Loss of CIB2 Causes Profound Hearing Loss and Abolishes Mechanoelectrical Transduction in Mice
 Yanfei Wang1,2†
Yanfei Wang1,2†  Jie Li3†
Jie Li3†  Xuerui Yao1,2†
Xuerui Yao1,2†  Wei Li1,2 Haibo Du1,2
Wei Li1,2 Haibo Du1,2  Mingliang Tang4,5
Mingliang Tang4,5  Wei Xiong3
Wei Xiong3  Renjie Chai4,5,6*
Renjie Chai4,5,6*  Zhigang Xu1,2*
Zhigang Xu1,2*- 1Shandong Provincial Key Laboratory of Animal Cells and Developmental Biology, School of Life Sciences, Shandong University, Jinan, China
- 2Shandong Provincial Collaborative Innovation Center of Cell Biology, Shandong Normal University, Jinan, China
- 3School of Life Sciences, IDG/McGovern Institute for Brain Research, Tsinghua University, Beijing, China
- 4Key Laboratory for Developmental Genes and Human Disease, Ministry of Education, Institute of Life Sciences, Southeast University, Nanjing, China
- 5Co-Innovation Center of Neuroregeneration, Nantong University, Nantong, China
- 6Jiangsu Province High-Tech Key Laboratory for Bio-Medical Research, Southeast University, Nanjing, China
Calcium and integrin-binding protein 2 (CIB2) belongs to a protein family with four known members, CIB1 through CIB4, which are characterized by multiple calcium-binding EF-hand domains. Among the family members, the Cib1 and Cib2 genes are expressed in mouse cochlear hair cells, and mutations in the human CIB2 gene have been associated with nonsyndromic deafness DFNB48 and syndromic deafness USH1J. To further explore the function of CIB1 and CIB2 in hearing, we established Cib1 and Cib2 knockout mice using the clustered regularly interspaced short palindromic repeat (CRISPR)-associated Cas9 nuclease (CRISPR/Cas9) genome editing technique. We found that loss of CIB1 protein does not affect auditory function, whereas loss of CIB2 protein causes profound hearing loss in mice. Further investigation revealed that hair cell stereocilia development is affected in Cib2 knockout mice. Noticeably, loss of CIB2 abolishes mechanoelectrical transduction (MET) currents in auditory hair cells. In conclusion, we show here that although both CIB1 and CIB2 are readily detected in the cochlea, only loss of CIB2 results in profound hearing loss, and that CIB2 is essential for auditory hair cell MET.
Introduction
Usher syndrome (USH) is the most frequent form of inherited sensory deaf-blindness. It is characterized by hearing loss and vision defects with or without balance problems (Boughman et al., 1983). Based on the clinical symptoms, USH is classified into three types, namely USH1, USH2 and USH3. USH1 is the most severe type, which shows profound congenital deafness combined with retinitis pigmentosa and vestibular areflexia. USH2 is less severe and shows moderate congenital hearing impairment combined with retinitis pigmentosa and some balance problems. USH3, the least severe form, shows progressive hearing loss combined with retinitis pigmentosa and slight vestibular problems.
To date, 10 genes have been associated with USH, including MYO7A (Weil et al., 1995), USH1C (Bitner-Glindzicz et al., 2000; Verpy et al., 2000), CDH23 (Bolz et al., 2001; Bork et al., 2001), PCDH15 (Ahmed et al., 2001; Alagramam et al., 2001), USH1G (Weil et al., 2003), CIB2 (Riazuddin et al., 2012), USH2A (Eudy et al., 1998), ADGRV1 (Weston et al., 2004), WHRN (Ebermann et al., 2007) and CLRN-1 (Joensuu et al., 2001). Furthermore, PDZD7 was suggested to be a USH modifier and a contributor to digenic USH (Ebermann et al., 2010). Mutations in the USH genes have also been shown to cause nonsyndromic hearing loss. In the inner ear sensory hair cells, USH proteins bind to one another and form multiprotein complexes, and they are indispensable for the development, maintenance and function of hair cell stereocilia (Mathur and Yang, 2015). For example, CDH23 and PCDH15 interact with each other and form the so-called tip links, which connect the tips of shorter stereocilia to the sides of their taller neighboring stereocilia and play a pivotal role in hair cell mechanoelectrical transduction (MET; Kazmierczak et al., 2007).
Calcium and integrin-binding protein 2 (CIB2) is a newly discovered member of the USH protein family. Its biological function as well as its role in hearing remain largely unknown. Besides USH, mutations in the CIB2 gene are also associated with nonsyndromic hearing loss DFNB48 (Riazuddin et al., 2012; Patel et al., 2015; Seco et al., 2016). CIB2 belongs to a family with four known members, CIB1 through CIB4, all of which contain multiple calcium-binding EF-hand domains (Gentry et al., 2005). As the prototype of this protein family, CIB1 has been intensively investigated, and it has been shown that CIB1 is involved in various cellular and physiological processes such as cell proliferation, apoptosis, cytoskeleton organization, angiogenesis and spermatogenesis (Leisner et al., 2005; White et al., 2006; Yuan et al., 2006; Naik et al., 2009; Heineke et al., 2010; Jarman et al., 2010; Zayed et al., 2010; Naik and Naik, 2011a,b; Kostyak et al., 2012). Mammalian CIB2 has been detected in skeletal muscle, the brain, the eye and the inner ear. In skeletal muscle, CIB2 colocalizes with the integrin 7B subunit at the sarcolemma as well as the neuromuscular junctions (NMJ) and the myotendinous junctions (MTJ; Häger et al., 2008). In the brain, CIB2 has mainly been detected in the hippocampus and cortex where it localizes together with the Golgi apparatus and dendrite markers (Blazejczyk et al., 2009). In the eye, CIB2 has been detected in the inner and outer segments of photoreceptor cells as well as retinal pigmented epithelium (RPE) cells (Riazuddin et al., 2012). In the inner ear, CIB2 has mainly been detected on hair cell stereocilia, where it binds to two other USH proteins—MYO7A and whirlin (Riazuddin et al., 2012).
The physiological function of CIB2 has been investigated using the zebrafish and fruit fly as models. In the zebrafish embryo, morpholinos against Cib2 result in developmental defects such as microphthalmia, curled tail, hypopigmentation and edematous heart (Riazuddin et al., 2012). Hair cell patches in lateral-line neuromasts of morphants are markedly reduced, and FM1-43 dye uptake and microphonic potential measurements suggested that the MET components are abnormal (Riazuddin et al., 2012). In the fly, knockdown of Cib2 expression through RNA interference (RNAi) causes significantly reduced photoresponse amplitude and impaired responses to visual stimuli (Riazuddin et al., 2012). Recently, during the preparation of our manuscript, two groups independently reported that Cib2 knockout mice are profoundly deaf (Giese et al., 2017; Zou et al., 2017). It was also shown that CIB2 binds TMC1/2 and the MET currents are completely absent in Cib2 knockout hair cells (Giese et al., 2017).
Among the four known Cib genes, the Cib1 and Cib2 genes are abundantly expressed in mouse cochlear hair cells. In the present work, Cib1 and Cib2 knockout mice were developed to examine their role in hearing transduction. Our data showed that Cib2 disruption indeed causes hearing loss and abolishes the MET currents. We also found that the voltage-gated currents in Cib2 knockout outer hair cells (OHCs) are slightly reduced. Furthermore, our results suggested that loss of CIB1 does not affect the auditory function in mice.
Materials and Methods
Generation of Cib1 and Cib2 Knockout Mice
Knockout mice were generated using the clustered regularly interspaced short palindromic repeat (CRISPR)-associated Cas9 nuclease (CRISPR/Cas9) genome editing technique as previously described (Yang et al., 2013). Briefly, C57BL/6 female mice (7–8 weeks old) were superovulated by intraperitoneally injecting pregnant mare serum gonadotropin (PMSG) and human chorionic gonadotrophin (hCG) and then mated to C57BL/6 male mice. The fertilized embryos (zygotes) were collected from the oviducts, and mixed Cas9 mRNA (50 ng/μl) and small guide RNA (sgRNA; 25 ng/μl) were injected into the cytoplasm of zygotes with visible pronuclei in Chatot-Ziomek-Bavister (CZB) medium. The injected zygotes were then cultured in Quinn’s Advantage cleavage medium (in vitro Fertilization, Inc.) for 24 h, at which time 18–20 2-cell–stage embryos were transferred into the oviduct of a pseudopregnant ICR female mouse at 0.5 day post coitus (dpc). The accession numbers of the Cib1 and Cib2 cDNAs used to design sgRNA are NM_011870 and NM_019686, respectively. To determine the nucleotide sequence of mutated alleles, genomic DNA of F0 mice was amplified using the following primers: Cib1 forward, 5′-GGA GGA TGA GGA GTT GGA-3′, Cib1 reverse, 5′-CTG TGT GGG AAG GAG TGG-3′, Cib2 forward, 5′-GGA TTG TGG AGG CTT TCT-3′ and Cib2 reverse, 5′-ATG GCT CAC AAG ATG CTC-3′. DNA sequencing was then performed after TA cloning into plasmid pMD19T. In order to obtain F1 knockout mice, F0 mice were crossed with C57BL/6 mice and newborns were examined by Sanger sequencing.
