V., Mastropasqua, C., Picazio, S., Bonnì, S., Di Lorenzo, F., et al. (2014). Theta burst stimulation modulates Cerebellar-cortical connectivity in patients with progressive supranuclear palsy. Brain Stimul. 7, 29–35. doi: 10.1016/j.brs.2013.07.003
Cantone, M., Di Pino, G., Capone, F., Piombo, M., Chiarello, D., Cheeran, B., et al. (2014). The contribution of transcranial magnetic stimulation in the diagnosis and in the management of dementia. Clin. Neurophysiol. 125, 1509–1532. doi: 10.1016/j.clinph.2014.04.010
Caviness, J. N., Driver-Dunckley, E., Connor, D. J., Sabbagh, M. N., Hentz, J. G., Noble, B., et al. (2007). Defining mild cognitive impairment in Parkinson’s disease. Mov. Disord. 22, 1272–1277. doi: 10.1002/mds.21453
Celebi, O., Temuçin, C. M., Elibol, B., and Saka, E. (2012). Short latency afferent inhibition in Parkinson’s disease patients with dementia. Mov. Disord. 27, 1052–1055. doi: 10.1002/mds.25040
Celebi, O., Temuçin, Ç. M., Elibol, B., and Saka, E. (2014). Cognitive profiling in relation to short latency afferent inhibition of frontal cortex in multiple system atrophy. Parkinsonism Relat. Disord. 20, 632–636. doi: 10.1016/j.parkreldis.2014.03.012
Chaudhuri, K. R., and Schapira, A. H. (2009). Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 8, 464–474. doi: 10.1016/S1474-4422(09)70068-7
Cheron, G., Piette, T., Thiriaux, A., Jacquy, J., and Godaux, E. (1994). Somatosensory evoked potentials at rest and during movement in Parkinson’s disease: evidence for a specific apomorphine effect on the frontal N30 wave. Electroencephalogr. Clin. Neurophysiol. 92, 491–501. doi: 10.1016/0168-5597(94)90133-3
Cho, H. J., Panyakaew, P., Thirugnanasambandam, N., Wu, T., and Hallett, M. (2016). Dynamic modulation of corticospinal excitability and short-latency afferent inhibition during onset and maintenance phase of selective finger movement. Clin. Neurophysiol. 127, 2343–2349. doi: 10.1016/j.clinph.2016.02.020
Classen, J., Steinfelder, B., Liepert, J., Stefan, K., Celnik, P., Cohen, L. G., et al. (2000). Cutaneomotor integration in humans is somatotopically organized at various levels of the nervous system and is task dependent. Exp. Brain Res. 130, 48–59. doi: 10.1007/s002210050005
Cohen, L. G., and Starr, A. (1987). Localization, timing and specificity of gating of somatosensory evoked potentials during active movement in man. Brain 110, 451–467. doi: 10.1093/brain/110.2.451
Conte, A., Khan, N., Defazio, G., Rothwell, J. C., and Berardelli, A. (2013). Pathophysiology of somatosensory abnormalities in Parkinson’s Disease. Nat. Rev. Neurol. 9, 687–697. doi: 10.1038/nrneurol.2013.224
Degardin, A., Devos, D., Defebvre, L., Destée, A., Plomhause, L., Derambure, P., et al. (2012). Effect of intermittent theta-burst stimulation on akinesia and sensorimotor integration in patients with Parkinson’s disease. Eur. J. Neurosci. 36, 2669–2678. doi: 10.1111/j.1460-9568.2012.08158.x
Demirci, M., Grill, S., McShane, L., and Hallett, M. (1997). A mismatch between kinesthetic and visual perception in Parkinson’s disease. Ann. Neurol. 41, 781–788. doi: 10.1002/ana.410410614
