Tau-tubulin kinase
 Seiko Ikezu1
Seiko Ikezu1  Tsuneya Ikezu1,2,3*
Tsuneya Ikezu1,2,3*- 1Department of Pharmacology and Experimental Therapeutics, Boston University School of Medicine, Boston, MA, USA
- 2Department of Neurology, Boston University School of Medicine, Boston, MA, USA
- 3Alzheimer’s Disease Center, Boston University School of Medicine, Boston, MA, USA
Tau-tubulin kinase (TTBK) belongs to casein kinase superfamily and phosphorylates microtubule-associated protein tau and tubulin. TTBK has two isoforms, TTBK1 and TTBK2, which contain highly homologous catalytic domains but their non-catalytic domains are distinctly different. TTBK1 is expressed specifically in the central nervous system and is involved in phosphorylation and aggregation of tau. TTBK2 is ubiquitously expressed in multiple tissues and genetically linked to spinocerebellar ataxia type 11. TTBK1 directly phosphorylates tau protein, especially at Ser422, and also activates cycline-dependent kinase 5 in a unique mechanism. TTBK1 protein expression is significantly elevated in Alzheimer’s disease (AD) brains, and genetic variations of the TTBK1 gene are associated with late-onset Alzheimer’s disease in two cohorts of Chinese and Spanish populations. TTBK1 transgenic mice harboring the entire 55-kilobase genomic sequence of human TTBK1 show progression of tau accumulation, neuroinflammation, and neurodegeneration when crossed with tau mutant mice. Our recent study shows that there is a striking switch in mononuclear phagocyte and activation phenotypes in the anterior horn of the spinal cord from alternatively activated (M2-skewed) microglia in P301L tau mutant mice to pro-inflammatory (M1-skewed) infiltrating peripheral monocytes by crossing the tau mice with TTBK1 transgenic mice. TTBK1 is responsible for mediating M1-activated microglia-induced neurotoxicity, and its overexpression induces axonal degeneration in vitro. These studies suggest that TTBK1 is an important molecule for the inflammatory axonal degeneration, which may be relevant to the pathobiology of tauopathy including AD.
Tauopathy and Tau Kinases
The microtubule-associated protein tau (MAPT) is a physiological component of the microtubule architecture of neuronal axons (Weingarten et al., 1975). Hyperphosphorylated tau protein (pTau) is a principal component of neurofibrillary tangles (NFTs), which are a hallmark of tauopathies, such as Alzheimer’s disease (AD) and frontotemporal dementia (FTD; Grundke-Iqbal et al., 1986; Wolozin et al., 1986; Iqbal et al., 1989). The human tau gene is segregated to a rare form of familial FTD with Parkinsonism linked to chromosome 17 (FTDP-17), demonstrating MAPT as a causative gene of neurodegenerative disorders (Hong et al., 1998; Hutton et al., 1998). The mechanism of NFT formation is centered around tau phosphorylation, mono-ubiquitination, and conformational changes of the protein from highly extended native form to paired helical filament (Ihara et al., 1986; Kosik et al., 1986). pTau found in NFT suggests that there is an imbalance between the tau kinase and phosphatase activities or some dysfunction in the clearance of pTau in affected human brain (Takashima et al., 1995; Gong et al., 2000; Jinwal et al., 2013). Tau protein is phosphorylated by multiple protein kinases, such as glycogen synthase kinase 3-β (GSK3β) and cycline-dependent kinase 5 (Cdk5; Ishiguro et al., 1991, 1992; Arioka et al., 1993; Takashima et al., 1993). GSK3β and Cdk5 are suspected to play a major role in pathological phosphorylation of tau in the brain. As such, large efforts have been made to develop inhibitors toward these kinases (Brunden et al., 2010). One of the main disadvantages of targeting such kinases is their ubiquitous expression in multiple peripheral tissues, which warrants serious unwanted side effects. Brain-specific tau kinases are attractive targets for ameliorating the phosphorylation status of tau protein. Through the complementary DNA library screening of brain-specific kinases, we have identified tau-tubulin kinase 1 (TTBK1), which is uniquely expressed in neurons in the central nervous system (CNS) and can phosphorylate tau protein (Sato et al., 2006).
Tau-Tubulin Kinase Family
The TTBK family consists of TTBK1 (1321 a.a.; Sato et al., 2006) and TTBK2 (1244 a.a.; Houlden et al., 2007). These two kinases belong to the casein kinase 1 (CK1) group, which contains CK1 (α1, α2, γ1, γ2, γ3, δ, and ε), TTBK (1 and 2), and vaccinia-related kinase (VRK) 1–3 (Figure 1A; Manning et al., 2002; Sato et al., 2006). CK1δ can phosphorylate tau in cultured cells and is known to be up-regulated in AD brain (Li et al., 2004). The alignment of the kinase domain of TTBK1 and CK1δ shows 38% identity and 52% similarity (Sato et al., 2006); therefore, TTBK1 is the closest relative of CK1, but not a member of CK1 family. The catalytic domain of TTBK1 is conserved in vertebrates, C. elegans, and D. melanogaster (Figure 1B). TTBK1 and TTBK2 amino acid sequences are 60% identical and 71% similar in the TTBK1 14–577 and TTBK2 1–589 regions. Their kinase domains (TTBK1 35–294 and TTBK2 21–280) are highly homologous (88% identity and 96% similarity). The rest of the sequences have no homology except a small domain (TTBK1 1053–1117 and TTBK2 942–1006 with 43% identity and 58% similarity). TTBK1 and TTBK2 are distinctly conserved among vertebrates from zebrafish (D. rerio) to human (Figure 1C). However, TTBK homologs in C. elegans (TTBK), C. capitata (TTBK1), and D. melanogaster (ASATOR) only conserve the catalytic domain of TTBK1 or TTBK2 (Figure 1C). Since the catalytic domain of TTBK1 and TTBK2 are highly homologous, this suggests that TTBK1 and TTBK2 genes are diversified from a common shorter TTBK gene during the evolution from invertebrates to vertebrates.
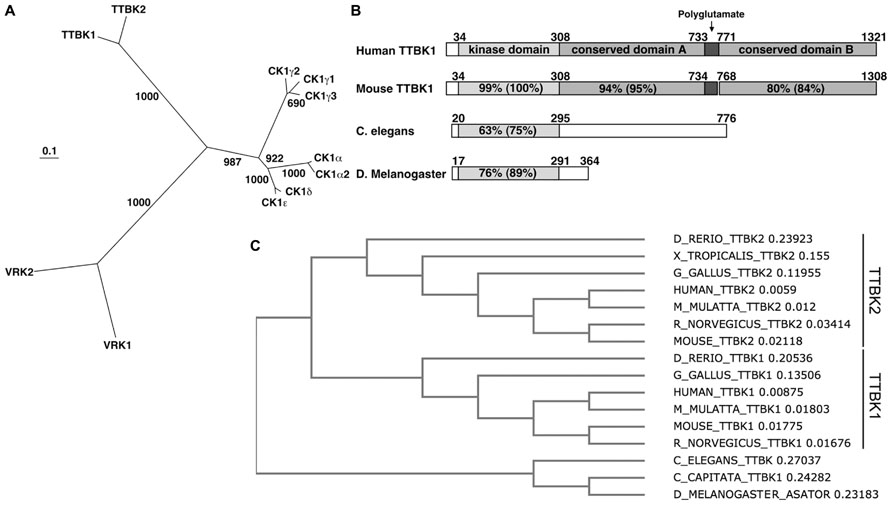
FIGURE 1. Dendrogram of CK1 group and sequence conservation of TTBK1. (A) Phylogenetic tree of human casein kinase family. The scale bar represents 0.1 amino acid substitution per site. Bootstrap values (1,000 bootstrap iterations) are indicated. VRK3 was removed due to low homology to other kinases in CK1 family. (B) Alignment of human and mouse TTBK1, and C. elegans and D. melanogaster TTBK amino acid sequences. TTBK1 34–308, dual-kinase domain; 309–732, conserved domain A; 733–770, polyglutamate domain; 771–1321, conserved domain. (C) Dendrogram of TTBK family among species. D_RERIO; zebrafish, X_TROPICALIS; Western clawed frog, G_GALLIS; chicken, M_MULATTA; rhesus macaque, R_NORVEGICUS; rat, C_ELEGANS; roundworm, C_CAPITATA; Mediterranean fruit fly, D_MELANOGASTER; common fruit fly. The number after each name indicates the branch length.
