The Emerging Role of microRNAs in Polyglutamine Diseases
 Xiaoyu Dong
Xiaoyu Dong Shuyan Cong
Shuyan Cong- Department of Neurology, Shengjing Hospital of China Medical University, Shenyang, China
MicroRNAs (miRNAs) are small non-coding molecules that regulate a large amount of post-transcriptional repressor genes by recognizing semi-complementary target sequences that are normally located in the 3′ UTR of the mRNA. Altered expression of miRNA has been related to several pathological processes, including polyglutamine (Poly Q) diseases. Specific expression patterns in the circulating fluids and brain parenchyma have been speculated as potential biomarkers for Poly Q disease diagnosis and prognosis. Several miRNAs have been consistently identified in diseases including Huntington’s disease (HD) and spinocerebellar ataxia (SCA). In our review, we describe the emerging role of miRNAs in Poly Q diseases and provide an overview on general miRNA biology, implications in pathophysiology, and their potential roles as future biomarkers and applications for therapy.
Introduction
Polyglutamine (Poly Q) diseases are characterized by trinucleotide repeat amplification within a gene, which results in the formation of Poly Q peptides, selective neuronal degeneration and death in neurodegenerative diseases (Liu et al., 2018). To date, there are at least nine known Poly Q diseases, including Huntington’s disease (HD), spinobulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy (DRPLA), and six spinocerebellar ataxias (SCAs; He et al., 2010). Although different Poly Q diseases involve different pathogenic genes, a large amount of studies have shown a common pathogenesis mechanism related to “gain of toxic functions.” That is, the pathogenic Poly Q proteins cause selective neuronal toxicity; accumulation of Poly Q-mutant proteins and formation of neuronal intranuclear inclusions (NIIs) are core elements of the pathogenic process (Yushchenko et al., 2018). Hypotheses regarding possible pathogenic mechanisms include: (1) protein misfolding; (2) gene transcription and expression imbalance; (3) intracellular protein homeostasis failure; and (4) mitochondrial dysfunction.
In recent years, microRNAs (miRNAs) have been confirmed to play important roles in both physiological and pathological conditions of many brain-related functions (Saraiva et al., 2017). Abnormal miRNA expression patterns have been proposed to be related to the etiology and progression of Poly Q diseases. Several studies have also shown that increases or decreases in expression levels can be used as potential diagnostic biomarkers or to ameliorate neurodegenerative processes and promote endogeneous regeneration.
In this review article, we introduce the canonical miRNA biogenesis pathway and miRNA function and describe the most relevant brain-specific miRNAs associated with neurodegenerative processes in Poly Q disease. Potential biomarkers involved in Poly Q disease diagnosis and prognosis and the advantages and limitations of using miRNA-based therapeutic strategies will also be highlighted.
MiRNA Basics
MiRNAs are a novel class of small (~21 nucleotides), single-stranded, non-coding RNAs that act as post-transcriptional regulators of gene expression (Goodall et al., 2013). MiRNAs function by binding to the 3′-UTR of mRNA targets and recruiting the RNA-induced silencing complex (RISC) to inhibit target expression (Lau and de Strooper, 2010). The mechanism of how RISC inhibits the expression of bound mRNA targets remains unclear (Filipowicz et al., 2008). Inhibition in translation at the initiation step (Mathonnet et al., 2007) or deadenylation of targets followed by mRNA decay may take part in the process (Wakiyama and Yokoyama, 2014). To date, more than 10,000 miRNA sequences have been included in miRBase1, of which 721 have been identified in the human genome, and approximately 30% are encoded within human protein-encoding genes (Carroll et al., 2011). Primary miRNA (pri-miRNA) genes are normally transcribed by RNA polymerase II and can generate an incomplete stem-loop structure of several thousand bases in length (Yin et al., 2015). Pri-miRNA is further processed by the microprocessor complex in the nucleus and can generate precursor miRNAs (pre-miRNAs), which are characterized by a stem-loop structure of ~70 nucleotides. After export from the nucleus by Exportin 5/Ran, pre-miRNAs are generally cleaved by Dicer to generate mature miRNAs (Winter et al., 2009; Lau and de Strooper, 2010).
MiRNAs play an important role in various physiological processes in the nervous system. They are involved in regulating neurodevelopment, proliferation, plasticity, and memory and are closely associated with the development of a variety of nervous system disorders (Snyman et al., 2017). MiRNA expression is usually regulated by epigenetic mechanisms, including histone modification, DNA methylation, or by enzymes that stabilize mature miRNAs (Femminella et al., 2015). Studies have shown that there may be more than 1,000 types of miRNAs expressed in the human brain and miRNA expression in the nervous system has regional, tissue and spatiotemporal specificity. In particular, miR-10b-5p, miR-145-5p, miR-378c, and miR-217 are mainly expressed in the prefrontal cortex, miR-205 in the frontal cortex and miR-21-3p, miR-224, and miR-373-3p are mainly expressed in the substantia nigra. MiR-134 is involved in the formation of dendritic spines, while miR-124 and miR-132 participate in axon growth. As some studies have pointed out, these small RNAs have been identified as stable in blood and cerebrospinal fluid (CSF; Wu et al., 2016). Techniques such as quantitative PCR and microarrays are being applied to measure miRNA levels in biofluids. Microarray analysis is generally used as a non-targeted approach to analyze multiple miRNAs simultaneously, while PCR is more accurate and sensitive for validating results obtained from microarray analysis (Huang, 2017).
Recently, aberrant expression or deregulation of miRNAs has been found to play a major role in the pathogenesis of several Poly Q diseases (Shah et al., 2018). Their potential as diagnostic and prognostic biomarkers is gradually being recognized and applied in clinical practice. Gene-silencing methods that target miRNAs have been used to treat several Poly Q diseases in animal models.
MiRNA in Poly Q Disease
MiRNAs in HD
In Europe and North America, the incidence of HD per 100,000 people is 5%–10% (Graham et al., 2018). CAG trinucleotide repeats within the huntingtin gene result in abnormal accumulation of the misfolded huntingtin (HTT) protein in intranuclear inclusions and lead to progressive loss of striatal neurons (Lau and de Strooper, 2010), which are the main causative factors of the disease (Hu et al., 2010). The clinical characteristics of HD are dystonia, chorea, and cognitive or psychiatric disorders. There is currently no effective treatment and patients usually die 10–20 years after illness onset (Jacobsen et al., 2010). By applying RNA sequencing, microarray, and qRT-PCR techniques, aberrant miRNA expression in HD has been reported in a large number of studies using different human samples. Chang et al. (2017) found the expression of a set of miRNAs was dysregulated in Brodmann’s area four in HD patients including miR-218, miR-196a, and miR-486, which are highly expressed in the pathological region, and miR-132 miR-9, miR-124a, miR-29b and miR-22, which show low expression in HD patients. Martí et al. (2010) also found that miR-100, miR-16, miR-151-3p, miR-219-2-3p, miR-451, miR-27b, and miR-92a were up-regulated in the striatum and frontal cortex of HD patients, while miR-128, miR-222, miR-139-3p, miR-382, miR-483-3p and miR-433 were down-regulated in the diseased tissues. In recent decades, a great number of HD cell model and animal studies have shown that miRNAs may affect the pathogenesis, progression, and prognosis of patients through various pathways (Johnson and Buckley, 2009). In a monkey model, Kocerha et al. (2014) pointed out that miR-128 was down-regulated in the brain of pre- and post-symptomatic HD monkeys; by suppressing HIP-1, HTT and SP-1, they concluded that miR-128 may play a pivotal role in HD pathogenesis. Repressor element-1 silencing transcription factor (REST) is a zinc finger protein that binds to the corresponding neuron-restricted silencing elements (NRSEs) and deacetylates histones to inhibit the expression of some neurogenic genes (Soldati et al., 2013). Poly Q expansion of mutant HTT protein can inhibit the interaction between REST and the HTT protein, thereby promoting REST aggregation in the nucleus of HD patients and inhibiting the expression of related genes (Wu and Xie, 2006). Several studies have found that REST is regulated by a variety of miRNAs including miR-29a, miR-29b, miR-132, and miR-135b (Reed et al., 2018). Johnson et al. (2008) demonstrated that miR-9, -29a, -29b, -124a, -132, -135b, -139, -203, -204, -212, -330, and -346 were also associated with REST expression. MiRNAs may also have neuroprotective functions. Jovicic et al. (2013) identified miR-22 as a potentially neuroprotective miRNA in HD by regulating several targets such as REST corepressor 1 (Rcor1), histone deacetylase 4 (HDAC4) and regulator of G-protein signaling 2 (Rgs2). MiR-196a may ameliorate cytotoxicity and the cellular phenotype in transgenic HD monkey neuronal cell models.