Di Cara, B., Panayi, F., Gobert, A., Dekeyne, A., Sicard, D., De Groote, L., et al. (2007). Activation of dopamine D1 receptors enhances cholinergic transmission and social cognition: a parallel dialysis and behavioural study in rats. Int. J. Neuropsychopharmacol. 10, 383–399. doi: 10.1017/s1461145706007103
Di Lazzaro, V., Oliviero, A., Pilato, F., Saturno, E., Dileone, M., Marra, C., et al. (2005a). Neurophysiological predictors of long term response to AChE inhibitors in AD patients. J. Neurol. Neurosurg. Psychiatry 76, 1064–1069. doi: 10.1136/jnnp.2004.051334
Di Lazzaro, V., Oliviero, A., Saturno, E., Dileone, M., Pilato, F., Nardone, R., et al. (2005b). Effects of lorazepam on short latency afferent inhibition and short latency intracortical inhibition in humans. J. Physiol. 564, 661–668. doi: 10.1113/jphysiol.2004.061747
Di Lazzaro, V., Pilato, F., Dileone, M., Tonali, P. A., and Ziemann, U. (2005c). Dissociated effects of diazepam and lorazepam on short-latency afferent inhibition. J. Physiol. 569, 315–323. doi: 10.1113/jphysiol.2005.092155
Di Lazzaro, V., Oliviero, A., Tonali, P. A., Marra, C., Daniele, A., Profice, P., et al. (2002). Noninvasive in vivo assessment of cholinergic cortical circuits in AD using transcranial magnetic stimulation. Neurology 59, 392–397. doi: 10.1212/wnl.59.3.392
Di Lazzaro, V., Oliviero, A., Pilato, F., Saturno, E., Dileone, M., Bentivoglio, A. R., et al. (2004). Normal or enhanced short-latency afferent inhibition in Parkinson’s disease? Brain 127:E8; author reply E9. doi: 10.1093/brain/awh089
Di Lazzaro, V., Oliviero, A., Profice, P., Pennisi, M. A., Di Giovanni, S., Zito, G., et al. (2000). Muscarinic receptor blockade has differential effects on the excitability of intracortical circuits in the human motor cortex. Exp. Brain Res. 135, 455–461. doi: 10.1007/s002210000543
Di Lazzaro, V., Pilato, F., Dileone, M., Profice, P., Ranieri, F., Ricci, V., et al. (2007). Segregating two inhibitory circuits in human motor cortex at the level of GABAA receptor subtypes: a TMS study. Clin. Neurophysiol. 118, 2207–2214. doi: 10.1016/j.clinph.2007.07.005
Di Lazzaro, V., Pilato, F., Dileone, M., Saturno, E., Oliviero, A., Marra, C., et al. (2006). In vivo cholinergic circuit evaluation in frontotemporal and Alzheimer dementias. Neurology 66, 1111–1113. doi: 10.1212/01.wnl.0000204183.26231.23
Di Lazzaro, V., and Ziemann, U. (2013). The contribution of transcranial magnetic stimulation in the functional evaluation of microcircuits in human motor cortex. Front. Neural Circuits 7:18. doi: 10.3389/fncir.2013.00018
Di Martino, A., Scheres, A., Margulies, D. S., Kelly, A. M. C., Uddin, L. Q., Shehzad, Z., et al. (2008). Functional connectivity of human striatum: a resting state fMRI study. Cereb. Cortex 18, 2735–2747. doi: 10.1093/cercor/bhn041
Dickson, D. W. (2012). Parkinson’ s disease and Parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2:a009258. doi: 10.1101/cshperspect.a009258
Dubbioso, R., de Rosa, A., Esposito, M., Peluso, S., Iodice, R., de Michele, G., et al. (2017a). Does motor cortex plasticity depend on the type of mutation in the leucine-rich repeat kinase 2 gene? Mov. Disord. 32, 947–948. doi: 10.1002/mds.27012