TTBK2
TTBK2 Gene and Protein Function
The human TTBK2 gene (Gene ID: 146057) is positioned on chromosome 15q15.2, and consisted with nine exons and 10 introns with a complementary DNA (cDNA) length of 6192bp. TTBK2 was previously extracted from bovine and mouse brain as a 36 kDa protein, and it phosphorylates tau protein at Ser208 and Ser210 (Takahashi et al., 1995; Tomizawa et al., 2001). TTBK2 is ubiquitously expressed in multiple tissues, such as placenta, liver, skeletal muscle, pancreas, heart, and brain. There is especially higher expression in the Purkinje cells and granular cell layer of the cerebellum, hippocampus, midbrain and substantia nigra (Houlden et al., 2007), on the other hand, lower expression is shown in the cortex. At a protein level, TTBK2 is highly expressed in brain and testis, which corresponds to the elevated kinase activity in these tissues (Bouskila et al., 2011).
A recombinant kinase domain of mouse TTBK2 corresponds to human TTBK2 1–316 residues (98% identity), and the kinase domain (residues 1–331) of human TTBK2 was expressed in insect cells with a baculovirus overexpression system and crystallized (Kitano-Takahashi et al., 2007). Diffraction data were collected to 2.9 Å resolution, but so far no publication is available for the structural information. Consensus phosphorylation sites for CK1 isoforms is S/Tp-X-X-S/T, where S/Tp refers to a phosphoserine or phosphothreonine: X refers to any amino acid and the underlined residues refer to the target site (Flotow and Roach, 1989; Flotow et al., 1990; Nakielny et al., 1991). The priming phosphorylation site is at the -3 position in the case of CK1. However, the preferred priming phosphorylation site is the tyrosine residue at the +2 position for TTBK2 (S/T-X-Yp), and either serine or threonine residue on the +2 position rather diminish the priming effect (Bouskila et al., 2011).
Multiple biological functions of TTBK2 have recently been identified: TTBK2 is critical in the initiation of ciliogenesis, which is important for sonic hedgehog developmental pathway (Goetz et al., 2012; Tanos et al., 2013; Ye et al., 2014). TTBK2 may also play a role in mitosis: Asator, a TTBK2 homolog in Drosophila, is localized in the mitotic spindle and directly interacts with the spindle matrix protein Megator (Qi et al., 2009). TTBK2 may also be involved in anti-cancer drug resistance: TTBK2 increases the cell surface number of sodium-coupled glucose transporter (SLC5A1) in cancer cells and confers cell survival against anti-cancer drugs (Almilaji et al., 2013). These studies indicate the diverse functions of TTBK2 in embryogenesis, mitosis and cell survival (Table 1). Further studies will be necessary for characterizing the function of TTBK2 in the CNS.
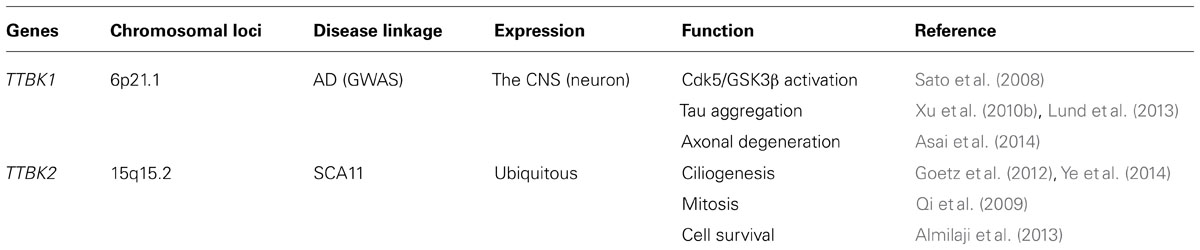
TABLE 1. Summary of TTBK family.
TTBK2 Mutation in Spinocerebellar Ataxia 11 (SCA11)
Spinocerebellar ataxias (SCAs) are a heterogeneous group of neurodegenerative disorders characterized by poor coordination, abnormal eye movements, impairment of speech and swallowing, and pyramidal signs (Schols et al., 2004). Almost all affected individuals show cerebellar atrophy when tested by MRI. SCA11 is a pure progressive cerebellar ataxia that has been genetically linked to human chromosome 15q14–21 (Worth et al., 1999). The mean age of onset for the disease is 24.2 ± 8.4 years, ranging from 15–43. SCA11 is an autosomal dominant form of cerebellar ataxia and so far two families have been identified: a Caucasian family that traces its earliest known British ancestry to the 19th century and another of Pakistani ancestry, with five affected individuals present over three generations (Figure 2A; Houlden et al., 2007; Johnson et al., 2008). Both families show genetically linked mutations in the TTBK2 gene, with the Caucasian family having a one-base insertion of an adenosine in exon 13 at nucleotide 1329 (codon 444), creating a premature stop site (TGA) in the mRNA at codon 450 and truncating the normal protein from 1,244 to 450 amino acids (Figure 2B, 1329InsA). The same mutation was also found in another case of SCA11 by next generation sequencing (Németh et al., 2013). The Pakistani family was associated with a frameshift deletion of two bases (GA) in exon 13 of TTBK2 at nucleotides 1284/1285, codons 428 and 429, creating a premature stop site (TGA) in the mRNA at codon 449 (Figure 2B, 1284/85DelAG). Transient expression study shows that SCA11-linked mutations of TTBK2 dramatically reduce the kinase activity to 10% of full-length TTBK2 and promote their nuclear localization (Bouskila et al., 2011). TTBK2-1329InsA-knockin mice also show reduced kinase activity in vivo (Bouskila et al., 2011). Since kinase-negative TTBK2 mutant also shows nuclear localization, the reduction of kinase activity and autophosphorylation may regulate its subcellular localization. The effect of TTBK2 nuclear localization on neurotoxicity is unknown. These mutations have no effect on the half-life of expressed TTBK2 proteins, which is approximately 24 h (Bouskila et al., 2011).
FIGURE 2. Pedigrees of SCA11 families 1 and 2. (A) The family trees are anonymized, as there are many at-risk individuals. Males or females are given as a diamond, blackened symbols indicate affected individuals, a slash through the symbol indicates the individual is deceased and an arrow indicates the family proband. Adapted from Houlden et al. (2007). (B) Sequence alignment of human TTBK1 and TTBK2 and sites of SCA11-linked TTBK2 mutations. Gray background shows putative catalytic domain (PHA02882). Accession number: BAE78660 (human TTBK1) and EAW92583 (human TTBK2). *Denotes translational termination.