RNA Extraction and RT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol, and 1 μg of total RNA was used as the template for reverse transcription (RT) using the PrimeScript RT reagent kit (Takara). Polymerase chain reaction (PCR) was then performed with the following primers: Cib1 RT forward, 5′-ATG GGA GGT TCG GGC AGT CG-3′, Cib1 RT reverse, 5′-TCA CAG GAC AAT CTT AAA GG-3′, Cib2 RT forward, 5′-AAG AGA GGA TTG TGG AGG-3′, Cib2 RT reverse, 5′-CAG ACT TGG TGA GTC GGG-3′, Cib3 RT forward, 5′-ATG AGG CTG TTC TAT CGA TA-3′, Cib3 RT reverse, 5′-GTC AGA TGC GGA TGT GGA AGG-3′, Cib4 RT forward, 5′-ATG GGG CAG TGT TTA AGG TA-3′, and Cib4 RT reverse, 5′-TCA GCA GCC CCA GAA GTG AA-3′, β-actin RT forward, 5′-ACG GCC AGG TCA TCA CTA TTG-3′, β-actin RT reverse, 5′-AGG GGC CGG ACT CAT CGT A-3′. The PCR products were separated by electrophoresis on agarose gels. Primers for quantitate-PCR are as follows: Cib1 forward, 5′- AGC CTG AGC TTT GAG GAC TTC-3′, Cib1 reverse, 5′- ACA TGC TGG AAC TCG GAA AGA-3′, Cib2 forward, 5′- TTT CTC CGA GGA TGG CGA-3′, Cib2 reverse, 5′- CAG ACT TGG TGA GTC GGG-3′, Cib3 forward, 5′- GGA TGG TCA CAT GAC CTT AGA G-3′, Cib3 reverse, 5′- GTC CCA TGC ACA GAT GTA GTT-3′, Cib4 forward, 5′- TGG TTT CAT TGA TGA GGA GGA-3′ and Cib4 reverse, 5′- AGA TCT GAC TCA CTC AGG ACA T-3′.
Auditory Brainstem Response (ABR) Measurements
Mice were anesthetized intraperitoneally with 8.4 mg pentobarbital/100 g body weight. Body temperature was maintained at 37°C by placing the mice on an isothermal pad during testing and recovery from anesthesia. Electrodes were inserted subcutaneously at the vertex and pinna as well as near the tail. The stimulus generation, presentation, ABR acquisition and data management were coordinated using a RZ6 workstation and BioSig software (Tucker Davis Technologies, Inc.). Specific acoustic stimuli (clicks or tone bursts) were generated using high-frequency transducers. At each sound level, 512 responses were sampled and averaged. ABR thresholds were obtained for each animal by reducing the stimulus intensity in 10 dB SPL steps to identify the lowest intensity at which all ABR waves were detectable.
Distortion Product Otoacoustic Emission (DPOAE) Measurement
Mice were anesthetized and maintained as mentioned above. Two sine wave tones of different frequencies (F2 = 1.2 × F1) were presented for 1 s durations ranging from 20 dB to 80 dB SPL in 10 dB steps. The emitted acoustic signal was picked up by the microphone and digitized, and the magnitude of the distortion product (2 × F1 − F2) was determined. The surrounding noise floor was calculated by averaging adjacent frequency bins around the distortion product frequency. Distortion product otoacoustic emission (DPOAE) thresholds were calculated when the signal was at least 5 dB SPL above the noise floor.
Whole-Mount Immunostaining
All steps were performed at room temperature unless otherwise indicated. Mouse basilar membrane was dissected and fixed with cold 4% paraformaldehyde (PFA) in PBS for 30 min, then permeabilized and blocked with PBT1 (0.1% Triton X-100, 1% BSA and 5% heat-inactivated goat serum in PBS, pH 7.3) for 30 min. Samples were incubated overnight at 4°C with mouse anti-CIB2 polyclonal antibody (Abnova, Cat. No. H00010518-A01, 1:100 diluted) in PBT1 followed by incubation with Alexa Fluor® 488 donkey anti-mouse secondary antibody (Invitrogen, Cat. No. A21202, 1:300 diluted) in PBT2 (0.1% Triton X-100 and 0.1% BSA in PBS) for 1 h. After that, samples were incubated with TRITC-conjugated phalloidin (Sigma-Aldrich, Cat. No. P1951) in PBS for 30 min, then mounted in PBS/glycerol (1:1) and imaged with a confocal microscope (LSM 700, Zeiss, Germany) and a 40-fold objective. Immunostaining was performed at least three times for each genotype and age of mice tested.
Scanning Electron Microscopy (SEM)
Mouse inner ears were dissected and fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer overnight at 4°C. Cochleae were dissected out of the temporal bone, post-fixed with 1% osmium tetroxide in 0.1 M phosphate buffer at 4°C for 2 h, and dehydrated in ethanol and critically point dried using a Leica EM CPD300 (Leica, Germany). Samples were then mounted and sputter coated with platinum (15 nm) using a Cressington 108 sputter coater (Cressington, United Kingdom) and imaged using a Quanta250 field-emission scanning electron microscope (FEI, Netherlands).
Electrophysiology
The apical-middle part of cochleae was acutely isolated and immobilized under a cross of dental floss. Whole cell recordings were carried out with an electrophysiology amplifier (HEKA, EPC-10 USB) running the PatchMaster software. Borosilicate glass filaments (Sutter) were made with a pipette puller (Sutter, P-2000) and polished with an MF-830 microforge (Narishige) to resistances of 3–5 MOhm. The recording solution contained 144 mM NaCl, 0.7 mM NaH2PO4, 5.8 mM KCl, 1.3 mM CaCl2, 0.9 mM MgCl2, 5.6 mM glucose and 10 mM H-HEPES (pH 7.4). The pipette solution contained 140 mM KCl, 1 mM MgCl2, 0.1 mM EGTA, 2 mM MgATP, 0.3 mM Na2GTP and 10 mM H-HEPES, pH7.2. The MET currents were evoked using a fluid jet from a pipette (tip diameter of 5–10 μm). The pipette tip delivering the fluid jet was positioned by facing the staircase side of the hair bundle at a distance of around 5 μm to elicit saturating MET currents. The 40 Hz sinusoidal force stimuli were generated by a 27-mm-diameter piezoelectric disc controlled by the HEKA amplifier and driven by a homemade piezo amplifier.
Endocochlear Potential (EP) Recording
For stable endocochlear potential (EP) recording, 1-month-old mice were used. The mice were anesthetized by injection intraperitoneally with 4% pentobarbital sodium in saline. The left cochlea was exposed at its ventrolateral side, then the bulla near the round window was carefully removed to expose the round window. The sharp glass electrodes were pulled by pipette puller (Sutter, P-2000) with a tip-diameter of 0.1–0.2 μm. The electrode filled with 150 mM KCl was insert into the scala media through the round window controlled by a micromanipulator (Luigs&Neumann, mini 25). An Ag-AgCl reference electrode was placed into the neck muscle. The baseline of potential was offset to zero when the pipette tip reached to the round window before penetration. The data were collected by a patch-clamp amplifier (Axon, 700B) and an I/O interface (ITC18) that were powered with an electrophysiology software (WinWCP, University of Strathclyde Glasgow).
Statistical Analysis
Data are shown as means ± standard deviations. Student’s t-test was used for statistical analysis, and p < 0.05 was considered statistically significant.
Results
Generation of Cib1 and Cib2 Knockout Mice
CIB2 belongs to a family with four known members, CIB1 through CIB4, which show significant similarity to each other (Figure 1A). In this protein family, CIB2 is more closely related to CIB3 (Figure 1B). RT-PCR results showed that Cib1 and Cib2 are ubiquitously expressed in various mouse tissues, whereas Cib3 and Cib4 have more restricted expression patterns. All Cib genes except Cib4 were detected in the inner ear (Figure 1C). Further examination revealed that in the inner ear, Cib1 and Cib2 are expressed in the vestibule, basilar membrane and spiral ganglion, whereas Cib3 is mainly detected in the vestibule (Figure 1D). The similar expression pattern of Cib1 and Cib2 in the inner ear suggests that both genes might play important roles in hearing, hence we focused on these two genes in the following work.
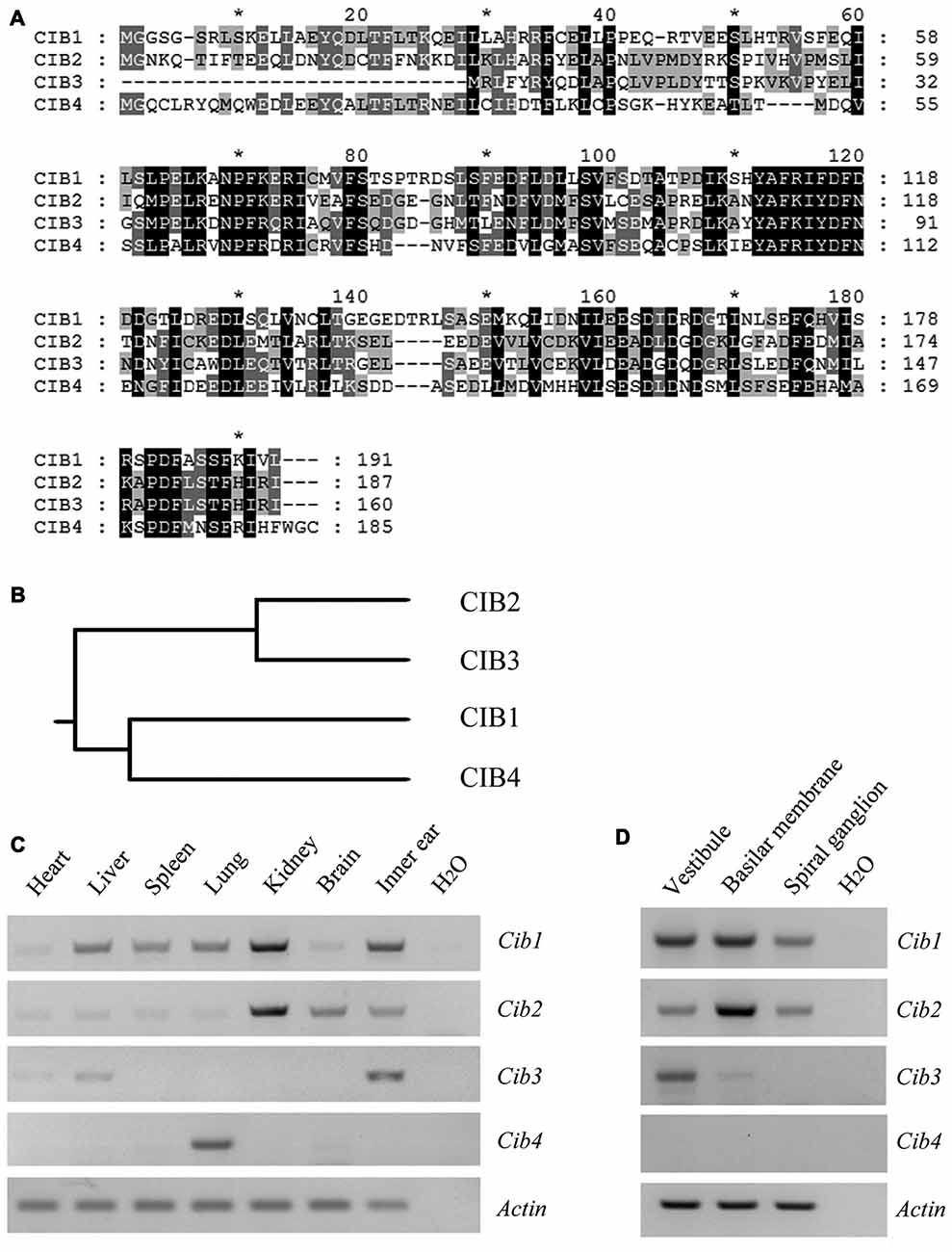
Figure 1. Expression of Cib1, Cib2, Cib3 and Cib4 in different mouse tissues. (A) Amino acid sequences of Mus musculus calcium and integrin-binding protein 1 (CIB1), CIB2, CIB3 and CIB4 were aligned using the CLUSTALW multiple sequence alignment program. (B) The phylogenic tree of Mus musculus CIB1, CIB2, CIB3 and CIB4 was constructed using the rooted phylogenetic tree with branch length (UPGMA) method. (C) Expression of Cib1, Cib2, Cib3 and Cib4 in different tissues of 6-month-old mice was determined by RT-PCR. (D) Expression of Cib1, Cib2, Cib3 and Cib4 in the vestibule, basilar membrane and spiral ganglion cells of P2 mice was determined by RT-PCR. β-actin was included as the internal control.