In the following section, we will review some of the miRNAs that may be associated with HD pathology and have been consistently identified as dysregulated in HD (Table 1; Chen J. et al., 2018).
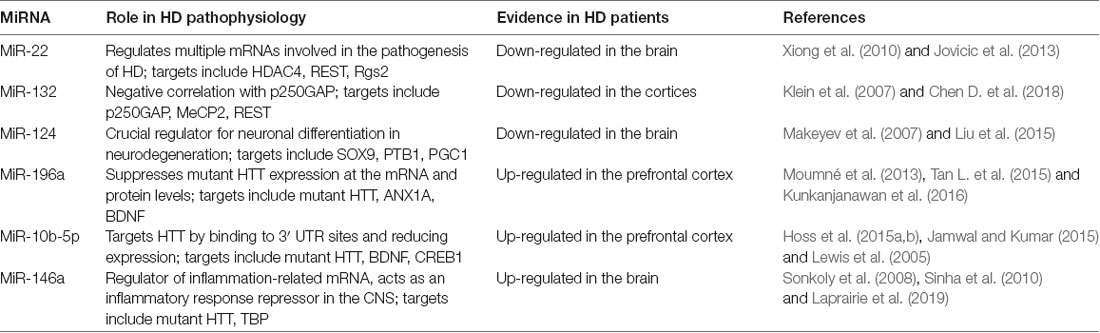
Table 1. MicroRNAs (miRNAs) most commonly associated with Huntington’s disease (HD).
MiR-22 was originally identified in HeLa cells (an immortal cell line derived from cervical cancer cells), but was later shown to be ubiquitously expressed in various tissues (Xiong et al., 2010) and down-regulated in HD brain tissue (Jovicic et al., 2013). Using TargetScan, miR-22 was found to target multiple mRNAs implicated in HD pathogenesis; among the targets is HDAC4, which has been shown to reverse histone acetylation and plays a neuroprotective effect in animal and cellular models of HD (Harding and Tong, 2018). The REST pathway is hyperactive in HD, which leads to large-scale repression of neuronal genes in affected neurons (Zuccato et al., 2007). Seredenina et al. (2011) reported that decreased expression of Rgs2 in striatal neurons has a protective effect in cell models of HD by reducing activation of extracellular-signal-regulated kinase (ERK). Although the specific mechanism is not yet elucidated, miR-22 could regulate HDAC4, REST corepressor 1 (Rcor1) and Rgs2 to achieve neuroprotection and inhibit neurodegeneration (Jovicic et al., 2013).
MiR-132 is a key component of the activity-dependent gene regulatory response in neuronal cells (Chen D. et al., 2018). Vo et al. (2006) determined that miR-132 promotes neuronal outgrowth and sprouting by decreasing p250GAP levels. Another target for miR-132 is methyl CpG binding protein 2 (MeCP2). Failure to regulate MeCP2 levels is connected to neurological disorders (Klein et al., 2007). As one of the most important growth factors that preserve neural cell growth and survival, BDNF has been identified dysregulation in HD patients. MeCP2 increases BDNF levels in the brain, which in turn increases transcription from the miR-132 cluster. Thus, miR-132 dysregulation might contribute to HD progression by impairing neurogenesis and affecting BDNF balance in the diseased brain (Conaco et al., 2006). A rise in miRNA-132 levels will decrease MeCP2 levels and restore the balance. Failure to regulate MeCP2 levels is connected to neurological disorders (Klein et al., 2007). REST has been reported to repress BDNF, which is a CREB-dependent activator of miR-132 and directly repress miR-132 transcription. Thus, REST could effectively repress the expression of miR-132 in two independent pathways by an effective repressive feed-forward loop (Johnson and Buckley, 2009; Bicker et al., 2014).
MiR-124 is a neuronal-specific miRNA and is thought to be a crucial regulator of neuronal differentiation in neurodegeneration (Liu et al., 2015). SRY-related HMG box transcription factor 9 (SOX9) is a physiological target of miR-124; repression of SOX9 mediated by miR-124 is essential for neuron formation from neural stem cells (Cheng et al., 2009). MiR-124 could promote the differentiation process by facilitating neuron-specific splicing of mRNAs through repression of polypyrimidine tract binding protein (PTB1) in developing neurons. Thus, a decrease in the level of miR-124 may promote the accumulation of non-neuronal splice isoforms in neurons (Makeyev et al., 2007; Johnson and Buckley, 2009). As a presumed target of miR-124, Cyclin A2 (CCNA2) is increased in mutant STHdh (Q121)/Hdh (Q121) cells. Predictably, decreased expression of miR-124 would increase CCNA2 levels in animal and cell models of HD and would be resulted in cell cycle deregulated in the HD cell model (Das et al., 2013). In contrast, miR-124 overexpression would up-regulate BDNF and peroxisome proliferator-activated receptor gamma coactivator 1 (PGC1) expression and down-regulate SOX9 expression in the striatum of R6/2 mouse brain, which implies that miR-124 slows down the HD progression (Meza-Sosa et al., 2012).
Several recent researches have confirmed the protective function of miR-196a in HD disease (Moumné et al., 2013). Tan L. et al. (2015) suggested that miR-196a suppresses mutant HTT expression at the protein and mRNA levels. Moreover, the inhibition was not resulted by binding of miR-196a to the 3′-UTR of mutated HTT directly but occurred through inhibiting the synthesis of protein and partly through enhancing the degradation of protein (Tan L. et al., 2015). MiR-196a also targets ANX1A to promote cell survival and reduce apoptosis. Over-expression of miR-196a could enhance BDNF expression, which benefits neural cell survival in HD animal models (Kunkanjanawan et al., 2016).
In recent years, MiR-10b-5p has been reported to be significantly up-regulated in the HD brain (prefrontal cortex) and data suggest that this change may occur pre-symptomatically. As one of the targets of miR-10b-5p (Jamwal and Kumar, 2015), BDNF down-regulation in HD brains might be mediated through up-regulation of miR-10b-5p, which has a binding target within the 3′-UTR of the BDNF transcript (Varendi et al., 2014). CREB1 is another target of this miRNA. The 3′-UTR of CREB1 also harbors a miR-10b-5p binding site and reduced CREB1 expression has been reported in HD (Lewis et al., 2005). Moreover, miR-10b-5p presumably targets HTT by binding to two 3′-UTR sites (both 7mer-1A seeds, positions 2,742–2,748 and 3,301–3,307) and could decrease HTT expression (Ritchie, 2017). Given the stability of miR-10b-5p in serum, it could be considered an accessible biomarker for stage of disease, rate of progression and other important clinical HD characteristics (Hoss et al., 2014).
MiR-146a is primarily involved in regulating inflammation and other processes that have important functions in the innate immune system (Sonkoly et al., 2008). Recruitment of the basal transcription factor TBP (tata binding protein) to disruption of TBP structures by HTT or mutant HTT aggregates, may compromise its function (Laprairie et al., 2019). MiR-146a could directly regulate TBP expression; decreased expression of miR-146a would lead to high TBP expression levels and further affect cellular responses in HD cell models and contribute to the pathogenesis of HD (Sinha et al., 2010).
MiRNA in SCAs
SCAs are chronic progressive neurological disorders in which cerebellar ataxia is the most prominent symptom (Roshan et al., 2014; van der Stijl et al., 2017). Of more than 40 different SCA types (Durr, 2010), there are at least six SCAs (SCA-1, 2, 3, 6, 7, and 17) that are associated with Poly Q diseases (Sinha et al., 2018). The expression of CAG repeats of abnormal length in the open reading frame (ORF) of the ataxin (ATXN) genes are the common denominator. Specific linkages between miRNA regulation and CAG repeat-dependent SCAs have been described in some studies (Chau and Kalsotra, 2015; Table 2).
