Hydrogen Sulfide Inhibits High Glucose-Induced Neuronal Senescence by Improving Autophagic Flux via Up-regulation of SIRT1
 Lei Wu1,2† Ying Chen1,2,3† Chun-Yan Wang2† Yi-Yun Tang1 Hong-Lin Huang2* Xuan Kang4 Xiang Li4 Yu-Rong Xie5
Lei Wu1,2† Ying Chen1,2,3† Chun-Yan Wang2† Yi-Yun Tang1 Hong-Lin Huang2* Xuan Kang4 Xiang Li4 Yu-Rong Xie5  Xiao-Qing Tang1,2,4*
Xiao-Qing Tang1,2,4*- 1Institute of Neuroscience, Hengyang Medical College, University of South China, Hengyang, China
- 2Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study, Institute of Pharmacy and Pharmacology, University of South China, Hengyang, China
- 3Department of Pharmacology, The Central Hospital of Hengyang, Hengyang, China
- 4Institute of Neurology, the First Affiliated Hospital, University of South China, Hengyang, China
- 5College of Chemistry and Chemical Engineering, University of South China, Hengyang, China
Hyperglycemia, a key characteristic and risk factor for diabetes mellitus (DM), causes neuronal senescence. Hydrogen sulfide (H2S) is a novel neuroprotectant. The present work was to investigate the potential effect of H2S on hyperglycemia-induced neuronal senescence and the underlying mechanisms. We found that NaHS, a donor of H2S, inhibited high glucose (HG)-induced cellular senescence in HT22 cells (an immortalized mouse hippocampal cell line), as evidenced by a decrease in the number of senescence associated-β-galactosidase (SA-β-gal) positive cells, increase in the growth of cells, and down-regulations of senescence mark proteins, p16INK4a and p21CIP1. NaHS improved the autophagic flux, which is judged by a decrease in the amount of intracellular autophagosome as well as up-regulations of LC3II/I and P62 in HG-exposed HT22 cells. Furthermore, blocked autophagic flux by chloroquine (CQ) significantly abolished NaHS-exerted improvement in the autophagic flux and suppression in the cellular senescence of GH-exposed HT22 cells, which indicated that H2S antagonizes HG-induced neuronal senescence by promoting autophagic flux. We also found that NaHS up-regulated the expression of silent mating type information regulation 2 homolog 1 (SIRT1), an important anti-aging protein, in HG-exposed HT22 cells. Furthermore, inhibition of SIRT1 by sirtinol reversed the protection of H2S against HG-induced autophagic flux blockade and cellular senescence in HT22 cells. These data indicated that H2S protects HT22 cells against HG-induced neuronal senescence by improving autophagic flux via up-regulation of SIRT1, suggesting H2S as a potential treatment strategy for hyperglycemia-induced neuronal senescence and neurotoxicity.
Introduction
Diabetes mellitus (DM) increases the risk of central nervous system disease leading to encephalopathy (Sima et al., 2004), because hyperglycemia, the mainly characterized of DM, triggers neuronal damage (Fan et al., 2016; Kumar et al., 2017). Increasing evidence demonstrated that hyperglycemia induces neuronal senescence (Reno et al., 2013; Kaur et al., 2017; Yerra et al., 2017; Zhang et al., 2019). Therefore, increasing the defenses against hyperglycemia-induced neuronal senescence represents a future strategy for diabetic encephalopathy. Hydrogen sulfide (H2S), the third gaseous signaling molecule (Wang, 2010), serves as an important neuroprotective agent in the central nervous system (Hu et al., 2009; Li et al., 2014; Wei et al., 2014). It has been confirmed that H2S protects neuronal cells against stress-induced senescence (Liu et al., 2013; Xie et al., 2014). Furthermore, H2S has emerged as an important therapeutic target for the treatment of neurological diseases (Suzuki et al., 2011; Si et al., 2013). Therefore, we speculated that H2S also protects neurons against hyperglycemia-induced cellular senescence.
Autophagic flux, which regulates cell survival, metabolism and senescence (Mizushima et al., 2008; Shen et al., 2015), includes the formation of autophagic bodies and autophagic lysosomes, and the degradation of autophagy lysosomal inclusions (Lim et al., 2014). Recent evidence suggests that blocked autophagic flux contributes to neurodegeneration while improving autophagic flux has antagonistic effects on neurodegeneration (Nixon, 2013; Menzies et al., 2015; Lumkwana et al., 2017). Moreover, it has been reported that autophagic flux impairment induced by hyperglycemia is an essential pathogenic factor contributing to several DM-associated complications (Xie et al., 2016; Li Y. et al., 2017). Therefore, we hypothesized that block of autophagic flux mediates high glucose (HG)-induced neuronal senescence. Notably, H2S plays an important role in regulating autophagic flux (Xie et al., 2015). Consequently, we speculated that the protection of H2S against HG-induced neuronal senescence is by improving autophagic flux.
Silent mating type information regulation 2 homolog 1 (SIRT1) is an NAD+-dependent deacetylase (Cohen et al., 2004; Bordone and Guarente, 2005). In the brain, SIRT1 is mainly expressed in neurons and has numerous biological functions, including regulation differentiation, metabolism, cell survival, and aging (Qadir and Anwar, 2017). It has been reported that SIRT1 activation attenuates hyperglycemia and plays an important mediatory role in treating diabetic complication (Li X. N. et al., 2017; Zhang Q. et al., 2018; Zhang Y. et al., 2018). Furthermore, SIRT1 improves autophagic flux (Zhang et al., 2016; Liu et al., 2017). Based on that H2S up-regulates the expression of SIRT1 in neuron (Li et al., 2014), we hypothesized that SIRT1 mediates the protection of H2S against HG-triggered neuronal senescence by improving autophagic flux.
The present work identified that H2S inhibits HG-induced cellular senescence in HT22 cells, an immortalized mouse hippocampal cell line (Liu et al., 2009), and that H2S significantly attenuated HG-induced autophagy flux obstruction in HT22 cells. Furthermore, Chloroquine (CQ), an autophagic flux inhibitor, reversed the inhibitory roles of H2S in HG-induced autophagic flux obstruction and cellular senescence in HT22 cells. We also found that H2S significantly up-regulated SIRT1 expression in HG-exposed HT22 cells. Furthermore, Sirtinol, a SIRT1 inhibitor, reversed the inhibitory roles of H2S in HG-induced autophagic flux obstruction and cellular senescence in HT22 cells. Taken together, we demonstrated the protection of H2S against HG-induced cellular senescence of HT22 cells, as a result of improvement of autophagic flux via up-regulation of SIRT1.
Materials and Methods
Materials
Glucose, Sodium hydrosulfide (NaHS, a donor of H2S), CQ (inhibitor of autophagic flux), 3-methyladenine (3-MA, inhibitor of autophagic flux), Sirtinol (the specific inhibitor of SIRT-1), and trypan blue were obtained from Sigma Chemical Company (St. Louis, MO, USA). Specific monoclonal antibodies p62 and LC3 were purchased from Cell Signaling Technology. Specific monoclonal anti-SIRT1 antibody was obtained from Abcam (Hong Kong, China). Specific antibodies of p16INK4a and p21CIP1 were purchased from OriGene Technologies.
Methods
Cell Cultures
The mouse hippocampal neuron HT22 cell line was obtained from China Center for Type Culture Collection (Wuhan, China) and was used for all experiments. The cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM), which contains 10% FBS, 100 IU/mL of penicillin and 100 mg /mL of streptomycin, at 37°C and 5% CO2, under humidified atmosphere. The culture medium was changed every 3 days to ensure stable nutritional level.