In order to investigate the functions of CIB1 and CIB2 in hearing, we developed knockout mice using the CRISPR/Cas9 genome editing technique. The mouse Cib1 gene contains seven exons encoding 191 amino acids, and two sgRNAs were designed to target exon 4 (Figure 2A). DNA sequencing revealed that a deletion of 131 bp was introduced in exon 4 in Cib1 knockout mice, which causes a premature translational stop and gives rise to a potentially truncated CIB1 protein of 71 amino acids (Figure 2B). The mouse Cib2 gene contains six exons encoding 187 amino acids, and two sgRNAs were designed to target exon 4 (Figure 2D). DNA sequencing revealed that two deletions (9 bp and 8 bp, respectively) were introduced into exon 4 in Cib2 knockout mice, and these cause a premature translational stop that gives rise to a potentially truncated protein of 109 amino acids (Figure 2E). Three Cib2 transcript variants have been reported that result from alternative splicing (Riazuddin et al., 2012). Given that exon 4 is a common exon for all three transcript variants, the deletions will affect all of the three known CIB2 isoforms. RT-PCR was then performed to confirm the disruption of Cib1 and Cib2 mRNA in knockout mice. A single PCR fragment was amplified in the wild type and heterozygous mice, whereas this fragment was not detected in the homozygous knockout mice (Figures 2C,F). A smaller fragment corresponding to the deletion was detected in Cib2 homozygous knockout mice at very low level (Figure 2F), whereas no such fragment was detected in Cib1 homozygous knockout mice (Figure 2C), possibly caused by nonsense-mediated mRNA decay (NMD). We then utilized a specific anti-CIB2 antibody to examine the expression of CIB2 protein in Cib2 knockout mice. This antibody recognizes the first 99 amino acids of CIB2, hence could detect both the wildtype as well as the potentially truncated CIB2 (Figure 2E). Although the antibody does not work for western blot, immunostaining result suggests that no truncated CIB2 protein is expressed in the Cib2 knockout mice (see below). Taken together, our results indicate that Cib1 and Cib2 expression is successfully disrupted in the Cib1 and Cib2 knockout mice, respectively.
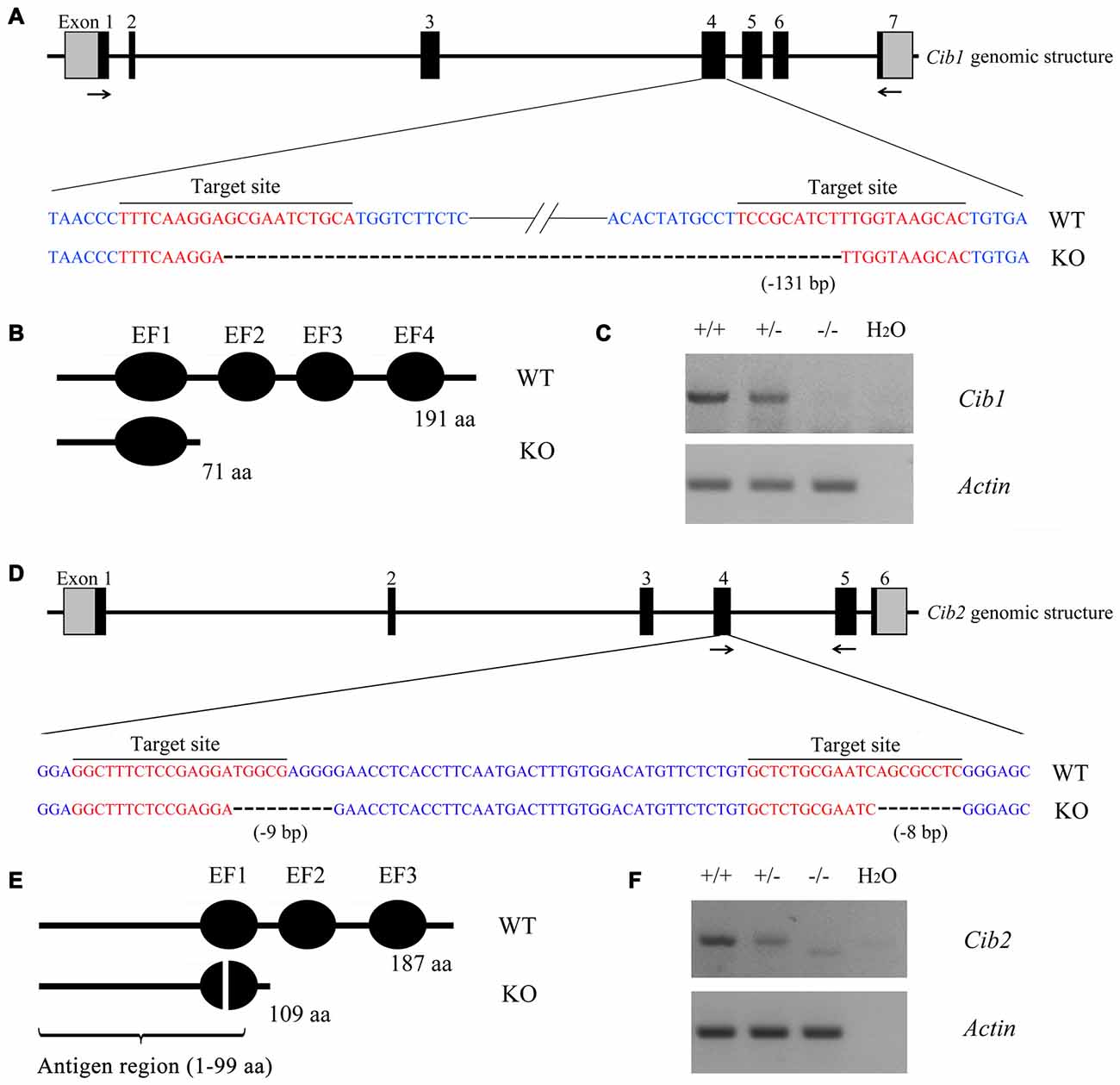
Figure 2. Construction of Cib1 and Cib2 knockout mice. (A) The schematic drawing of the strategy for Cib1 gene disruption. The target sites of clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 small guide RNAs (sgRNAs) in the Cib1 gene are indicated in red, and the deleted region in the Cib1 gene of knockout mice is indicated by dashes. The positions of RT-PCR primers are indicated by arrows. (B) The schematic drawing of the domain structure of CIB1 in wildtype and knockout mice. (C) The expression of Cib1 mRNA in the inner ear of P60 mice was determined by RT-PCR. β-actin was included as the internal control. (D) The schematic drawing of the strategy for Cib2 gene disruption. The target sites of CRISPR-Cas9 sgRNAs in the Cib2 gene are indicated in red, and the deleted regions in the Cib2 gene of knockout mice are indicated by dashes. The positions of RT-PCR primers are indicated by arrows. (E) The schematic drawing of the domain structure of CIB2 in wildtype and knockout mice. (F) The expression of Cib2 mRNA in the inner ear of P60 mice was determined by RT-PCR. β-actin was included as the internal control.
Loss of CIB2 but Not CIB1 Causes Profound Hearing Loss in Mice
We evaluated the auditory function of Cib1 and Cib2 knockout mice by performing ABR measurements. The homozygous Cib1 knockout mice had normal hearing thresholds in response to click stimuli at all ages examined up to 6 months (Figure 3A). Similar results were obtained when pure tone stimuli of different frequencies were used, suggesting that CIB1 is not necessary for auditory function in mice (Figure 3B). We then examined the expression of Cib members in the inner ear of Cib1 knockout mice by performing quantitate PCR. The result showed that Cib3 is upregulated in the inner ear of Cib1 knockout mice, suggesting that CIB3 might compensate for the loss of CIB1 (Supplementary Figure S1).