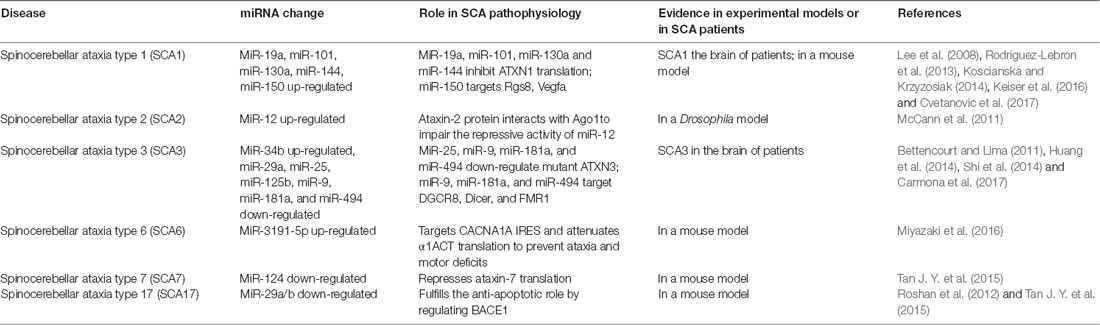
Table 2. Summary of current links between miRNA regulation and spinocerebellar ataxia (SCAs).
SCA1 is a dominantly inherited neurodegenerative disease caused by repeated expansions of Poly Q in ataxin-1 (Rodriguez-Lebron et al., 2013). Lee et al. (2008) revealed that miR-19a, miR-101, and miR-130a co-regulate the 3′-UTR of ATXN1 by inhibiting ATXN1 translation. Using a reporter assay, miR-19a, -101, and -130a were shown to down-regulate ATXN1 expression (Lee et al., 2008). MiRNA-mediated post-transcriptional regulation of the ATXN1 gene may modulate SCA1-associated neuropathology by affecting levels of ATXN1 protein (Koscianska and Krzyzosiak, 2014).
MiR-144 was predicted to be related to SCA1 in a genome-wide microarray analysis (Persengiev et al., 2011). MiR-144 considerably decreased endogenous ATXN1 protein levels. In addition, activation of miR-144 reduced the cytotoxic effects of the expanded mutATXN1 and miRNA deregulation may be involved in SCA1 development. It was also demonstrated that increased miR-150 levels influenced SCA1 pathogenesis by suppressing Rgs8 expression and contributed to the reduction of Vascular Endothelial Growth Factor A (VEGFA) in cerebellar Purkinje neurons in a mouse model of SCA1 (Rodriguez-Lebron et al., 2013; Cvetanovic et al., 2017).
SCA2 is an autosomal dominant hereditary neurodegenerative disorder. It is caused by a repeated amplification of CAG in the coding region of the ATXN2 gene, resulting in an abnormally long poly Q tract expression in the ataxin-2 protein and acquired neurotoxicity leading to neuronal loss in the brainstem, cerebellum, brain cortex and spinal cord. At present, little research has been done on the role of miRNAs in the pathogenesis of SCA2. McCann et al. (2011) showed that ataxin-2 might be required for miRNA functioning in Drosophila; it was also shown the ataxin-2 protein impairs the repressive activity of miR-12 by interacting with Argonaute 1 (Ago1).
SCA3/Machado-Joseph disease (MJD) is caused by CAG repeated amplification of the ATXN3 gene (Shi et al., 2014). Using miRCURY™ LNA Array followed by qRT-PCR validation, Shi et al. (2014) found that miR-34b expression was significantly elevated, whereas miR-25, miR-29a and, miR-125b were down-regulated in SCA3 patient serum. Regarding molecular mechanisms, miR-25 and miR-125b may bind the 3′-UTR of ATXN3 and regulate the ATXN3 expression. Moreover, miR-25 and miR-125b expression were also associated with disease progression (Shi et al., 2014). MiR-25 reduces mutant ATXN3 protein levels by interacting with the 3′-UTR of the ATXN3 mRNA, thereby decreasing early apoptosis, increasing cell viability and alleviating the accumulation of mutant ATXN3 protein aggregates in SCA3/MJD cells (Huang et al., 2014). Carmona et al. (2017) revealed that miR-9, miR-181a, and miR-494 were significantly down-regulated in SCA3/MJD based on in vivo and in vitro models and gene expression analysis. Down-regulation of DGCR8, Dicer, and FMR1 may be responsible for miRNA dysregulation. Dual luciferase assay confirmed that miR-9, miR-181a and miR-494 bind directly to the ATXN3 3′-UTR and down-regulate its expression. Furthermore, miR-181a and miR-494 down-regulate muttATXN3 expression either by inhibiting translation or by inducing the degradation of its mRNA, while miR-9 acts mainly through translational inhibition in vitro (Carmona et al., 2017).
SCA6 is a dominant hereditary neurodegenerative disorder characterized by slowly progressive ataxia and Purkinje cell degeneration. It is caused by repeated amplification of Poly Q in the second CACNA1A gene product, α1ACT; expression of α1ACT is controlled by the internal ribosomal entry site (IRES) present within the coding region of CACNA1A (Miyazaki et al., 2016). MiR-3191-5p targets the CACNA1A IRES and preferentially attenuates IRES-driven α1ACT translation, in an Ago4-dependent manner, to cause Purkinje cell degeneration. Viral delivery of miR-3191-5p can rescue the disease phenotype by modulating the cellular IRES activity in a mouse model (Miyazaki et al., 2016).
SCA7 is a rare inherited neurodegenerative disorder caused by an in-frame repeated CAG expansion in the first exon of the ataxin-7 gene (ATXN7, David et al., 1997). Translation of the mutated ATXN7 allele results in expansion of poly Q tracts, the formation of protein aggregates and decreases the protein activity (Martin et al., 1999). MiR-124 was determined to repress ataxin-7 mRNA translation by modulation of a conserved long-ncRNA-mediated negative feedback loop. By inhibiting STAGA activity, the loop is disrupted by the poly Q-expanded ataxin-7, resulting in decreased miR-124 transcription, and followed by increased poly Q-expanded ataxin-7 translation in the retina and cerebellum (Das et al., 2013; Tan J. Y. et al., 2015; van der Stijl et al., 2017).
SCA17 is caused by the expansion of a repeated Poly Q in the TBP gene and is characterized by intranuclear protein aggregation and selective loss of cerebellar neurons (Roshan et al., 2017). MiR-29a/b has been found down-regulation in a SCA17 cellular model by qRT PCR. MiR-29a/b down-regulation may lead to an increased expression of beta-site amyloid precursor protein cleaving enzyme 1 (BACE1), Bcl 2 homologous antagonist (BAK), p53 upregulated modulator of apoptosis (PUMA), increased cytochrome c release and apoptosis restoration of miR-29a/b in the pathogenic poly Q background reduced expression of BACE1 (Das et al., 2013). The miR-29a/b-BACE1 regulatory interaction may be part of the molecular mechanism that leads to neuronal cell death in SCA17 (Roshan et al., 2012).
MiRNAs in SBMA
SBMA is an adult-onset neuromuscular disease caused by repeated expansion of poly Q in the androgen receptor (AR; La Spada et al., 1991). MiR-298 downregulates the levels of AR mRNA and protein when transfected into cells overexpressing wild and mutant type AR and fibroblasts derived from patients with SBMA. Furthermore, miR-298 could counteract AR toxicity by binding directly to the 3′-UTR of the human AR transcript in vitro (Pourshafie et al., 2016).
MiRNAs in DRPLA
A poly Q expanded mutant Atrophin-1 protein is the pathogenic protein of DRPLA. Atrophin-1 mRNA is one of the direct targets of miR-8. MiR-8 mutant phenotypes can be attributed to an increase in atrophin activity, which leads to an increase in brain apoptosis and behavioral defects (Huang et al., 2010). Deletion of miR-8 was found to increase expression of Atrophin-1 in a Drosophila model, thereby causing degeneration of nerve cells and impaired motor function. In addition, miR-200b and miR-429 are identical to miR-8 in the seed region, which permits miR-200b and miR-429 to recruit HDACs and serve as a transcriptional corepressor. Therefore, they could target the 3′-UTR of human Atrophin-1 to contribute to the pathogenesis of DRPLA (Karres et al., 2007).