Senescence Associated-β-Galactosidase (SA-β-gal) Assay
HT22 cells were fixed with 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) for 10 min. After three washes with PBS, HT22 cells were dyed with senescence associated-β-galactosidase (SA-β-gal) incubation overnight at 37°C under dry environment. HT22 cells were observed under an optical microscope (CKX41 SF, OLYMPUS, Japan). Living cells displayed normal nuclear size and morphology, whereas senescent cells with the characteristic morphology (enlarged and flattened) and positivity or SA-β-gal staining (turquoise color). The number of SA-β-gal-positive cell was counted to determine the percentage of senescent cells.
HT22 Cells Growth Curve Draw by Trypan Blue Counting
HT22 cells in logarithmic growth phase were seeded in 24-well plate with 1 × 104 cells at each well. After the density of HT22 cells at 80%, cells were exposed to different experimental treatment. Three holes were randomly sampled for trypan blue count every day and counted for 7 days. For trypan blue staining, cell suspension was incubated with trypan blue in equal volume for 2 min and counting the unstained living cells on hemocytometer. The growth curve of the cell was plotted with the mean value of cell density per day as the ordinate and the growing days as the abscissa.
Transmission Electron Microscope
HT22 cells in logarithmic phase growth were seeded in a culture dish and were exposed to different experimental treatment for 48 h. After the treatment, cells were washed twice with PBS and fixed with 2.5% glutaraldehyde solution for 30 min at 4°C. Cells were harvested in a 1.5 ml of EP tube with 2.5% glutaraldehyde solution and conserved at 4°C. Samples were tested in Shanghai Fu cheng biology co., Ltd. Briefly, cells were dehydrated, embedded, sliced into 60 nm sections and stained with uranyl acetate at room temperature for 15 min, followed by lead citrate at room temperature for 15 min. Autophagosome and autolysosome formation were observed by transmission electron microscope.
Western Blots Analysis
HT22 cell lysates were used to detect the protein expressions of p16INK4a, p21CIPL, SIRT1, LC3, and p62. After the exposure was terminated, cells were washed with PBS and then lysed in an ice-cold lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM phenyl methyl sulphonyl fluoride (PMSF), 1 mM Na3VO4, leupeptin, and EDTA) for 30 min. Liquid supernatant was collected after centrifugation for 10 min at 5,000 g. The protein concentration was determined by BCA Protein Assay Kit (Beyotime, Shanghai, China). The samples were diluted with sample buffer (Beyotime, Shanghai, China) at 100°C for 5 min and were separated by 10% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE). And then, the proteins were transferred electrophoretically to polyvinylidene fluoride (PVDF) membranes. Non-specific protein binding was incubated with 5% non-fat dried milk in TBST buffer (pH 7.6, 3.03 g Tris base, 18.8 g glycine, 1 g SDS, plus 1 ml Tween-20) for 2 h at room temperature. The membranes were incubated overnight at 4°C with diluted primary antibody (anti-p16INK4a, 1:2,000; anti-p21CIPL, 1:2,000; anti-SIRT1, 1:2,000; anti-p62, 1:1,000 and anti-LC3, 1:1,000). The membranes were then washed three times for 5 min each time with TBST, and incubated with HRP-conjugated secondary antibody (1:5,000, Protein Tech, SA00001-2) at room temperature for 2 h. Protein bands were analyzed using the enhanced chemiluminescence detection system (BeyoECL Plus kit, Beyotime, P0018). Integrated optical densities were analyzed by using ImageJ software.
Statistical Analysis
Statistical analysis of all data was performed by SPSS 18.0 software. Data are displayed as the mean ± SEM. Umbers of the biological replicates (n) are noted in the figure legends. The significance of intergroup differences was evaluated by LSD-t to compare the data of different experimental groups with multiple comparisons. Differences were considered significant at P < 0.05.
Results
H2S Antagonizes HG-Induced Cellular Senescence in HT22 Cells
We investigated the effect of H2S on HG-induced cellular senescence in HT22 cells. As illustrated in Figure 1A, the cell growth curves show that the growth of HT22 cell exposed by HG (27 mg/ml, for 48 h) was increased by pretreatment with 100, 200, or 400 μM of NaHS for 30 min (Day 1, F(5,12)= 39.718, P < 0.001; Day 2, F(5,12)= 83.543, P < 0.001; Day 3, F(5,12) = 235.790, P < 0.001; Day 4, F(5,12) = 149.725, P < 0.001; Day 5, F(5,12)= 65.619, P < 0.001; Day 6, F(5,12) = 10.661, P < 0.001; Day 7, F(5,12) = 6.735, P < 0.01), respectively. In addition, the number of SA-β-gal positive cell was increased in HT22 cells treated with 27 mg/ml of HG for 48 h (F(3,16) = 393.481, P < 0.001); However, pretreated with NaHS (200 μM for 30 min) reduced the number of SA-β-gal positives cell in HT22 cells exposed to 27 mg/ml of HG for 48 h (F(3,16) = 393.481, P < 0.001, Figure 1B). Furthermore, pretreatment of HT22 cells with 200 μM NaHS for 30 min significantly reversed the up-regulation of p16INK4a (F(3,8) = 4.257, P < 0.05; Figure 1C) and p21CIP1 (F(3,8) = 3.559, P < 0.05; Figure 1D) protein expression induced by HG (27 mg/ml, for 48 h). Taken together, these data indicated that H2S inhibits HG-induced cellular senescence in HT22 cells.
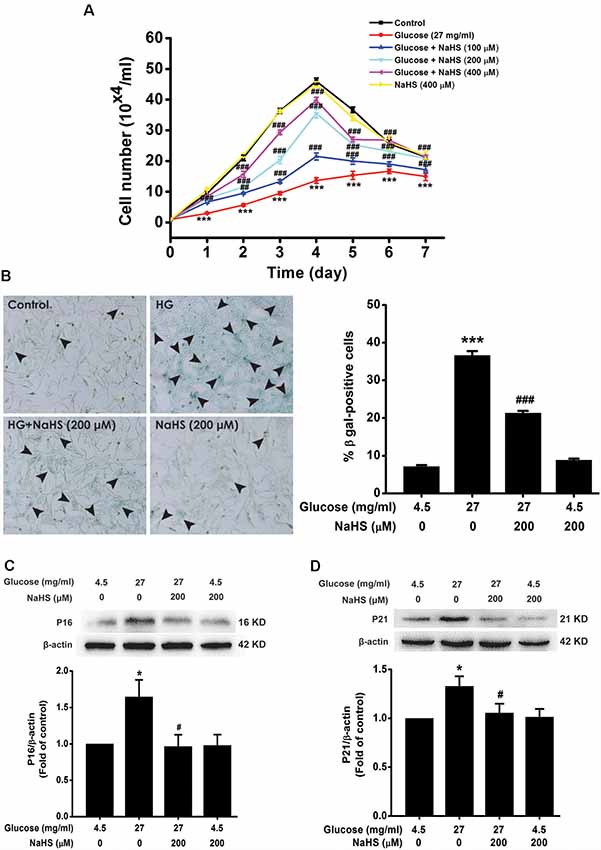
Figure 1. Effect of hydrogen sulfide (H2S) on high glucose (HG)-elicited cellular senescence in HT22 cells. (A) Cell growth curves were generated using Trypan blue stain assays. (B) Representative images of senescent cells that were stained using senescence associated-β-galactosidase (SA-β-gal; Left, Magnification ×100) and quantitative analysis of the SA-β-gal positive cells (Right). The black arrows indicate the senescent cells, (C,D) The expressions of p16INK4a and p21CIP1 in HT22 cells were detected by Western blotting. Values were expressed as the mean ± SEM, n = 3. *P < 0.05, ***P < 0.001, vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. HG-treated alone group.