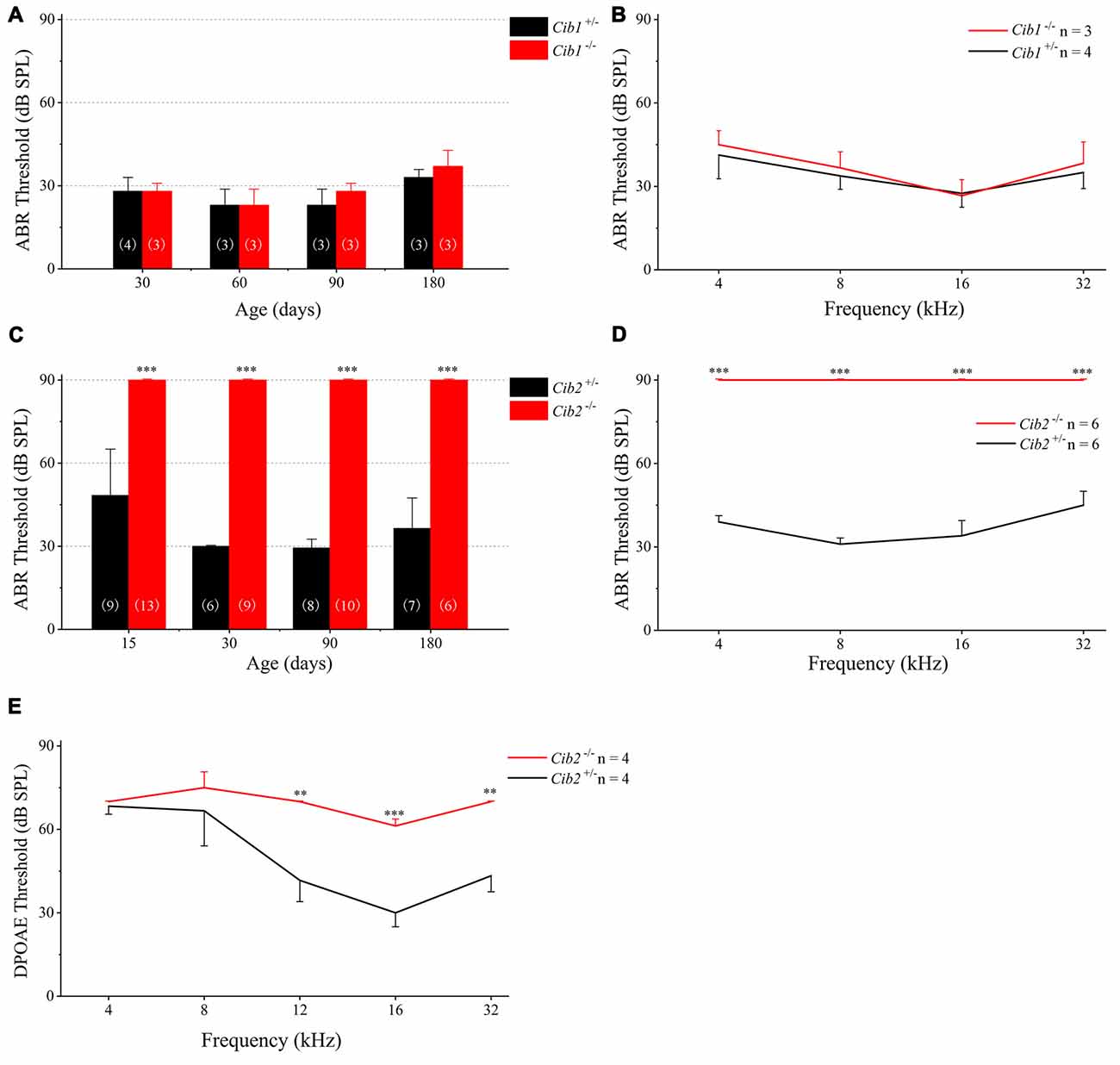
Figure 3. Hearing threshold is elevated in Cib2 knockout mice, but not in Cib1 knockout mice. (A) The auditory brainstem response (ABR) thresholds for click stimuli in Cib1+/− and Cib1−/− mice at different ages. (B) The ABR thresholds for pure tone stimuli in 1-month-old Cib1+/− and Cib1−/− mice. (C) The ABR thresholds for click stimuli in Cib2+/− and Cib2−/− mice at different ages. (D) The ABR thresholds for pure tone stimuli in 1-month-old Cib2+/− and Cib2−/− mice. (E) The distortion product otoacoustic emission (DPOAE) thresholds for pure tone stimuli in 1-month-old Cib2+/− and Cib2−/− mice. The numbers of animals for each group used in the experiments are indicated. The differences were evaluated by Student’s t-test (**p < 0.01; ***p < 0.001).
In contrast to Cib1, homozygous Cib2 knockout mice showed significant hearing threshold elevation to click stimuli when examined as early as postnatal day 14 (P14; Figure 3C). In fact, the loudest sound stimuli (90 dB) used in our experiment could not evoke any response in homozygous Cib2 knockout mice. Significant threshold elevation was also observed when pure-tone sound stimuli of different frequencies were used (Figure 3D). DPOAE measurements were then performed to examine the function of OHCs in Cib2 knockout mice. The DPOAE threshold of homozygous Cib2 knockout mice was significantly elevated compared to heterozygous mice, suggesting that there are OHC function deficits in Cib2 knockout mice (Figure 3E). Taken together, our data suggest that hearing function is severely affected by loss of CIB2 but not by loss of CIB1. Hence we focused on CIB2 in the following work.
Loss of CIB2 Affects Stereocilia Development in Mice
We examined stereocilia morphology by performing whole-mount staining. The OHC stereociliary bundles of homozygous Cib2 knockout mice are disorganized at P7, the earliest age examined, whereas inner hair cell (IHC) stereociliary bundles are largely unaffected. In some Cib2−/− OHCs, stereociliary bundle fragmentation was observed (Figures 4A,B). At P30, the disorganization of Cib2−/− OHC stereociliary bundles is further exacerbated (Figures 4C,D). Consistent with previous report (Riazuddin et al., 2012), CIB2 immunoreactivity is detected in the stereociliary bundles of heterozygous mice but is undetectable in Cib2 homozygous knockout mice (Figures 4A–D).
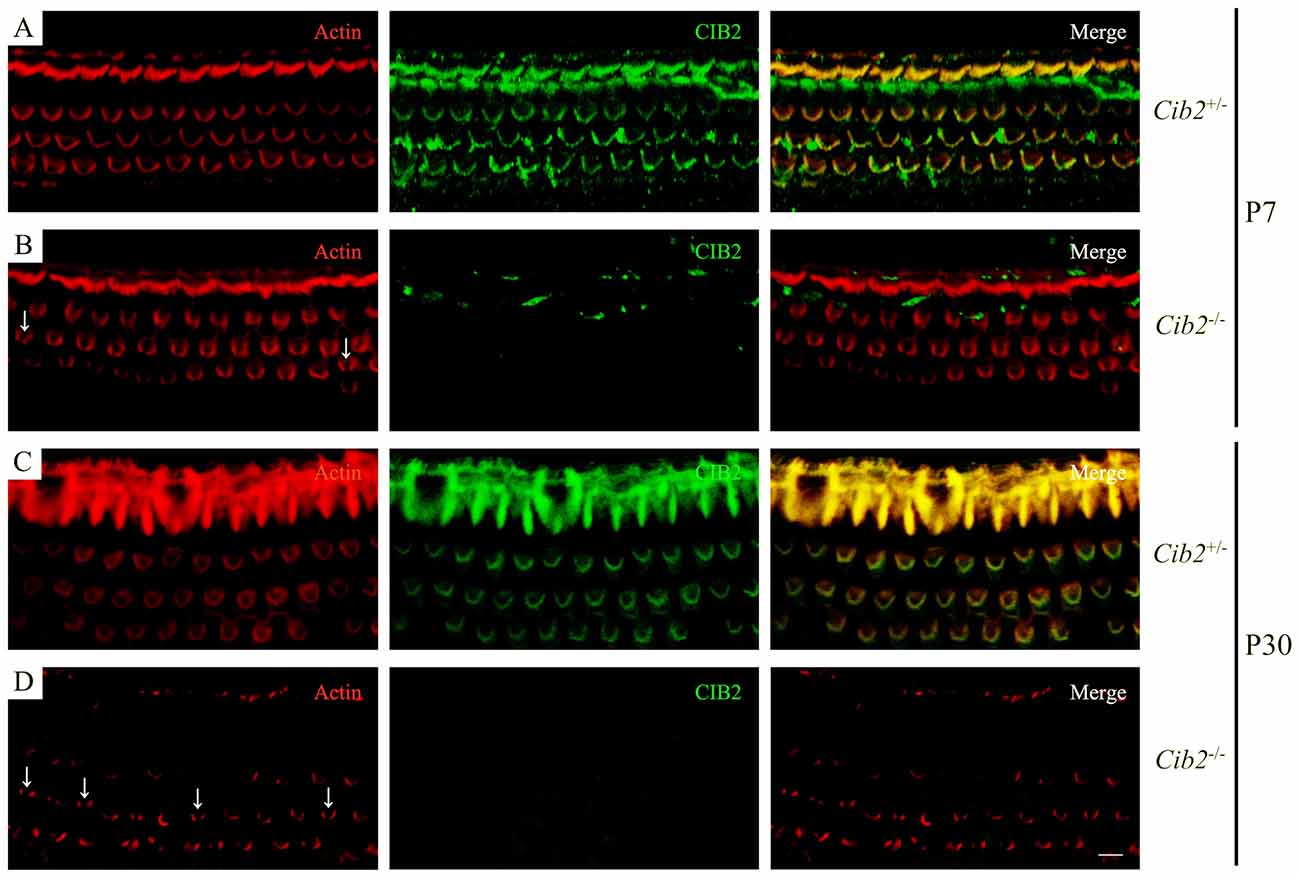
Figure 4. Whole-mount immunostaining in Cib2 knockout mice. Shown are single confocal sections. CIB2 immunoreactivity was visualized with FITC-conjugated secondary antibody, and F-actin was visualized with rhodamine-conjugated phalloidin. (A) P7 Cib2+/− mice. (B) P7 Cib2−/− mice. (C) P30 Cib2+/− mice. (D) P30 Cib2−/− mice. The images were taken at the middle turn of the cochlea. Stereocilia bundle fragmentation was indicated by arrows. Scale bars: 5 μm.
