Neuroligins and Neurodevelopmental Disorders: X-Linked Genetics
 Thien A. Nguyen1,2*
Thien A. Nguyen1,2*  Katherine W. Roche
Katherine W. Roche- 1Receptor Biology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, United States
- 2Department of Pharmacology and Physiology, Georgetown University, Washington, DC, United States
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that results in social-communication impairments, as well as restricted and repetitive behaviors. Moreover, ASD is more prevalent in males, with a male to female ratio of 4 to 1. Although the underlying etiology of ASD is generally unknown, recent advances in genome sequencing have facilitated the identification of a host of associated genes. Among these, synaptic proteins such as cell adhesion molecules have been strongly linked with ASD. Interestingly, many large genome sequencing studies exclude sex chromosomes, which leads to a shift in focus toward autosomal genes as targets for ASD research. However, there are many genes on the X chromosome that encode synaptic proteins, including strong candidate genes. Here, we review findings regarding two members of the neuroligin (NLGN) family of postsynaptic adhesion molecules, NLGN3 and NLGN4. Neuroligins have multiple isoforms (NLGN1-4), which are both autosomal and sex-linked. The sex-linked genes, NLGN3 and NLGN4, are both on the X chromosome and were among the first few genes to be linked with ASD and intellectual disability (ID). In addition, there is a less studied human neuroligin on the Y chromosome, NLGN4Y, which forms an X-Y pair with NLGN4X. We will discuss recent findings of these neuroligin isoforms regarding function at the synapse in both rodent models and human-derived differentiated neurons, and highlight the exciting challenges moving forward to a better understanding of ASD/ID.
Introduction
Autism spectrum disorder (ASD) is a highly prevalent neurodevelopmental disorder affecting one in 54 children in the United States. ASD is characterized by deficits in communication and social interaction (Miles, 2011; Fombonne, 2013). Intellectual disability (ID) is characterized by deficits in intellectual functioning and adaptive behavior thus limiting an individual’s ability to thrive independently (Raymond, 2006; Lubs et al., 2012; Ellison et al., 2013). Interestingly, both ASD and ID are more prevalent in males (Geschwind, 2011; Miles, 2011; Werling and Geschwind, 2013; Werling et al., 2016), although this strong sex bias in ASD remains unclear. It is notable that a subset of ASD-associated genes are located on the X chromosome indicating that the sex chromosomes may play a role in at least some of the sexual dimorphism in these disorders.
Autism spectrum disorder is divided into two categories: syndromic and nonsyndromic. Syndromic ASD is defined as a condition in patients who already have an existing neurological disorder. For example, a subset of patients with Fragile-X syndrome, tuberous sclerosis, or Rett syndrome display phenotypes that are attributed to ASD (Singh and Eroglu, 2013; Geschwind and State, 2015). Nonsyndromic ASD accounts for ASD cases that are not linked to any neurological disorders. Before the advancement of next-generation sequencing (NGS), genetic researchers focused on finding rare genetic variants in ASD and ID pedigrees to link genes to these disorders, which led to the association of the neuroligins NLGN3 and NLGN4X to ASD/ID (Jamain et al., 2003; Laumonnier et al., 2004). Other notable genes identified through rare de novo mutations and recessive inheritance mutations include SHANK3, CNTNAP2, NRXN1, PTEN, FMR1, and TSC1 (Geschwind and State, 2015). Although these cases are rare, functional and genetic studies definitively showed their link with ASD and ID. With NGS becoming cheaper and easier to access, genome wide association studies (GWAS) and whole exome sequencing (WES) studies became the major approaches used to identify common and rare variants for ASD/ID. Large cohort studies continue to identify more genes associated with ASD/ID, including genes that are important in chromatin modification, transcriptional regulation, or are FMRP-associated, embryonically expressed, or affect synaptic function (Sanders et al., 2012; Yu et al., 2013; De Rubeis et al., 2014; Iossifov et al., 2014). Although NGS has dramatically accelerated the identification of new risk genes, it is important to mention that NGS studies often ignore the sex chromosomes due to the limitations for statistical analysis (Wise et al., 2013; No Author List, 2017).
The neuroligin (NLGN) family of postsynaptic cell adhesion molecules have emerged as important factors regulating neuronal development and synaptic transmission. There are five members of the NLGN family in humans and other primates: NLGN1, 2, 3, 4X, and 4Y (Bemben et al., 2015b; Jeong et al., 2017; Südhof, 2017, 2018). However, in rodents, there are only four members: NLGN1, 2, 3, and 4-like (Bolliger et al., 2001, 2008). NLGNs have an isoform-specific localization: NLGN1 is localized to excitatory synapses, NLGN2 at inhibitory synapses, and NLGN3 is at both (Chih et al., 2005; Chubykin et al., 2007; Bemben et al., 2015b). Interestingly human NLGN4X is localized at excitatory synapses, whereas mouse NLGN4-like is at glycinergic synapses (Hoon et al., 2011; Bemben et al., 2015a; Chanda et al., 2016; Marro et al., 2019). NLGN4X and NLGN4Y were historically grouped together and assumed to have the same function due to their almost identical sequence identity. However, recent findings show that a single amino acid difference in NLGN4Y results in a trafficking deficit leading to decreased surface expression and synaptic function (Nguyen et al., 2020).
Neuroligins are highly dynamic, regulated via posttranslational modifications and protein–protein interactions. NLGN1 is phosphorylated by calcium/calmodulin-dependent protein kinase 2 (CaMKII), protein kinase A (PKA), and tyrosine kinases to regulate its function at excitatory synapses (Bemben et al., 2013; Giannone et al., 2013; Letellier et al., 2018; Jeong et al., 2019). Furthermore, a recent paper established that NLGN1-mediated synaptogenic properties are mediated by interacting with Kalirin7, a Rho guanine nucleotide exchange factor (GEF) (Paskus et al., 2019, 2020). Phosphorylation of NLGN2 affects binding with inhibitory scaffolding proteins, thus regulating its function at inhibitory synapses (Poulopoulos et al., 2009; Antonelli et al., 2014; Nguyen et al., 2016). NLGN3 can be cleaved by proteases to reduce its function at synapses (Bemben et al., 2019). Interestingly, the extracellular cleaved fragment of NLGN3 has been identified as a potent mitogen in brain cancer (Venkatesh et al., 2015, 2017). Lastly, NLGN4X can be phosphorylated by protein kinase C (PKC) to enhance excitatory synapses (Bemben et al., 2015a). Together, NLGNs comprise an important class of proteins that are dynamic and have multiple functions at synapses.
Of the NLGN family, NLGN3, NLGN4X, and NLGN4Y are sex-linked with NLGN3 and NLGN4X on the X-chromosome and NLGN4Y on the Y-chromosome. Early genetic studies using a family pedigree of ASD/ID probands associated NLGN3 and NLGN4X with ASD/ID (Jamain et al., 2003; Laumonnier et al., 2004) (Tables 1, 2). Interestingly, the majority of cases for NLGN3- and NLGN4X-associated ASD/ID are males. In this review, we provide an overview of the current literature of sex-linked NLGNs functions and their links to ASD/ID.