H2S Rescues HG-Impaired Autophagic Flux in HT22 Cells
To explore whether autophagic flux involves in the protection of H2S against HG-induced cellular senescence in HT22 cells, we investigated the effect of NaHS on the autophagic flux in HG-exposed HT22 cells. The number of autophagosomes in HT22 cells exposed to HG (40.5 mg/ml, for 48 h) was higher than that of the control group, which was suppressed by pretreatment with NaHS (200 μM, for 30 min; Figure 2A). In addition, after exposure to HG (27, 40.5 mg/ml) for 48 h, the expressions of LC3 II/I (F(3,8) = 25.027, P < 0.001; Figure 2B) and P62 (F(3,8) = 16.177, P < 0.01; Figure 2C) were significantly increased in HT22 cells. However, pretreatment with NaHS (200 μM, for 30 min) significantly down-regulated the expressions of LC3 II/I (F(3,8) = 4.452, P < 0.05; Figure 2D) and P62 (F(3,8) = 11.547, P < 0.05; Figure 2E) in HG-exposed HT22 cells. Collectively, these results demonstrated that H2S reverses HG-induced autophagic flux block in HT22 cells.
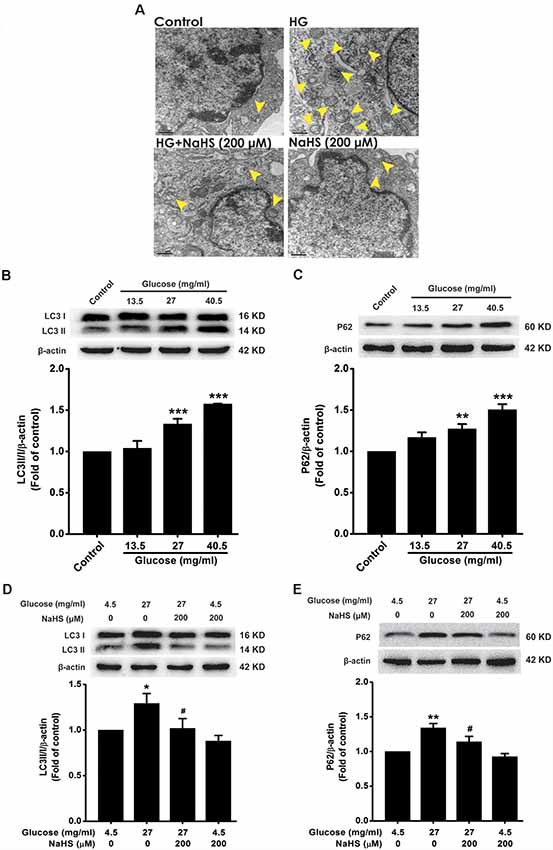
Figure 2. Effect of H2S on HG-elicited disruption of autophagic flux in HT22 cells. (A) Transmission electron microscope observed the number of autophagosome. The yellow arrows indicate autophagosome. (B–E) The expressions of LC3II/LC3I and P62 in HT22 cells were detected by Western blotting. Values were expressed as the mean ± SEM, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, vs. control group; #P < 0.05, vs. HG-treated alone group.
Blockage of Autophagic Flux Reverses the Improving Effect of H2S on Autophagic Flux in HG-Exposed HT22 Cells
3-MA and CQ are the specific autophagic flux inhibitors, which inhibit initial and late stage of autophagic flux (Caro et al., 1988; Ye et al., 2016), respectively. Next, we explored whether 3-MA and CQ abolish the improving effect of H2S on autophagic flux in HG-exposed HT22 cells. We found that CQ (10 μM) reversed the suppression of NaHS (200 μM) in HG (40.5 mg/ml, for 48 h)-induced increase in the number of autophagosome (Figure 3A) as well as upregulations of LC3II/LC3I (F(4,10) = 6.151, P < 0.05; Figure 3B) and P62 (F(4,10) = 13.498, P < 0.01; Figure 3C) in HT22 cells. However, 3-MA (10 mM) had no significant effect on the suppressive effect of NaHS (200 μM) on HG (40.5 mg/ml, for 48 h)-induced increase in the number of autophagosome (Figure 3D) as well as upregulations of LC3II/LC3I (F(4,10) = 4.189, P > 0.05; Figure 3E) and P62 (F(4,10) = 5.900, P > 0.05; Figure 3F) in HT22 cells. Taken together, these results indicated that H2S overcomes HG-induced disturbance in autophagic flux via restoring the late stage of autophagic flux.
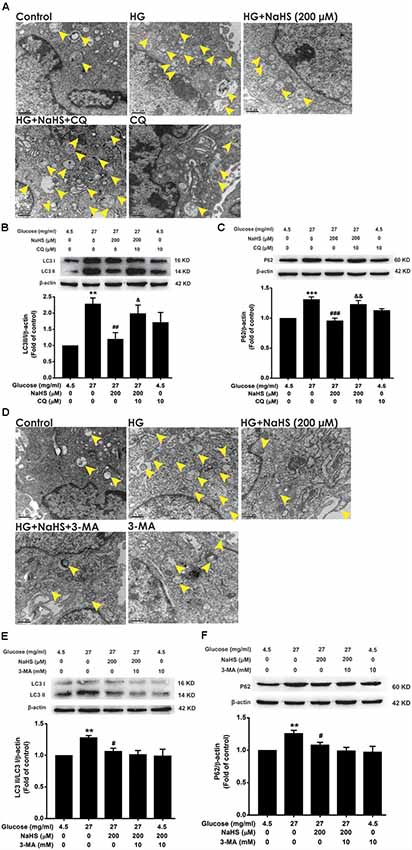
Figure 3. Effects of chloroquine (CQ) and 3-methyladenine (3-MA) on the improving role of H2S in autophagic flux in HG-exposed HT22 cells. (A,D) Transmission electron microscope images of HT22 cells showing the change in the number of autophagosome. The yellow arrows indicate autophagosome. (B,C,E,F) The expressions of LC3II/LC3I and P62 in HT22 cells were detected by Western blotting. Values were expressed as the mean ± SEM, n = 3. **P < 0.01, ***P < 0.001, vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. HG-treated alone group; &P < 0.05, &&P < 0.01, vs. co-treated with NaHS and HG group.
Blockage of Autophagic Flux by CQ Reverses the Protection of H2S Against HG-Induced Cellular Senescence in HT22 Cells
We further examined the effects of CQ and 3-MA on the protection of H2S against HG-induced cellular senescence in HT22 cells. Pretreatment with CQ (10 μM) reversed the suppression of NaHS (200 μM) in HG (40.5 mg/ml, for 48 h)-induced inhibition of the cell growth (Day 1, F(4,10) = 32.703, P < 0.001; Day 2, F(4,10) = 42.562, P < 0.001; Day 3, F(4,10) = 94.889, P < 0.001; Day 4, F(4,10)= 58.601, P < 0.001; Day 5, F(4,10) = 42.892, P < 0.001; Day 6, F(4,10) = 26.198, P < 0.01; Day 7, F(4,10) = 23.688, P < 0.01; Figure 4A), increase in the number of SA-β-gal positive cells (F(4,20) = 330.457, P < 0.001; Figure 4B), as well as up-regulations of p16INK4a (F(4,10) = 4.161, P < 0.05; Figure 4C) and p21CIP1 (F(4,10) = 11.886, P < 0.05; Figure 4D) in HT22 cells. However, pretreated HT22 cells with 3-MA (10 mM) had no significant effect on the suppressive effect of NaHS (200 μM) on HG (40.5 mg/ml, for 48 h)-induced inhibition of the cell growth (Day 1, F(4,10) = 16.885, P > 0.05; Day 2, F(4,10) = 170.863, P < 0.05; Day 3, F(4,10) = 91.809, P > 0.05; Day 4, F(4,10) = 94.120, P > 0.05; Day 5, F(4,10) = 68.969, P > 0.05; Day 6, F(4,10) = 44.611, P > 0.05; Day 7, F(4,10) = 20.872, P > 0.05; Figure 4E), increase in the number of SA-β-gal positive cells (F(4,20) = 185.297, P > 0.05; Figure 4F), as well as up-regulations of p16INK4a (F(4,10) = 4.841, P > 0.05; Figure 4G) and p21CIP1 (F(4,10) = 5.801, P > 0.05; Figure 4H). These results indicated that improving autophagic flux contributes to H2S-exerted protection against HG-induced cellular senescence in HT22 cells.