Dubbioso, R., Esposito, M., Peluso, S., Iodice, R., De Michele, G., Santoro, L., et al. (2017b). Disruption of GABAA-mediated intracortical inhibition in patients with chorea-acanthocytosis. Neurosci. Lett. 654, 107–110. doi: 10.1016/j.neulet.2017.06.032
Dubbioso, R., Raffin, E., Karabanov, A., Thielscher, A., and Siebner, H. R. (2017c). Centre-surround organization of fast sensorimotor integration in human motor hand area. Neuroimage 158, 37–47. doi: 10.1016/j.neuroimage.2017.06.063
Dubbioso, R., Pellegrino, G., Antenora, A., De Michele, G., Filla, A., Santoro, L., et al. (2015). The effect of cerebellar degeneration on human sensori-motor plasticity. Brain Stimul. 8, 1144–1150. doi: 10.1016/j.brs.2015.05.012
Guerra, A., Pogosyan, A., Nowak, M., Tan, H., Ferreri, F., Di Lazzaro, V., et al. (2016). Phase dependency of the human primary motor cortex and cholinergic inhibition cancelation during β tACS. Cereb. Cortex 26, 3977–3990. doi: 10.1093/cercor/bhw245
Gulledge, A. T., and Stuart, G. J. (2005). Cholinergic inhibition of neocortical pyramidal neurons. J. Neurosci. 25, 10308–10320. doi: 10.1523/jneurosci.2697-05.2005
Haber, S. N., and Calzavara, R. (2009). The cortico-basal ganglia integrative network: the role of the thalamus. Brain Res. Bull. 78, 69–74. doi: 10.1016/j.brainresbull.2008.09.013
Hillen, M. E., and Sage, J. I. (1996). Nonmotor fluctuations in patients with Parkinson’s disease. Neurology 47, 1180–1183. doi: 10.1212/WNL.47.5.1180
Hutchison, W. D., Allan, R. J., Opitz, H., Levy, R., Dostrovsky, J. O., Lang, A. E., et al. (1998). Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson’s disease. Ann. Neurol. 44, 622–628. doi: 10.1002/ana.410440407
Insola, A., Le Pera, D., Restuccia, D., Mazzone, P., and Valeriani, M. (2004). Reduction in amplitude of the subcortical low- and high-frequency somatosensory evoked potentials during voluntary movement: an intracerebral recording study. Clin. Neurophysiol. 115, 104–111. doi: 10.1016/j.clinph.2003.08.003
Jobst, E. E., Melnick, M. E., Byl, N. N., Dowling, G. A., and Aminoff, M. J. (1997). Sensory perception in Parkinson’s Disease. Arch. Neurol. 54, 450–454. doi: 10.1001/archneur.1997.00550160080020
Kleine, B. U., Praamstra, P., Stegeman, D. F., and Zwarts, M. J. (2001). Impaired motor cortical inhibition in Parkinson’s disease: motor unit responses to transcranial magnetic stimulation. Exp. Brain Res. 138, 477–483. doi: 10.1007/s002210100731
Klockgether, T., Borutta, M., Rapp, H., Spieker, S., and Dichgans, J. (1995). A defect of kinesthesia in Parkinson’s disease. Mov. Disord. 10, 460–465. doi: 10.1002/mds.870100410
Kojovic, M., Bologna, M., Kassavetis, P., Murase, N., Palomar, F. J., Berardelli, A., et al. (2012). Functional reorganization of sensorimotor cortex in early Parkinson’s Disease. Neurology 78, 1441–1448. doi: 10.1212/wnl.0b013e318253d5dd
Künzle, H. (1977). Projections from the primary somatosensory cortex to basal ganglia and thalamus in the monkey. Exp. Brain Res. 30, 481–492. doi: 10.1007/bf00237639