The effect of Cib2 disruption on stereocilia development was further examined by scanning electron microscopy (SEM). At P0, the stereocilia morphology of Cib2−/− mice is indistinguishable from that of Cib2+/− mice (Figures 5A–B″). However, at P7 Cib2−/− OHC stereocilia are largely disorganized, and fragmentation was observed in some Cib2−/− OHC stereocilia bundles (Figures 5C–D″). At P30, the disorganization of Cib2−/− OHC stereocilia is increased, and some Cib2−/− OHC completely lose their stereocilia (Figures 5E–F″). Additionally, the IHCs of Cib2−/− mice at this age also show morphological abnormalities, with stereocilia fusion and occasional stereocilia loss (Figures 5E–F″).
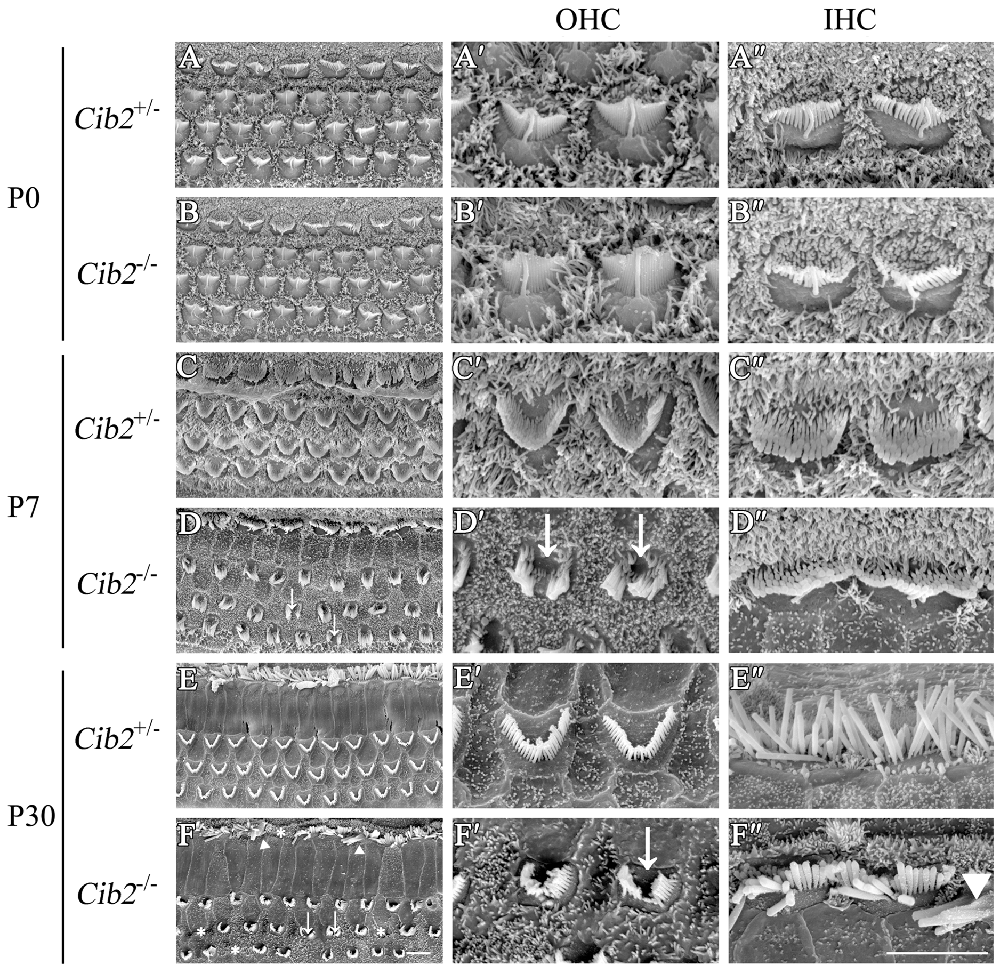
Figure 5. Stereocilia are disorganized in Cib2 knockout mice. (A–F) Low-magnification scanning electron microscopy (SEM) images of cochlear hair bundles from mice of different genotypes and ages as indicated. (A′–F′) High-magnification SEM images of outer hair cell (OHC) hair bundles from mice of different genotypes and ages. (A″–F″) High-magnification SEM images of inner hair cell (IHC) hair bundles from mice of different genotypes and ages. The images were taken at the apical-middle turn of the cochlea. Stereociliary bundle fragmentation is indicated by arrows, stereocilia loss is indicated by asterisks, and stereocilia fusion is indicated by triangles. Scale bars, 5 μm.
Further SEM examination at higher magnification showed that at P8 the second row stereocilia of Cib2−/– OHCs are over-grown to the height close to the first row, whereas the third row are largely retracted, resulting in the loss of staircase architecture (Figures 6A,B). In contrast, the staircase architecture of Cib2−/− IHC stereocilia maintains despite that the second and third row stereocilia are over-grown, which is more obvious at P20 (Figures 6C–F). Furthermore, the kinocilium is not retracted properly in Cib2−/− IHC (Figure 6F). Taken together, our data suggest that loss of CIB2 affects stereocilia development in mice.
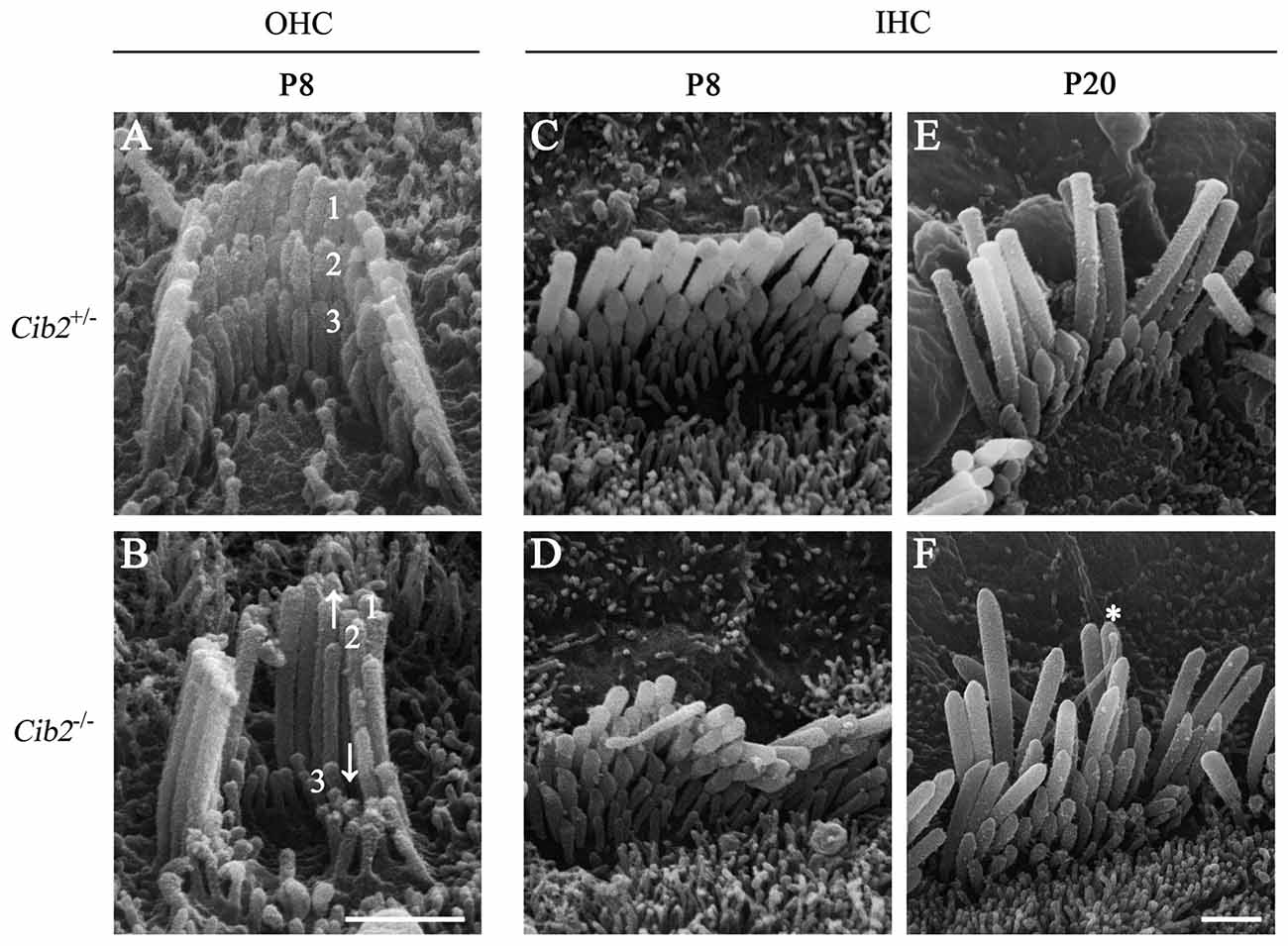
Figure 6. Hair bundle development is affected in Cib2 knockout mice. (A–F) High-magnification SEM images of cochlear hair bundles from mice of different genotypes and ages as indicated. The images were taken at the apical-middle turn of the cochlea. Three rows of stereocilia are indicated by numbers. Over-grown and retracted stereocilia are indicated by arrows up and down, respectively. The kinocilium that does not regress properly is indicated by asterisk. Scale bars, 1 μm.