MiRNAs as Biomarkers in Poly Q
The diagnosis of probable Poly Q diseases is based on clinical and neuropsychological assessments and can be confirmed by genetic testing. However, early treatment is crucial in these diseases and there is an urgent need for easily accessible biomarkers to aid in early diagnosis. Moreover, with several therapeutic approaches in development for Poly Q disease, biomarkers could also be utilized to monitor disease progression and responses to therapy (Table 3).
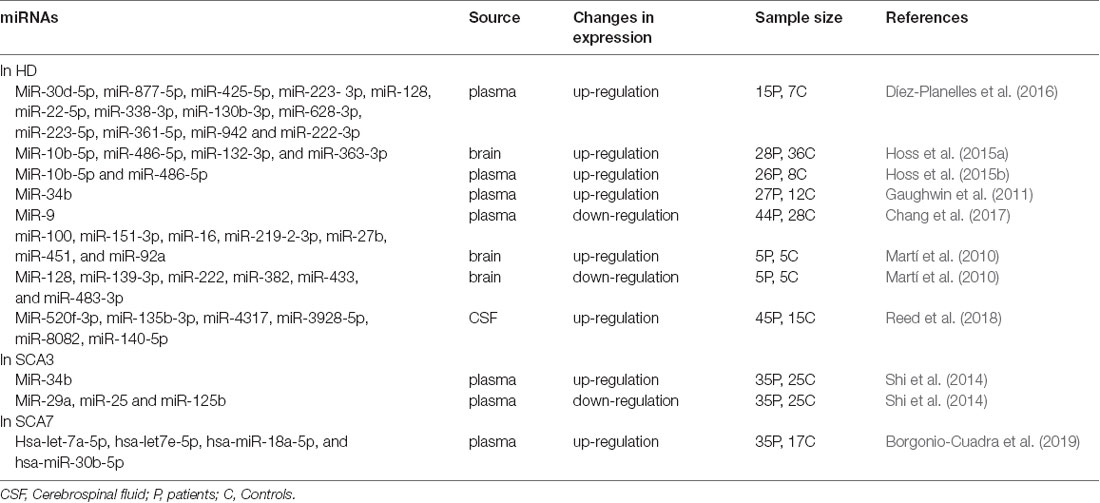
Table 3. Aberrant expression of miRNAs as biomarkers in Poly Q diseases.
It has been reported that miRNAs are present in various biofluids, such as saliva, blood, urine, tears, breast milk, bronchial secretions, amniotic fluid and CSF (Weber et al., 2010). The stability and abundance of circulatory miRNAs in biofluids are the main factors in clinical application as potential diagnostic and progression biomarkers (Li et al., 2018). There may be five possible miRNA transportation mechanisms into the extracellular circulation. MiRNAs may: (1) form a complex with the Ago2 protein; (2) be packaged within exosomes; (3) be encapsulated within micro-vesicles; (4) be bound to high-density lipoprotein (HDL) particles in non-vesicle form; or (5) accumulate in apoptotic bodies (Kumar et al., 2017). However, the details of these mechanisms need to be further clarified (Chen et al., 2012).
In this section, we review the findings of the most significant studies to investigate the potential role of miRNAs as biomarkers in Poly Q diseases.
Mature miRNA levels were determined from 752 plasma samples obtained from 15 symptomatic patients with 40–45 CAG repeats in the HTT gene and seven healthy matched controls (Kumar et al., 2017). Alterations in a total of 168 plasma miRNAs were identified in symptomatic patients with a significant up-regulation of 13 miRNAs (miR-30d-5p, miR-877-5p, miR-425-5p, miR-223-3p, miR-128, miR-22-5p, miR-338-3p, miR-130b-3p, miR-628-3p, miR-223-5p, miR-361-5p, miR-942 and miR-222-3p; Díez-Planelles et al., 2016). In addition, four miRNAs (miR-132-3p, miR-363-3p, miR-10b-5p and miR-486-5p) showed significant genome-wide changes in HD brain tissue and were abundant in both brain and blood. Plasma samples from 26 HD patients (compared to eight controls) had increased miR-10b-5p and miR-486-5p levels, whereas the levels of miR132-3p, though not significant, were lower (Hoss et al., 2015a). MiR-10b-5p was subsequently confirmed to be related to the age of onset of the disease (Hoss et al., 2015b; Ghatak and Raha, 2018). Similar findings were reported, in which 11 pre-manifest HD, eight early HD, eight moderate HD, and 12 control subjects were included (Gaughwin et al., 2011). The results revealed that miR-34b and miR-1285 were up-regulated in the plasma of HD patients and, most interestingly, the levels of miR-34b in the plasma of pre-manifest HD patients were significantly increased (Ridolfi and Abdel-Haq, 2017).
In recent years, an increasing number of miRNA profiles have been used in SCA research. Shi et al. (2014) searched for different plasma levels of miRNAs from 35 SCA3 patients and 25 controls. Plasma miR-34b levels were significantly elevated, whereas miR-25, miR-29a, and miR-125b levels was down-regulated. Additional research showed that miR-25 and miR-125b expression levels were related to disease progression (Langfelder et al., 2018). In SCA7 patients, a signature of four miRNAs (hsa-let-7a-5p, hsa-let7e-5p, hsa-miR-18a-5p, and hsa-miR-30b-5p) were correlated with disease severity and stage. These miRNAs could help to distinguish early-onset from adult-onset disease and to identify novel cellular processes associated with SCA7 (Borgonio-Cuadra et al., 2019).
There is increasing evidence that changes in miRNA levels in serum/plasma, CSF and other biological fluids are associated with certain biological conditions, such as HD and SCA3. Novel informative biomarkers could provide more information about the development and progression of these diseases and facilitate observation of disease progression and the effects of treatment. However, a more refined understanding of the mechanisms regarding how circulating miRNAs change as disease development and progression will be required to effectively use miRNAs in biomarker applications (Kumar et al., 2017). Additionally, clinical studies that include more patients at different stages of the disease will be essential.
MiRNAs play important roles in regulating gene expression in different cellular processes and disease pathogenesis. By interacting with complementary mRNAs at the post-transcriptional level to affect most mechanisms, deregulation of miRNAs is becoming a contributor to neurodegeneration (Miniarikova et al., 2016). Studying miRNAs associated with neurodegenerative diseases might also provide strategies for innovative therapies. Since the first phase 1 miRNA replacement therapy clinical trial was posted on Clinical Trials.gov (Wen, 2016), miRNAs have been extensively studied as promising therapeutic tools in a variety of diseases, including neurodegenerative diseases.
Poly Q diseases are monogenic dominantly inherited disorders with complex etiologies. To date, a large variety of potential therapeutic strategies have been applied in HD and other Poly Q diseases. Attempts include delivery of neurotrophic factors, mitochondrial function normalization, activation of neuronal stem cells and mutant gene targeting (Miniarikova et al., 2016). The most promising treatment methods at present are directed at lowering mutant mRNA levels.
Interfering with the expression of pathogenic mRNA by synthesizing antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), single-stranded RNAs (ssRNAs), the expressions of short hairpin RNAs (shRNAs) or artificial miRNAs have been widely explored. Although ASO and siRNA strategies have made progress in the treatment of some Poly Q diseases (Stiles et al., 2012; Southwell et al., 2014), efficacy and safety still need to be confirmed when sufficient ASOs and siRNAs are applied to the human brain. Moreover, the half-lives of ASOs and siRNAs are relatively short, requiring repeated administration, which impose a clinical burden on patients.
Previous studies have indicated that siRNA, shRNA, or miRNA expression can significantly silence the HTT gene by delivering with adeno-associated viral (AAV) vectors in murine HD models. Consistently, a miRNA-based expression system was suggested as a promising approach for HD treatment due to lower toxicity (Borel et al., 2011). Miniarikova et al. (2016) evaluated three different approaches for HTT gene silencing (including mtHTT and wtHTT) using expressed artificial miHTTs (miRNAs targeting HTT) in human and mouse HD models. Their findings considered miHTTs to be the best treatment strategy for further development. Sufficient inhibition of mtHTT, while maintaining wtHTT at appropriate levels, was achieved in several researches (Boudreau et al., 2009; McBride et al., 2011). Furthermore, bilateral striatal injections of AAV9-GFP-miRHtt vectors lowered mRNA expression of HTT by >50% in an HD mouse model. This treatment was well tolerated; it did not appear to eliminate HTT expression completely and reduced HTT levels may be sufficient to significantly slow disease progression (Holmans et al., 2017).