Neuropathological examination with TTBK2 mutated brain also had been carried out (Houlden et al., 2007). The brain macroscopically showed intense atrophy of the cerebellum. Microscopically, severe and almost complete loss of Purkinje cells and a significant loss of cerebellar granule cells have been shown. The medullary tegmentum, nigra, midbrain tegmentum and putamen presented NFTs, neuropil threads and tau-positive neuritis. Pre-tangles and NFTs, as well as oligodendrocyte tau-positive filamentous inclusions, were found in the globus pallidus. There also exist pathological aging, sparse tangles and β-amyloid-positive plaques in the neocortex, hippocampus, and transentorhinal, entorhinal, and insular cortices. No plaques were present in the cerebellum. Although the brain that has the TTBK2 mutation (codon 444) showed cell loss in the cerebellum, there is no other visible pathology (Houlden et al., 2007). Therefore, the degree of TTBK2 gene expression did not associate with pathology in the cerebellum although the gene was highly expressed.
TTBK2 truncation mutations found in the SCA11 families resulted in a shorter half-life of mRNA as compared to wild-type TTBK2 mRNA, suggesting the reduction in expression of TTBK2 by the mutations (Houlden et al., 2007). Kraemer et al. (2006) reported the role of TTBK2 in the tau pathway using RNA interference experiments in C. elegans. Treating worms with short interfering RNA to a C. elegans homolog of TTBK2 (TTBK) in the presence of an FTDP-17 mutant tau transgene enhanced the uncoordinated movement phenotype (dyskinesia). Taken together, these data suggest that reduction in TTBK2 expression levels may accelerate tau-related pathogenesis. In accord, TTBK2-knockin mice expressing an SCA11 disease-causing mutation show the reduction of endogenous TTBK2 kinase activity (Bouskila et al., 2011). The SCA11 mutation homozygosity causes lethality at embryonic day 10. These studies demonstrate that SCA11-linked TTBK2 mutation is a loss-of-function mutation and the TTBK2 gene is essential in embryogenesis. This is consistent with the recent series of ciliogenesis studies on TTBK2 knockout mice (Goetz et al., 2012; Tanos et al., 2013; Ye et al., 2014).
In addition to the two reported mutations on TTBK2 in SCA11 cases, a recent study also identified 1306_1307delGA mutation in exon 2 of one German and one French SCA11 cases (Bauer et al., 2010). This also leads to the premature stop codon after the catalytic domain sequence. Three additional missense mutations were found in SCA11 cases (L8P, E842G, and R1110H; Edener et al., 2009). However, these mutations are outside of the catalytic domain and also reported in the 1000 Genomes project, suggesting them as common variants. In addition, recent 1000 Genomes project identified a total of 12,708 somatic variations in human TTBK2 gene, including 301 missense variants, 11 stop-gained variants, 17 frameshift variants, 32 splicing variants, 30 5′UTR variants, and 83 3′UTR variants. This results in seven different splicing variants of TTBK2 genes (TTBK2-1, TTBK2-2, TTBK2-3, TTBK2-7, TTBK2-6, TTBK-8, and TTBK2-5) and 6 different amino acid sequences (TTBK2-5 has no protein product), whose lengths are 1244, 478, 443, 117, 143 and 84, respectively. Only TTBK2-1, TTBK2-2, and TTBK2-3 genes encode the catalytic domain. Interestingly, among the 11 stop-gained variants, six are terminated at Glu19 (MSGGGEQPDILSVGILVKE/*), suggesting the somatic loss-of-function mutation. Although none of the somatic loss-of-function mutations is linked to human diseases, these data suggest that TTBK2 loci are highly susceptible to somatic mutations and may be associated with human diseases related to cancer or neurodegenerative disorders.
TTBK1
TTBK1 Gene Expression and Distribution
Human TTBK1 gene (Gene ID: 84630), which we originally cloned, is positioned on chromosome 6p21.1 and consists of 14 exons and 15 introns with a cDNA length of 6932bp (Sato et al., 2006). The TTBK1 protein (UniProt ID: Q5TCY1) consists of 1,321 amino acids and has a kinase domain and a polyglutamate domain in the middle region of the sequence (Figure 1B). TTBK1 isoform2 was also recently identified (TTBK1–2, UniProt ID: Q5TCY1–2), which consists of 621 amino acids and share amino acids 52–540 and 1194–1229 of isoform1. Since isoform2 lacks in the N-terminal region of the consensus kinase domain, it likely has no kinase activity. TTBK1 mRNA was detected in cortex and cerebellum, and in fetal brain (Sato et al., 2006). In situ hybridization study on mouse brains detected strong expression of TTBK1 gene expression in the CA1 pyramidal cell layer of the hippocampus, perinuclear, and cytoplasmic regions of the large cortical pyramidal cell in the temporal cortex, and the Purkinje cell layer of the cerebellum (Sato et al., 2006). Recent independent studies also confirm the specific expression of TTBK1 mRNA in neuronal layers of human brain as determined by in situ hybridization (Lund et al., 2013). Immunohistochemistry of human brain with several TTBK1 antibodies produced particularly strong granular staining in the apical and basal dendrites and in deposits in the neuronal cell soma.
Tau-tubulin kinase 1 protein expression is significantly upregulated in AD brains (Sato et al., 2008). Immunohistochemical analysis of AD brains shows that TTBK1 co-localizes with pSer422-positive pre-tangles but not with thioflavin-S-positive NFTs (Lund et al., 2013). pSer422 antibody identifies a rare population of AT8-unlabeled neurons, which scatter throughout the hippocampal formation and the entorhinal cortex. These data suggest that TTBK1 may play a significant role in pre-tangle formation by tau phosphorylation at Ser422 in the entorhinal cortex and hippocampus. This is in line with a previous report claiming that Ser422 becomes phosphorylated earlier in the development of an NFT, preferentially marking intracellular NFTs (Augustinack et al., 2002).
In addition, genetic variations of the TTBK1 gene (SNPs rs2651206, rs10807287, and rs7764257) are associated with late-onset Alzheimer’s disease (LOAD) in two large cohorts of Spanish and Chinese populations (Vazquez-Higuera et al., 2011; Yu et al., 2011), further validating the importance of the TTBK1 gene in the development of tauopathy and AD pathogenesis (Cuny, 2009). These findings are summarized in Table 1. Recent 1000 Genomes project identified a total of 1,650 variations in human TTBK1 gene, including 194 missense variants, five stop-gained variants, nine frameshift variants, 23 splicing variants, 8 5′UTR variants, and 32 3′UTR variants. The number of TTBK1 variants was much smaller than that of TTBK2 variants. The five stop variants are terminated at Arg650, Arg599, Gln686, Glu753, and Ser1067. All of them still encode the catalytic domain and are possibly gain-of-function mutations, since shorter amino acid sequences are generally more efficient in protein translations. These data suggest that more genetic associations to TTBK1 gene may be found in human diseases.
Tau and Tubulin Phosphorylation
Tau-tubulin kinase 1 directly phosphorylates itself, tau and tubulin. Furthermore, we determined the exact human tau 40 (htau40) phosphorylation sites in vitro by phosphopeptide mapping with liquid chromatography and tandem mass-spectrometry (LC/MS/MS; Sato et al., 2006). The htau40 phosphorylation at Tyr197, Ser198, Ser199, Ser202 and Ser422, which have all been reported as phosphorylation sites in PHF-tau (Iqbal et al., 1989; Morishima-Kawashima et al., 1995a, b; Hanger et al., 1998; Vega et al., 2005). These results show that TTBK1 is a critical tau protein kinase for the phosphorylation of PHF-specific sites. Direct phosphorylation of Ser422 and AT8 epitopes (Ser202 and Thr205) by TTBK1 was also independently demonstrated (Lund et al., 2013). pSer422 is a very specific marker for pathological PHF-tau, since little phosphorylation is present in the normal adult brain (Hasegawa et al., 1996). TTBK1 is one of the few kinases, along with the microtubule-associated protein kinases, that can phosphorylate tau protein at Ser422 (Reynolds et al., 1997; Sato et al., 2002, 2006).