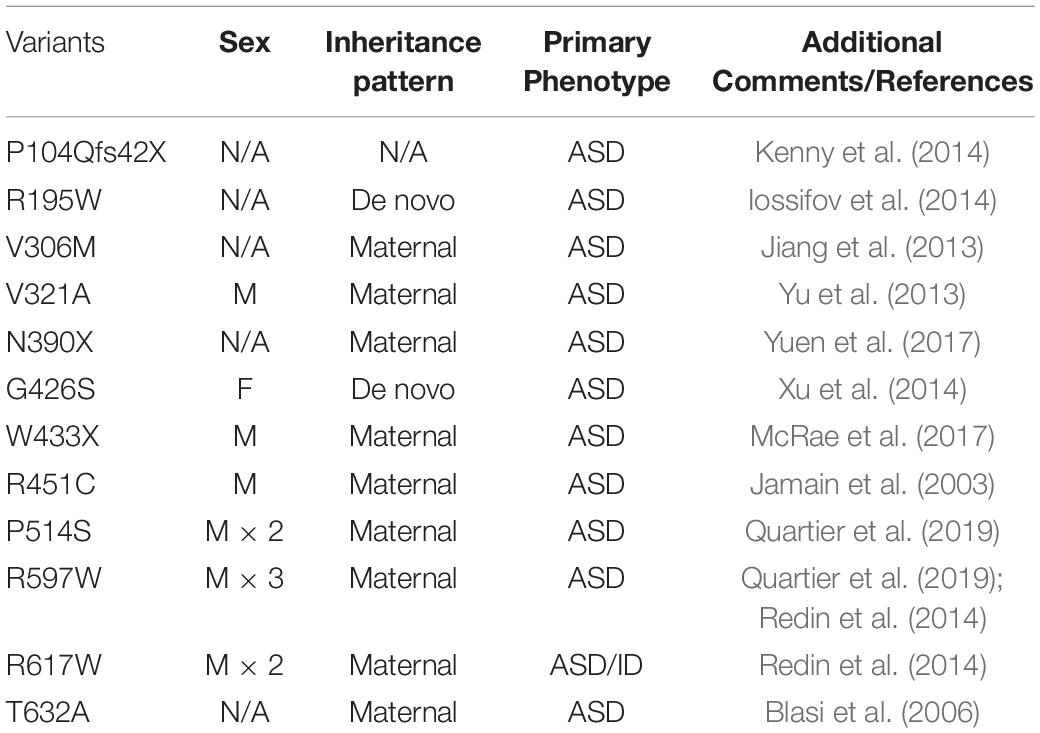
Table 1. ASD-associated NLGN3 variants.
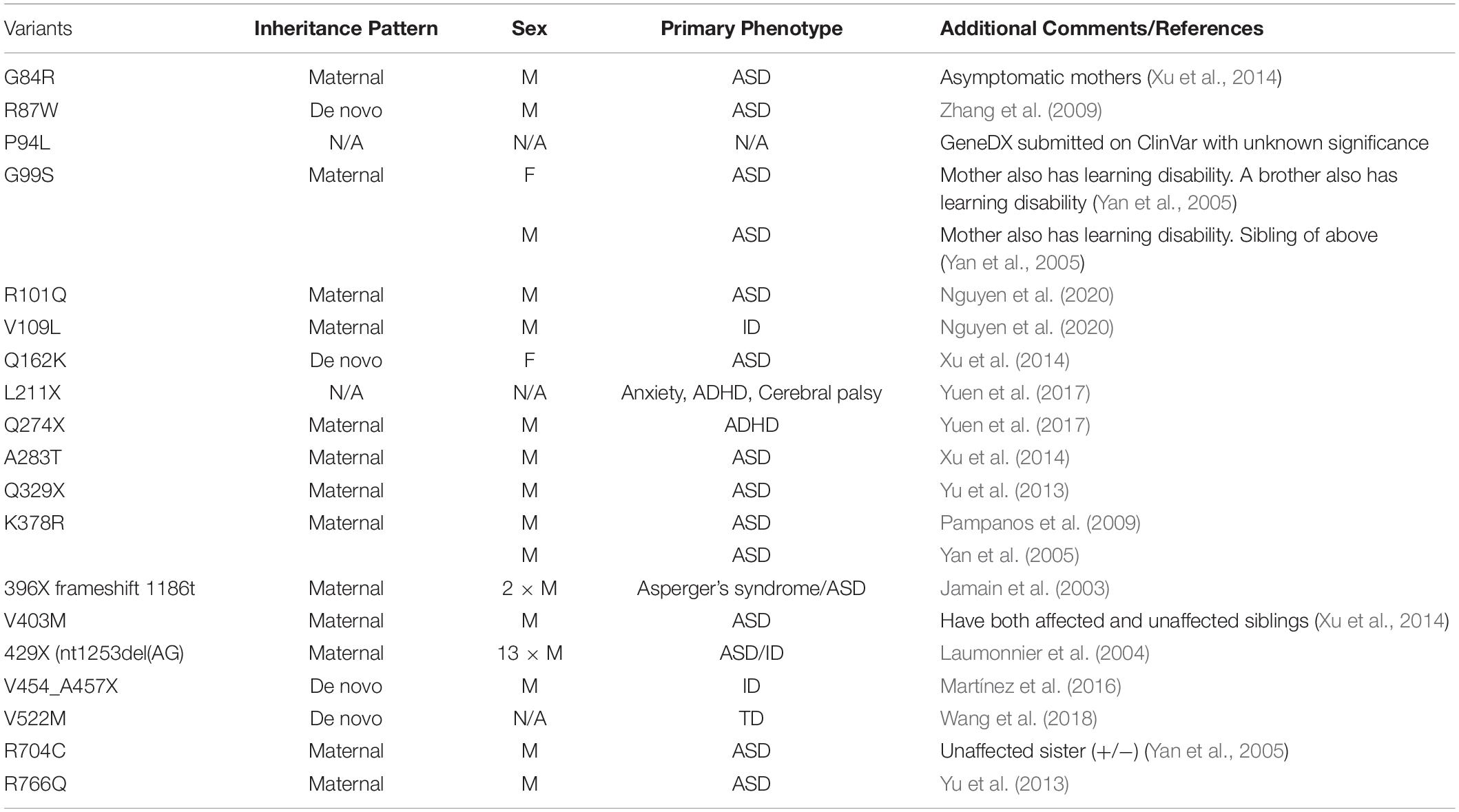
Table 2. ASD-associated NLGN4X variants.
NLGN3 and ASD
The first link between ASD and NLGN3 was revealed from a case study of ASD patients. Jamain et al. (2003) identified a missense mutation in a Swedish family with two affected brothers, one with ASD and the other with Asperger’s syndrome. They showed that both probands contain a missense mutation in NLGN3 (NLGN3 R451C), which encodes an arginine instead of a cysteine at amino acid 451 within the extracellular domain (ECD) of NLGN3. The NLGN3 R451C mutant displays a decrease in surface expression compared to WT, and is retained in the ER by binding to the chaperone protein BiP (Chih et al., 2004; Comoletti et al., 2004; De Jaco et al., 2006). Unlike the human specific NLGN4X, NLGN3 is highly conserved across mammals, facilitating the development of knock-in (KI) mouse models to study how NLGN3 mutations affect behavior.
In agreement with molecular studies, the NLGN3 R451C KI mouse displays a significant (∼90%) decrease in protein levels compared to WT. Interestingly, the NLGN3 R451C mutant demonstrated a synaptic transmission gain-of-function phenotype, and these effects are synapse specific. Although the NLGN3 R451C KI mice have reduced protein levels, NLGN3 R451C mice, but not NLGN3 KO mice, display an increase in inhibitory synapses as measured by VGAT and gephyrin immunoreactivity. Furthermore, a concomitant increase in mIPSCs frequency in somatosensory cortex was observed in NLGN3 R451C mice, but not NLGN3 KO mice (Tabuchi et al., 2007). In addition, NLGN3 R451C leads to impaired inhibitory synaptic transmission in PV-neurons in KI animals, unlike the NLGN3 KO; however, both mouse lines show enhanced inhibitory synaptic transmission in cholecystokinin basket cells (Földy et al., 2013). Horn and Nicoll (2018) also provide additional evidence of the synapse-specific function of NLGN3 by showing that knocking down NLGN3 using miRNA specifically affected IPSCs recorded from somatostatin neurons, but not from PV-neurons. In addition, NLGN3 R451C mice, but not NLGN3 KO mice, have a striking phenotype at glutamatergic synapses. In the CA1 region of the hippocampus, NLGN3 R451C mice display an increase in excitatory synaptic transmission (Etherton et al., 2011). Along with this observation, Etherton et al. (2011) saw an increase in dendritic complexity and NMDAR protein levels in NLGN3 R451C mice. In contrast, NLGN3 R451C mice display impaired synaptic transmission at the calyx of Held synapses. Furthermore, Zhang et al. (2017) elegantly demonstrated that the synaptic effect of NLGN3 on the calyx of Held synapses is only observed when NLGN3 is deleted late, but not early, in development. Lastly, NLGN3 R451C KI mice and NLGN3 KO mice share a common synaptic defect at striatal synapses; the deletion or KI of NLGN3 in D1 neurons, but not D2 neurons, results in a decrease in mIPSCs frequency (Rothwell et al., 2014). Taken together, the NLGN3 R451C mutation differentially alter synaptic function depending on neuron and synapse type.