Similar treatment strategies have also been studied in SCA1. Keiser et al. (2016) tested an artificial miRNA targeting the mouse ATXN1 gene (in a knock-in model) and human ATXN1 (in a transgenic model) utilizing AAV serotypes 2/1 and 2/5, respectively. Delivery of AAVs encoding RNA interference (RNAi) sequences to the cerebella of SCA1 mice resulted in widespread cerebellar Purkinje cell transduction and improved behavioral and histological phenotypes. This approach induced inhibition of ataxin-1 in the cerebellar cortex and brainstem neurons and may be sufficient for SCA1 treatment.
SCA3/MJD is the most common of the SCAs. Recently, two studies used artificial miRNA mimics to target the 3′-UTR of human ATXN3. A recombinant AAV was used to deliver the mimics to the cerebellum of transgenic mice expressing the full human disease gene (SCA3/MJD84.2 mice) and it was shown that anti- ATXN3 miRNAs effectively phenocopy the suppression of human ATXN3 expression in SCA3/MJD84.2 mice (Rodríguez-Lebrón et al., 2014). However, despite significant knockdown of cerebellar ATXN3 and apparent tolerance of the constructs, the mice still developed motor phenotypes (Costa et al., 2013).
In addition to cerebellar neurodegeneration and ataxic symptoms, SCA7 patients usually lose visions (Garden and La Spada, 2008). Using non-allele-specific artificial miRNAs targeting ataxin-7 (ATXN7) injected into either the dorsal cochlear nucleus (DCN) or the eye of SCA7 mice, Ramachandran et al. (2014a) delivered AAV2/1 to the DCN and found relief of the motor phenotypes, and improvement of cerebellar Purkinje cell dendrites. Subsequently, it was verified that injection of AAV2/1 to the retina of SCA7 mice could achieve more than 50% inhibition of ATXN7 in the retina without side effects in retinal functioning and further confirmed the safety of long-term knockout of ATXN7 in the cerebellum and brainstem (Ramachandran et al., 2014a,b).
Some progress has also been made in the treatment of SBMA. Expression of miR-196a enhanced the decay of the AR mRNA by silencing CUGBP (an Elav-like family member 2 encoded by the CELF2 gene), which directly acted on AR mRNA and enhanced its stability. Furthermore, the early intervention of the AAV vector on miR-196a improved the SBMA phenotypes in a mouse model (Miyazaki et al., 2012). Moreover, overexpression of miR-298 in SBMA mice by using a recombinant AAV9, delivered by just a single tail-vein injection, could induce sustained and extensive overexpression of skeletal muscle and motor neurons and lead to improvement of the neuromuscular phenotype in mice (Pourshafie et al., 2018).
We conclude that suppression of expanded mutant proteins in Poly Q diseases can have a significant effect on disease progression in animal models (Keiser et al., 2016). However, more researches should be conducted on the pharmacokinetics of miRNA to understand the threshold copies of miRNA that should be replaced or inhibited in each disease state. Specific miRNA carriers also should be designed for long term gene expression, as knockout in the CNS remains a tremendous challenge, that is as yet to be overcome (Wen, 2016).
Conclusions
MiRNAs have emerged as a novel class of RNA molecules that play a crucial role in the pathological events of neurodegenerative diseases. It is important to note that many miRNAs have been identified to be significantly deregulated in Poly Q diseases, some of which have been consistently identified as Poly Q disease-specific. Circulating miRNAs are promising biomarkers for Poly Q diseases and for monitoring disease progression and therapy response. Although miRNAs are considered stable in biological fluids, variability still exists for numerous reasons. A more refined understanding of the mechanisms regarding how circulating miRNAs change as the disease development and progression will be required for accurate miRNA biomarker applications. Since in vitro and in vivo researches have indicated a dramatic effect on suppression of mutant proteins, developing efficient miRNA delivery techniques holds great promise for potential therapeutic approaches in the treatment of poly Q diseases.
Author Contributions
This manuscript was primarily written by XD. Tables were produced by XD and SC. SC contributed to editing the review and contributed to the review revision. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81371271) and was also sponsored by the “Liaoning BaiQianWan Talents Program.”
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Bettencourt, C., and Lima, M. (2011). Machado-Joseph Disease: from first descriptions to new perspectives. Orphanet J. Rare Dis. 6:35. doi: 10.1186/1750-1172-6-35
Bicker, S., Lackinger, M., Weiss, K., and Schratt, G. (2014). MicroRNA-132,-134, and -138: a microRNA troika rules in neuronal dendrites. Cell. Mol. Life Sci. 71, 3987–4005. doi: 10.1007/s00018-014-1671-7
Borel, F., van Logtenstein, R., Koornneef, A., Maczuga, P., Ritsema, T., Petry, H., et al. (2011). In vivo knock-down of multidrug resistance transporters ABCC1 and ABCC2 by AAV-delivered shRNAs and by artificial miRNAs. J. RNAi Gene Silencing 7, 434–442.
Borgonio-Cuadra, V. M., Valdez-Vargas, C., Romero-Córdoba, S., Hidalgo-Miranda, A., Tapia-Guerrero, Y., Cerecedo-Zapata, C. M., et al. (2019). Wide profiling of circulating MicroRNAs in spinocerebellar ataxia type 7. Mol. Neurobiol. doi: 10.1007/s12035-019-1480-y [Epub ahead of print]
Boudreau, R. L., Mcbride, J. L., Martins, I., Shen, S., Xing, Y., Carter, B. J., et al. (2009). Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 17, 1053–1063. doi: 10.1038/mt.2009.17
Carmona, V., Cunha-Santos, J., Onofre, I., Simoes, A. T., Vijayakumar, U., Davidson, B. L., et al. (2017). Unravelling endogenous microRNA system dysfunction as a new pathophysiological mechanism in Machado-Joseph disease. Mol. Ther. 25, 1038–1055. doi: 10.1016/j.ymthe.2017.01.021
Carroll, J. B., Warby, S. C., Southwell, A. L., Doty, C. N., Greenlee, S., Skotte, N., et al. (2011). Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the huntington disease gene/allele-specific silencing of mutant huntingtin. Mol. Ther. 19, 2178–2185. doi: 10.1038/mt.2011.201
Chang, K.-H., Wu, Y.-R., and Chen, C.-M. (2017). Down-regulation of miR-9*in the peripheral leukocytes of Huntington’s disease patients. Orphanet J. Rare Dis. 12:185. doi: 10.1186/s13023-017-0742-x
Chau, A., and Kalsotra, A. (2015). Developmental insights into the pathology of and therapeutic strategies for DM1: back to the basics. Dev. Dyn. 244, 377–390. doi: 10.1002/dvdy.24240
Chen, D., Hu, S., Wu, Z., Liu, J., and Li, S. (2018). The role of MiR-132 in regulating neural stem cell proliferation, differentiation and neuronal maturation. Cell. Physiol. Biochem. 47, 2319–2330. doi: 10.1159/000491543