Loss of CIB2 Abolishes MET Currents
As CIB2 is largely detected in the hair bundles and contributes to stereocilia development, we asked whether CIB2 plays a role in MET in hair cells. By applying fluid jet stimulation that generally deflects hair bundles in a physiological manner, we recorded the saturating MET currents of OHCs. Strikingly, the MET current is completely abolished in P7 Cib2 knockout OHCs. In contrast, there is a large MET current around 800 pA recorded in control OHCs (Figures 7A,B). Lack of MET current in Cib2 knockout OHCs is not caused by stereocilia disorganization, since we always chose OHCs with relatively normal hair bundle morphology for recordings. Furthermore, we measured MET currents in P1 and P4 Cib2 knockout OHCs whose stereocilia are less affected by Cib2 disruption, and obtained similar results (Supplementary Figures S2A,B). Cib2 knockout IHCs have more intact stereocilia compared to OHCs, and we found that MET currents are also absent in P7 Cib2 knockout IHCs (data not shown). Taken together, our data suggest that CIB2 disruption abolishes MET currents in auditory hair cells. In contrast, reverse-polarity currents could be recorded in P4 Cib2 knockout OHCs (Supplementary Figure S3), suggesting that CIB2 is not necessary for Piezo2-dependent reverse-polarity currents (Wu et al., 2017).
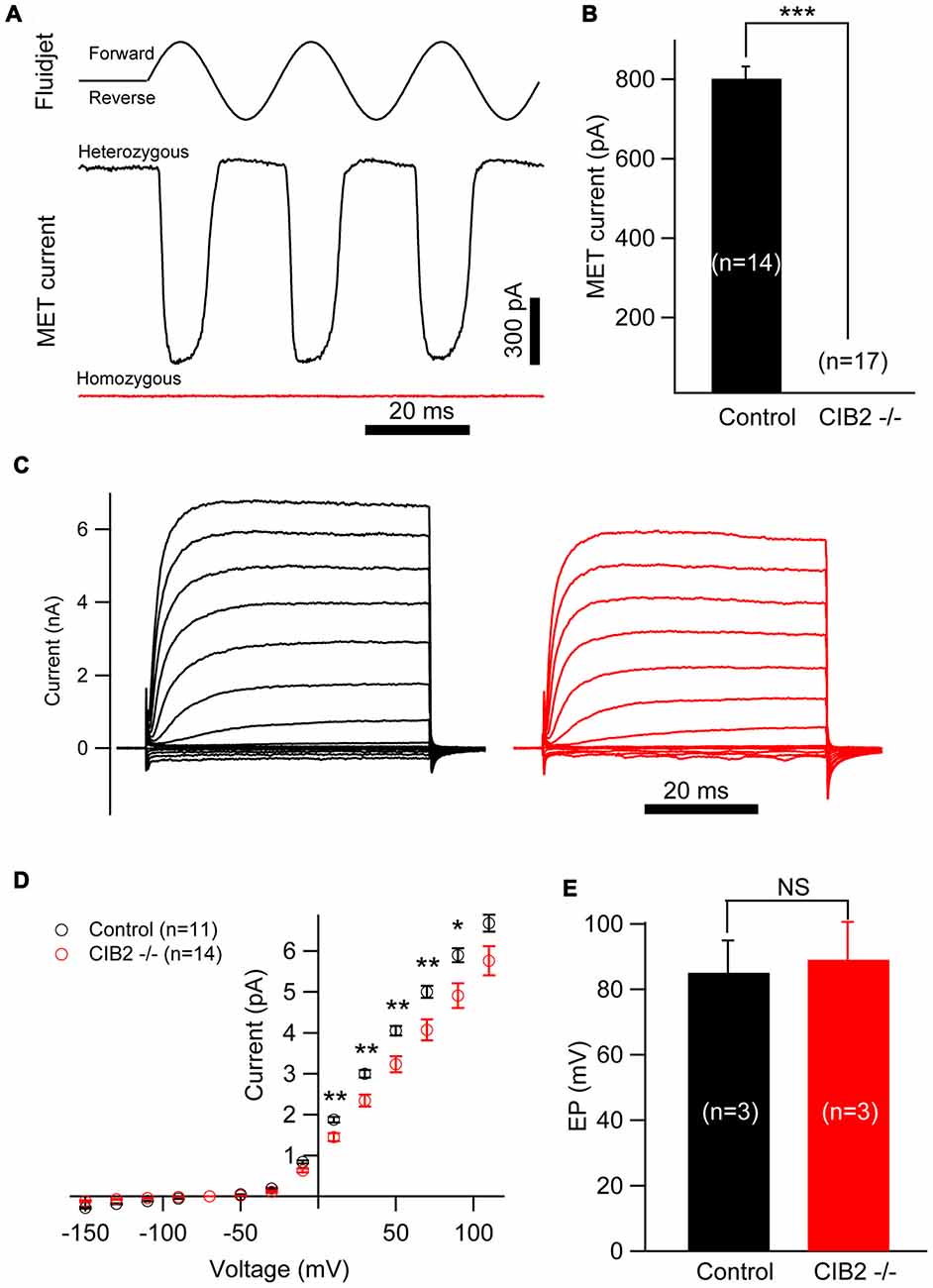
Figure 7. Mechanosensitivity of hair cells is lost in Cib2 knockout mice. (A) Mechanoelectrical transduction (MET) current was examined in OHCs from control and Cib2 knockout mice. A fluid jet system that drives a sinusoidal deflection of hair bundles was used to evaluate the saturating MET current from hair cells. (B) The average peak current was 829.2 pA in control OHCs but absent in knockout OHCs. (C) Voltage-gated current was recorded from control and Cib2 knockout OHCs. The membrane potential was changed from −150 mV to +110 mV in 20 mV steps. (D) Current–voltage (I-V) curves were drawn from data similar to (C), indicating reduced membrane potassium currents in Cib2 knockout OHCs. (E) The average endocochlear potential (EP) was around 85 mV in control and 89 mV in Cib2 knockout mice. In all panels, data were collected from three control heterozygous mice (shown in black) and three Cib2 knockout mice (shown in red). The age of mice is P7 (A–D) or P30 (E). The numbers of cells used are shown in each panel. The differences were evaluated by Student’s t-test (*p < 0.05; **p < 0.01; ***p < 0.001).
Interestingly, we found that the voltage-gated currents in P7 Cib2 knockout OHCs are slightly reduced when compared to the OHCs collected from the littermate controls in the same cage (Figures 7C,D). The reduction of voltage-gated currents was not observed in P1 and P4 Cib2 knockout OHCs (Supplementary Figure S4). Accordingly, we also examined the EP of Cib2 knockout mice, which indicated that Cib2 knockout mice maintain a normal potential in the scala media (Figure 7E).
Discussion
CIB2 is an important deafness gene whose mutations are associated with nonsyndromic deafness DFNB48 and syndromic deafness USH1J (Riazuddin et al., 2012; Patel et al., 2015; Seco et al., 2016), but its biological function and the mechanism through which CIB2 mutations cause hearing loss remain elusive. Here we show that loss of CIB2 in mice affects MET as well as stereocilia development and results in profound hearing loss.
CIB2 belongs to a protein family with four known members, CIB1 through CIB4, that are characterized by multiple calcium-binding EF-hand domains (Gentry et al., 2005). RT-PCR experiments revealed that Cib1 and Cib2 are expressed in the vestibule, basilar membrane and spiral ganglion, whereas Cib3 is mainly expressed in the vestibule. The fourth member Cib4 was not detected in the inner ear. In all the mouse tissues examined, Cib4 expression was only detected in the lung. The absence of Cib4 expression in the mouse inner ear contradicts the recent report that Cib4 is expressed in mouse cochlea and vestibule (Giese et al., 2017). We used two different pairs of primers to examine Cib4 expression, both of which could not detect the expression of Cib4 in mouse inner ear. Consistent with our finding, the RNA transcriptome sequencing result revealed extremely low level of Cib4 expression in mouse inner ear (SHIELD1; Shen et al., 2015). It is worth noticing that in the present work we examined the expression of CIBs only at the mRNA level, which might not faithfully represent the protein expression level. Nevertheless, our data suggest that Cib1, Cib2 and Cib3 are expressed abundantly in mouse inner ear, whereas Cib4 is expressed at very low level, if at all, in the inner ear.
The expression of Cib1 and Cib2 in the cochlea is consistent with the RNA transcriptome sequencing result, which also suggests that the expression level of Cib2 in cochlear hair cells is about 10-fold greater than that of Cib1 (SHIELD1; Shen et al., 2015). The relatively low expression level of Cib1 in the cochlear hair cells might explain the fact that disruption of the Cib1 gene does not affect auditory function in mice, since loss of CIB1 might be compensated for by other CIB members. Indeed, quantitate PCR result showed that Cib3 expression is upregulated in the inner ear of Cib1 knockout mice, suggesting that CIB3 might compensate for loss of CIB1. Similar scenario has been reported in platelets of Cib1 knockout mice with Cib1 exon 4 and 5 deleted through homologous recombination (Yuan et al., 2006; Denofrio et al., 2008).
In contrast to Cib1, Cib2 disruption results in profound hearing loss in mice, suggesting an indispensable role of CIB2 in hearing. Similar phenotype was recently reported by two other groups using different Cib2 mutant mice (Giese et al., 2017; Zou et al., 2017). Cib2tm1a mice contain a gene trap cassette between Cib2 exon 3 and 4 that consists of lacZ and neomycin resistance genes (Giese et al., 2017; Zou et al., 2017). Crossing Cib2tm1a mice with Cre-expressing mice results in the deletion of the neomycin cassette and exon 4, giving rise to Cib2tm1b mice (Giese et al., 2017). A deafness-associated missense mutation, p.F91S, was introduced into mice to give rise to Cib2F91S knockin mice (Giese et al., 2017). Similar to our Cib2 knockout mice, all these Cib2 mutant mice show profound hearing loss, strongly suggesting that CIB2 plays an important role in hearing.