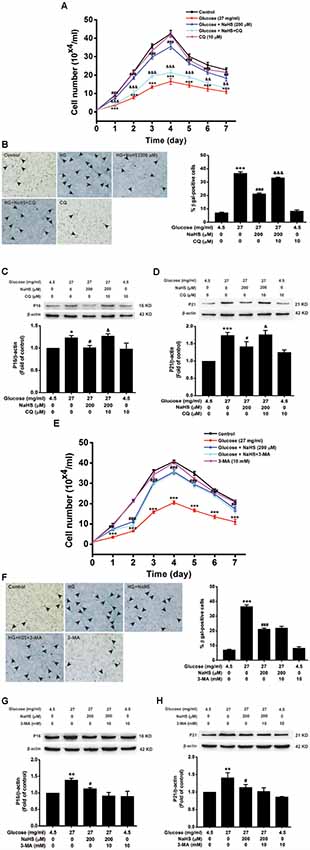
Figure 4. Effects of CQ and 3-MA on the protection of H2S against HG-induced cellular senescence in HT22 cells. (A,E) Cell growth curves were generated by using Trypan blue stain assays. (B,F) Representative images of senescent cells that were stained for SA-β-gal (Left, Magnification ×10) and quantitative analysis of the SA-β-gal positive cells (Right). The black arrows indicate the senescent cells, n = 5. (C,D,G,H) The expressions of p16INK4a and p21CIP1 in HT22 cells were detected by Western blotting. Values were expressed as the mean ± SEM, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. HG-treated alone group. &P < 0.05, &&P < 0.01, &&&P < 0.001, vs. co-treated with NaHS and HG group.
H2S Up-Regulates SIRT1 Expression in HG-Treated HT22 Cells
In order to ascertain whether the protective effect of H2S is dependent on SIRT1, we first investigated the effect of H2S on the expression of SIRT1. As showed in Figure 5A, the expression of SIRT1 in HT22 cells were increased by treatment with NaHS (100, 200 or 400 μM) for 24 h (F(3,8) = 169.526, P < 0.001). Furthermore, the down-regulation of SIRT1 caused by HG (40.5 mg/ml, for 48 h) was reversed by pretreatment with NaHS (200 μM, for 30 min; F(3,8) = 37.762, P < 0.001; Figure 5B). These data indicated that H2S up-regulates the expression of SIRT1 in HT22 cells.
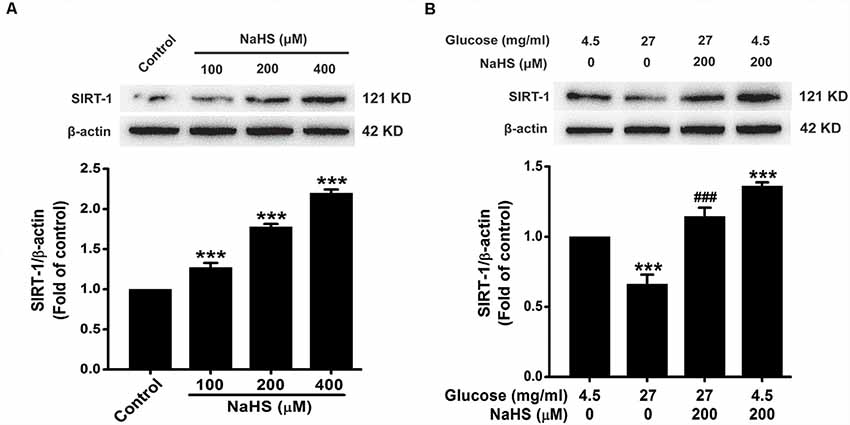
Figure 5. Effect of H2S on the expression of silent mating type information regulation 2 homolog 1 (SIRT1) in HT22 cells. (A) HT22 cells were treated with NaHS (100, 200 or 400 mM) for 24 h. (B) after pretreatment with NaHS (200 mM) for 30 min, HT22 cells were co-treated with HG (40.5 mg/ml) for 48 h. The expressions of SRT1 in HT22 cells were detected by Western blotting. Values were expressed as the mean ± SEM, n = 3. ***P < 0.001, vs. control group; ###P < 0.001 vs. HG-treated alone group.
Sirtinol, an Inhibitor of SIRT1, Reverses the Improving Effect of H2S on Autophagic Flux in HG-Exposed HT22 Cells
To confirm whether SIRT1 mediates the improving effect of H2S on autophagic flux in HG-exposed HT22 cells, we explored whether the SIRT1 inhibitor, Sirtinol, reverses this improving effect of H2S on autophagic flux. Pretreatment with Sirtinol (15 μM) abolished the suppression of NaHS (200 μM) in HG (40.5 mg/ml, for 48 h)-induced increase in the number of autophagosome (Figure 6A) as well as up-regulations of LC3II/LC3I (F(5,12) = 7.733, P < 0.05; Figure 6B) and P62 protein (F(5,12) = 29.863, P < 0.001; Figure 6C) in HT22 cells, which indicated that inhibited SIRT1 by Sirtinol reverses the improving effect of H2S on autophagic flux in HG-exposed HT22 cells.
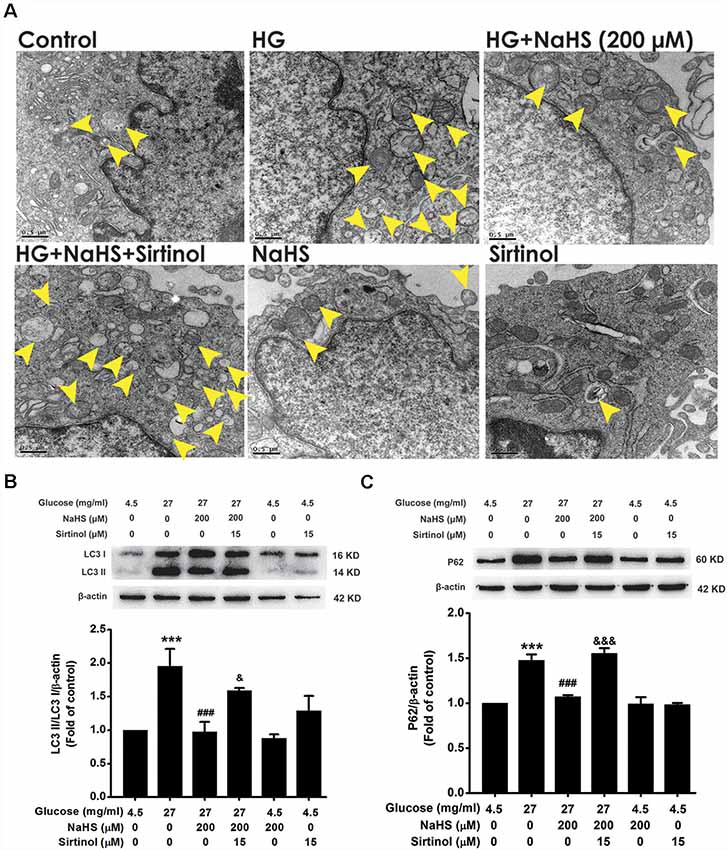
Figure 6. Effect of Sirtinol on the improving effect of H2S on autophagic flux in HG-exposed HT22 cells. (A) Representative transmission electron microscope images of HT22 cells showing the change in the number of autophagosome. The yellow arrows indicate autophagosome. (B,C) The expressions of LC3II/LC3I and P62 in HT22 cells were detected by Western blotting. Values were expressed as the mean ± SEM, n = 3. ***P < 0.001, vs. control group; ###P < 0.001, vs. HG-treated alone group. &P < 0.05, &&&P < 0.001, vs. co-treated with NaHS and HG group.