Behavioral analyses of NLGN3 R451C KI mice revealed a deficit in social interaction and an enhancement in spatial learning; however, these findings were not reproduced in a separate independent study, likely due to differences in mouse strains or behavioral protocols (Tabuchi et al., 2007; Chadman et al., 2008; Jaramillo et al., 2014; Lakhani et al., 2019). Another phenotype of ASD is repetitive behavior; and, interestingly, the NLGN3 R451C KI and NLGN3 KO mice share this phenotype despite differences in social interaction and spatial memory paradigms (Rothwell et al., 2014; Burrows et al., 2015). Indeed, NLGN3 R451C KI and NLGN3 KO mice both have an enhanced ability to stay on an accelerated rod (Chadman et al., 2008; Rothwell et al., 2014). Importantly, the repetitive behavior of NLGN3 mutants is due to dysfunction of D1-dopamine receptor-expressing medium spiny neurons, but not D2 neurons. Taken together, the ASD phenotypes of NLGN3 R451C KI and NLGN3 KO mice are circuit- and neuron-specific. Further investigations into which circuits affect the social interaction, spatial memory, and social memory phenotypes in NLGN3 R451C and NLGN3 KO are needed to better understand the mechanisms driving these behavioral deficits in ASD.
Studies in NLGN3 R451C KI and NLGN3 KO mice highlighted a need to better understand the physiological function of NLGN3. For example, a striking observation in NLGN3 R451C KI mice is a ∼90% reduction in protein levels, while displaying both gain-of-function and loss-of-function phenotypes depending on the type of synapses. Different synaptic phenotypes induced by the single point mutation, NLGN3 R451C, suggest that WT NLGN3 normally functions in a context-dependent manner. Indeed, context-dependent function of NLGNs has been reported in which excitatory synapses are regulated by the relative expression of NLGN1. For example, NLGN1 KO mice display similar spine density as WT animals, but when NLGN1 KO neurons are co-cultured with WT neurons, the NLGN1 KO neurons show a reduction in spine density (Kwon et al., 2012). Applying this model of competition to NLGN3 R451C KI mice to explain the gain-of-function observed in this animal is worthy of investigation. It is also important to carefully study NLGN3 function throughout development. Zhang et al. (2017) demonstrated reduced synaptic transmission at the calyx of Held synapse when NLGN3 is deleted late, but not early, in development. They further showed that when NLGN3 is conditionally knocked out in early development, cerebellin-1 can compensate for the lack of NLGN3.
NLGN4X and Its Link to ASD
Divergence of NLGN4
Of the ASD-associated genes identified from human genetic screens, NLGNs are of particular interest due to their important function at synapses. Early genetic studies on the X chromosome indicated that a deletion at Xp22.3 was found in ASD/ID probands (Thomas et al., 1999; Zinn et al., 2007). Interestingly, NLGN4X is located within this region. Although disease-associated mutations in NLGNs are relatively rare, rigorous genetic studies using probands’ pedigrees have established a causal link between NLGN4X and ASD/ID (Table 2).
Because NLGN4X is a human-specific gene, the discovery of mouse NLGN4-like was exciting because it allowed the study of NLGN4 in rodents to probe its role in ASD/ID. Although, there have been enormous advances in the field regarding the synaptic function of NLGN1-3, there are still many gaps in our understanding of the NLGN4 isoforms, which is complicated due to their unusually rapid divergence in humans and rodents. In humans, NLGN4 is sex-linked, and NLGN4X and NLGN4Y combine to form an X-Y gene pair. However, in mice, NLGN4 exists as a pseudo-autosomal gene often referred to as NLGN4-like. In addition, Maxeiner et al. (2020) observed that mouse NLGN4-like undergoes rapid evolution resulting in changes in protein sequence. Sequence alignment of NLGN4X with NLGN4-like shows seven insertions in NLGN4-like across both the ECD and intracellular domain (ICD). Interestingly, NLGN4 from the rodent infra-orders castorimorpha, hystricomorpha, and sciuromorpha retains similarity to human NLGN4X, whereas the rodent infra-order myomorpha, which includes mice, do not. Thus far NLGN4 has not been identified in rats (Bolliger et al., 2008; Maxeiner et al., 2020). Sequence alignment of mouse NLGN4-like, human NLGN4X, and NLGN4Y shows that NLGN4-like only shares ∼60% sequence identity with NLGN4X/4Y, whereas NLGN4X shares ∼97% sequence identity with NLGN4Y (Figure 1). A decade of research later, it is now clear that the human and rodent NLGN4 genes do not share the same function as previously assumed.

Figure 1. Alignment of NLGN4. Alignment of mouse and human NLGN4s and their conservation.
Human and Mouse NLGN4
Human NLGN4X was first cloned almost two decades ago. In the initial studies, NLGN4X was shown to be expressed and processed in a similar fashion to that of NLGN1. NLGN4X, like NLGN1, is glycosylated, traffics to the cell surface, and can bind to PSD-95 (Bolliger et al., 2001). Furthermore, NLGN4X is found at excitatory synapses. NLGN4X overexpression in mouse hippocampal neurons increases dendritic spine density, but it decreases mEPSCs frequency and amplitude (Chanda et al., 2016; Zhang et al., 2009). However, exogenously expressed human NLGN4X in rat hippocampal slices in combination with NLGN1-3 microRNA to knockdown endogenous NLGN1-3 showed an increase in spine density and a concomitant increase in both AMPAR- and NMDAR-mediated EPSCs (Bemben et al., 2015a). The difference between these two sets of experiments is the presence of endogenous NLGN1-3. It is unclear whether NLGN4X can form heterodimers with NLGN1-3 in vivo, although NLGN4X has been shown to form heterodimers with NLGN1 (Poulopoulos et al., 2012). Further investigation into this subject can provide a better understanding of the function of endogenous NLGN4X at synapses.
Using differentiated neurons from human induced pluripotent stem cells (iPS cells), NLGN4X was shown to colocalize with VGLUT and PSD-95, revealing NLGN4X localization at excitatory synapses (Marro et al., 2019). However, in NLGN4X KO differentiated neurons, Marro et al. (2019) did not observe any changes in either EPSCs or IPSCs. It is important to note that although differentiated human neurons from iPS cells can be useful, these differentiated neurons are not fully mature and are lacking NMDARs, a key component of excitatory synapses (Zhang et al., 2013; Quadrato et al., 2017; Marro et al., 2019).
In contrast to NLGN4X, mouse NLGN4-like functions at inhibitory synapses. Localization experiments in mice show that NLGN4-like is at glycinergic inhibitory synapses where it colocalizes with glycine receptors and gephyrin, but not PSD-95 in brainstem, spinal cord, and retina. Moreover, NLGN4-like KO mice were shown to have deficits in glycinergic synaptic transmission (Jamain et al., 2008; Hoon et al., 2011; Zhang et al., 2018). In addition, NLGN4-like also functions at GABAergic synapses (Hammer et al., 2015; Unichenko et al., 2018). In KO NLGN4-like mice, GABAergic synaptic transmission is impaired in hippocampal CA3 region (Hammer et al., 2015). Together, NLGN4-like primarily acts at inhibitory synapses, either glycinergic or GABAergic, whereas human NLGN4X acts at excitatory synapses.