Kuo, H. I., Paulus, W., Batsikadze, G., Jamil, A., Kuo, M. F., and Nitsche, M. A. (2017). Acute and chronic noradrenergic effects on cortical excitability in healthy humans. Int. J. Neuropsychopharmacol. 20, 634–643. doi: 10.1093/ijnp/pyx026
Lange, K. W., Wells, F. R., Jenner, P., and Marsden, C. D. (1993). Altered muscarinic and nicotinic receptor densities in cortical and subcortical brain regions in Parkinson’s disease. J. Neurochem. 60, 197–203. doi: 10.1111/j.1471-4159.1993.tb05838.x
Lee, K. D., Koo, J. H., Song, S. H., Jo, K. D., Lee, M. K., and Jang, W. (2015). Central cholinergic dysfunction could be associated with oropharyngeal dysphagia in early Parkinson’s disease. J. Neural Transm. 122, 1553–1561. doi: 10.1007/s00702-015-1427-z
Lidsky, T. I., Manetto, C., and Schneider, J. S. (1985). A consideration of sensory factors involved in motor functions of the basal ganglia. Brain Res. Rev. 9, 133–146. doi: 10.1016/0165-0173(85)90010-4
Litvan, I., Goldman, J. G., Tröster, A. I., Schmand, B. A., Weintraub, D., Petersen, R. C., et al. (2012). Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov. Disord. 27, 349–356. doi: 10.1002/mds.24893
Macerollo, A., Brown, M. J. N., Kilner, J. M., and Chen, R. (2018). Neurophysiological changes measured using somatosensory evoked potentials. Trends Neurosci. 41, 294–310. doi: 10.1016/j.tins.2018.02.007
Macerollo, A., Chen, J. C., Korlipara, P., Foltynie, T., Rothwell, J., Edwards, M. J., et al. (2016). Dopaminergic treatment modulates sensory attenuation at the onset of the movement in Parkinson’s disease: a test of a new framework for bradykinesia. Mov. Disord. 31, 143–146. doi: 10.1002/mds.26493
Magill, P. J., Bolam, J. P., and Bevan, M. D. (2001). Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus-globus pallidus network. Neuroscience 106, 313–330. doi: 10.1016/s0306-4522(01)00281-0
Manganelli, F., Dubbioso, R., Iodice, R., Topa, A., Dardis, A., Russo, C. V., et al. (2014). Central cholinergic dysfunction in the adult form of Niemann Pick disease type C: a further link with Alzheimer’s disease? J. Neurol. 261, 804–808. doi: 10.1007/s00415-014-7282-2
Manganelli, F., Dubbioso, R., Pisciotta, C., Antenora, A., Nolano, M., De Michele, G., et al. (2013). Somatosensory temporal discrimination threshold is increased in patients with cerebellar atrophy. Cerebellum 12, 456–459. doi: 10.1007/s12311-012-0435-x
Manganelli, F., Vitale, C., Santangelo, G., Pisciotta, C., Iodice, R., Cozzolino, A., et al. (2009). Functional involvement of central cholinergic circuits and visual hallucinations in Parkinsons disease. Brain 132, 2350–2355. doi: 10.1093/brain/awp166
Marra, C., Quaranta, D., Profice, P., Pilato, F., Capone, F., Iodice, F., et al. (2012). Central cholinergic dysfunction measured “in vivo” correlates with different behavioral disorders in Alzheimer’s disease and dementia with Lewy body. Brain Stimul. 5, 533–538. doi: 10.1016/j.brs.2011.08.009
Martin-Rodriguez, J. F., and Mir, P. (2018). Short-afferent inhibition and cognitive impairment in Parkinson’s disease: a quantitative review and challenges. Neurosci. Lett. doi: 10.1016/j.neulet.2018.06.048 [Epub ahead of print].