Sirtinol Abolishes the Protection of H2S Against HG-Induced Cellular Senescence in HT22 Cells
Next, we explored whether inhibited SIRT1 abolishes the protection of H2S against HG-induced cellular senescence in HT22 cells. Pretreatment with Sirtinol (15 μM) reversed the suppression of NaHS (200 μM) in HG (40.5 mg/ml, for 48 h)-induced inhibition of the cell growth (Day 1, F(5,12) = 29.984, P < 0.01; Day 2, F(5,12) = 50.438, P < 0.001; Day 3, F(5,12) = 181.255, P < 0.001; Day 4, F(5,12) = 256.034, P < 0.001; Day 5, F(5,12) = 155.338, P < 0.001; Day 6, F(5,12) = 65.111, P < 0.001; Day 7, F(5,12) = 16.613, P < 0.05; Figure 7A), increase in the number of SA-β-gal positive cells (F(5,24) = 368.555, P < 0.001; Figure 7B), as well as upregulations of p16INK4a (F(5,12) = 5.609, P < 0.05; Figure 7C) and p21CIP1 (F(5,12) = 6.522, P < 0.05; Figure 7D) in HT22 cells, which indicated that inhibited SIRT1 by sirtinol reverses the protection of H2S against HG-induced cellular senescence in HT22 cells.
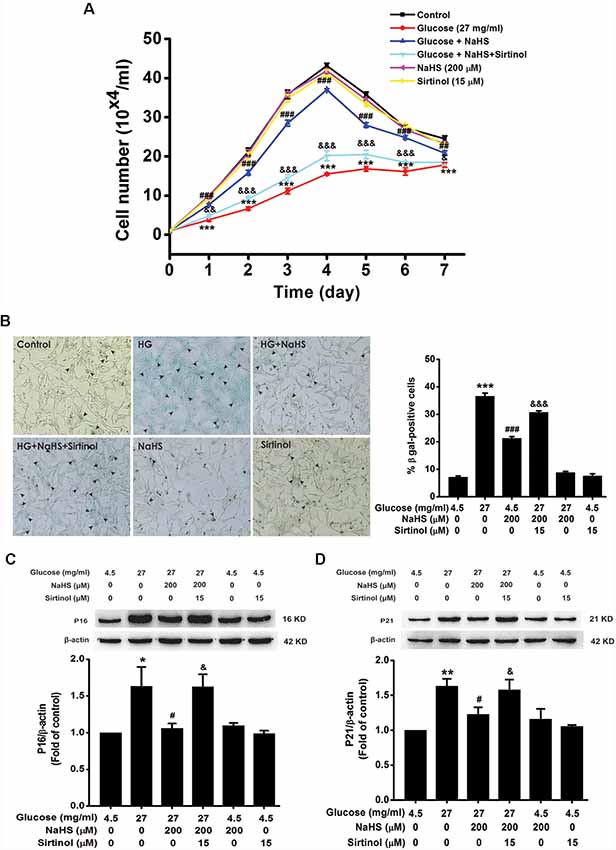
Figure 7. Sirtinol reversed H2S-inhibited cellular senescence in HG-exposed HT22 cells. (A) Cell growth curves were generated by using Trypan blue stain assays. (B) Representative images of senescent cells that were stained for SA-β-gal (Left, Magnification ×10) and quantitative analysis of the SA-β-gal positive cell (Right). The black arrows indicate the senescent cells. (C,D) The expressions of LC3II/LC3I and P62 in HT22 cells were detected by Western blotting. Values were expressed as the mean ± SEM, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. HG-treated alone group. &P < 0.05, &&P < 0.001, &&&P < 0.001, vs. co-treated with NaHS and HG group.
Discussion
Neuronal senescence plays key risk factors in hyperglycemia-induced neurotoxicity (Allen et al., 2005; Tomlinson and Gardiner, 2008). Our present work was to investigate whether H2S prevents hyperglycemia-induced neuronal senescence and the underlying mechanisms. The main findings of the present study are as followings: (1) NaHS (a donor of H2S) inhibits HG-induced cellular senescence of HT22 cells; (2) NaHS restores HG-induced autophagic flux dysfunction in HT22 cells; (3) inhibition of autophagic flux by CQ reversed H2S-restored autophagic flux in HG-exposed HT22 cells; (4) CQ abolishes the protective effects of H2S on HG-induced cellular senescence in HT22 cells; (5) H2S upregulates the expression of SIRT1 protein in HG-exposed HT22 cells; and (6) sirtinol, an inhibitor of SIRT-1, not only reverses the improvement of H2S in HG-induced autophagic flux disturbance in HT22 cells but also blocks the protection of H2S against HG-induced cellular senescence in HT22 cells. This is the first report that H2S rescues HG-induced neuronal senescence by improving autophagic flux through up-regulation of SIRT1.
Accumulating evidence demonstrates that long-term hyperglycemia causes neurotoxicity (Liu et al., 2014; Bahniwal et al., 2017; Li Y. et al., 2017; Renaud et al., 2018). It is well established that neuronal senescence plays a crucial role in the neurotoxicity of hyperglycemia (Sinadinos et al., 2014; Song et al., 2016; Zhu et al., 2018). Therefore, strategies to preserve neuronal cells by increasing the defenses of hyperglycemia-induced neuronal senescence are emerging as promising therapeutic approaches to prevent hyperglycemia-induced neurotoxicity. Due to their higher metabolic demands, HT22 cells require higher levels of glucose in normal growth media (25 mM glucose; Ward and Ergul, 2016). We have previously confirmed that treatment of HT22 cells with HG (150 mM) for 48 h, the cell viability was decreased to 70% compared with the control group (Zhu et al., 2018). In order to eliminate the effect of HG concentration on alteration of osmolarity, we have previously found that equal concentration of mannitol (150 mM) had no significant effect on the cell viability compared with the control group (Zhu et al., 2018). Similarly, Song et al. (2016) used 200 mM glucose to explore the effect of HG on senescence of neuronal cells. Therefore, in the present work, HT22 cells were exposed by 13.5–40.5 mg/ml of glucose to effectuate hyperglycemia-induced neuronal senescence in vitro. H2S, as a neuroprotector, has universal anti-senescence effects (Qi et al., 2012; Yang et al., 2013; Panthi et al., 2016), identifying as a potent preventive and therapeutic agent for senescence-associated disease (Zhang et al., 2013). Therefore, we explored whether H2S prevents hyperglycemia-induced neuronal senescence. The SA-β-gal (Dimri et al., 1995; Debacq-Chainiaux et al., 2009) and trypan blue (Tennant, 1964) are main staining methods for identifying senescence cell. The p16INK4a (Baker et al., 2011) and p21CIP1 (Carreira et al., 2005) are the main cellular senescence biomarker protein. We examined these senescent makers to explore whether pretreatment with NaHS prevents HG-induced senescence in HT22 cells. Our results showed that NaHS decreased the number of SA-β-gal positive cells, increased the growth of cells, and down-regulated the expressions of senescence marker, p16INK4a and p21CIP1, in HG-exposed HT22 cells. Our results indicated that H2S antagonizes HG-induced cellular senescence in HT22 cells. It has been reported that H2S reverses H2O2-induced senescence in SH-SY5Y cells (Xie et al., 2014). Thus, it is reasonable to believe H2S as a potentially promising therapeutic approach to the treatment of HG-induced neuronal senescence.