NLGN4-like KO mice were generated over a decade ago and have been characterized extensively. However, the behavioral data have been complicated. For instance, NLGN4-like KO mice were first characterized as having a deficit in social interaction and vocalization (Jamain et al., 2008; El-Kordi et al., 2013; Ju et al., 2014); however, another study using the same NLGN4-like KO mice did not find any deficit in social interaction or vocalization (Ey et al., 2012). Although NLGN4-like KO mice provide insights into how this protein may function at synapses, because human NLGN4X and mouse NLGN4-like are divergent, there should be caution in linking mouse NLGN4-like studies with NLGN4X-associated neurodevelopmental disorders.
Lastly, NLGNs are dynamically regulated through posttranslational modifications (Bemben et al., 2015b; Jeong et al., 2017). Similar to NLGN1 and NLGN2, posttranslational modifications have an important role in regulating NLGN4X function (Bemben et al., 2015b; Jeong et al., 2017). NLGN4X is phosphorylated by PKC at T707 (Bemben et al., 2015a). Unlike CaMKII phosphorylation of NLGN1, PKC phosphorylation of NLGN4X does not increase its trafficking to the surface. However, phosphorylated NLGN4X T707 does lead to increases in spine density and aggregation of the excitatory synapse markers VGLUT and PSD-95 (Bemben et al., 2013; Bemben et al., 2015a). In addition, analyses of the NLGN4X phospho-mimetic mutation, T707D, reveal significant enhancement of both AMPAR and NMDAR EPSCs compared to WT (Bemben et al., 2015a). How phosphorylated NLGN4X is able to increase excitatory synaptic strength will require additional investigation to reveal the precise mechanisms underlying synaptic potentiation. This topic would benefit from techniques that allow the characterization of spatiotemporal dynamics of PKC phosphorylation of NLGN4X in vivo. Furthermore, NLGN4X T707 is conserved in mouse NLGN4-like, but it is unclear whether this residue is phosphorylated in mouse NLGN4-like. Would the phosphorylation of this conserved threonine residue in mouse NLGN4-like enhance synaptic transmission as it does in human NLGN4X? Investigation on the mechanism of phosphorylation and the enhancement of synaptic transmission is a worthy topic to study.
NLGN4X and ASD/ID
Jamain et al. (2003) first established NLGN4X as causative genes for ASD/ID through screening patients with ASD and Asperger’s syndrome, and identified a frameshift mutation (1186insT) in NLGN4X, which leads to a premature stop codon at amino acid 396. Interestingly, in addition to the two probands, their mother also carries the mutation, but does not display any autistic symptoms (Jamain et al., 2003). The most convincing case for NLGN4X as an ASD/ID risk gene is from a study following a French family with a nonsense mutation in NLGN4X. Laumonnier et al. (2004) observed a 2-base-pair deletion in NLGN4X that resulted in a stop codon at position 429. By documenting the clinical data from this large family, Laumonnier et al. (2004) observed that 13 males with the nonsense mutation were diagnosed with ASD, ID, or pervasive neurodevelopmental disorders, whereas female carriers were unaffected. This finding is remarkable in showing that this mutation in NLGN4X follows an X-linked recessive pattern. Many subsequent studies have linked NLGN4X with neurodevelopmental disorders, and the recurrent theme is that the majority of affected probands are males (Table 2).
Along with frameshift and nonsense mutations, many disease-associated missense mutations have been identified in NLGN4X. How might these missense mutations affect NLGN4X function? A missense mutation was identified in two ASD probands resulting in a substitution of an arginine residue to tryptophan (NLGN4X R87W). The NLGN4X R87W variant displays a profound deficit in NLGN4X surface expression, which leads to hypofunction of the protein due to decreased synaptogenesis. Furthermore, expression of NLGN4X R87W results in increased synaptic strength when overexpressed in neurons on a WT background (Zhang et al., 2009). It is puzzling why a variant that failed to induce synaptogenesis on a null background can still enhance synaptic function. Interestingly, a cluster of NLGN4X-associated variants has been identified near the NLGN4X R87W that also display a deficit in surface expression (Nguyen et al., 2020). Because these NLGN4X-associated variants are in the ECD, it is of interest to investigate their ability to bind to neurexin. Using the solved structure of NLGN4X, it was shown that ASD-associated mutations, such as NLGN4X G99S, are located outside of the neurexin binding domain (Fabrichny et al., 2007). These data suggest the observed phenotype from the cluster of NLGN4X-associated mutations is due to a deficit in trafficking.
Another NLGN4X rare variant that has garnered much attention is a substitution in the ICD from arginine to cysteine, NLGN4X R704C (Yan et al., 2005). As discussed above, NLGN4X is phosphorylated by PKC at T707 resulting in an increase in spine numbers and EPSCs (Bemben et al., 2015a). Interestingly, there were significant deficits in phosphorylation of NLGN4X T707 in the NLGN4X R704C variant, and the effects mediated by phosphorylation were abolished (Bemben et al., 2015a). In a separate study, Chanda et al. (2016) expressed NLGN4X R704C in cultured mouse neurons on a WT background and observed an increase in both NMDAR and AMPAR EPSCs compared to WT. Interestingly, neither study observed a change in surface trafficking. The discrepancy in these studies likely results from differences in experimental design, chiefly whether to include or exclude endogenous NLGN1-3. Taken together, NLGN4X R704C displays profound differences, compared to WT, in regulation of excitatory synapses. Using human differentiated neurons from NLGN4X R704C KI hiPSCs, Marro et al. (2019) observed an increase in EPSCs compared to WT. In addition, NLGN4X R704C was shown to increase binding with GluA1, but not PSD-95 (Marro et al., 2019), again revealing that this rare variant has multiple functional effects.
With the advances in stem cell research, it is now possible to study how different NLGN4X variants function in human neurons. Although studies taking this approach provide attractive new tools to study endogenous NLGN4X and its variants, there are pitfalls that needs to be addressed. Use of differentiated neurons from hiPSCs is still in its infancy and synaptic activity from these neurons does not represent the full endogenous nature of a synapse. For instance, it has been shown that differentiated neurons using single transcription expression models lack NMDA receptors (Zhang et al., 2013; Quadrato et al., 2017; Wang et al., 2017; Nehme et al., 2018). These neurons can express NMDARs if, and only if, they are allowed to grow for a long period of time (35+ days). Even so, to date, there is little biochemical evidence that NMDARs are present under these differentiation protocols. For the study of neuroligins, this is particularly problematic as they have been shown to directly interact with NMDARs via their ECDs (Budreck et al., 2013). Thus, although stem cell and differentiation technology are attractive and can be a powerful tool to study human neurons and diseases, a better understanding of the PSD in these neurons is needed before it can be used with great confidence as a model to study synaptic transmission.
NLGN4X and NLGN4Y
Until recently, the studies on human specific NLGN4s have focused on NLGN4X. However, it is important to explore the function of NLGN4Y as well. NLGN4X and NLGN4Y are remarkably conserved with only 19 amino acid differences between them. Due to this high sequence conservation, the two proteins have been assumed to have the same function (Bemben et al., 2015b; Südhof, 2018); however, this hypothesis had not been experimentally examined until recently. Because NLGN4X/Y are sex-linked genes, an important consideration is the sex-bias in the expression of NLGN4X. Outside of the pseudo autosomal regions (PARs), some genes on the X chromosome can escape X-inactivation thus providing an imbalance of gene dosage between males and females (Carrel and Willard, 2005; Skuse, 2005; Helena Mangs and Morris, 2007; Tukiainen et al., 2017). Interestingly, there are Y-linked genes that are homologs to X-linked genes that escaped X-inactivation in order to balance the gene dosage in males. Furthermore, these X-Y gene pairs have been shown to have an important function in transcription, translation and protein stability (Bellott et al., 2014; Cortez et al., 2014; Hughes and Page, 2015). Together, these studies reveal an important role for genes on the Y chromosome other than sex determining genes. Indeed, comparison of NLGN4X and NLGN4Y expression in males and females reveals interesting differences. In a large transcriptomic study, NLGN4Y was shown to express only in males, as expected; however, NLGN4X was shown to express at similar level between males and females (Kang et al., 2011; Trabzuni et al., 2013). To complicate the issue further, a separate study reported that incomplete X-inactivation exists in mammals, and NLGN4X partially escapes (Carrel and Willard, 2005; Berletch et al., 2011). Interestingly, in a study using different tissues to study X-inactivation, NLGN4X expression is higher in the cortex in female vs. male (Tukiainen et al., 2017). Although gene expression of NLGN4X and NLGN4Y has been compared, research comparing NLGN4X and NLGN4Y protein function has lagged behind.