Martorana, A., Mori, F., Esposito, Z., Kusayanagi, H., Monteleone, F., Codecà, C., et al. (2009). Dopamine modulates cholinergic cortical excitability in Alzheimer’s disease patients. Neuropsychopharmacology 34, 2323–2328. doi: 10.1038/npp.2009.60
Millan, M. J., Di Cara, B., Dekeyne, A., Panayi, F., De Groote, L., Sicard, D., et al. (2007). Selective blockade of dopamine D3 versus D2 receptors enhances frontocortical cholinergic transmission and social memory in rats: a parallel neurochemical and behavioural analysis. J. Neurochem. 100, 1047–1061. doi: 10.1111/j.1471-4159.2006.04262.x
Mima, T., Terada, K., Maekawa, M., Nagamine, T., Ikeda, A., and Shibasaki, H. (1996). Somatosensory evoked potentials following proprioceptive stimulation of finger in man. Exp. Brain Res. 111, 233–245. doi: 10.1007/bf00227300
Mirdamadi, J. L., Suzuki, L. Y., and Meehan, S. K. (2017). Attention modulates specific motor cortical circuits recruited by transcranial magnetic stimulation. Neuroscience 359, 151–158. doi: 10.1016/j.neuroscience.2017.07.028
Nardone, R., Bergmann, J., Brigo, F., Christova, M., Kunz, A., Seidl, M., et al. (2013). Functional evaluation of central cholinergic circuits in patients with Parkinson’s disease and REM sleep behavior disorder: a TMS study. J. Neural Transm. 120, 413–422. doi: 10.1007/s00702-012-0888-6
Nardone, R., Bergmann, J., Kronbichler, M., De Blasi, P., Caleri, F., Tezzon, F., et al. (2010). Functional involvement of the cerebral cortex following paramedian bithalamic infarction. Neurocase 16, 286–292. doi: 10.1080/13554790903463593
Nardone, R., Florio, I., Lochner, P., and Tezzon, F. (2005). Cholinergic cortical circuits in Parkinson’s disease and in progressive supranuclear palsy: a transcranial magnetic stimulation study. Exp. Brain Res. 163, 128–131. doi: 10.1007/s00221-005-2228-7
Nardone, R., Höller, Y., Thomschewski, A., Kunz, A. B., Lochner, P., Golaszewski, S., et al. (2014). Dopamine differently modulates central cholinergic circuits in patients with Alzheimer disease and CADASIL. J. Neural Transm. 121, 1313–1320. doi: 10.1007/s00702-014-1195-1
Nelson, A. J., Hoque, T., Gunraj, C., and Chen, R. (2018). Altered somatosensory processing in Parkinson’s disease and modulation by dopaminergic medications. Parkinsonism Relat. Disord. 53, 76–81. doi: 10.1016/j.parkreldis.2018.05.002
Newman, E. L., Gupta, K., Climer, J. R., Monaghan, C. K., and Hasselmo, M. E. (2012). Cholinergic modulation of cognitive processing: insights drawn from computational models. Front. Behav. Neurosci. 6:24. doi: 10.3389/fnbeh.2012.00024
Oh, E., Park, J., Youn, J., Kim, J. S., Park, S., and Jang, W. (2017). Olfactory dysfunction in early Parkinson’s disease is associated with short latency afferent inhibition reflecting central cholinergic dysfunction. Clin. Neurophysiol. 128, 1061–1068. doi: 10.1016/j.clinph.2017.03.011
Oliviero, A., León, A. M., Holler, I., Vila, J. F., Siebner, H. R., Della Marca, G., et al. (2005). Reduced sensorimotor inhibition in the ipsilesional motor cortex in a patient with chronic stroke of the paramedian thalamus. Clin. Neurophysiol. 116, 2592–2598. doi: 10.1016/j.clinph.2005.07.015
Pallis, C. A. (1971). Parkinsonism—natural history and clinical features. Br. Med. J. 3, 683–690. doi: 10.1136/bmj.3.5776.683