For the crystallography of TTBK1, TTBK1 (1–313) and (14–313) fragments were expressed in E. coli and insect cells, which were found as multi-phosphorylated forms (Xue et al., 2013). To avoid this problem, TTBK1 protein was co-expressed with lambda phosphatase, by which TTBK1 was dephosphorylated and was subsequently co-crystallized with two high-affinity ATP-competitive inhibitors: 3-[(6,7-dimethoxyquinazolin-4-yl)amino]phenol (compound 1) and methyl 2-bromo-5-(7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)benzoate (compound 2). Two positive clusters identified on the surface of TTBK1 indicate putative binding sites for the “primed” substrate, suggesting that TTBK1 favors “primed” pre-phosphorylated substrates. The consensus phosphorylation sites of TTBK1 have not been characterized. Considering the very high homology in the kinase domain of TTBK1 and TTBK2 (Figure 2B), they may share the same consensus phosphorylation sites (S/T-X-Yp) of the substrate. The crystallography shows that compound 1 enters the selectivity pocket, while compound 2 does not reach the selectivity pocket and shows slow binding kinetics as determined by surface plasmon resonance analysis (Xue et al., 2013). The distinct structural-kinetic behavior could be used for the design of selective TTBK1 inhibitors. The other group also just published a crystal structure of TTBK1 (14–343), which was purified from baculovirus-infected sf9 insect cells (Kiefer et al., 2014). They report co-crystalization with compound 3, 3-({5-[(4-amino-4-methylpiperidin-1-yl)methyl]pyrrolo[2,1-f][1,2,4]triazin-4-yl}amino)-5-bromophenol, a TTBK1 inhibitor with IC50 of 170 nM. Due to the structural and sequence similarity of TTBK1 and TTBK2, TTBK1/2 selectivity may be still difficult to achieve.
TTBK1 Transgenic Mouse Study
A TTBK1 transgenic mouse model harbors the entire human TTBK1 genomic DNA, which consists of a CpG island in the 5′ non-coding region, 13 introns and 14 exons, spanning 57 kb in chromosome 6p21.1 (Sato et al., 2008). Transgenic human TTBK1 mRNA is expressed in cerebellum, cortex and spinal cord, and TTBK1 protein is expressed in a similar pattern in TTBK1 transgenic mice (Sato et al., 2008).
Immunohistochemistry of TTBK1 transgenic mice exhibited very specific expression of TTBK1 protein in the perforant path region of subiculum, layer II/III of the entorhinal cortex and the external pyramidal layer of the visual cortex as compared to the age-matched non-Tg mice (Sato et al., 2008). TTBK1 transgenic mice showed strong phospho-neurofilament staining in the entorhinal cortex, visual cortex and subiculum, suggesting neuronal dysfunction in these regions. Enhanced microgliosis was observed in the hippocampus and visual cortex, as well as astrogliosis in the visual cortex of the TTBK1 transgenic mouse brains (Sato et al., 2008).
The most striking phenotype of the TTBK1 transgenic mice is the loss of spatial learning at 9–10 months of age as determined by the 6-arm radial arm water maze test (Sato et al., 2008). TTBK1 transgenic mice showed increased levels of the Cdk5 co-activators p35 and p25; enhanced calpain-1 activity, which cleaves p35 into p25; and enhanced p35-associated Cdk5 activity. Cdk5 and calpain-1 closely regulate the post-synaptic levels of the NMDA receptor (NR) subunits, especially NR2B, and affect hippocampal long-term potentiation and spatial learning (Hawasli et al., 2007; Zhang et al., 2008). Calpain-1 also cleaves NR2B directly and reduces its cell surface expression (Simpkins et al., 2003; Wu et al., 2005, 2007). Consistently, we observed significantly reduced expression of NR2B in the hippocampus of TTBK1 transgenic mice as compared to age-matched non-Tg mice. We further confirmed that transient overexpression of TTBK1 downregulated NR2B expression in primary cultured mouse cortical neurons, which were sensitive to a calpain inhibitor or the silencing of endogenous Cdk5 by Cdk5-targeting siRNA transfection. We have also found that G-actin but not F-actin directly bind to Cdk5 and prevent its activation by p35 or p25 (Xu et al., 2010a).
Collectively, we have developed the following hypothesis in the scheme (Figure 3): inactive μ-calpain forms a stable complex with calpastatin, and TTBK1 may dissociate the complex via direct or indirect phosphorylation of calpastatin, leading to the activation of calpain (Figure 3A). FAA (F-actin and alpha-actinin complex) forms a stable complex with p35 and G-actin is associated with Cdk5, preventing the Cdk5/p35 complex formation in the normal state (Figure 3B). TTBK1 may phosphorylate alpha-actinin, leading to dissociation of p35 from FAA and the formation of the Cdk5/p35 complex via dissociation of Cdk5 from G-actin. The Cdk5/p35 complex serves as scaffolding molecules to generate the NR2B/Cdk5/p35/calpain complex as reported (Hawasli et al., 2007) and mediate the cleavage of the C-terminal region of NR2B, which is a critical region for post-synaptic membrane retention of NR2B. The loss of the C-terminal region leads to endocytosis and degradation of NR2B (Figure 3C). This multi-step signal is composed of a new calpain activation mechanism and a new regulation of Cdk5/p35 complex formation by FAA, and is consistent with the recent report on calpain-mediated NR2B processing (Hawasli et al., 2007). In addition, activated calpain cleaves the N-terminal of p35 to generate p25, a more potent Cdk5 activator. p25, however, lacks the N-terminal region of p35 necessary for the association with alpha-actinin (Dhavan et al., 2002) and thus can form a Cdk5/p25 complex independent of FAA or G-actin association with Cdk5, leading to dysregulated activation of Cdk5. This will further enhance NR2B degradation.
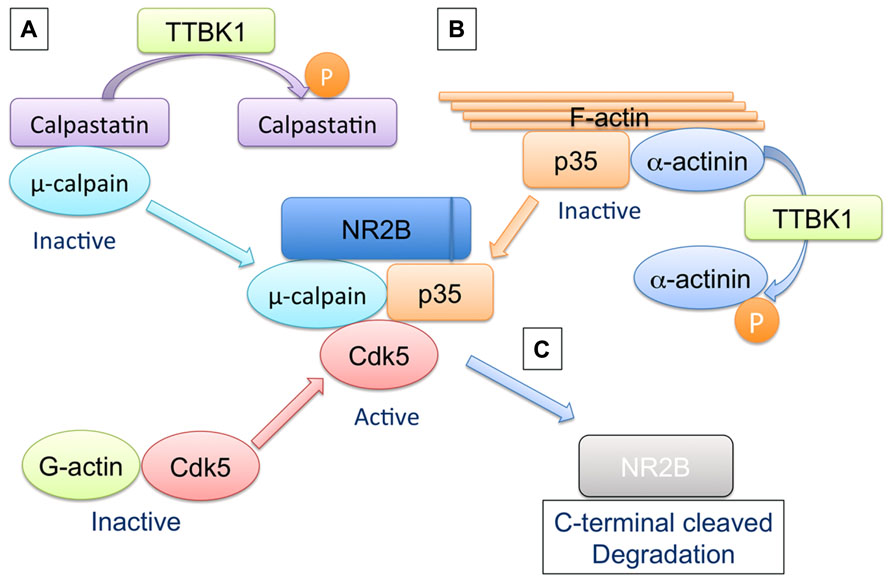
FIGURE 3. Hypothetical regulation of μ-calpain activation, Cdk5/p35 complex formation, and NR2B degradation by TTBK1. (A) Inactive μ-calpain forms a stable complex with calpastatin, and TTBK1 dissociates the complex via direct or indirect phosphorylation of either μ-calpain or calpastatin, leading to the activation of μ-calpain. (B) FAA (F-actin and α-actinin complex) forms a stable complex with p35. G-actin is associated with Cdk5. These two statuses prevent the Cdk5/p35 complex formation in the normal state. TTBK1 may phosphorylate α-actinin, leading to dissociation of p35 from FAA and the formation of the Cdk5/p35 complex via dissociation of G-actin from p35. (C) The Cdk5/p35 complex serves as scaffolding molecules to generate the NR2B/Cdk5/p35/μ-calpain complex and mediates the cleavage of the C-terminal region of NR2B. The C-terminus-lacking NR2B fails to be retained in post-synaptic membrane and undergoes endocytic degradation pathway.