Although it was reasonable to hypothesize that NLGN4X and NLGN4Y served the same function due to their high sequence homology (97%), this hypothesis had never been tested. Interestingly, many ASD/ID variants have been identified in NLGN4X (Jamain et al., 2003; Laumonnier et al., 2004; Yan et al., 2005; Volaki et al., 2013; Xu et al., 2014; Bemben et al., 2015a; Chanda et al., 2016), whereas only one missense mutation has been identified in NLGN4Y (Yan et al., 2008). Furthermore, ASD/ID-associated mutations in NLGN4X selectively affect more males than females, and the reason for this male bias is unknown. This strong male bias observation in NLGN4X-associated diseases, prompted us to focus on NLGN4Y. If NLGN4Y and NLGN4X are functionally redundant, then there should not be a male bias in NLGN4X-associated diseases.
To explore the function of NLGN4Y, in a recent study, we directly compared NLGN4X and NLGN4Y and found that NLGN4Y cannot traffic to the surface to induce synapses (Nguyen et al., 2020). Furthermore, the differential trafficking observed between NLGN4X and NLGN4Y is due to an amino acid difference at position 93, with proline for NLGN4X and serine for NLGN4Y. Indeed, the NLGN4Y S93P mutant was able to efficiently traffic to the surface and induce synapses. Interestingly, there is a cluster of disease-associated NLGN4X variants surrounding the critical amino acid in NLGN4X. Upon analysis, these variants phenocopied the NLGN4Y trafficking deficit and cannot induce synapses (Figure 2).
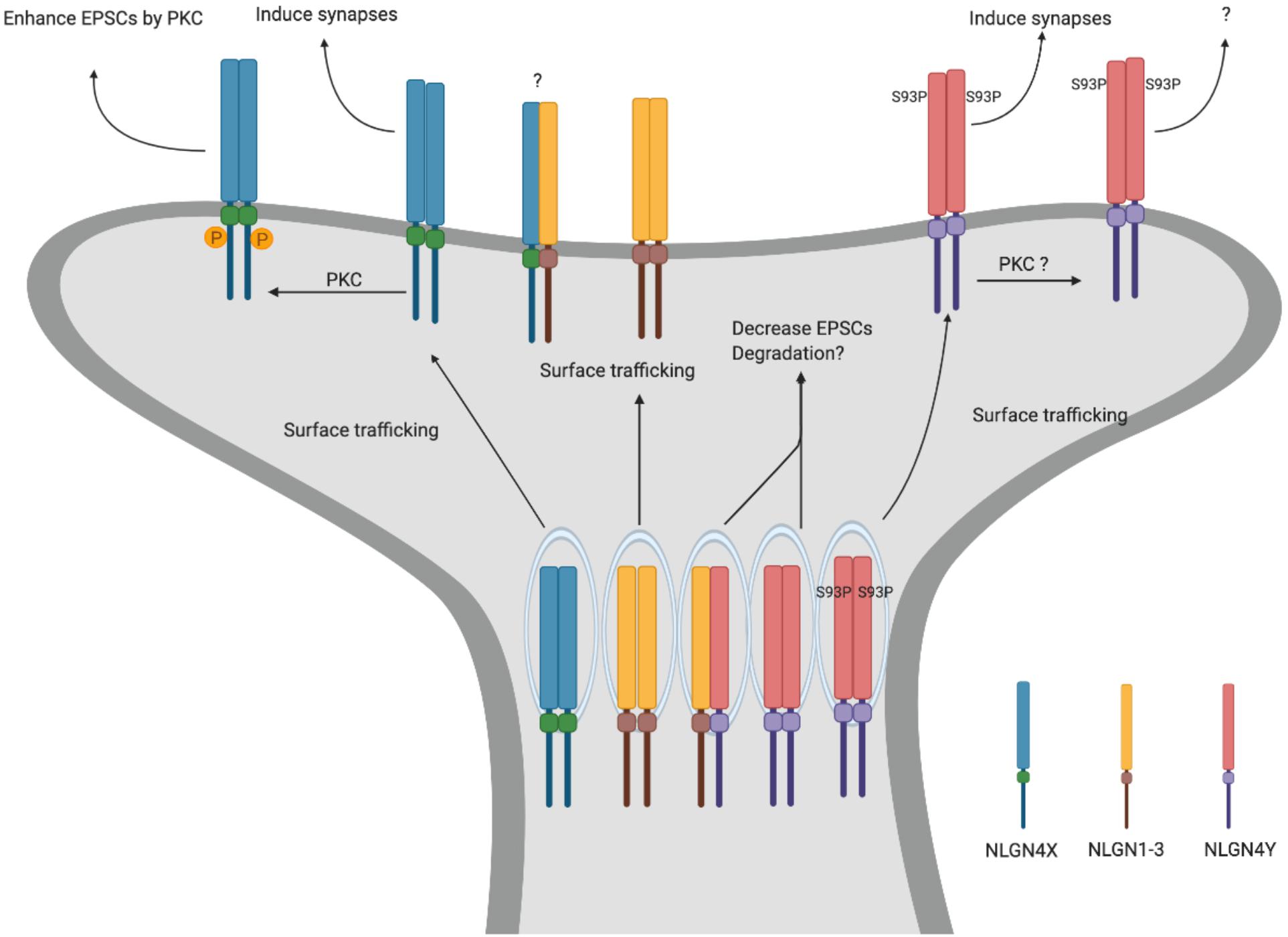
Figure 2. NLGN4X and NLGN4Y function. Schematic for differential trafficking of NLGN4X vs NLGN4Y. NLGN4X can traffic to the surface and induce excitatory synapses. Furthermore, phosphorylation of NLGN4X by PKC drastically enhances excitatory postsynaptic currents (EPSCs). In contrast, NLGN4Y cannot traffic to the surface, thus decreasing EPSCs through binding with other NLGNs.
What is the function of NLGN4Y if it cannot get to the surface? Nguyen et al. (2020) demonstrated that NLGN4Y can oligomerize with NLGN1, 2, 3, and 4X and reduce their surface trafficking. In addition, exogenously expressed NLGN4Y on a WT background decreased mEPSCs suggesting NLGN4Y acts to inhibit NLGN1-3 function. However, this study relies on exogenously expressed NLGNs in heterologous cells or rat hippocampal neurons. What the role is for endogenous NLGN4Y in human neurons is an important lingering question.
Conclusion
With the advances in NGS technologies, a wide variety of genes have been associated with ASD/ID. However, many of these studies have ignored the sex chromosomes due to the additional expense and a lack of statistical power. However, historically many genes on the X-chromosome have been linked to ASD/ID by evaluating proband pedigrees. NLGN3 and NLGN4X, both on the X chromosome, were among the first genes associated with ASD/ID. Although NLGN3 and NLGN4X variants only occur in a small population of ASD/ID cases, studies using NLGN3 and NLGN4 mouse models have provided many insights into how disruptions in NLGN3 and NLGN4 function contribute to ASD/ID phenotypes. With advances in stem cell and neuronal differentiation, it is now possible to study NLGN3 and NLGN4X variants using human iPSCs to explore the causality between disruption in sex-linked NLGNs and ASD/ID by examining endogenous human neuroligins. Although neuronal differentiation is an exciting new technology to further our understanding of the human brain, differentiated neurons from human iPSCs are still relatively immature. Further improvement in the technologies to develop reliable mature neurons will be of paramount importance going forward. In addition, the unexpected revelations from the study of NLGN4X and NLGN4Y highlight the need to investigate the often-ignored Y-chromosome. Although many facets of the sex-linked NLGNs have been characterized, many important questions remain unanswered and provide a fertile topic for future investigation into synaptic regulation and to develop therapeutic treatments.