Pelosin, E., Ogliastro, C., Lagravinese, G., Bonassi, G., Mirelman, A., Hausdorff, J. M., et al. (2016). Attentional control of gait and falls: is cholinergic dysfunction a common substrate in the elderly and Parkinson’s disease? Front. Aging Neurosci. 8:104. doi: 10.3389/fnagi.2016.00104
Petersen, R. C., and Morris, J. C. (2005). Mild Cognitive Impairment as a Clinical Entity and Treatment Target. Arch. Neurol. 62, 1160–1163. doi: 10.1001/archneur.62.7.1160
Perez-Lloret, S., and Barrantes, F. J. (2016). Deficits in cholinergic neurotransmission and their clinical correlates in Parkinson’s disease. NPJ Park. Dis. 2:16001. doi: 10.1038/npjparkd.2016.1
Picillo, M., Dubbioso, R., Iodice, R., Iavarone, A., Pisciotta, C., Spina, E., et al. (2015). Short-latency afferent inhibition in patients with Parkinson’s disease and freezing of gait. J. Neural Transm. 122, 1533–1540. doi: 10.1007/s00702-015-1428-y
Ponzo, V., Di Lorenzo, F., Brusa, L., Schirinzi, T., Battistini, S., Ricci, C., et al. (2017). Impaired intracortical transmission in G2019S leucine rich-repeat kinase Parkinson patients. Mov. Disord. 32, 750–756. doi: 10.1002/mds.26931
Praamstra, P., and Plat, F. M. (2001). Failed suppression of direct visuomotor activation in Parkinson’s disease. J. Cogn. Neurosci. 13, 31–43. doi: 10.1162/089892901564153
Quartarone, A., Rizzo, V., Bagnato, S., Morgante, F., Sant’Angelo, A., Girlanda, P., et al. (2006). Rapid-rate paired associative stimulation of the median nerve and motor cortex can produce long-lasting changes in motor cortical excitability in humans. J. Physiol. 575, 657–670. doi: 10.1113/jphysiol.2006.114025
Restuccia, D., Valeriani, M., Barba, C., Le Pera, D., Tonali, P., and Mauguière, F. (1999). Different contribution of joint and cutaneous inputs to early scalp somatosensory evoked potentials. Muscle Nerve 22, 910–919. doi: 10.1002/(sici)1097-4598(199907)22:7<910::aid-mus15>3.0.co;2-v
Riedel, O., Klotsche, J., Spottke, A., Deuschl, G., Förstl, H., Henn, F., et al. (2010). Frequency of dementia, depression and other neuropsychiatric symptoms in 1,449 outpatients with Parkinson’s disease. J. Neurol. 257, 1073–1082. doi: 10.1007/s00415-010-5465-z
Rizzo, V., Aricò, I., Liotta, G., Ricciardi, L., Mastroeni, C., Morgante, F., et al. (2010). Impairment of sensory-motor integration in patients affected by RLS. J. Neurol. 257, 1979–1985. doi: 10.1007/s00415-010-5644-y
Rochester, L., Yarnall, A. J., Baker, M. R., David, R. V., Lord, S., Galna, B., et al. (2012). Cholinergic dysfunction contributes to gait disturbance in early Parkinson’s disease. Brain 135, 2779–2788. doi: 10.1093/brain/aws207
Rodriguez-Oroz, M. C., Rodriguez, M., Guridi, J., Mewes, K., Chockkman, V., Vitek, J., et al. (2001). The subthalamic nucleus in Parkinson’s disease: somatotopic organization and physiological characteristics. Brain 124, 1777–1790. doi: 10.1055/b-0034-89968
Rossini, P. M., Babiloni, F., Bernardi, G., Cecchi, L., Johnson, P. B., Malentacca, A., et al. (1989). Abnormalities of short-latency somatosensory evoked potentials in parkinsonian patients. Electroencephalogr. Clin. Neurophysiol. 74, 277–289. doi: 10.1016/0168-5597(89)90058-0
Ruberg, M., Ploska, A., Javoy-Agid, F., and Agid, Y. (1982). Muscarinic binding and choline acetyltransferase activity in Parkinsonian subjects with reference to dementia. Brain Res. 232, 129–139. doi: 10.1016/0006-8993(82)90615-1