CIB2 contains three EF-hand domains, and the last two EF-hands could bind calcium (Blazejczyk et al., 2009). It has been shown that calcium plays important roles in hair cell MET (Mammano et al., 2007; Ceriani and Mammano, 2012). Calcium enters hair cells through MET channels and is necessary for the adaptation of the channels. CIB2 has been shown to inhibit ATP-induced calcium release in transiently transfected COS-7 cells, which might result from its calcium-buffering ability (Riazuddin et al., 2012). Moreover, CIB2 might regulate calcium release by interacting with IP3 receptors, just as its paralog CIB1 does (White et al., 2006; Hennigs et al., 2008). Loss of CIB2 might therefore result in dysregulation of intracellular calcium in hair cells, which in turn would affect MET. Interestingly, our data showed that MET currents are completely abolished in mice lacking functional CIB2, which was also observed in mice lacking potential MET components such as TMC1/TMC2 and TMIE (Kawashima et al., 2011; Kim et al., 2013; Pan et al., 2013; Zhao et al., 2014). This suggests that CIB2 might play important roles in MET in addition to intracellular calcium regulation. In line with this, CIB2 immunoreactivity localizes near the tips of shorter stereocilia (Riazuddin et al., 2012; Giese et al., 2017), a place where MET channels reside (Beurg et al., 2009). Giese et al. (2017) reported that CIB2 could interact with TMC1 and TMC2, and MET currents are absent in Cib2tm1a/Cib2tm1b and Cib2F91S mice, raising the possibility that CIB2 might be directly involved in MET activity. The association between CIB2 and MET warrants further investigation.
Interestingly, the voltage-gated current is slightly reduced in the OHCs of Cib2 knockout mice, whereas the EP was unaffected. Several voltage-regulated ion channels have been shown to play important roles in hair cells, including the voltage-gated K+ channel KCNQ4 and the Ca2+-activated K+ channel (BK channel; Ruttiger et al., 2004; Kharkovets et al., 2006). Mutations or disruptions in genes coding for KCNQ4 or BK channel cause hearing loss in both humans and mice (Coucke et al., 1999; Kubisch et al., 1999; Ruttiger et al., 2004; Kharkovets et al., 2006). Given the fact that CIB2 has Ca2+ buffering ability, loss of CIB2 might increase intracellular calcium concentration. On the other hand, CIB2 disruption abolishes MET currents, hence decreases calcium influx into hair cells. Although the net effect is unclear, loss of CIB2 might change intracellular calcium concentration, thus affect voltage-gated current through Ca2+- or voltage-gated channels. Furthermore, our data show that the voltage-gated current is reduced in P7 Cib2 knockout OHCs, but not in P1 or P4 Cib2 knockout OHCs, suggesting that CIB2 modulation on voltage-gated current is developmental stage-dependent. The significance of the role of CIB2 in this process remains elusive.
We also found that stereocilia development is affected in Cib2 knockout mice. Stereocilia bundle fragmentation is seen in many Cib2 knockout OHCs, and stereocilia fusion could be observed in some Cib2 knockout IHCs. Strikingly, over-growth of shorter row stereocilia is observed in Cib2 knockout OHCs, and similar scenario happens in Cib2 knockout IHCs to a lesser extent. This results in the loss of staircase architecture in Cib2 knockout OHC stereocilia bundle, but not IHC stereocilia bundle. Intracellular calcium has been shown to affect actin cytoskeleton dynamics in many cell types such as neutrophils and neurons (Downey et al., 1990; Welnhofer et al., 1999; Hutchins et al., 2013), and it was recently suggested that calcium influx through the MET channels controls the remodeling of shorter stereocilia (Vélez-Ortega et al., 2017). Hence, the potential changes of intracellular Ca2+ caused by loss of CIB2 is possibly responsible for the defects in stereocilia development seen in the knockout mice. Alternatively, CIB2 might regulate stereocilia development through interaction with other stereociliary proteins, such as whirlin and MYO7A (Riazuddin et al., 2012). Mutations in the genes encoding whirlin and MYO7A have been shown to affect stereocilia development, although the stereocilia deficits in these mutant mice are different from those of Cib2 knockout mice (Self et al., 1998; Holme et al., 2002). At present it remains unclear why loss of CIB2 affects OHC and IHC stereocilia differently, and why it affects the second row and third row OHC stereocilia differently, too. Further investigation is needed to clarify the mechanism by which CIB2 regulates stereocilia development.
Ethics Statement
All animal experiments were approved by the Ethics Committee of Shandong University (Permit Number: LL-201502028) and conducted accordingly.
Author Contributions
RC and ZX conceived the project and designed the experiments. YW, JL, XY, WL and HD performed the experiments. YW, JL, XY, MT and WX analyzed the data. WX, RC and ZX wrote the manuscript.
Funding
This work was supported by grants from the National Key R&D Program of China (2013CB967700, 2017YFA0103900, 2017YFA0103903), the National Natural Science Foundation of China (31371355, 31401007, 31571080, 31522025, 81622013, 81470692, 31500852), the Jiangsu Province Natural Science Foundation (BK20150022, BK20140620, BK20150598, BK20160125), the Yingdong Huo Education Foundation and the Fundamental Research Funds for the Central Universities. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2017.00401/full#supplementary-material
References
Ahmed, Z. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z., Khan, S., Griffith, A. J., et al. (2001). Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am. J. Hum. Genet. 69, 25–34. doi: 10.1086/321277
Alagramam, K. N., Yuan, H. J., Kuehn, M. H., Murcia, C. L., Wayne, S., Srisailpathy, C. R. S., et al. (2001). Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum. Mol. Genet. 10, 1709–1718. doi: 10.1093/hmg/10.16.1709
Beurg, M., Fettiplace, R., Nam, J. H., and Ricci, A. J. (2009). Localization of inner hair cell mechanotransducer channels using high-speed calcium imaging. Nat. Neurosci. 12, 553–558. doi: 10.1038/nn.2295
Bitner-Glindzicz, M., Lindley, K. J., Rutland, P., Blaydon, D., Smith, V. V., Milla, P. J., et al. (2000). A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat. Genet. 26, 56–60. doi: 10.1038/79178
Blazejczyk, M., Sobczak, A., Debowska, K., Wisniewska, M. B., Kirilenko, A., Pikula, S., et al. (2009). Biochemical characterization and expression analysis of a novel EF-hand Ca2+ binding protein calmyrin2 (Cib2) in brain indicates its function in NMDA receptor mediated Ca2+ signaling. Arch. Biochem. Biophys. 487, 66–78. doi: 10.1016/j.abb.2009.05.002
Bolz, H., von Brederlow, B., Ramirez, A., Bryda, E. C., Kutsche, K., Nothwang, H. G., et al. (2001). Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 27, 108–112. doi: 10.1038/83667
Bork, J. M., Peters, L. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z. M., Ness, S. L., et al. (2001). Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68, 26–37. doi: 10.1086/316954
Boughman, J. A., Vernon, M., and Shaver, K. A. (1983). Usher syndrome: definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 36, 595–603. doi: 10.1016/0021-9681(83)90147-9
Ceriani, F., and Mammano, F. (2012). Calcium signaling in the cochlea—molecular mechanisms and physiopathological implications. Cell Commun. Signal. 10:20. doi: 10.1186/1478-811X-10-20
Coucke, P. J., Van Hauwe, P., Kelley, P. M., Kunst, H., Schatteman, I., Van Velzen, D., et al. (1999). Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum. Mol. Genet. 8, 1321–1328. doi: 10.1093/hmg/8.7.1321
Denofrio, J. C., Yuan, W., Temple, B. R., Gentry, H. R., and Parise, L. V. (2008). Characterization of calcium- and integrin-binding protein 1 (CIB1) knockout platelets: potential compensation by CIB family members. Thromb. Haemost. 100, 847–856. doi: 10.1160/th08-06-0351
Downey, G. P., Chan, C. K., Trudel, S., and Grinstein, S. (1990). Actin assembly in electropermeabilized neutrophils: role of intracellular calcium. J. Cell Biol. 110, 1975–1982. doi: 10.1083/jcb.110.6.1975
Ebermann, I., Phillips, J. B., Liebau, M. C., Koenekoop, R. K., Schermer, B., Lopez, I., et al. (2010). PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Invest. 120, 1812–1823. doi: 10.1172/jci39715
Ebermann, I., Scholl, H. P., Charbel Issa, P., Becirovic, E., Lamprecht, J., Jurklies, B., et al. (2007). A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum. Genet. 121, 203–211. doi: 10.1007/s00439-006-0304-0
Eudy, J. D., Weston, M. D., Yao, S., Hoover, D. M., Rehm, H. L., Ma-Edmonds, M., et al. (1998). Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 280, 1753–1757. doi: 10.1126/science.280.5370.1753
Gentry, H. R., Singer, A. U., Betts, L., Yang, C., Ferrara, J. D., Sondek, J., et al. (2005). Structural and biochemical characterization of CIB1 delineates a new family of EF-hand-containing proteins. J. Biol. Chem. 280, 8407–8415. doi: 10.1074/jbc.m411515200
Giese, A. P. J., Tang, Y. Q., Sinha, G. P., Bowl, M. R., Goldring, A. C., Parker, A., et al. (2017). CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat. Commun. 8:43. doi: 10.1038/s41467-017-00061-1