Author Contributions
TN and KR wrote the manuscript. AL helped to create table for variants.
Funding
This review was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program (1ZIANS003140-06).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Antonelli, R., Pizzarelli, R., Pedroni, A., Fritschy, J.-M., Del Sal, G., Cherubini, E., et al. (2014). Pin1-dependent signalling negatively affects GABAergic transmission by modulating neuroligin2/gephyrin interaction. Nat. Commun. 5:5066. doi: 10.1038/ncomms6066
Bellott, D. W., Hughes, J. F., Skaletsky, H., Brown, L. G., Pyntikova, T., Cho, T. J., et al. (2014). Mammalian y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508, 494–499. doi: 10.1038/nature13206
Bemben, M. A., Nguyen, Q. A., Wang, T., Li, Y., Nicoll, R. A., and Roche, K. W. (2015a). Autism-associated mutation inhibits protein kinase C-mediated neuroligin-4X enhancement of excitatory synapses. Proc. Natl. Acad. Sci. U.S.A. 112, 2551–2556. doi: 10.1073/pnas.1500501112
Bemben, M. A., Nguyen, T. A., Li, Y., Wang, T., Nicoll, R. A., and Roche, K. W. (2019). Isoform-specific cleavage of neuroligin-3 reduces synapse strength. Mol. Psychiatry 24, 145–160. doi: 10.1038/s41380-018-0242-y
Bemben, M. A., Shipman, S. L., Hirai, T., Herring, B. E., Li, Y., Badger, J. D., et al. (2013). CaMKII phosphorylation of neuroligin-1 regulates excitatory synapses. Nat. Neurosci. 17, 56–64. doi: 10.1038/nn.3601
Bemben, M. A., Shipman, S. L., Nicoll, R. A., and Roche, K. W. (2015b). The cellular and molecular landscape of neuroligins. Trends Neurosci. 38, 496–505. doi: 10.1016/j.tins.2015.06.004
Berletch, J. B., Yang, F., Xu, J., Carrel, L., and Disteche, C. M. (2011). Genes that escape from X inactivation. Hum. Genet. 130, 237–245. doi: 10.1007/s00439-011-1011-z
Blasi, F., Bacchelli, E., Pesaresi, G., Carone, S., Bailey, A. J., and Maestrini, E. (2006). Absence of coding mutations in the X-linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am. J. Med. Genet. Neuropsychiatr. Genet. 141B, 220–221. doi: 10.1002/ajmg.b.30287
Bolliger, M. F., Frei, K., Winterhalter, K. H., and Gloor, S. M. (2001). Identification of a novel neuroligin in humans which binds to PSD-95 and has a widespread expression. Biochem. J. 356(Pt 2), 581–588. doi: 10.1042/0264-6021:3560581
Bolliger, M. F., Pei, J., Maxeiner, S., Boucard, A. A., Grishin, N. V., and Südhof, T. C. (2008). Unusually rapid evolution of Neuroligin-4 in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 6421–6426. doi: 10.1073/pnas.0801383105
Budreck, E. C., Kwon, O.-B., Jung, J. H., Baudouin, S., Thommen, A., Kim, H.-S., et al. (2013). Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc. Natl. Acad. Sci. U.S.A. 110, 725–730. doi: 10.1073/pnas.1214718110
Burrows, E. L., Laskaris, L., Koyama, L., Churilov, L., Bornstein, J. C., Hill-Yardin, E. L., et al. (2015). A neuroligin-3 mutation implicated in autism causes abnormal aggression and increases repetitive behavior in mice. Mol. Autism 6:62. doi: 10.1186/s13229-015-0055-7
Carrel, L., and Willard, H. F. (2005). X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434, 400–404. doi: 10.1038/nature03479
Chadman, K. K., Gong, S., Scattoni, M. L., Boltuck, S. E., Gandhy, S. U., Heintz, N., et al. (2008). Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 1, 147–158. doi: 10.1002/aur.22
Chanda, S., Aoto, J., Lee, S. J., Wernig, M., and Südhof, T. C. (2016). Pathogenic mechanism of an autism-associated neuroligin mutation involves altered AMPA-receptor trafficking. Mol. Psychiatry 21, 169–177. doi: 10.1038/mp.2015.20
Chih, B., Afridi, S. K., Clark, L., and Scheiffele, P. (2004). Disorder-associated mutations lead to functional inactivation of neuroligins. Hum. Mol. Genet. 13, 1471–1477. doi: 10.1093/hmg/ddh158
Chih, B., Engelman, H., and Scheiffele, P. (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328. doi: 10.1126/science.1107470
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., et al. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. doi: 10.1016/j.neuron.2007.05.029
Comoletti, D., De Jaco, A., Jennings, L. L., Flynn, R. E., Gaietta, G., Tsigelny, I., et al. (2004). The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J. Neurosci. 24, 4889–4893. doi: 10.1523/JNEUROSCI.0468-04.2004
Cortez, D., Marin, R., Toledo-Flores, D., Froidevaux, L., Liechti, A., Waters, P. D., et al. (2014). Origins and functional evolution of y chromosomes across mammals. Nature 508, 488–493. doi: 10.1038/nature13151
De Jaco, A., Comoletti, D., Kovarik, Z., Gaietta, G., Radiæ, Z., Lockridge, O., et al. (2006). A mutation linked with autism reveals a common mechanism of endoplasmic reticulum retention for theα,β-hydrolase fold protein family. J. Biol. Chem. 281, 9667–9676. doi: 10.1074/jbc.M510262200
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
El-Kordi, A., Winkler, D., Hammerschmidt, K., Kästner, A., Krueger, D., Ronnenberg, A., et al. (2013). Development of an autism severity score for mice using Nlgn4 null mutants as a construct-valid model of heritable monogenic autism. Behav. Brain Res. 251, 41–49. doi: 10.1016/j.bbr.2012.11.016
Ellison, J. W., Rosenfeld, J. A., and Shaffer, L. G. (2013). Genetic Basis of Intellectual Disability. Annu. Rev. Med. 64, 441–450. doi: 10.1146/annurev-med-042711-140053
Etherton, M., Földy, C., Sharma, M., Tabuchi, K., Liu, X., and Shamloo, M. (2011). Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. U.S.A. 108, 13764–13769. doi: 10.1073/pnas.1111093108
Ey, E., Yang, M., Katz, A. M., Woldeyohannes, L., Silverman, J. L., Leblond, C. S., et al. (2012). Absence of deficits in social behaviors and ultrasonic vocalizations in later generations of mice lacking neuroligin4. Genes Brain Behav. 11, 928–941. doi: 10.1111/j.1601-183X.2012.00849.x
Fabrichny, I. P., Leone, P., Sulzenbacher, G., Comoletti, D., Miller, M. T., Taylor, P., et al. (2007). Structural analysis of the synaptic protein neuroligin and its β-neurexin complex: determinants for folding and cell adhesion. Neuron 56, 979–991. doi: 10.1016/j.neuron.2007.11.013
Földy, C., Malenka, R. C., and Südhof, T. C. (2013). Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 78, 498–509. doi: 10.1016/j.neuron.2013.02.036
Fombonne, E. (2013). The epidemiology of autism: a review. Sci. Ment. Health 2, 65–82. doi: 10.1017/S0033291799008508
Geschwind, D. H. (2011). Genetics of autism spectrum disorders. Trends Cogn. Sci. 15, 409–416. doi: 10.1016/j.tics.2011.07.003
Geschwind, D. H., and State, M. W. (2015). Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 14, 1109–1120. doi: 10.1016/S1474-4422(15)00044-7
Giannone, G., Mondin, M., Grillo-Bosch, D., Tessier, B., Saint-Michel, E., Czöndör, K., et al. (2013). Neurexin-1β binding to neuroligin-1 triggers the preferential recruitment of PSD-95 versus gephyrin through tyrosine phosphorylation of neuroligin-1. Cell Rep. 3, 1996–2007. doi: 10.1016/j.celrep.2013.05.013