Next, we explored the mechanisms underlying this inhibitory role of H2S on HG-induced cellular senescence. Emerging evidence suggests that blockage of autophagic flux results in the decrease in degradation of misfolded proteins or damaged organelles, which in turn leads to cellular senescence (Lin et al., 2015; Han et al., 2016; Yin et al., 2017). In the present study, we found that accumulation of autophagosomes, increase in LC3II/I, and upregulation of P62 were displayed in HG-exposed HT22 cell. P62 connects between LC3 and ubiquitination substrate, which is degraded in autolysosomes (Bjørkøy et al., 2005). The aggregation of P62 is associated with autophagic degradation dysfunction (Komatsu and Ichimura, 2010). Thus, our results indicated that HG induces blockage of autophagic flux in HT22 cells. There is evidence that HG exposure reduces autophagic flux in cardiomyocyte (Hou et al., 2018). Therefore, we suggested that blockage of autophagic flux plays an important role in HG-induced senescence of HT22 cells. It has been reported that activating autophagic flux by H2S prevents HG-induced injury in H9C2 cells (Yang et al., 2017). Moreover, H2S-mediated autophagic flux reduces serum triglyceride in rat liver (Sun et al., 2015). Therefore, we investigated whether improving autophagic flux plays a key role in the protection of H2S against HG-induced cellular senescence in HT22 cells. Notably, NaHS reduced the number of autophagosomes, decreased the ratio of LC3II/I, and downregulated the expression of P62 in HG-exposed HT22 cell, which indicated the improving role of H2S in autophagic flux. To further validate whether improving autophagic flux mediates the protection of H2S against HG-induced cellular senescence, we explored whether the inhibition of autophagic flux abolishes the protection of H2S. Thus, two autophagy inhibitors, CQ and 3-MA, were used in our research. We found that blockage of autophagic flux by CQ reverses the protection of H2S against HG-induced cellular senescence in HT22 cells. However, 3-MA could not reverse the above effects. Consistent with this notion, prolonged treatment with 3-MA promotes autophagic flux under nutrient-rich conditions (Wu et al., 2010). CQ impairs lysosome function that restrains fusion of autophagosome and lysosome, while 3-MA is a type III phosphatidylinositol 3-kinase (PI3KCIII) inhibitor that inhibits autophagy from the initiation step (Pliyev and Menshikov, 2012). These results thus indicated that H2S may block lysosome to rescue HG-impaired autophagic flux in HT22 cells. Taken together, our outcomes indicated that improving autophagic flux mediates the antagonistic action of H2S in HG-induced cellular senescence in HT22 cells. It has been demonstrated that blocking autophagic flux reverses the protective effect of H2S against acrylonitrile-induced neurotoxicity (Yang et al., 2018). Therefore, we suggest that improving autophagic flux contributes to the protective effect of H2S against HG-induced neuronal senescence.
SIRT1 is a NAD+-dependent deacetylase. Increasing evidence confirms the anti-senescence effect of SIRT1 (Hwang et al., 2013; Herskovits and Guarente, 2014). While, SIRT1 deficiency induces cellular senescence (Furukawa et al., 2007). Furthermore, it has been reported that SIRT1 prevents oxidative stress-induced cellular senescence via restoration of autophagic flux (Han et al., 2016) and that up-regulation of SIRT1 by resveratrol enhances autophagic flux in human umbilical vein endothelial cell (Zhang et al., 2016). It is reasonable to believe that SIRT1 suppresses cellular senescence via improving autophagic flux. Does SIRT1 mediate the protection of H2S against HG-induced neuronal senescence in HT22 cells? It has been documented that SIRT1 mediates the roles of H2S in attenuating chronic restraint stress-induced cognitive impairment (Li X. N. et al., 2017) and inhibiting homocysteine-induced endoplasmic reticulum stress in PC12 cells (Wang et al., 2017). Therefore, it seems advisable to conjecture that SIRT1 mediates the protection of H2S against HG-triggered neuronal senescence. In the present work, we found that H2S up-regulated the expression of SIRT1 in HG-exposed HT22 cells and that Sirtinol, an inhibitor of SIRT1 abolished the protective effect of H2S on HG-induced cellular senescence in HT22 cells. These results indicated that SIRT1 mediates the protection of H2S against HG-triggered neuronal senescence in HT22 cell. This finding is consistent with the previous report that stimulating of SIRT1 by H2S antagonizes senescence in human fibroblasts (Sanokawa-Akakura et al., 2016) and H2O2-induced senescence in human umbilical vein endothelial cells (Suo et al., 2013). Furthermore, we found that inhibition of SIRT1 by sirtinol reversed the improving role of H2S in the autophagic flux of HG-exposed HT22 cells. Taken together, we suggested that SIRT1 mediates the protective effect of H2S against HG-induced senescence in HT22 cells via improving autophagic flux.
In summary, our research indicated that H2S increases autophagic flux by up-regulation of SIRT1 expression and thereby antagonizes HG-induced cellular senescence in HT22 cells. These findings provide a deeper mechanism explanation for the protection of H2S against hyperglycemia-induced neuronal senescence and suggest that H2S may be a prospective strategy for prevention of hyperglycemia-induced neurotoxicity.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
X-QT and H-LH conceived and designed the experiments. YC, LW, C-YW, Y-YT, XL, XK, and Y-RX performed the experiments. X-QT, YC, and LW analyzed the data. C-YW, Y-YT, XL, XK, and Y-RX contributed reagents, materials and analysis tools. X-QT and LW wrote and revised the manuscript.
Funding
This study was supported by Natural Science Foundation of China (81671057) and Natural Science Foundation of Hunan province (2019JJ50546, 2019JJ80101).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allen, D. A., Yaqoob, M. M., and Harwood, S. M. (2005). Mechanisms of high glucose-induced apoptosis and its relationship to diabetic complications. J. Nutr. Biochem. 16, 705–713. doi: 10.1016/j.jnutbio.2005.06.007
Bahniwal, M., Little, J. P., and Klegeris, A. (2017). High glucose enhances neurotoxicity and inflammatory cytokine secretion by stimulated human astrocytes. Curr. Alzheimer Res. 14, 731–741. doi: 10.2174/1567205014666170117104053
Baker, D. J., Wijshake, T., Tchkonia, T., LeBrasseur, N. K., Childs, B. G., van de Sluis, B., et al. (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. doi: 10.1038/nature10600
Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., et al. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614. doi: 10.1083/jcb.200507002
Bordone, L., and Guarente, L. (2005). Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat. Rev. Mol. Cell Biol. 6, 298–305. doi: 10.1038/nrm1616
Caro, L. H., Plomp, P. J., Wolvetang, E. J., Kerkhof, C., and Meijer, A. J. (1988). 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur. J. Biochem. 175, 325–329. doi: 10.1111/j.1432-1033.1988.tb14200.x