Chen, X., Liang, H., Zhang, J., Zen, K., and Zhang, C.-Y. (2012). Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 22, 125–132. doi: 10.1016/j.tcb.2011.12.001
Chen, J., Qi, Y., Liu, C.-F., Lu, J.-M., Shi, J., and Shi, Y. (2018). MicroRNA expression data analysis to identify key miRNAs associated with Alzheimer’s disease. J. Gene Med. 20:e3014. doi: 10.1002/jgm.3014
Cheng, L.-C., Pastrana, E., Tavazoie, M., and Doetsch, F. (2009). miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 12, 399–408. doi: 10.1038/nn.2294
Conaco, C., Otto, S., Han, J. J., and Mandel, G. (2006). Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl. Acad. Sci. U S A 103, 2422–2427. doi: 10.1073/pnas.0511041103
Costa, M. D. C., Luna-Cancalon, K., Fischer, S., Ashraf, N. S., Ouyang, M., Dharia, R. M., et al. (2013). Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol. Ther. 21, 1898–1908. doi: 10.1038/mt.2013.144
Cvetanovic, M., Hu, Y.-S., and Opal, P. (2017). Mutant Ataxin-1 inhibits neural progenitor cell proliferation in SCA1. Cerebellum 16, 340–347. doi: 10.1007/s12311-016-0794-9
Das, E., Jana, N. R., and Bhattacharyya, N. P. (2013). MicroRNA-124 targets CCNA2 and regulates cell cycle in STHdh(Q121)/Hdh(Q121) cells. Biochem. Biophys. Res. Commun. 437, 217–224. doi: 10.1016/j.bbrc.2013.06.041
David, G., Abbas, N., Stevanin, G., Durr, A., Yvert, G., Cancel, G., et al. (1997). Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70. doi: 10.1038/ng0997-65
Díez-Planelles, C., Sánchez-Lozano, P., Crespo, M. C., Gil-Zamorano, J., Ribacoba, R., Gonzalez, N., et al. (2016). Circulating microRNAs in Huntington’s disease: emerging mediators in metabolic impairment. Pharmacol. Res. 108, 102–110. doi: 10.1016/j.phrs.2016.05.005
Durr, A. (2010). Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 9, 885–894. doi: 10.1016/s1474-4422(10)70183-6
Femminella, G. D., Ferrara, N., and Rengo, G. (2015). The emerging role of microRNAs in Alzheimer’s disease. Front. Physiol. 6:40. doi: 10.3389/fphys.2015.00040
Filipowicz, W., Bhattacharyya, S. N., and Sonenberg, N. (2008). Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 9, 102–114. doi: 10.1038/nrg2290
Garden, G. A., and La Spada, A. R. (2008). Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum 7, 138–149. doi: 10.1007/s12311-008-0027-y
Gaughwin, P. M., Ciesla, M., Lahiri, N., Tabrizi, S. J., Brundin, P., and Bjorkqvist, M. (2011). Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 20, 2225–2237. doi: 10.1093/hmg/ddr111
Ghatak, S., and Raha, S. (2018). β catenin is regulated by its subcellular distribution and mutant huntingtin status in Huntington’s disease cell STHdhQ111/HdhQ111. Biochem. Biophys. Res. Commun. 503, 359–364. doi: 10.1016/j.bbrc.2018.06.034
Goodall, E. F., Heath, P. R., Bandmann, O., Kirby, J., and Shaw, P. J. (2013). Neuronal dark matter: the emerging role of microRNAs in neurodegeneration. Front. Cell. Neurosci. 7:178. doi: 10.3389/fncel.2013.00178
Graham, S. F., Pan, X., Yilmaz, A., Macias, S., Robinson, A., Mann, D., et al. (2018). Targeted biochemical profiling of brain from Huntington’s disease patients reveals novel metabolic pathways of interest. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2430–2437. doi: 10.1016/j.bbadis.2018.04.012
Harding, R. J., and Tong, Y.-F. (2018). Proteostasis in Huntington’s disease: disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 39, 754–769. doi: 10.1038/aps.2018.11
He, X.-H., Lin, F., and Qin, Z.-H. (2010). Current understanding on the pathogenesis of polyglutamine diseases. Neurosci. Bull. 26, 247–256. doi: 10.1007/s12264-010-0113-2
Holmans, P. A., Massey, T. H., and Jones, L. (2017). Genetic modifiers of Mendelian disease: huntington’s disease and the trinucleotide repeat disorders. Hum. Mol. Genet. 26, R83–R90. doi: 10.1093/hmg/ddx261
Hoss, A. G., Kartha, V. K., Dong, X., Latourelle, J. C., Dumitriu, A., Hadzi, T. C., et al. (2014). MicroRNAs located in the Hox gene clusters are implicated in huntington’s disease pathogenesis. PLoS Genet. 10:e1004188. doi: 10.1371/journal.pgen.1004188
Hoss, A. G., Labadorf, A., Latourelle, J. C., Kartha, V. K., Hadzi, T. C., Gusella, J. F., et al. (2015a). miR-10b-5p expression in Huntington’s disease brain relates to age of onset and the extent of striatal involvement. BMC Med. Genomics 8:10. doi: 10.1186/s12920-015-0083-3
Hoss, A. G., Lagomarsino, V. N., Frank, S., Hadzi, T. C., Myers, R. H., and Latourelle, J. C. (2015b). Study of plasma-derived miRNAs mimic differences in huntington’s disease brain. Mov. Disord. 30, 1961–1964. doi: 10.1002/mds.26457
Hu, J., Liu, J., and Corey, D. R. (2010). Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem. Biol. 17, 1183–1188. doi: 10.1016/j.chembiol.2010.10.013
Huang, W. (2017). “MicroRNAs: biomarkers, diagnostics, and therapeutics,” in Bioinformatics in MicroRNA Research, eds J. Huang, G. M. Borchert, D. Dou, J. Huan, W. Lan, M. Tan and B. Wu (New York, NY: Humana Press), 57–67.
Huang, Y., Shen, X. J., Zou, Q., and Zhao, Q. L. (2010). Biological functions of microRNAs. Bioorg. Khim. 36, 747–752. doi: 10.1134/s1068162010060026
Huang, F., Zhang, L., Long, Z., Chen, Z., Hou, X., Wang, C., et al. (2014). miR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett. 588, 4791–4798. doi: 10.1016/j.febslet.2014.11.013
Jacobsen, J. C., Bawden, C. S., Rudiger, S. R., Mclaughlan, C. J., Reid, S. J., Waldvogel, H. J., et al. (2010). An ovine transgenic Huntington’s disease model. Hum. Mol. Genet. 19, 1873–1882. doi: 10.1093/hmg/ddq063
Jamwal, S., and Kumar, P. (2015). Antidepressants for neuroprotection in huntington’s disease: a review. Eur. J. Pharmacol. 769, 33–42. doi: 10.1016/j.ejphar.2015.10.033
Johnson, R., and Buckley, N. J. (2009). Gene dysregulation in huntington’s disease: REST, microRNAs and beyond. Neuromolecular Med. 11, 183–199. doi: 10.1007/s12017-009-8063-4
Johnson, R., Zuccato, C., Belyaev, N. D., Guest, D. J., Cattaneo, E., and Buckley, N. J. (2008). A microRNA-based gene dysregulation pathway in huntington’s disease. Neurobiol. Dis. 29, 438–445. doi: 10.1016/j.nbd.2007.11.001
Jovicic, A., Zaldivar Jolissaint, J. F., Moser, R., Silva Santos Mde, F., and Luthi-Carter, R. (2013). MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific huntington’s disease-related mechanisms. PLoS One 8:e54222. doi: 10.1371/journal.pone.0054222
Karres, J. S., Hilgers, V., Carrera, I., Treisman, J., and Cohen, S. M. (2007). The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell 131, 136–145. doi: 10.1016/j.cell.2007.09.020
Keiser, M. S., Kordasiewicz, H. B., and Mcbride, J. L. (2016). Gene suppression strategies for dominantly inherited neurodegenerative diseases: lessons from huntington’s disease and spinocerebellar ataxia. Hum. Mol. Genet. 25, R53–R64. doi: 10.1093/hmg/ddv442