Hammer, M., Krueger-Burg, D., Tuffy, L. P., Cooper, B. H., Taschenberger, H., Goswami, S. P., et al. (2015). Perturbed hippocampal synaptic inhibition and γ-oscillations in a neuroligin-4 knockout mouse model of autism. Cell Rep. 13, 516–523. doi: 10.1016/j.celrep.2015.09.011
Helena Mangs, A., and Morris, B. J. (2007). The human pseudoautosomal region (PAR): origin, function and future. Curr. Genom. 8, 129–136. doi: 10.2174/138920207780368141
Hoon, M., Soykan, T., Falkenburger, B., Hammer, M., Patrizi, A., Schmidt, K.-F. K.-F., et al. (2011). Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc. Natl. Acad. Sci. U.S.A. 108, 3053–3058. doi: 10.1073/pnas.1006946108
Horn, M. E., and Nicoll, R. A. (2018). Somatostatin and parvalbumin inhibitory synapses onto hippocampal pyramidal neurons are regulated by distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 115, 589–594. doi: 10.1073/pnas.1719523115
Hughes, J. F., and Page, D. C. (2015). The Biology and Evolution of Mammalian Y Chromosomes. Annu. Rev. Genet. 49, 507–527. doi: 10.1146/annurev-genet-112414-055311
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Jamain, S., Radyushkin, K., Hammerschmidt, K., Granon, S., Boretius, S., Varoqueaux, F., et al. (2008). Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. U.S.A. 105, 1710–1715. doi: 10.1073/pnas.0711555105
Jaramillo, T. C., Liu, S., Pettersen, A., Birnbaum, S. G., and Powell, C. M. (2014). Autism-related neuroligin-3 mutation alters social behavior and spatial learning. Autism Res. 7, 264–272. doi: 10.1002/aur.1362
Jeong, J., Pandey, S., Li, Y., Badger, J. D., Lu, W., and Roche, K. W. (2019). PSD-95 binding dynamically regulates NLGN1 trafficking and function. Proc. Natl. Acad. Sci. U.S.A. 116, 12035–12044. doi: 10.1073/pnas.1821775116
Jeong, J., Paskus, J. D., and Roche, K. W. (2017). Posttranslational modifications of neuroligins regulate neuronal and glial signaling. Curr. Opin. Neurobiol. 45, 130–138. doi: 10.1016/j.conb.2017.05.017
Jiang, Y. H., Yuen, R. K. C., Jin, X., Wang, M., Chen, N., Wu, X., et al. (2013). Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93, 249–263. doi: 10.1016/j.ajhg.2013.06.012
Ju, A., Hammerschmidt, K., Tantra, M., Krueger, D., Brose, N., and Ehrenreich, H. (2014). Juvenile manifestation of ultrasound communication deficits in the neuroligin-4 null mutant mouse model of autism. Behav. Brain Res. 270, 159–164. doi: 10.1016/j.bbr.2014.05.019
Kang, H. J., Kawasawa, Y. I., Cheng, F., Zhu, Y., Xu, X., Li, M., et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489. doi: 10.1038/nature10523
Kenny, E. M., Cormican, P., Furlong, S., Heron, E., Kenny, G., Fahey, C., et al. (2014). Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol. Psychiatry 19, 872–879. doi: 10.1038/mp.2013.127
Kwon, H.-B., Kozorovitskiy, Y., Oh, W.-J., Peixoto, R. T., Akhtar, N., Saulnier, J. L., et al. (2012). Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nat. Neurosci. 15, 1667–1674. doi: 10.1038/nn.3256
Lakhani, C. M., Manrai, A. K., Yang, J., Visscher, P. M., Patel, C. J., and Department, B. T. T. (2019). Genetic background effects in neuroligin-3 mutant mice: minimal behavioral abnormalities on C57 background. Physiol. Behav. 176, 139–148. doi: 10.1016/j.physbeh.2017.03.040
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M.-P., et al. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557. doi: 10.1086/382137
Letellier, M., Szíber, Z., Chamma, I., Saphy, C., Papasideri, I., Tessier, B., et al. (2018). A unique intracellular tyrosine in neuroligin-1 regulates AMPA receptor recruitment during synapse differentiation and potentiation. Nat. Commun. 9:3979. doi: 10.1038/s41467-018-06220-2
Lubs, H. A., Stevenson, R. E., and Schwartz, C. E. (2012). Fragile X and X-linked intellectual disability: four decades of discovery. Am. J. Hum. Genet. 90, 579–590. doi: 10.1016/j.ajhg.2012.02.018
Marro, S. G., Chanda, S., Yang, N., Janas, J. A., Valperga, G., Trotter, J., et al. (2019). Neuroligin-4 regulates excitatory synaptic transmission in human neurons. Neuron 103, 617.e6–626.e6. doi: 10.1016/j.neuron.2019.05.043
Martínez, F., Caro-Llopis, A., Roselló, M., Oltra, S., Mayo, S., Monfort, S., et al. (2016). High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J. Med. Genet. 54, 87–92. doi: 10.1136/jmedgenet-2016-103964
Maxeiner, S., Benseler, F., Krasteva-Christ, G., Brose, N., and Südhof, T. C. (2020). Evolution of the autism-associated neuroligin-4 gene reveals broad erosion of pseudoautosomal regions in rodents. Mol. Biol. Evol. 37, 1243–1258. doi: 10.1093/molbev/msaa014
McRae, J. F., Clayton, S., Fitzgerald, T. W., Kaplanis, J., Prigmore, E., Rajan, D., et al. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062
Miles, J. H. (2011). Autism spectrum disorders-A genetics review. Genet. Med. 13, 278–294. doi: 10.1097/GIM.0b013e3181ff67ba
Nehme, R., Zuccaro, E., Ghosh, S. D., Li, C., Sherwood, J. L., Pietilainen, O., et al. (2018). Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep. 23, 2509–2523. doi: 10.1016/j.celrep.2018.04.066
Nguyen, Q. A., Horn, M. E., and Nicoll, R. A. (2016). Distinct roles for extracellular and intracellular domains in neuroligin function at inhibitory synapses. eLife 5, 1–21. doi: 10.7554/eLife.19236
Nguyen, T. A., Wu, K., Pandey, S., Lehr, A. W., Li, Y., Bemben, M. A., et al. (2020). A cluster of autism-associated variants on X-Linked NLGN4X functionally resemble NLGN4Y. Neuron 106, 759.e7–768.e7. doi: 10.1016/j.neuron.2020.03.008
Pampanos, A., Volaki, K., Kanavakis, E., Papandreou, O., Youroukos, S., Thomaidis, L., et al. (2009). A substitution involving the NLGN4 gene associated with autistic behavior in the greek population. Genet. Test. Mol. Biomark. 13, 611–615. doi: 10.1089/gtmb.2009.0005
Paskus, J. D., Herring, B. E., and Roche, K. W. (2020). Kalirin and trio: RhoGEFs in synaptic transmission, plasticity, and complex brain disorders. Trends Neurosci. 43, 505–518. doi: 10.1016/j.tins.2020.05.002
Paskus, J. D., Tian, C., Fingleton, E., Shen, C., Chen, X., Li, Y., et al. (2019). Synaptic kalirin-7 and trio interactomes reveal a gef protein-dependent neuroligin-1 mechanism of action. Cell Rep. 29, 2944.e5–2952.e5. doi: 10.1016/j.celrep.2019.10.115
Poulopoulos, A., Aramuni, G., Meyer, G., Soykan, T., Hoon, M., Papadopoulos, T., et al. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642. doi: 10.1016/j.neuron.2009.08.023
Poulopoulos, A., Soykan, T., Tuffy, L. P., Hammer, M., Varoqueaux, F., and Brose, N. (2012). Homodimerization and isoform-specific heterodimerization of neuroligins. Biochem. J. 446, 321–330. doi: 10.1042/BJ20120808