Carreira, S., Goodall, J., Aksan, I., La Rocca, S. A., Galibert, M. D., Denat, L., et al. (2005). Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 433, 764–769. doi: 10.1038/nature03269
Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., et al. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392. doi: 10.1126/science.1099196
Debacq-Chainiaux, F., Erusalimsky, J. D., Campisi, J., and Toussaint, O. (2009). Protocols to detect senescence-associated β-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806. doi: 10.1038/nprot.2009.191
Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U S A 92, 9363–9367. doi: 10.1073/pnas.92.20.9363
Fan, F., Liu, T., Wang, X., Ren, D., Liu, H., Zhang, P., et al. (2016). ClC-3 expression and its association with hyperglycemia induced HT22 hippocampal neuronal cell apoptosis. J. Diabetes Res. 2016:2984380. doi: 10.1155/2016/2984380
Furukawa, A., Tada-Oikawa, S., Kawanishi, S., and Oikawa, S. (2007). H2O2 accelerates cellular senescence by accumulation of acetylated p53 via decrease in the function of SIRT1 by NAD+ depletion. Cell. Physiol. Biochem. 20, 45–54. doi: 10.1159/000104152
Han, X., Tai, H., Wang, X., Wang, Z., Zhou, J., Wei, X., et al. (2016). AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD+ elevation. Aging Cell 15, 416–427. doi: 10.1111/acel.12446
Herskovits, A. Z., and Guarente, L. (2014). SIRT1 in neurodevelopment and brain senescence. Neuron 81, 471–483. doi: 10.1016/j.neuron.2014.01.028
Hou, J., Zheng, D., Xiao, W., Li, D., Ma, J., and Hu, Y. (2018). Mangiferin enhanced autophagy via inhibiting mTORC1 pathway to prevent high glucose-induced cardiomyocyte injury. Front. Pharmacol. 9:383. doi: 10.3389/fphar.2018.00383
Hu, L. F., Lu, M., Wu, Z. Y., Wong, P. T., and Bian, J. S. (2009). Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol. Pharmacol. 75, 27–34. doi: 10.1124/mol.108.047985
Hwang, J. W., Yao, H., Caito, S., Sundar, I. K., and Rahman, I. (2013). Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 61, 95–110. doi: 10.1016/j.freeradbiomed.2013.03.015
Kaur, N., Kishore, L., and Singh, R. (2017). Chromane isolated from leaves of Dillenia indica improves the neuronal dysfunction in STZ-induced diabetic neuropathy. J. Ethnopharmacol. 206, 19–30. doi: 10.1016/j.jep.2017.05.018
Komatsu, M., and Ichimura, Y. (2010). Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 584, 1374–1378. doi: 10.1016/j.febslet.2010.02.017
Kumar, P., Raman, T., Swain, M. M., Mishra, R., and Pal, A. (2017). Hyperglycemia-induced oxidative-nitrosative stress induces inflammation and neurodegeneration via augmented tuberous sclerosis complex-2 (TSC-2) activation in neuronal cells. Mol. Neurobiol. 54, 238–254. doi: 10.1007/s12035-015-9667-3
Li, X. N., Chen, L., Luo, B., Li, X., Wang, C. Y., Zou, W., et al. (2017). Hydrogen sulfide attenuates chronic restrain stress-induced cognitive impairment by upreglulation of Sirt1 in hippocampus. Oncotarget 8, 100396–100410. doi: 10.18632/oncotarget.22237
Li, Y., Zhang, Y., Wang, L., Wang, P., Xue, Y., Li, X., et al. (2017). Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 13, 1145–1160. doi: 10.1080/15548627.2017.1320467
Li, X., Zhang, K. Y., Zhang, P., Chen, L. X., Wang, L., Xie, M., et al. (2014). Hydrogen sulfide inhibits formaldehyde-induced endoplasmic reticulum stress in PC12 cells by upregulation of SIRT-1. PLoS One 9:e89856. doi: 10.1371/journal.pone.0089856
Lim, J., Lee, Y., Jung, S., Youdim, M. B., and Oh, Y. J. (2014). Impaired autophagic flux is critically involved in drug-induced dopaminergic neuronal death. Parkinsonism Relat. Disord. 20, S162–S166. doi: 10.1016/s1353-8020(13)70039-7
Lin, J. R., Shen, W. L., Yan, C., and Gao, P. J. (2015). Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler. Thromb. Vasc. Biol. 35, 1413–1422. doi: 10.1161/atvbaha.115.305706
Liu, J., Li, L., and Suo, W. Z. (2009). HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 84, 267–271. doi: 10.1016/j.lfs.2008.12.008
Liu, Y. Y., Nagpure, B. V., Wong, P. T., and Bian, J. S. (2013). Hydrogen sulfide protects SH-SY5Y neuronal cells against d-galactose induced cell injury by suppression of advanced glycation end products formation and oxidative stress. Neurochem. Int. 62, 603–609. doi: 10.1016/j.neuint.2012.12.010
Liu, C., Song, Z., Wang, L., Yu, H., Liu, W., Shang, Y., et al. (2017). Sirt1 regulates acrosome biogenesis by modulating autophagic flux during spermiogenesis in mice. Development 144, 441–451. doi: 10.1242/dev.147074
Liu, R. Y., Wang, J. J., Qiu, X., and Wu, J. M. (2014). Acute hyperglycemia together with hematoma of high-glucose blood exacerbates neurological injury in a rat model of intracerebral hemorrhage. Neurosci. Bull. 30, 90–98. doi: 10.1007/s12264-013-1371-6
Lumkwana, D., du Toit, A., Kinnear, C., and Loos, B. (2017). Autophagic flux control in neurodegeneration: progress and precision targeting-Where do we stand? Prog. Neurobiol. 153, 64–85. doi: 10.1016/j.pneurobio.2017.03.006
Menzies, F. M., Fleming, A., and Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. doi: 10.1038/nrn3961
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Panthi, S., Chung, H. J., Jung, J., and Jeong, N. Y. (2016). Physiological importance of hydrogen sulfide: emerging potent neuroprotector and neuromodulator. Oxid. Med. Cell. Longev. 2016:9049782. doi: 10.1155/2016/9049782
Pliyev, B. K., and Menshikov, M. (2012). Differential effects of the autophagy inhibitors 3-methyladenine and chloroquine on spontaneous and TNF-α-induced neutrophil apoptosis. Apoptosis 17, 1050–1065. doi: 10.1007/s10495-012-0738-x
Qadir, M. I., and Anwar, S. (2017). Sirtuins in brain aging and neurological disorders. Crit. Rev. Eukaryot. Gene Expr. 27, 321–329. doi: 10.1615/critreveukaryotgeneexpr.2017019532
Qi, H. N., Cui, J., Liu, L., Lu, F. F., Song, C. J., Shi, Y., et al. (2012). Exogenous hydrogen sulfide delays the senescence of human umbilical vein endothelial cells by lessening oxidative stress. Sheng Li Xue Bao 64, 425–432, doi: 10.1016/j.jacc.2014.06.211
Renaud, J., Bassareo, V., Beaulieu, J., Pinna, A., Schlich, M., Lavoie, C., et al. (2018). Dopaminergic neurodegeneration in a rat model of long-term hyperglycemia: preferential degeneration of the nigrostriatal motor pathway. Neurobiol. Aging 69, 117–128. doi: 10.1016/j.neurobiolaging.2018.05.010
Reno, C. M., Tanoli, T., Bree, A., Daphna-Iken, D., Cui, C., Maloney, S. E., et al. (2013). Antecedent glycemic control reduces severe hypoglycemia-induced neuronal damage in diabetic rats. Am. J. Physiol. Endocrinol. Metab. 304, E1331–E1337. doi: 10.1152/ajpendo.00084.2013
Sanokawa-Akakura, R., Akakura, S., and Tabibzadeh, S. (2016). Replicative senescence in human fibroblasts is delayed by hydrogen sulfide in a NAMPT/SIRT1 dependent manner. PLoS One. 11:e0164710. doi: 10.1371/journal.pone.0164710
Shen, D. N., Zhang, L. H., Wei, E. Q., and Yang, Y. (2015). Autophagy in synaptic development, function, and pathology. Neurosci. Bull. 31, 416–426. doi: 10.1007/s12264-015-1536-6