Tau/TTBK1 Double Transgenic Mouse Study
Crossing of TTBK1 transgenic mice with JNPL3 [P301L tau mutant mice (Lewis et al., 2000)] results in enhanced intraneuronal accumulation of phosphorylated tau at multiple sites (AT8, 12E8, PHF-1, and pS422) in cortical and hippocampal regions. Testing of TTBK1/JNPL3 and JNPL3 mice at 5–11 months of age with accelerated rotarod tests demonstrated that TTBK1/JNPL3 mice showed significantly shorter retention time compared to JNPL3 mice as early as 7 months of age (Xu et al., 2010b). We have also detected the significant reduction in forelimb grip strength in TTBK1/JNPL3 mice as compared to JNPL3 littermates starting at 6 months of age, suggesting the sensorimotor or neuromuscular dysfunction in TTBK1/JNPL3 mice. These findings prompted us to examine the neuropathology of motor neurons in the anterior horn of spinal cords at the L4–L5 region. TTBK1/JNPL3 mice also show increased accumulation of oligomeric tau protein in the forebrain and spinal cord, and enhanced reduction in motor neurons in the anterior horn of the lumbar spinal cord, which are associated with neuroinflammation. This suggests that TTBK1 is significantly involved in progression of tauopathy.
Tau And Neuroinflammaiton
Recent studies demonstrated that neuroinflammation may play a significant role in the pathophysiology of tauopathies (Gebicke-Haerter, 2001; Ishizawa and Dickson, 2001; Gerhard et al., 2006). In the LOAD brains, glial activation (both astrocytes and microglia) has a significant linear positive correlation with disease course and NFT formation (Serrano-Pozo et al., 2011). Enhanced pro-inflammatory activation of microglia by disruption of CX3CR1, an anti-inflammatory fractalkine receptor, accelerates tangle formation in tau mouse models (Bhaskar et al., 2010). In accord, immunosuppressant drug FK506 can attenuate microglial activation and delayed the tau-related neuropathology in P301S tau mice (Yoshiyama et al., 2007). These studies suggest that pro-inflammatory activation of microglia plays a critical role in the onset and progression of tauopathy. The neuroinflammation is orchestrated by both resident microglia and infiltrated peripheral macrophages, as they can be recruited by chemokines such as CCL2, which are highly up-regulated in the LOAD brain (Xia and Hyman, 1999; Grammas and Ovase, 2001; Lue et al., 2001; Sun et al., 2003; Pola et al., 2004; Janelsins et al., 2005; Yamamoto et al., 2005; Galimberti et al., 2006; El Khoury et al., 2007; Hickman et al., 2008; Semple et al., 2010). However, the mechanism of how neuroinflammation accelerates tauopathy development is poorly understood.
Neuroinflammation is triggered by the innate immune response, in which mononuclear phagocytes play a major role. When these phagocytes recognize pathogen- or damage-associated molecular pattern molecules (PAMPs and DAMPs), they become “activated,” and this activation can be classified into two phenotypes: classical/pro-inflammatory (M1) and alternative/anti-inflammatory activation (M2; Gordon and Martinez, 2010). M1-skewed activation of mononuclear phagocytes causes the release of pro-inflammatory cytokines, such as interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-12, IL-1β, and IL-23, and reactive oxygen/nitrogen intermediates induced by the expression of nitric oxide synthase (NOS) and NADPH oxidases (NOXes; Mantovani et al., 2002; Brown, 2007; Benoit et al., 2008). In contrast, M2-skewed activation is characterized by abundant levels of non-opsonic receptors (e.g., the mannose receptor) and production of high levels of anti-inflammatory cytokines (Mantovani et al., 2004). However, there is no comprehensive characterization of CNS mononuclear phagocytes and their activation status (M1 or M2) in tauopathy-related neurodegenerative disorders.
Our recent study demonstrated that (1) TTBK1 accelerates motor neuron loss in the spinal cord of JNPL3 mice; (2) JNPL3 mice show mostly M2-skewed microglial accumulation in the spinal cord ventral horn, whereas TTBK1/JNPL3 mice show M1-skewed infiltrating monocytes in the same region. These cell can be regognized as CD169+ (sialoadhesin+) cells; (3) TTBK1 upregulation reduces the axonal length, whereas resting microglia enhance neurite extension; and (4) motor neurons are sensitive to neurotoxicity induced by M1-activated microglia, and this is dependent on endogenous TTBK1 gene expression (Asai et al., 2014). While these are multifaceted findings and it is difficult to draw a simple conclusion, Figure 4 depicts our proposed mechanism of how TTBK1 initiates axonal degeneration and subsequent inflammatory status and neurodegeneration: in JNPL3 mice, motor neuron loss is evident in the ventral horn of the lumbar spinal cord, which is accompanied by accumulation of CD11c+ neuroprotective microglia, thereby limiting neurodegeneration. TTBK1/JNPL3 mice, on the other hand, show a dramatic conversion of the cell population from CD11c+ microglia to M1-skewed infiltrating CD169+ monocytes. This could be due to the accelerated neurodegeneration by TTBK1 upregulation and enhanced generation of DAMPs (M1-skewing ligands) from degenerating motor neurons, which leads to pro-inflammatory activation, generation of CCL2 chemokine and infiltration of peripheral monocytes. This may lead to the acceleration of neurodegeneration via bystander killing of neurons. Overall, these data support the idea that TTBK1 plays a significant role in facilitating inflammatory mononuclear phagocyte-mediated neurotoxicity and subsequent infiltration of peripheral monocytes into the CNS. More study will be necessary to understand the exact molecular mechanism.
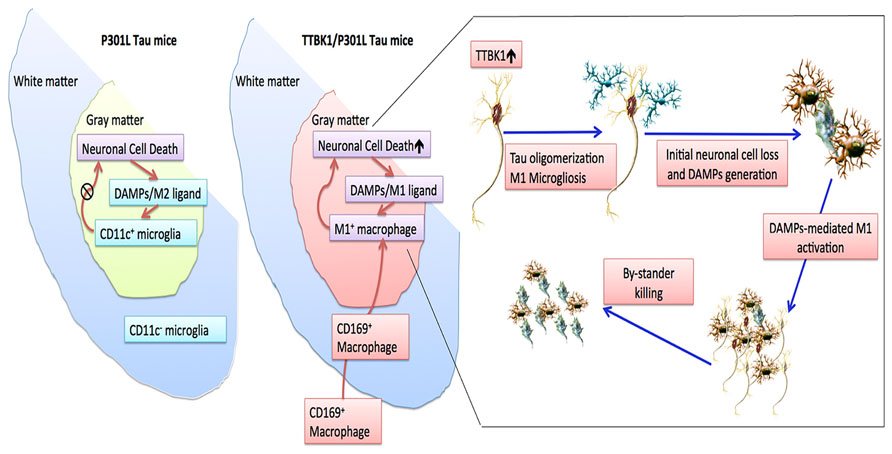
FIGURE 4. Scheme of neuroinflammation and neurodegeneration in JNPL3 and TTBK1/JNPL3 mice. In JNPL3 mice, CD11c+ neuroprotective microglia accumulate in the gray matter of the ventral horn, thereby protecting from accelerated neurodegeneration. Conversely, in TTBK1/JNPL3 mice, the cell population is shifted from CD11c+ microglia to M1-skewed infiltrating NOS2+CD169+ monocytes, resulting in enhanced neuroinflammation and accelerated motor neuron loss. Adapted from Asai et al. (2014).