Rubin, J. E., and Terman, D. (2004). High frequency stimulation of the subthalamic nucleus eliminates pathological thalamic rhythmicity in a computational model. J. Comput. Neurosci. 16, 211–235. doi: 10.1023/b:jcns.0000025686.47117.67
Sailer, A., Cunic, D. I., Paradiso, G. O., Gunraj, C. A., Wagle-Shukla, A., Moro, E., et al. (2007). Subthalamic nucleus stimulation modulates afferent inhibition in Parkinson’s Disease. Neurology 68, 356–363. doi: 10.1212/01.wnl.0000252812.95774.aa
Sailer, A., Molnar, G. F., Paradiso, G., Gunraj, C. A., Lang, A. E., and Chen, R. (2003). Short and long latency afferent inhibition in Parkinson’s disease. Brain 126, 1883–1894. doi: 10.1093/brain/awg183
Schneider, J. S., Diamond, S. G., and Markham, C. H. (1987). Parkinson’s disease: sensory and motor problems in arms and hands. Neurology 37, 951–956. doi: 10.1212/wnl.37.6.951
Seiss, E., and Praamstra, P. (2004). The basal ganglia and inhibitory mechanisms in response selection: evidence from subliminal priming of motor responses in Parkinson’s disease. Brain 127, 330–339. doi: 10.1093/brain/awh043
Seki, K., and Fetz, E. E. (2012). Gating of sensory input at spinal and cortical levels during preparation and execution of voluntary movement. J. Neurosci. 32, 890–902. doi: 10.1523/JNEUROSCI.4958-11.2012
Sirviö, J., Rinne, J. O., Valjakka, A., Rinne, U. K., Riekkinen, P. J., and Paljärvi, L. (1989). Different forms of brain acetylcholinesterase and muscarinic binding in Parkinson’s disease. J. Neurol. Sci. 90, 23–32. doi: 10.1016/0022-510x(89)90042-7
Snider, S. R., Fahn, S., Isgreen, W. P., and Cote, L. J. (1976). Primary sensory symptoms in parkinsonism. Neurology 26, 423–429. doi: 10.1212/wnl.26.5.423
Suzuki, L. Y., and Meehan, S. K. (2018). Verbal working memory modulates afferent circuits in motor cortex. Eur. J. Neurosci. 48, 3117–3125. doi: 10.1111/ejn.14154
Theodosopoulos, P. V., Marks, W. J. Jr., Christine, C., and Starr, P. A. (2003). Locations of movement-related cells in the human subthalamic nucleus in Parkinson’s disease. Mov. Disord. 18, 791–798. doi: 10.1002/mds.10446
Tokimura, H., Di Lazzaro, V., Tokimura, Y., Oliviero, A., Profice, P., Insola, A., et al. (2000). Short latency inhibition of human hand motor cortex by somatosensory input from the hand. J. Physiol. 523, 503–513. doi: 10.1111/j.1469-7793.2000.t01-1-00503.x
Tsang, P., Bailey, A. Z., and Nelson, A. J. (2015). Rapid-rate paired associative stimulation over the primary somatosensory cortex. PLoS One 10:e0120731. doi: 10.1371/journal.pone.0120731
Tsang, P., Jacobs, M. F., Lee, K. G. H., Asmussen, M. J., Zapallow, C. M., and Nelson, A. J. (2014). Continuous theta-burst stimulation over primary somatosensory cortex modulates short-latency afferent inhibition. Clin. Neurophysiol. 125, 2253–2259. doi: 10.1016/j.clinph.2014.02.026
Turco, C. V., El-Sayes, J., Locke, M. B., Chen, R., Baker, S., and Nelson, A. J. (2018a). Effects of lorazepam and baclofen on short- and long-latency afferent inhibition. J. Physiol. 596, 5267–5280. doi: 10.1113/jp276710
Turco, C. V., El-Sayes, J., Savoie, M. J., Fassett, H. J., Locke, M. B., and Nelson, A. J. (2018b). Short- and long-latency afferent inhibition; uses, mechanisms and influencing factors. Brain Stimul. 11, 59–74. doi: 10.1016/j.brs.2017.09.009