Si, Y. F., Wang, J., Guan, J., Zhou, L., Sheng, Y., and Zhao, J. (2013). Treatment with hydrogen sulfide alleviates streptozotocin-induced diabetic retinopathy in rats. Br. J. Pharmacol. 169, 619–631. doi: 10.1111/bph.12163
Sima, A. A., Kamiya, H., and Li, Z. G. (2004). Insulin, C-peptide, hyperglycemia, and central nervous system complications in diabetes. Eur. J. Pharmacol. 490, 187–197. doi: 10.1016/j.ejphar.2004.02.056
Sinadinos, C., Valles-Ortega, J., Boulan, L., Solsona, E., Tevy, M. F., Marquez, M., et al. (2014). Neuronal glycogen synthesis contributes to physiological aging. Aging Cell 13, 935–945. doi: 10.1111/acel.12254
Song, J., Lee, B., Kang, S., Oh, Y., Kim, E., Kim, C. H., et al. (2016). Agmatine ameliorates high glucose-induced neuronal cell senescence by regulating the p21 and p53 signaling. Exp. Neurobiol. 25, 24–32. doi: 10.5607/en.2016.25.1.24
Sun, L., Zhang, S., Yu, C., Pan, Z., Liu, Y., Zhao, J., et al. (2015). Hydrogen sulfide reduces serum triglyceride by activating liver autophagy via the AMPK-mTOR pathway. Am. J. Physiol. Endocrinol. Metab. 309, E925–E935. doi: 10.1152/ajpendo.00294.2015
Suo, R., Zhao, Z. Z., Tang, Z. H., Ren, Z., Liu, X., Liu, L. S., et al. (2013). Hydrogen sulfide prevents H2O2-induced senescence in human umbilical vein endothelial cells through SIRT1 activation. Mol. Med. Rep. 7, 1865–1870. doi: 10.3892/mmr.2013.1417
Suzuki, K., Olah, G., Modis, K., Coletta, C., Kulp, G., Gerö, D., et al. (2011). Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc. Natl. Acad. Sci. U S A 108, 13829–13834. doi: 10.1073/pnas.1105121108
Tennant, J. R. (1964). Evaluation of the trypan blue technique for determination of cell viabilty. Transplantation 2, 685–694. doi: 10.1097/00007890-196411000-00001
Tomlinson, D. R., and Gardiner, N. J. (2008). Glucose neurotoxicity. Nat. Rev. Neurosci. 9, 36–45. doi: 10.1038/nrn2294
Wang, R. (2010). Hydrogen sulfide: the third gasotransmitter in biology and medicine. Antioxid. Redox Signal. 12, 1061–1064. doi: 10.1089/ars.2009.2938
Wang, C. Y., Zou, W., Liang, X. Y., Jiang, Z. S., Li, X., Wei, H. J., et al. (2017). Hydrogen sulfide prevents homocysteineinduced endoplasmic reticulum stress in PC12 cells by upregulating SIRT1. Mol. Med. Rep. 16, 3587–3593. doi: 10.3892/mmr.2017.7004
Ward, R., and Ergul, A. (2016). Relationship of endothelin-1 and NLRP3 inflammasome activation in HT22 hippocampal cells in diabetes. Life Sci. 159, 97–103. doi: 10.1016/j.lfs.2016.02.043
Wei, H. J., Xu, J. H., Li, M. H., Tang, J. P., Zou, W., Zhang, P., et al. (2014). Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol. Sin. 35, 707–715. doi: 10.1038/aps.2013.197
Wu, Y. T., Tan, H. L., Shui, G., Bauvy, C., Huang, Q., Wenk, M. R., et al. (2010). Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 285, 10850–10861. doi: 10.1074/jbc.M109.080796
Xie, J., Cui, K., Hao, H., Zhang, Y., Lin, H., Chen, Z., et al. (2016). Acute hyperglycemia suppresses left ventricular diastolic function and inhibits autophagic flux in mice under prohypertrophic stimulation. Cardiovasc. Diabetol. 15:136. doi: 10.1186/s12933-016-0452-z
Xie, Z. Z., Shi, M. M., Xie, L., Wu, Z. Y., Li, G., Hua, F., et al. (2014). Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid. Redox Signal. 21, 2531–2542. doi: 10.1089/ars.2013.5604
Xie, H., Xu, Q., Jia, J., Ao, G., Sun, Y., Hu, L., et al. (2015). Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem. Biophys. Res. Commun. 458, 632–638. doi: 10.1016/j.bbrc.2015.02.017
Yang, B., Bai, Y., Yin, C., Qian, H., Xing, G., Wang, S., et al. (2018). Activation of autophagic flux and the Nrf2/ARE signaling pathway by hydrogen sulfide protects against acrylonitrile-induced neurotoxicity in primary rat astrocytes. Arch. Toxicol. 92, 2093–2108. doi: 10.1007/s00204-018-2208-x
Yang, F., Zhang, L., Gao, Z., Sun, X., Yu, M., Dong, S., et al. (2017). Exogenous H2S protects against diabetic cardiomyopathy by activating autophagy via the AMPK/mTOR pathway. Cell. Physiol. Biochem. 43, 1168–1187. doi: 10.1159/000481758
Yang, G., Zhao, K., Ju, Y., Mani, S., Cao, Q., Puukila, S., et al. (2013). Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 18, 1906–1919. doi: 10.1089/ars.2012.4645
Ye, H., Chen, M., Cao, F., Huang, H., Zhan, R., and Zheng, X. (2016). Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 16:178. doi: 10.1186/s12883-016-0700-6
Yerra, V. G., Areti, A., and Kumar, A. (2017). Erratum to: adenosine monophosphate-activated protein kinase abates hyperglycaemia-induced neuronal injury in experimental models of diabetic neuropathy: effects on mitochondrial biogenesis, autophagy and neuroinflammation. Mol. Neurobiol. 54, 2313–2314. doi: 10.1007/s12035-016-0183-x
Yin, Y., Sun, G., Li, E., Kiselyov, K., and Sun, D. (2017). ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev. 34, 3–14. doi: 10.1016/j.arr.2016.08.008
Zhang, Y., Cao, X., Zhu, W., Liu, Z., Liu, H., Zhou, Y., et al. (2016). Resveratrol enhances autophagic flux and promotes Ox-LDL degradation in HUVECs via upregulation of SIRT1. Oxid. Med. Cell. Longev. 2016:7589813. doi: 10.1155/2016/7589813
Zhang, L., Chen, Z. W., Yang, S. F., Shaer, M., Wang, Y., Dong, J. J., et al. (2019). MicroRNA-219 decreases hippocampal long-term potentiation inhibition and hippocampal neuronal cell apoptosis in type 2 diabetes mellitus mice by suppressing the NMDAR signaling pathway. CNS Neurosci. Ther. 25, 69–77. doi: 10.1111/cns.12981
Zhang, Q., Deng, Q., Zhang, J., Ke, J., Zhu, Y., Wen, R. W., et al. (2018). Activation of the Nrf2-ARE pathway ameliorates hyperglycemia-mediated mitochondrial dysfunction in podocytes partly through Sirt1. Cell. Physiol. Biochem. 48, 1–15. doi: 10.1159/000491658
Zhang, Y., Tang, Z. H., Ren, Z., Qu, S. L., Liu, M. H., Liu, L. S., et al. (2013). Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol. Cell. Biol. 33, 1104–1113. doi: 10.1128/mcb.01215-12
Zhang, Y., Thai, K., Jin, T., Woo, M., and Gilbert, R. E. (2018). SIRT1 activation attenuates α cell hyperplasia, hyperglucagonaemia and hyperglycaemia in STZ-diabetic mice. Sci. Rep. 8:13972. doi: 10.1038/s41598-018-32351-z
Keywords: hydrogen sulfide, high glucose, SIRT1, autophagic flux, neuronal senescence
Citation: Wu L, Chen Y, Wang C-Y, Tang Y-Y, Huang H-L, Kang X, Li X, Xie Y-R and Tang X-Q (2019) Hydrogen Sulfide Inhibits High Glucose-Induced Neuronal Senescence by Improving Autophagic Flux via Up-regulation of SIRT1. Front. Mol. Neurosci. 12:194. doi: 10.3389/fnmol.2019.00194
Received: 01 May 2019; Accepted: 25 July 2019;
Published: 20 August 2019.
Edited by:
Claudio Grassi, Catholic University of the Sacred Heart, ItalyReviewed by:
Salvatore Fusco, Fondazione Policlinico Universitario A. Gemelli, Università Cattolica del Sacro Cuore, ItalyEva Maria Jimenez-Mateos, Trinity College Dublin, Ireland
Copyright © 2019 Wu, Chen, Wang, Tang, Huang, Kang, Li, Xie and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong-Lin Huang, huanghl@usc.edu.cn; Xiao-Qing Tang, tangxq-usc@qq.com; tangxq-usc@usc.edu.cn
† These authors have contributed equally to this work