Häger, M., Bigotti, M. G., Meszaros, R., Carmignac, V., Holmberg, J., Allamand, V., et al. (2008). Cib2 binds integrin α7Bβ1D and is reduced in laminin α2 chain-deficient muscular dystrophy. J. Biol. Chem. 283, 24760–24769. doi: 10.1074/jbc.m801166200
Heineke, J., Auger-Messier, M., Correll, R. N., Xu, J., Benard, M. J., Yuan, W., et al. (2010). CIB1 is a regulator of pathological cardiac hypertrophy. Nat. Med. 16, 872–879. doi: 10.1038/nm.2181
Hennigs, J. K., Burhenne, N., Stähler, F., Winnig, M., Walter, B., Meyerhof, W., et al. (2008). Sweet taste receptor interacting protein CIB1 is a general inhibitor of InsP3-dependent Ca2+ release in vivo. J. Neurochem. 106, 2249–2262. doi: 10.1111/j.1471-4159.2008.05563.x
Holme, R. H., Kiernan, B. W., Brown, S. D., and Steel, K. P. (2002). Elongation of hair cell stereocilia is defective in the mouse mutant whirler. J. Comp. Neurol. 450, 94–102. doi: 10.1002/cne.10301
Hutchins, B. I., Klenke, U., and Wray, S. (2013). Calcium release-dependent actin flow in the leading process mediates axophilic migration. J. Neurosci. 33, 11361–11371. doi: 10.1523/JNEUROSCI.3758-12.2013
Jarman, K. E., Moretti, P. A., Zebol, J. R., and Pitson, S. M. (2010). Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium- and integrin-binding protein 1. J. Biol. Chem. 285, 483–492. doi: 10.1074/jbc.M109.068395
Joensuu, T., Hämäläinen, R., Yuan, B., Johnson, C., Tegelberg, S., Gasparini, P., et al. (2001). Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am. J. Hum. Genet. 69, 673–684. doi: 10.1086/324290
Kawashima, Y., Geleoc, G. S., Kurima, K., Labay, V., Lelli, A., Asai, Y., et al. (2011). Mechanotransduction in mouse inner ear hair cells requires transmembrane channel-like genes. J. Clin. Invest. 121, 4796–4809. doi: 10.1172/JCI60405
Kazmierczak, P., Sakaguchi, H., Tokita, J., Wilson-Kubalek, E. M., Milligan, R. A., Muller, U., et al. (2007). Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449, 87–91. doi: 10.1038/nature06091
Kharkovets, T., Dedek, K., Maier, H., Schweizer, M., Khimich, D., Nouvian, R., et al. (2006). Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 25, 642–652. doi: 10.1038/sj.emboj.7600951
Kim, K. X., Beurg, M., Hackney, C. M., Furness, D. N., Mahendrasingam, S., and Fettiplace, R. (2013). The role of transmembrane channel-like proteins in the operation of hair cell mechanotransducer channels. J. Gen. Physiol. 142, 493–505. doi: 10.1085/jgp.201311068
Kostyak, J. C., Naik, M. U., and Naik, U. P. (2012). Calcium- and integrin-binding protein 1 regulates megakaryocyte ploidy, adhesion, and migration. Blood 119, 838–846. doi: 10.1182/blood-2011-04-346098
Kubisch, C., Schroeder, B. C., Friedrich, T., Lütjohann, B., El-Amraoui, A., Marlin, S., et al. (1999). KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–446. doi: 10.1016/s0092-8674(00)80556-5
Leisner, T. M., Liu, M., Jaffer, Z. M., Chernoff, J., and Parise, L. V. (2005). Essential role of CIB1 in regulating PAK1 activation and cell migration. J. Cell Biol. 170, 465–476. doi: 10.1083/jcb.200502090
Mammano, F., Bortolozzi, M., Ortolano, S., and Anselmi, F. (2007). Ca2+ signaling in the inner ear. Physiology 22, 131–144. doi: 10.1152/physiol.00040.2006
Mathur, P., and Yang, J. (2015). Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 1852, 406–420. doi: 10.1016/j.bbadis.2014.11.020
Naik, M. U., and Naik, U. P. (2011a). Calcium- and integrin-binding protein 1 regulates microtubule organization and centrosome segregation through polo like kinase 3 during cell cycle progression. Int. J. Biochem. Cell Biol. 43, 120–129. doi: 10.1016/j.biocel.2010.10.003
Naik, M. U., and Naik, U. P. (2011b). Contra-regulation of calcium- and integrin-binding protein 1-induced cell migration on fibronectin by PAK1 and MAP kinase signaling. J. Cell. Biochem. 112, 3289–3299. doi: 10.1002/jcb.23255
Naik, M. U., Nigam, A., Manrai, P., Millili, P., Czymmek, K., Sullivan, M., et al. (2009). CIB1 deficiency results in impaired thrombosis: the potential role of CIB1 in outside-in signaling through integrin α IIb β 3. J. Thromb. Haemost. 7, 1906–1914. doi: 10.1111/j.1538-7836.2009.03581.x
Pan, B., Geleoc, G. S., Asai, Y., Horwitz, G. C., Kurima, K., Ishikawa, K., et al. (2013). TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron 79, 504–515. doi: 10.1016/j.neuron.2013.06.019
Patel, K., Giese, A. P., Grossheim, J. M., Hegde, R. S., Delio, M., Samanich, J., et al. (2015). A novel C-terminal CIB2 (calcium and integrin binding protein 2) mutation associated with non-syndromic hearing loss in a hispanic family. PLoS One 10:e0133082. doi: 10.1371/journal.pone.0141259
Riazuddin, S., Belyantseva, I. A., Giese, A. P., Lee, K., Indzhykulian, A. A., Nandamuri, S. P., et al. (2012). Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat. Genet. 44, 1265–1271. doi: 10.1038/ng.2426
Ruttiger, L., Sausbier, M., Zimmermann, U., Winter, H., Braig, C., Engel, J., et al. (2004). Deletion of the Ca2+-activated potassium (BK) α-subunit but not the BKβ1-subunit leads to progressive hearing loss. Proc. Natl. Acad. Sci. U S A 101, 12922–12927. doi: 10.1073/pnas.0402660101
Seco, C. Z., Giese, A. P., Shafique, S., Schraders, M., Oonk, A. M., Grossheim, M., et al. (2016). Novel and recurrent CIB2 variants, associated with nonsyndromic deafness, do not affect calcium buffering and localization in hair cells. Eur. J. Hum. Genet. 24, 542–549. doi: 10.1038/ejhg.2015.157
Self, T., Mahony, M., Fleming, J., Walsh, J., Brown, S. D., and Steel, K. P. (1998). Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 125, 557–566.
Shen, J., Scheffer, D. I., Kwan, K. Y., and Corey, D. P. (2015). SHIELD: an integrative gene expression database for inner ear research. Database 2015:bav071. doi: 10.1093/database/bav071
Vélez-Ortega, A. C., Freeman, M. J., Indzhykulian, A. A., Grossheim, J. M., and Frolenkov, G. I. (2017). Mechanotransduction current is essential for stability of the transducing stereocilia in mammalian auditory hair cells. Elife 6:e24661. doi: 10.7554/eLife.24661
Verpy, E., Leibovici, M., Zwaenepoel, I., Liu, X. Z., Gal, A., Salem, N., et al. (2000). A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet. 26, 51–55. doi: 10.1038/79171
Weil, D., Blanchard, S., Kaplan, J., Guilford, P., Gibson, F., Walsh, J., et al. (1995). Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374, 60–61. doi: 10.1038/374060a0
Weil, D., El-Amraoui, A., Masmoudi, S., Mustapha, M., Kikkawa, Y., Lainé, S., et al. (2003). Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum. Mol. Genet. 12, 463–471. doi: 10.1093/hmg/ddg051
Welnhofer, E. A., Zhao, L., and Cohan, C. S. (1999). Calcium influx alters actin bundle dynamics and retrograde flow in Helisoma growth cones. J. Neurosci. 19, 7971–7982.
Weston, M. D., Luijendijk, M. W., Humphrey, K. D., Möller, C., and Kimberling, W. J. (2004). Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet. 74, 357–366. doi: 10.1086/381685
White, C., Yang, J., Monteiro, M. J., and Foskett, J. K. (2006). CIB1, a ubiquitously expressed Ca2+-binding protein ligand of the InsP3 receptor Ca2+ release channel. J. Biol. Chem. 281, 20825–20833. doi: 10.1074/jbc.M602175200
Wu, Z., Grillet, N., Zhao, B., Cunningham, C., Harkins-Perry, S., Coste, B., et al. (2017). Mechanosensory hair cells express two molecularly distinct mechanotransduction channels. Nat. Neurosci. 20, 24–33. doi: 10.1038/nn.4449
Yang, H., Wang, H., Shivalila, C. S., Cheng, A. W., Shi, L., and Jaenisch, R. (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379. doi: 10.1016/j.cell.2013.08.022
Yuan, W., Leisner, T. M., McFadden, A. W., Clark, S., Hiller, S., Maeda, N., et al. (2006). CIB1 is essential for mouse spermatogenesis. Mol. Cell. Biol. 26, 8507–8514. doi: 10.1128/mcb.01488-06
Zayed, M. A., Yuan, W., Chalothorn, D., Faber, J. E., and Parise, L. V. (2010). Tumor growth and angiogenesis is impaired in CIB1 knockout mice. J. Angiogenes Res. 2:17. doi: 10.1186/2040-2384-2-17
Zhao, B., Wu, Z., Grillet, N., Yan, L., Xiong, W., Harkins-Perry, S., et al. (2014). TMIE is an essential component of the mechanotransduction machinery of cochlear hair cells. Neuron 84, 954–967. doi: 10.1016/j.neuron.2014.10.041
Keywords: hearing loss, Usher syndrome, CIB2, knockout mice, stereocilia, mechanoelectrical transduction
Citation: Wang Y, Li J, Yao X, Li W, Du H, Tang M, Xiong W, Chai R and Xu Z (2017) Loss of CIB2 Causes Profound Hearing Loss and Abolishes Mechanoelectrical Transduction in Mice. Front. Mol. Neurosci. 10:401. doi: 10.3389/fnmol.2017.00401
Received: 10 August 2017; Accepted: 20 November 2017;
Published: 04 December 2017.
Edited by:
David Z. He, Creighton University School of Medicine, United StatesReviewed by:
Gregory I. Frolenkov, University of Kentucky, United StatesJun Yang, University of Utah, United States
Inna Belyantseva, National Institute on Deafness and Other Communication Disorders (NIH), United States
Copyright © 2017 Wang, Li, Yao, Li, Du, Tang, Xiong, Chai and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renjie Chai, renjiec@seu.edu.cn
Zhigang Xu, xuzg@sdu.edu.cn
† These authors have contributed equally to this work.