Summary
Large efforts have been made to develop inhibitors toward GSK3β and Cdk5 as primary tau kinases (Brunden et al., 2010). One of the main disadvantages of targeting the two kinases is their ubiquitous expression in multiple peripheral tissues, which warrants serious unwanted off-target side effects in multiple tissues. TTBK1 therefore is a potentially attractive candidate for pharmacological inhibition because of its apparent restricted expression to neurons in the CNS. Overexpression of TTBK1 was sufficient in inducing spatial learning impairment in mice, which is associated with enhanced Cdk5 activity and reduction in cell surface level of NR2B. Over-expression of TTBK1 transgene in JNPL3 mice resulted in accumulation of pre-tangle forms of tau. In addition, TTBK1 expression plays a unique role in accelerating motor neuron degeneration and neuroinflammation in JNPL3 mice. One would therefore expect that an enhanced expression or activation of TTBK1 would lead to increased tau phosphorylation and predispose tau to pre-tangle formation. Indeed, genetic variations in TTBK1, which are thought to result in decreased TTBK1 activity, can decrease the risk of AD (Vazquez-Higuera et al., 2011; Yu et al., 2011). Thus, TTBK1 is a novel therapeutic target of neuroinflammation-associated neurodegeneration, such as AD, FTD, and tauopathy-related ALS complex. Further studies are necessary to characterize the association of TTBK1 polymorphism, TTBK1 expression, pre-tangle formation and neuroinflammation in AD cases, and to see if reduction in TTBK1 level by TTBK1 gene targeting or silencing delays the progression of tau pathology in established tau mouse models for pre-clinical validations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Drs. Shinji Sato, Satoshi Okuyama, Jixing Xu, Kaitlin Ingraham, Libin Cui, Maya Woodbury, and Hirohide Asai for your work on these studies.
References
Almilaji, A., Munoz, C., Hosseinzadeh, Z., and Lang, F. (2013). Upregulation of Na+,Cl(-)-coupled betaine/gamma-amino-butyric acid transporter BGT1 by Tau tubulin kinase 2. Cell. Physiol. Biochem. 32, 334–343. doi: 10.1159/000354441
Arioka, M., Tsukamoto, M., Ishiguro, K., Kato, R., Sato, K., Imahori, K., et al. (1993). Tau protein kinase II is involved in the regulation of the normal phosphorylation state of tau protein. J. Neurochem. 60, 461–468. doi: 10.1111/j.1471-4159.1993.tb03173.x
Asai, H., Ikezu, S., Woodbury, M. E., Yonemoto, G. M., Cui, L., and Ikezu, T. (2014). Accelerated neurodegeneration and neuroinflammation in transgenic mice expressing P301L tau mutant and tau-tubulin kinase 1. Am. J. Pathol. 184, 808–818. doi: 10.1016/j.ajpath.2013.11.026
Augustinack, J. C., Schneider, A., Mandelkow, E. M., and Hyman, B. T. (2002). Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 103, 26–35. doi: 10.1007/s004010100423
Bauer, P., Stevanin, G., Beetz, C., Synofzik, M., Schmitz-Hubsch, T., Wullner, U., et al. (2010). Spinocerebellar ataxia type 11 (SCA11) is an uncommon cause of dominant ataxia among French and German kindreds. J. Neurol. Neurosurg. Psychiatry 81, 1229–1232. doi: 10.1136/jnnp.2009.202150
Benoit, M., Desnues, B., and Mege, J. L. (2008). Macrophage polarization in bacterial infections. J. Immunol. 181, 3733–3739. doi: 10.1097/QCO.0b013e328344b73e
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi: 10.1016/j.neuron.2010.08.023
Bouskila, M., Esoof, N., Gay, L., Fang, E. H., Deak, M., Begley, M. J., et al. (2011). TTBK2 kinase substrate specificity and the impact of spinocerebellar-ataxia-causing mutations on expression, activity, localization and development. Biochem. J. 437, 157–167. doi: 10.1042/BJ20110276
Brown, G. C. (2007). Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem. Soc. Trans. 35, 1119–1121. doi: 10.1042/BST0351119
Brunden, K. R., Ballatore, C., Crowe, A., Smith, A. B. III, Lee, V. M., and Trojanowski, J. Q. (2010). Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: a focus on tau assembly inhibitors. Exp. Neurol. 223, 304–310. doi: 10.1016/j.expneurol.2009.08.031
Cuny, G. D. (2009). Kinase inhibitors as potential therapeutics for acute and chronic neurodegenerative conditions. Curr. Pharm. Des. 15, 3919–3939. doi: 10.2174/138161209789649330
Dhavan, R., Greer, P. L., Morabito, M. A., Orlando, L. R., and Tsai, L. H. (2002). The cyclin-dependent kinase 5 activators p35 and p39 interact with the alpha-subunit of Ca2+/calmodulin-dependent protein kinase II and alpha-actinin-1 in a calcium-dependent manner. J. Neurosci. 22, 7879–7891.
Edener, U., Kurth, I., Meiner, A., Hoffmann, F., Hubner, C. A., Bernard, V., et al. (2009). Missense exchanges in the TTBK2 gene mutated in SCA11. J. Neurol. 256, 1856–1859. doi: 10.1007/s00415-009-5209-0
El Khoury, J., Toft, M., Hickman, S. E., Means, T. K., Terada, K., Geula, C., et al. (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438. doi: 10.1038/nm1555
Flotow, H., Graves, P. R., Wang, A. Q., Fiol, C. J., Roeske, R. W., and Roach, P. J. (1990). Phosphate groups as substrate determinants for casein kinase I action. J. Biol. Chem. 265, 14264–14269.
Flotow, H., and Roach, P. J. (1989). Synergistic phosphorylation of rabbit muscle glycogen synthase by cyclic AMP-dependent protein kinase and casein kinase I. implications for hormonal regulation of glycogen synthase. J. Biol. Chem. 264, 9126–9128.
Galimberti, D., Fenoglio, C., Lovati, C., Venturelli, E., Guidi, I., Corra, B., et al. (2006). Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol. Aging 27, 1763–1768. doi: 10.1016/j.neurobiolaging.2005.10.007
Gebicke-Haerter, P. J. (2001). Microglia in neurodegeneration: molecular aspects. Microsc. Res. Tech. 54, 47–58. doi: 10.1002/jemt.1120
Gerhard, A., Trender-Gerhard, I., Turkheimer, F., Quinn, N. P., Bhatia, K. P., and Brooks, D. J. (2006). In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov. Disord. 21, 89–93. doi: 10.1002/mds.20668
Goetz, S. C., Liem, K. F. Jr., and Anderson, K. V. (2012). The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell 151, 847–858. doi: 10.1016/j.cell.2012.10.010
Gong, C. X., Lidsky, T., Wegiel, J., Zuck, L., Grundke-Iqbal, I., and Iqbal, K. (2000). Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. implications for neurofibrillary degeneration in Alzheimer’s disease. J. Biol. Chem. 275, 5535–5544. doi: 10.1074/jbc.275.8.5535
Gordon, S., and Martinez, F. O. (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604. doi: 10.1016/j.immuni.2010.05.007
Grammas, P., and Ovase, R. (2001). Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 22, 837–842. doi: 10.1016/S0197-4580(01)00276-7
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Hanger, D. P., Betts, J. C., Loviny, T. L., Blackstock, W. P., and Anderton, B. H. (1998). New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J. Neurochem. 71, 2465–2476. doi: 10.1046/j.1471-4159.1998.71062465.x
Hasegawa, M., Jakes, R., Crowther, R. A., Lee, V. M., Ihara, Y., and Goedert, M. (1996). Characterization of mAb AP422, a novel phosphorylation-dependent monoclonal antibody against tau protein. FEBS Lett. 384, 25–30. doi: 10.1016/0014-5793(96)00271-2
Hawasli, A. H., Benavides, D. R., Nguyen, C., Kansy, J. W., Hayashi, K., Chambon, P., et al. (2007). Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat. Neurosci. 10, 880–886. doi: 10.1038/nn1914
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Hong, M., Zhukareva, V., Vogelsberg-Ragaglia, V., Wszolek, Z., Reed, L., Miller, B. I., et al. (1998). Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282, 1914–1917. doi: 10.1126/science.282.5395.1914
Houlden, H., Johnson, J., Gardner-Thorpe, C., Lashley, T., Hernandez, D., Worth, P., et al. (2007). Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat. Genet. 39, 1434–1436. doi: 10.1038/ng.2007.43
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Ihara, Y., Nukina, N., Miura, R., and Ogawara, M. (1986). Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 99, 1807–1810.