Quadrato, G., Nguyen, T., Macosko, E. Z., Sherwood, J. L., Min Yang, S., Berger, D. R., et al. (2017). Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545, 48–53. doi: 10.1038/nature22047
Quartier, A., Courraud, J., Thi Ha, T., McGillivray, G., Isidor, B., Rose, K., et al. (2019). Novel mutations in NLGN3 causing autism spectrum disorder and cognitive impairment. Hum. Mutat. 40, 2021–2032. doi: 10.1002/humu.23836
Raymond, F. L. (2006). X linked mental retardation: a clinical guide. J. Med. Genet. 43, 193–200. doi: 10.1136/jmg.2005.033043
Redin, C., Gérard, B., Lauer, J., Herenger, Y., Muller, J., Quartier, A., et al. (2014). Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 51, 724–736. doi: 10.1136/jmedgenet-2014-102554
Rothwell, P. E., Fuccillo, M. V., Maxeiner, S., Hayton, S. J., Gokce, O., Lim, B. K., et al. (2014). Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158, 198–212. doi: 10.1016/j.cell.2014.04.045
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Singh, S. K., and Eroglu, C. (2013). Neuroligins provide molecular links between syndromic and nonsyndromic autism. Sci. Signal. 6:re4. doi: 10.1126/scisignal.2004102
Skuse, D. H. (2005). X-linked genes and mental functioning. Hum. Mol. Genet. 14, 27–32. doi: 10.1093/hmg/ddi112
Südhof, T. C. (2017). Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171, 745–769. doi: 10.1016/j.cell.2017.10.024
Südhof, T. C. (2018). Towards an understanding of synapse formation. Neuron 100, 276–293. doi: 10.1016/j.neuron.2018.09.040
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., et al. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76. doi: 10.1126/science.1146221
Thomas, N. S., Sharp, A. J., Browne, C. E., Skuse, D., Hardie, C., and Dennis, N. R. (1999). Xp deletions associated with autism in three females. Hum. Genet. 104, 43–48. doi: 10.1007/s004390050908
Trabzuni, D., Ramasamy, A., Imran, S., Walker, R., Smith, C., Weale, M. E., et al. (2013). Widespread sex differences in gene expression and splicing in the adult human brain. Nat. Commun. 4:2771. doi: 10.1038/ncomms3771
Tukiainen, T., Villani, A. C., Yen, A., Rivas, M. A., Marshall, J. L., Satija, R., et al. (2017). Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248. doi: 10.1038/nature24265
Unichenko, P., Yang, J. W., Kirischuk, S., Kolbaev, S., Kilb, W., Hammer, M., et al. (2018). Autism related neuroligin-4 knockout impairs intracortical processing but not sensory inputs in mouse barrel cortex. Cereb. Cortex 28, 2873–2886. doi: 10.1093/cercor/bhx165
Venkatesh, H. S., Johung, T. B., Caretti, V., Noll, A., Tang, Y., Nagaraja, S., et al. (2015). Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 161, 803–816. doi: 10.1016/j.cell.2015.04.012
Venkatesh, H. S., Tam, L. T., Woo, P. J., Lennon, J., Nagaraja, S., Gillespie, S. M., et al. (2017). Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537. doi: 10.1038/nature24014
Volaki, K., Pampanos, A., Kitsiou-Tzeli, S., Vrettou, C., Oikonomakis, V., Sofocleous, C., et al. (2013). Mutation screening in the Greek population and evaluation of NLGN3 and NLGN4X genes causal factors for autism. Psychiatr. Genet. 23, 198–203. doi: 10.1097/YPG.0b013e3283643644
Wang, C., Ward, M. E., Chen, R., Liu, K., Tracy, T. E., Chen, X., et al. (2017). Scalable production of iPSC-derived human neurons to identify tau-lowering compounds by high-content screening. Stem Cell Rep. 9, 1221–1233. doi: 10.1016/j.stemcr.2017.08.019
Wang, S., Mandell, J. D., Kumar, Y., Sun, N., Morris, M. T., Arbelaez, J., et al. (2018). De novo sequence and copy number variants are strongly associated with tourette disorder and implicate cell polarity in pathogenesis. Cell Rep. 24, 3441.e12–3454.e12. doi: 10.1016/j.celrep.2018.08.082
Werling, D. M., and Geschwind, D. H. (2013). Understanding sex bias in autism spectrum disorder. Curr. Opin. Neurol. 26, 146–153. doi: 10.1097/WCO.0b013e32835ee548
Werling, D. M., Parikshak, N. N., and Geschwind, D. H. (2016). Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat. Commun. 7:10717. doi: 10.1038/ncomms10717
Wise, A. L., Gyi, L., and Manolio, T. A. (2013). EXclusion: toward integrating the X chromosome in genome-wide association analyses. Am. J. Hum. Genet. 92, 643–647. doi: 10.1016/j.ajhg.2013.03.017
Xu, X., Xiong, Z., Zhang, L., Liu, Y., Lu, L., Peng, Y., et al. (2014). Variations analysis of NLGN3 and NLGN4X gene in Chinese autism patients. Mol. Biol. Rep. 41, 4133–4140. doi: 10.1007/s11033-014-3284-5
Yan, J., Feng, J., Schroer, R., Li, W., Skinner, C., Schwartz, C. E., et al. (2008). Analysis of the neuroligin 4Y gene in patients with autism. Psychiatr. Genet. 18, 204–207. doi: 10.1097/YPG.0b013e3282fb7fe6
Yan, J., Oliveira, G., Coutinho, A., Yang, C., Feng, J., Katz, C., et al. (2005). Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 10, 329–332. doi: 10.1038/sj.mp.4001629
Yu, T. W., Chahrour, M. H., Coulter, M. E., Jiralerspong, S., Okamura-Ikeda, K., Ataman, B., et al. (2013). Using whole-exome sequencing to identify inherited causes of autism. Neuron 77, 259–273. doi: 10.1016/j.neuron.2012.11.002
Yuen, R. K. C., Merico, D., Bookman, M., Howe, J. L., Thiruvahindrapuram, B., Patel, R. V., et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611. doi: 10.1038/nn.4524
Zhang, B., Gokce, O., Hale, W. D., Brose, N., and Südhof, T. C. (2018). Autism-associated neuroligin-4 mutation selectively impairs glycinergic synaptic transmission in mouse brainstem synapses. J. Exp. Med. 215, 1543–1553. doi: 10.1084/jem.20172162
Zhang, B., Seigneur, E., Wei, P., Gokce, O., Morgan, J., and Südhof, T. C. (2017). Developmental plasticity shapes synaptic phenotypes of autism-associated neuroligin-3 mutations in the calyx of Held. Mol. Psychiatry 22, 1483–1491. doi: 10.1038/mp.2016.157
Zhang, C., Milunsky, J. M., Newton, S., Ko, J., Zhao, G., Maher, T. A., et al. (2009). A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J. Neurosci. 29, 10843–10854. doi: 10.1523/JNEUROSCI.1248-09.2009
Zhang, Y., Pak, C. H., Han, Y., Ahlenius, H., Zhang, Z., Chanda, S., et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. doi: 10.1016/j.neuron.2013.05.029
Keywords: autism, intellectual disabililties, NLGN3, NLGN4X, neuroligin
Citation: Nguyen TA, Lehr AW and Roche KW (2020) Neuroligins and Neurodevelopmental Disorders: X-Linked Genetics. Front. Synaptic Neurosci. 12:33. doi: 10.3389/fnsyn.2020.00033
Received: 20 May 2020; Accepted: 22 July 2020;
Published: 11 August 2020.
Edited by:
Kimberly M. Huber, University of Texas Southwestern Medical Center, United StatesReviewed by:
Dilja Krueger-Burg, University Medical Center Göttingen, GermanyMichele H. Jacob, Tufts University School of Medicine, United States
Copyright © 2020 Nguyen, Lehr and Roche. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thien A. Nguyen, tan30@georgetown.edu; Katherine W. Roche, rochek@ninds.nih.gov