Klein, M. E., Lioy, D. T., Ma, L., Impey, S., Mandel, G., and Goodman, R. H. (2007). Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 10, 1513–1514. doi: 10.1038/nn2010
Kocerha, J., Xu, Y., Prucha, M. S., Zhao, D., and Chan, A. W. S. (2014). microRNA-128a dysregulation in transgenic huntington’s disease monkeys. Mol. Brain 7:46. doi: 10.1186/1756-6606-7-46
Koscianska, E., and Krzyzosiak, W. J. (2014). Current understanding of the role of microRNAs in spinocerebellar ataxias. Cerebellum Ataxias 1:7. doi: 10.1186/2053-8871-1-7
Kumar, S., Vijayan, M., Bhatti, J. S., and Reddy, P. H. (2017). MicroRNAs as peripheral biomarkers in aging and age-related diseases. Prog. Mol. Biol. Transl. Sci. 146, 47–94. doi: 10.1016/bs.pmbts.2016.12.013
Kunkanjanawan, T., Carter, R. L., Prucha, M. S., Yang, J., Parnpai, R., and Chan, A. W. S. (2016). miR-196a ameliorates cytotoxicity and cellular phenotype in transgenic Huntington’s disease monkey neural cells. PLoS One 11:e0162788. doi: 10.1371/journal.pone.0162788
La Spada, A. R., Wilson, E. M., Lubahn, D. B., Harding, A. E., and Fischbeck, K. H. (1991). Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79. doi: 10.1038/352077a0
Langfelder, P., Gao, F., Wang, N., Howland, D., Kwak, S., Vogt, T. F., et al. (2018). MicroRNA signatures of endogenous Huntingtin CAG repeat expansion in mice. PLoS One 13:e0190550. doi: 10.1371/journal.pone.0190550
Laprairie, R. B., Petr, G. T., Sun, Y., Fischer, K. D., Denovan-Wright, E. M., and Rosenberg, P. A. (2019). Huntington’s disease pattern of transcriptional dysregulation in the absence of mutant huntingtin is produced by knockout of neuronal GLT-1. Neurochem. Int. 123, 85–94. doi: 10.1016/j.neuint.2018.04.015
Lau, P., and de Strooper, B. (2010). Dysregulated microRNAs in neurodegenerative disorders. Semin Cell Dev. Biol. 21, 768–773. doi: 10.1016/j.semcdb.2010.01.009
Lee, Y., Samaco, R. C., Gatchel, J. R., Thaller, C., Orr, H. T., and Zoghbi, H. Y. (2008). miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat. Neurosci. 11, 1137–1139. doi: 10.1038/nn.2183
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Li, M., Zhou, Y., Xia, T., Zhou, X., Huang, Z., Zhang, H., et al. (2018). Circulating microRNAs from the miR-106a-363 cluster on chromosome X as novel diagnostic biomarkers for breast cancer. Breast Cancer Res. Treat. 170, 257–270. doi: 10.1007/s10549-018-4757-3
Liu, T., Im, W., Mook-Jung, I., and Kim, M. (2015). MicroRNA-124 slows down the progression of Huntington’s disease by promoting neurogenesis in the striatum. Neural Regen. Res. 10, 786–791. doi: 10.4103/1673-5374.156978
Liu, H., Tang, T. S., and Guo, C. (2018). Epigenetic profiles in polyglutamine disorders. Epigenomics 10, 9–25. doi: 10.2217/epi-2017-0089
Makeyev, E. V., Zhang, J., Carrasco, M. A., and Maniatis, T. (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative Pre-mRNA splicing. Mol. Cell 27, 435–448. doi: 10.1016/j.molcel.2007.07.015
Martí, E., Pantano, L., Bañez-Coronel, M., Llorens, F., Miñones-Moyano, E., Porta, S., et al. (2010). A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 38, 7219–7235. doi: 10.1093/nar/gkq575
Martin, J.-J., Van Regemorter, N., Del-Favero, J., Löfgren, A., and Van Broeckhoven, C. (1999). Spinocerebellar ataxia type 7 (SCA7)—correlations between phenotype and genotype in one large Belgian family. J. Neurol. Sci. 168, 37–46. doi: 10.1016/s0022-510x(99)00176-8
Mathonnet, G., Fabian, M. R., Svitkin, Y. V., Parsyan, A., Huck, L., Murata, T., et al. (2007). MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex elF4F. Science 317, 1764–1767. doi: 10.1126/science.1146067
McBride, J. L., Pitzer, M. R., Boudreau, R. L., Dufour, B., Hobbs, T., Ojeda, S. R., et al. (2011). Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther. 19, 2152–2162. doi: 10.1038/mt.2011.219
McCann, C., Holohan, E. E., Das, S., Dervan, A., Larkin, A., Lee, J. A., et al. (2011). The Ataxin-2 protein is required for microRNA function and synapse-specific long-term olfactory habituation. Proc. Natl. Acad. Sci. U S A 108, E655–E662. doi: 10.1073/pnas.1107198108
Meza-Sosa, K. F., Valle-García, D., Pedraza-Alva, G., and Pérez-Martinez, L. (2012). Role of microRNAs in central nervous system development and pathology. J. Neurosci. Res. 90, 1–12. doi: 10.1002/jnr.22701
Miniarikova, J., Zanella, I., Huseinovic, A., van der Zon, T., Hanemaaijer, E., Martier, R., et al. (2016). Design, characterization, and lead selection of therapeutic miRNAs targeting huntingtin for development of gene therapy for Huntington’s disease. Mol. Ther. Nucleic Acids 5:e297. doi: 10.1038/mtna.2016.7
Miyazaki, Y., Adachi, H., Katsuno, M., Minamiyama, M., Jiang, Y. M., Huang, Z., et al. (2012). Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat. Med. 18, 1136–1141. doi: 10.1038/nm.2791
Miyazaki, Y., Du, X., Muramatsu, S., and Gomez, C. M. (2016). An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci. Transl. Med. 8:347ra394. doi: 10.1126/scitranslmed.aaf5660
Moumné, L., Betuing, S., and Caboche, J. (2013). Multiple aspects of gene dysregulation Huntington’s disease. Front. Neurol. 4:127. doi: 10.3389/fneur.2013.00127
Persengiev, S., Kondova, I., Otting, N., Koeppen, A. H., and Bontrop, R. E. (2011). Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol. Aging 32, 2316.e17–2316.e27. doi: 10.1016/j.neurobiolaging.2010.03.014
Pourshafie, N., Lee, P. R., Chen, K. L., Harmison, G. G., Bott, L. C., Fischbeck, K. H., et al. (2018). Systemic delivery of MicroRNA using recombinant adeno-associated virus serotype 9 to treat neuromuscular diseases in rodents. J. Vis. Exp. 138:e55724. doi: 10.3791/55724
Pourshafie, N., Lee, P. R., Chen, K.-L., Harmison, G. G., Bott, L. C., Katsuno, M., et al. (2016). MiR-298 counteracts mutant androgen receptor toxicity in spinal and bulbar muscular atrophy. Mol. Ther. 24, 937–945. doi: 10.1038/mt.2016.13
Ramachandran, P. S., Bhattarai, S., Singh, P., Boudreau, R. L., Thompson, S., Laspada, A. R., et al. (2014a). RNA interference-based therapy for spinocerebellar ataxia type 7 retinal degeneration. PLoS One 9:e95362. doi: 10.1371/journal.pone.0095362
Ramachandran, P. S., Boudreau, R. L., Schaefer, K. A., La Spada, A. R., and Davidson, B. L. (2014b). Nonallele specific silencing of ataxin-7 improves disease phenotypes in a mouse model of SCA7. Mol. Ther. 22, 1635–1642. doi: 10.1038/mt.2014.108
Reed, E. R., Latourelle, J. C., Bockholt, J. H., Bregu, J., Smock, J., Paulsen, J. S., et al. (2018). MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology 90, E264–E272. doi: 10.1212/wnl.0000000000004844
Ridolfi, B., and Abdel-Haq, H. (2017). Neurodegenerative disorders treatment: the MicroRNA role. Curr. Gene Ther. 17, 327–363. doi: 10.2174/1566523218666180119120726
Ritchie, W. (2017). “microRNA target prediction,” in Cancer Gene Networks, eds U. Kasid and R. Clarke (Totowa, NJ: Humana Press), 193–200.