Turco, C. V., Locke, M. B., El-Sayes, J., Tommerdahl, M., and Nelson, A. J. (2018c). Exploring behavioral correlates of afferent inhibition. Brain Sci. 8:E64. doi: 10.3390/brainsci8040064
Udupa, K., Bahl, N., Ni, Z., Gunraj, C., Mazzella, F., Moro, E., et al. (2016). Cortical plasticity induction by pairing subthalamic nucleus deep-brain stimulation and primary motor cortical transcranial magnetic stimulation in Parkinson’s disease. J. Neurosci. 36, 396–404. doi: 10.1523/JNEUROSCI.2499-15.2016
Ulivelli, M., Rossi, S., Pasqualetti, P., Rossini, P. M., Ghiglieri, O., Passero, S., et al. (1999). Time course of frontal somatosensory evoked potentials: relation to L-dopa plasma levels and motor performance in PD. Neurology 53, 1451–1457. doi: 10.1212/wnl.53.7.1451
Versace, V., Langthaler, P. B., Sebastianelli, L., Höller, Y., Brigo, F., Orioli, A., et al. (2017). Impaired cholinergic transmission in patients with Parkinson’s disease and olfactory dysfunction. J. Neurol. Sci. 377, 55–61. doi: 10.1016/j.jns.2017.03.049
Wagle Shukla, A., Moro, E., Gunraj, C., Lozano, A., Hodaie, M., Lang, A., et al. (2013). Long-term subthalamic nucleus stimulation improves sensorimotor integration and proprioception. J. Neurol. Neurosurg. Psychiatry 84, 1020–1028. doi: 10.1136/jnnp-2012-304102
Wichmann, T., Bergman, H., and DeLong, M. R. (1994). The primate subthalamic nucleus. I. Functional properties in intact animals. J. Neurophysiol. 72, 494–506. doi: 10.1152/jn.1994.72.2.494
Wylie, S. A., van den Wildenberg, W. P. M., Ridderinkhof, K. R., Bashore, T. R., Powell, V. D., Manning, C. A., et al. (2009). The effect of Parkinson’s disease on interference control during action selection. Neuropsychologia 47, 145–157. doi: 10.1016/j.neuropsychologia.2008.08.001
Yarnall, A. J., Rochester, L., Baker, M. R., David, R., Khoo, T. K., Duncan, G. W., et al. (2013). Short latency afferent inhibition: a biomarker for mild cognitive impairment in Parkinson’s disease? Mov. Disord. 28, 1285–1288. doi: 10.1002/mds.25360
Zamir, O., Gunraj, C., Ni, Z., Mazzella, F., and Chen, R. (2012). Clinical neurophysiology effects of theta burst stimulation on motor cortex excitability in Parkinson’s disease. Clin. Neurophysiol. 123, 815–821. doi: 10.1016/j.clinph.2011.07.051
Keywords: short-latency afferent inhibition, cholinergic neuromodulation, cortical oscillations, dopaminergic dysfunction, Parkinson’s disease, movement disorder, neurophysiological biomarker
Citation: Dubbioso R, Manganelli F, Siebner HR and Di Lazzaro V (2019) Fast Intracortical Sensory-Motor Integration: A Window Into the Pathophysiology of Parkinson’s Disease. Front. Hum. Neurosci. 13:111. doi: 10.3389/fnhum.2019.00111
Received: 29 January 2019; Accepted: 13 March 2019;
Published: 08 April 2019.
Edited by:
Matt J. N. Brown, California State University, Sacramento, United StatesReviewed by:
Aimee J. Nelson, McMaster University, CanadaSean Kevin Meehan, University of Waterloo, Canada
Copyright © 2019 Dubbioso, Manganelli, Siebner and Di Lazzaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raffaele Dubbioso, rafdubbioso@gmail.com
† These authors have contributed equally to this work
‡ Co-senior authors