Iqbal, K., Grundke-Iqbal, I., Smith, A. J., George, L., Tung, Y. C., and Zaidi, T. (1989). Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 5646–5650. doi: 10.1073/pnas.86.14.5646
Ishiguro, K., Omori, A., Sato, K., Tomizawa, K., Imahori, K., and Uchida, T. (1991). A serine/threonine proline kinase activity is included in the tau protein kinase fraction forming a paired helical filament epitope. Neurosci. Lett. 128, 195–198. doi: 10.1016/0304-3940(91)90259-V
Ishiguro, K., Omori, A., Takamatsu, M., Sato, K., Arioka, M., Uchida, T., et al. (1992). Phosphorylation sites on tau by tau protein kinase I, a bovine derived kinase generating an epitope of paired helical filaments. Neurosci. Lett. 148, 202–206. doi: 10.1016/0304-3940(92)90839-Y
Ishizawa, K., and Dickson, D. W. (2001). Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J. Neuropathol. Exp. Neurol. 60, 647–657.
Janelsins, M. C., Mastrangelo, M. A., Oddo, S., LaFerla, F. M., Federoff, H. J., and Bowers, W. J. (2005). Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. J. Neuroinflammation 2, 23. doi: 10.1186/1742-2094-2-23
Jinwal, U. K., Akoury, E., Abisambra, J. F., O’Leary, J. C. III, Thompson, A. D., Blair, L. J., et al. (2013). Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 27, 1450–1459. doi: 10.1096/fj.12-220889
Johnson, J., Wood, N., Giunti, P., and Houlden, H. (2008). Clinical and genetic analysis of spinocerebellar ataxia type 11. Cerebellum 7, 159–164. doi: 10.1007/s12311-008-0022-3
Kiefer, S. E., Chang, C. J., Kimura, S. R., Gao, M., Xie, D., Zhang, Y., et al. (2014). The structure of human tau-tubulin kinase 1 both in the apo form and in complex with an inhibitor. Acta Crystallogr. F Struct. Biol. Commun. 70, 173–181. doi: 10.1107/S2053230X14000144
Kitano-Takahashi, M., Morita, H., Kondo, S., Tomizawa, K., Kato, R., Tanio, M., et al. (2007). Expression, purification and crystallization of a human tau-tubulin kinase 2 that phosphorylates tau protein. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63, 602–604. doi: 10.1107/S1744309107028783
Kosik, K. S., Joachim, C. L., and Selkoe, D. J. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 83, 4044–4048. doi: 10.1073/pnas.83.11.4044
Kraemer, B. C., Burgess, J. K., Chen, J. H., Thomas, J. H., and Schellenberg, G. D. (2006). Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 15, 1483–1496. doi: 10.1093/hmg/ddl067
Lewis, J., McGowan, E., Rockwood, J., Melrose, H., Nacharaju, P., Van Slegtenhorst, M., et al. (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405. doi: 10.1038/78078
Li, G., Yin, H., and Kuret, J. (2004). Casein kinase 1 delta phosphorylates tau and disrupts its binding to microtubules. J. Biol. Chem. 279, 15938–15945. doi: 10.1074/jbc.M314116200
Lue, L. F., Rydel, R., Brigham, E. F., Yang, L. B., Hampel, H., Murphy, G. M. Jr., et al. (2001). Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 35, 72–79. doi: 10.1002/glia.1072
Lund, H., Cowburn, R. F., Gustafsson, E., Stromberg, K., Svensson, A., Dahllund, L., et al. (2013). Tau-tubulin kinase 1 expression, phosphorylation and co-localization with phospho-Ser422 tau in the Alzheimer’s disease brain. Brain Pathol. 23, 378–389. doi: 10.1111/bpa.12001
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. doi: 10.1126/science.1075762
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. doi: 10.1016/j.it.2004.09.015
Mantovani, A., Sozzani, S., Locati, M., Allavena, P., and Sica, A. (2002). Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555. doi: 10.1016/S1471-4906(02)02302-5
Morishima-Kawashima, M., Hasegawa, M., Takio, K., Suzuki, M., Yoshida, H., Titani, K., et al. (1995a). Proline-directed and non-proline-directed phosphorylation of PHF-tau. J. Biol. Chem. 270, 823–829. doi: 10.1074/jbc.270.2.823
Morishima-Kawashima, M., Hasegawa, M., Takio, K., Suzuki, M., Yoshida, H., Watanabe, A., et al. (1995b). Hyperphosphorylation of tau in PHF. Neurobiol. Aging 16, 365–371; discussion 371–380. doi: 10.1016/0197-4580(95)00027-C
Nakielny, S., Campbell, D. G., and Cohen, P. (1991). The molecular mechanism by which adrenalin inhibits glycogen synthesis. Eur. J. Biochem. 199, 713–722. doi: 10.1111/j.1432-1033.1991.tb16175.x
Németh, A. H., Kwasniewska, A. C., Lise, S., Parolin Schnekenberg, R., Becker, E. B., Bera, K. D., et al. (2013). Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 136, 3106–3118. doi: 10.1093/brain/awt236
Pola, R., Flex, A., Gaetani, E., Proia, A. S., Papaleo, P., Giorgio, A. D., et al. (2004). Monocyte chemoattractant protein-1 (MCP-1) gene polymorphism and risk of Alzheimer’s disease in Italians. Exp. Gerontol. 39, 1249–1252. doi: 10.1016/j.exger.2004.05.001
Qi, H., Yao, C., Cai, W., Girton, J., Johansen, K. M., and Johansen, J. (2009). Asator, a tau-tubulin kinase homolog in Drosophila localizes to the mitotic spindle. Dev. Dyn. 238, 3248–3256. doi: 10.1002/dvdy.22150
Reynolds, C. H., Nebreda, A. R., Gibb, G. M., Utton, M. A., and Anderton, B. H. (1997). Reactivating kinase/p38 phosphorylates tau protein in vitro. J. Neurochem. 69, 191–198. doi: 10.1046/j.1471-4159.1997.69010191.x
Sato, S., Cerny, R. L., Buescher, J. L., and Ikezu, T. (2006). Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J. Neurochem. 98, 1573–1584. doi: 10.1111/j.1471-4159.2006.04059.x
Sato, S., Tatebayashi, Y., Akagi, T., Chui, D. H., Murayama, M., Miyasaka, T., et al. (2002). Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J. Biol. Chem. 277, 42060–42065. doi: 10.1074/jbc.M202241200
Sato, S., Xu, J., Okuyama, S., Martinez, L. B., Walsh, S. M., Jacobsen, M. T., et al. (2008). Spatial learning impairment, enhanced CDK5/p35 activity, and downregulation of NMDA receptor expression in transgenic mice expressing tau-tubulin kinase 1. J. Neurosci. 28, 14511–14521. doi: 10.1523/JNEUROSCI.3417-08.2008
Schols, L., Bauer, P., Schmidt, T., Schulte, T., and Riess, O. (2004). Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 3, 291–304. doi: 10.1016/S1474-4422(04)00737-9
Semple, B. D., Kossmann, T., and Morganti-Kossmann, M. C. (2010). Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 30, 459–473. doi: 10.1038/jcbfm.2009.240
Serrano-Pozo, A., Mielke, M. L., Gomez-Isla, T., Betensky, R. A., Growdon, J. H., Frosch, M. P., et al. (2011). Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 179, 1373–1384. doi: 10.1016/j.ajpath.2011.05.047
Simpkins, K. L., Guttmann, R. P., Dong, Y., Chen, Z., Sokol, S., Neumar, R. W., et al. (2003). Selective activation induced cleavage of the NR2B subunit by calpain. J. Neurosci. 23, 11322–11331.