Rodríguez-Lebrón, E., Costa Mdo, C., Luna-Cancalon, K., Peron, T. M., Fischer, S., Boudreau, R. L., et al. (2014). Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice (vol 21, pg 1909, 2013). Mol. Ther. 22, 891–891. doi: 10.1038/mt.2013.152
Rodriguez-Lebron, E., Liu, G., Keiser, M., Behlke, M. A., and Davidson, B. L. (2013). Altered Purkinje cell miRNA expression and SCA1 pathogenesis. Neurobiol. Dis. 54, 456–463. doi: 10.1016/j.nbd.2013.01.019
Roshan, R., Choudhary, A., Bhambri, A., Bakshi, B., Ghosh, T., and Pillai, B. (2017). microRNA dysregulation in polyglutamine toxicity of TATA-box binding protein is mediated through STAT1 in mouse neuronal cells. J. Neuroinflammation 14:155. doi: 10.1186/s12974-017-0925-3
Roshan, R., Ghosh, T., Gadgil, M., and Pillai, B. (2012). Regulation of BACE1 by miR-29a/b in a cellular model of spinocerebellar ataxia 17. RNA Biol. 9, 891–899. doi: 10.4161/rna.19876
Roshan, R., Shridhar, S., Sarangdhar, M. A., Banik, A., Chawla, M., Garg, M., et al. (2014). Brain-specific knockdown of miR-29 results in neuronal cell death and ataxia in mice. RNA 20, 1287–1297. doi: 10.1261/rna.044008.113
Saraiva, C., Esteves, M., and Bernardino, L. (2017). MicroRNA: basic concepts and implications for regeneration and repair of neurodegenerative diseases. Biochem. Pharmacol. 141, 118–131. doi: 10.1016/j.bcp.2017.07.008
Seredenina, T., Gokce, O., and Luthi-Carter, R. (2011). Decreased striatal RGS2 expression is neuroprotective in Huntington’s disease (HD) and exemplifies a compensatory aspect of HD-induced gene regulation. PLoS One 6:e22231. doi: 10.1371/journal.pone.0022231
Shah, S. Z. A., Zhao, D., Hussain, T., Sabir, N., and Yang, L. (2018). Regulation of MicroRNAs-mediated autophagic flux: a new regulatory avenue for neurodegenerative diseases with focus on prion diseases. Front. Aging Neurosci. 10:139. doi: 10.3389/fnagi.2018.00139
Shi, Y., Huang, F., Tang, B., Li, J., Wang, J., Shen, L., et al. (2014). MicroRNA profiling in the serums of SCA3/MJD patients. Int. J. Neurosci. 124, 97–101. doi: 10.3109/00207454.2013.827679
Sinha, M., Ghose, J., Das, E., and Bhattarcharyya, N. P. (2010). Altered microRNAs in STHdh(Q121)/Hdh(Q121) cells: miR-146a targets TBP. Biochem. Biophys. Res. Commun. 396, 742–747. doi: 10.1016/j.bbrc.2010.05.007
Sinha, S., Verma, S., Singh, A., Somvanshi, P., and Grover, A. (2018). Simulation based investigation of deleterious nsSNPs in ATXN2 gene and its structural consequence toward spinocerebellar ataxia. J. Cell. Biochem. 119, 499–510. doi: 10.1002/jcb.26209
Snyman, M. C., Solofoharivelo, M.-C., Souza-Richards, R., Stephan, D., Murray, S., and Burger, J. T. (2017). The use of high-throughput small RNA sequencing reveals differentially expressed microRNAs in response to aster yellows phytoplasma-infection in Vitis vinifera cv. ‘Chardonnay’. PLoS One 12:e0182629. doi: 10.1371/journal.pone.0182629
Soldati, C., Bithell, A., Johnston, C., Wong, K.-Y., Stanton, L. W., and Buckley, N. J. (2013). Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington’s disease. J. Neurochem. 124, 418–430. doi: 10.1111/jnc.12090
Sonkoly, E., Ståhle, M., and Pivarcsi, A. (2008). MicroRNAs and immunity: novel players in the regulation of normal immune function and inflammation. Semin. Cancer Biol. 18, 131–140. doi: 10.1016/j.semcancer.2008.01.005
Southwell, A. L., Skotte, N. H., Kordasiewicz, H. B., Østergaard, M. E., Watt, A. T., Carroll, J. B., et al. (2014). In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol. Ther. 22, 2093–2106. doi: 10.1038/mt.2014.153
Stiles, D. K., Zhang, Z., Ge, P., Nelson, B., Grondin, R., Ai, Y., et al. (2012). Widespread suppression of huntingtin with convection-enhanced delivery of siRNA. Exp. Neurol. 233, 463–471. doi: 10.1016/j.expneurol.2011.11.020
Tan, J. Y., Vance, K. W., Varela, M. A., Sirey, T., Watson, L. M., Curtis, H. J., et al. (2015). Cross-talking noncoding RNAs contribute to cell-specific neurodegeneration in SCA7 (vol 21, pg 955, 2014). Nat. Struct. Mol. Biol. 22, 272–272. doi: 10.1038/nsmb0315-272b
Tan, L., Yu, J.-T., and Tan, L. (2015). Causes and consequences of MicroRNA dysregulation in neurodegenerative diseases. Mol. Neurobiol. 51, 1249–1262. doi: 10.1007/s12035-014-8803-9
van der Stijl, R., Withoff, S., and Verbeek, D. S. (2017). Spinocerebellar ataxia: miRNAs expose biological pathways underlying pervasive Purkinje cell degeneration. Neurobiol. Dis. 108, 148–158. doi: 10.1016/j.nbd.2017.08.003
Varendi, K., Kumar, A., Harma, M.-A., and Andressoo, J.-O. (2014). miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell. Mol. Life Sci. 71, 4443–4456. doi: 10.1007/s00018-014-1628-x
Vo, N., Klein, M. E., Varlamova, O., Keller, D. M., Yamamoto, T., Goodman, R. H., et al. (2006). A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis (vol 102, pg 16426, 2005). Proc. Natl. Acad. Sci. U S A 103, 825–825. doi: 10.1073/pnas.0508448102
Wakiyama, M., and Yokoyama, S. (2014). “MicroRNA-mediated deadenylation in a mammalian cell-free system,” in Polyadenylation: Methods and Protocols, eds J. Rorbach and A. J. Bobrowicz (Totowa, NJ: Humana Press), 341–351.
Weber, J. A., Baxter, D. H., Zhang, S., Huang, D. Y., Huang, K. H., Lee, M. J., et al. (2010). The MicroRNA spectrum in 12 body fluids. Clin. Chem. 56, 1733–1741. doi: 10.1373/clinchem.2010.147405
Wen, M. M. (2016). Getting miRNA therapeutics into the target cells for neurodegenerative diseases: a mini-review. Front. Mol. Neurosci. 9:129. doi: 10.3389/fnmol.2016.00129
Winter, J., Jung, S., Keller, S., Gregory, R. I., and Diederichs, S. (2009). Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 11, 228–234. doi: 10.1038/ncb0309-228
Wu, H. Z. Y., Ong, K. L., Seeher, K., Armstrong, N. J., Thalamuthu, A., Brodaty, H., et al. (2016). Circulating microRNAs as biomarkers of Alzheimer’s disease: a systematic review. J. Alzheimers Dis. 49, 755–766. doi: 10.3233/JAD-150619
Wu, J., and Xie, X. (2006). Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 7:R85. doi: 10.1186/gb-2006-7-9-r85
Xiong, J., Yu, D., Wei, N., Fu, H., Cai, T., Huang, Y., et al. (2010). An estrogen receptor α suppressor, microRNA-22, is downregulated in estrogen receptor α-positive human breast cancer cell lines and clinical samples. FEBS J. 277, 1684–1694. doi: 10.1111/j.1742-4658.2010.07594.x
Yin, S., Yu, Y., and Reed, R. (2015). Primary microRNA processing is functionally coupled to RNAP II transcription in vitro. Sci. Rep. 5:11992. doi: 10.1038/srep11992
Yushchenko, T., Deuerling, E., and Hauser, K. (2018). Insights into the aggregation mechanism of PolyQ proteins with different glutamine repeat lengths. Biophys. J. 114, 1847–1857. doi: 10.1016/j.bpj.2018.02.037
Zuccato, C., Belyaev, N., Conforti, P., Ooi, L., Tartari, M., Papadimou, E., et al. (2007). Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. 27, 6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007
Keywords: microRNAs, polyglutamine diseases, biomarker, neurodegeneration, therapy
Citation: Dong X and Cong S (2019) The Emerging Role of microRNAs in Polyglutamine Diseases. Front. Mol. Neurosci. 12:156. doi: 10.3389/fnmol.2019.00156
Received: 30 March 2019; Accepted: 04 June 2019;
Published: 19 June 2019.
Edited by:
Baojin Ding, University of Louisiana at Lafayette, United StatesReviewed by:
Ashwin S. Shetty, Harvard University, United StatesLi Zeng, National Neuroscience Institute (NNI), Singapore
Copyright © 2019 Dong and Cong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuyan Cong, congshuyan@hotmail.com