Sun, Y. X., Minthon, L., Wallmark, A., Warkentin, S., Blennow, K., and Janciauskiene, S. (2003). Inflammatory markers in matched plasma and cerebrospinal fluid from patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 16, 136–144. doi: 10.1159/000071001
Takahashi, M., Tomizawa, K., Sato, K., Ohtake, A., and Omori, A. (1995). A novel tau-tubulin kinase from bovine brain. FEBS Lett. 372, 59–64. doi: 10.1016/0014-5793(95)00955-9
Takashima, A., Noguchi, K., Sato, K., Hoshino, T., and Imahori, K. (1993). Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 90, 7789–7793. doi: 10.1073/pnas.90.16.7789
Takashima, A., Yamaguchi, H., Noguchi, K., Michel, G., Ishiguro, K., Sato, K., et al. (1995). Amyloid beta peptide induces cytoplasmic accumulation of amyloid protein precursor via tau protein kinase I/glycogen synthase kinase-3 beta in rat hippocampal neurons. Neurosci. Lett. 198, 83–86. doi: 10.1016/0304-3940(95)11964-X
Tanos, B. E., Yang, H. J., Soni, R., Wang, W. J., Macaluso, F. P., Asara, J. M., et al. (2013). Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 27, 163–168. doi: 10.1101/gad.207043.112
Tomizawa, K., Omori, A., Ohtake, A., Sato, K., and Takahashi, M. (2001). Tau-tubulin kinase phosphorylates tau at Ser-208 and Ser-210, sites found in paired helical filament-tau. FEBS Lett. 492, 221–227. doi: 10.1016/S0014-5793(01)02256-6
Vazquez-Higuera, J. L., Martinez-Garcia, A., Sanchez-Juan, P., Rodriguez-Rodriguez, E., Mateo, I., Pozueta, A., et al. (2011). Genetic variations in tau-tubulin kinase-1 are linked to Alzheimer’s disease in a Spanish case-control cohort. Neurobiol. Aging 32, 550.e5–550.e9. doi: 10.1016/j.neurobiolaging.2009.12.021
Vega, I. E., Cui, L., Propst, J. A., Hutton, M. L., Lee, G., and Yen, S. H. (2005). Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Brain Res. Mol. Brain Res. 138, 135–144. doi: 10.1016/j.molbrainres.2005.04.015
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Wolozin, B. L., Pruchnicki, A., Dickson, D. W., and Davies, P. (1986). A neuronal antigen in the brains of Alzheimer patients. Science 232, 648–650. doi: 10.1126/science.3083509
Worth, P. F., Giunti, P., Gardner-Thorpe, C., Dixon, P. H., Davis, M. B., and Wood, N. W. (1999). Autosomal dominant cerebellar ataxia type III: linkage in a large British family to a 7.6-cM region on chromosome 15q14-21.3. Am. J. Hum. Genet. 65, 420–426. doi: 10.1086/302495
Wu, H. Y., Hsu, F. C., Gleichman, A. J., Baconguis, I., Coulter, D. A., and Lynch, D. R. (2007). Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J. Biol. Chem. 282, 20075–20087. doi: 10.1074/jbc.M700624200
Wu, H. Y., Yuen, E. Y., Lu, Y. F., Matsushita, M., Matsui, H., Yan, Z., et al. (2005). Regulation of N-methyl-D-aspartate receptors by calpain in cortical neurons. J. Biol. Chem. 280, 21588–21593. doi: 10.1074/jbc.M501603200
Xia, M. Q., and Hyman, B. T. (1999). Chemokines/chemokine receptors in the central nervous system and Alzheimer’s disease. J. Neurovirol. 5, 32–41. doi: 10.3109/13550289909029743
Xu, J., Tsutsumi, K., Tokuraku, K., Estes, K. A., Hisanaga, S. I., and Ikezu, T. (2010a). Actin interaction and regulation of cyclin-dependent kinase 5/p35 complex activity. J. Neurochem. 116, 192–204. doi: 10.1111/j.1471-4159.2010.06824.x
Xu, J., Sato, S., Okuyama, S., Swan, R. J., Jacobsen, M. T., Strunk, E., et al. (2010b). Tau-tubulin kinase 1 enhances prefibrillar tau aggregation and motor neuron degeneration in P301L FTDP-17 tau-mutant mice. FASEB J. 24, 2904–2915. doi: 10.1096/fj.09-150144
Xue, Y., Wan, P. T., Hillertz, P., Schweikart, F., Zhao, Y., Wissler, L., et al. (2013). X-ray structural analysis of tau-tubulin kinase 1 and its interactions with small molecular inhibitors. ChemMedChem 8, 1846–1854. doi: 10.1002/cmdc.201300274
Yamamoto, M., Horiba, M., Buescher, J. L., Huang, D., Gendelman, H. E., Ransohoff, R. M., et al. (2005). Overexpression of monocyte chemotactic protein-1/CCL2 in beta-amyloid precursor protein transgenic mice show accelerated diffuse beta-amyloid deposition. Am. J. Pathol. 166, 1475–1485. doi: 10.1016/S0002-9440(10)62364-4
Ye, X., Zeng, H., Ning, G., Reiter, J. F., and Liu, A. (2014). C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc. Natl. Acad. Sci. U.S.A. 111, 2164–2169. doi: 10.1073/pnas.1318737111
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Yu, N. N., Yu, J. T., Xiao, J. T., Zhang, H. W., Lu, R. C., Jiang, H., et al. (2011). Tau-tubulin kinase-1 gene variants are associated with Alzheimer’s disease in Han Chinese. Neurosci. Lett. 491, 83–86. doi: 10.1016/j.neulet.2011.01.011
Keywords: tau, kinase, Alzheimer’s disease, tauopathy, neuroinflammation, Cdk5, GSK3, SCA11
Citation: Ikezu S and Ikezu T (2014) Tau-tubulin kinase. Front. Mol. Neurosci. 7:33. doi: 10.3389/fnmol.2014.00033
Received: 09 March 2014; Accepted: 07 April 2014;
Published online: 28 April 2014.
Edited by:
Shin-ichi Hisanaga, Tokyo Metropolitan University, JapanReviewed by:
Daniele Bottai, University of Milan, ItalyNaruhiko Sahara, National Institute of Radiological Sciences, Japan
Copyright © 2014 Ikezu and Ikezu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tsuneya Ikezu, Department of Pharmacology and Experimental Therapeutics, Boston University School of Medicine, 72 East Concord Street, L-606, Boston, MA 02118, USA e-mail: tikezu@bu.edu