 Gioacchino P. Marceca
Gioacchino P. Marceca Giovanni Nigita
Giovanni Nigita Federica Calore
Federica Calore Carlo M. Croce
Carlo M. Croce- 1Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 2Department of Cancer Biology and Genetics and Comprehensive Cancer Center, The Ohio State University, Columbus, OH, United States
Cancer-associated cachexia is a heterogeneous, multifactorial syndrome characterized by systemic inflammation, unintentional weight loss, and profound alteration in body composition. The main feature of cancer cachexia is represented by the loss of skeletal muscle tissue, which may or may not be accompanied by significant adipose tissue wasting. Such phenotypic alteration occurs as the result of concomitant increased myofibril breakdown and reduced muscle protein synthesis, actively contributing to fatigue, worsening of quality of life, and refractoriness to chemotherapy. According to the classical view, this condition is primarily triggered by interactions between specific tumor-induced pro-inflammatory cytokines and their cognate receptors expressed on the myocyte membrane. This causes a shift in gene expression of muscle cells, eventually leading to a pronounced catabolic condition and cell death. More recent studies, however, have shown the involvement of regulatory non-coding RNAs in the outbreak of cancer cachexia. In particular, the role exerted by microRNAs is being widely addressed, and several mechanistic studies are in progress. In this review, we discuss the most recent findings concerning the role of microRNAs in triggering or exacerbating muscle wasting in cancer cachexia, while mentioning about possible roles played by long non-coding RNAs and ADAR-mediated miRNA modifications.
Introduction
In humans, the skeletal muscle represents the most substantial fraction of fat-free body mass and is highly relevant to physiology. It constitutes ~40% of total body mass, encloses 50%–75% of all body proteins, and accounts for about 30%–50% of total protein turnover (1, 2), with precise percentages depending on variables like genetic factors, age, health status and nutrition (3–5). Notoriously, skeletal muscles function as effectors of the locomotor system, serve as storage for amino acids and carbohydrates, and have a central role in thermogenesis (1, 2). Also, skeletal muscles function as secretory organs since they physiologically express and release cytokines and other regulatory peptides, exerting important hormone-like effects (6).
The function and integrity of skeletal muscles can be severely impaired by increased concentrations of reactive oxygen species (ROS) or adverse conditions related to chronic diseases. These include cardiovascular diseases, cancer, chronic respiratory diseases, and diabetes (7–10). In particular, several cancer types can determine a condition of chronic systemic inflammation, which eventually leads toward the onset of cancer cachexia. From a clinical standpoint, cancer cachexia is a multifactorial metabolic syndrome that can develop progressively through different stages. It is primarily characterized by ongoing depletion of the skeletal muscle and uncontrolled weight loss (11). Other usual manifestations include loss of fat mass (at various degrees), lower energy intake, increased resting energy expenditure (REE), loss of appetite, fatigue, and resistance to chemotherapy (12).
In patients with cancer cachexia, survival is directly related to both total weight loss and rate of weight loss. Based on this, it was estimated that ~20%–30% of mortalities in these patients are due to such debilitating condition rather than the tumor itself (13, 14). The incidence and prevalence of cancer cachexia vary depending on the tumor type and stage. Recent statistical analysis on large cohorts of advanced tumor patients let emerge that pancreatic and liver cancers are malignancies at highest risk of developing cachexia (80-90%), followed by lung, gastro-esophageal, colorectal, and head-and-neck cancers (60%–80%) (12, 15). Differently, thyroid, breast, prostate, and skin cancers represent the groups at lowest risk (20%–30%) (15). Further confirmations about these statements come from a wide-transcriptome analysis of >4,500 tumor samples including 12 cancer types, which revealed a strong correlation between tumor-specific expression of cachexia-inducing factors and prevalence of the syndrome (16).
According to the knowledge acquired over the past three decades, we now know that the development of cancer cachexia is mostly driven by an aberrant tumor-induced inflammatory response. Here, the persistent release of pro-inflammatory cytokines along with immune-suppressive factors leads to systemic inflammation, with subsequent immunosuppression, debilitation, and metabolic dysfunctions (17–19). Among the most relevant factors, interleukin (IL)-6, IL-1α, tumor necrosis factor-alpha (TNFα), and interferon-gamma (INFγ) have long been recognized as mediators of cancer cachexia, though several other potential mediators have been identified (12, 16, 20, 21). In the context of skeletal muscle, these factors function as triggers of the ubiquitin-proteasome and autophagy–lysosomes pathways, which are the main responsible for the high proteolysis rate observed in muscles of cachectic individuals. Moreover, interactions of these factors with their cognate receptors at the myocyte membrane level cause a shift in gene expression toward enhanced thermogenesis and exacerbation of the inflammatory state (12, 16, 20, 21). Following this reasoning, several therapeutic agents have been developed in the attempt to prevent or block the triggering of pathways downstream of such mediators. However, despite their proven efficacy at the molecular level, no conclusive results have been obtained in terms of the effectiveness of the treatment, except for very few drugs (20–23). This is probably attributable to the multifactorial etiology of cancer cachexia, which may be more appropriately treated by the exploitation of balanced multimodal treatments (24, 25). Nevertheless, several discrepancies have been observed between experimental models of cancer cachexia and the corresponding human condition, and inferences derived from such models likely failed to faithfully recapitulate mechanistic insights related to human cachexia (21, 26). Thus, cancer cachexia still represents a challenging issue that needs to be more accurately defined, and remains underdiagnosed in many instances.
More recently, the role of non-coding RNAs (ncRNAs) in the physiopathology of striated muscle has received more considerable attention, as these molecules are involved in processes like regulation of gene expression, translational control, chromatin remodeling, and cell-to-cell communication. In particular, microRNAs (miRNAs) represent the most studied and best-characterized class of small regulatory ncRNAs to the present day. Several studies have focused on their involvement in skeletal muscle decay following the onset of specific myopathies, including cancer cachexia. As a result, it seems that miRNAs may represent valuable diagnostic/prognostic tools for cancer cachexia, as well as new potential targets for therapeutic intervention.
After illustrating their role in skeletal muscle physiology, in the present article, we give an overview of the involvement of microRNAs in mechanisms of muscle wasting during cancer cachexia. Moreover, we briefly discuss some hints on possible roles of long non-coding RNAs and A-to-I microRNA editing in the same context.
MicroRNAs
In the current literature, ~4.6% of miRNAs are intragenic and excised from introns by the spliceosome, while >95% of the remaining are located into intergenic regions and transcribed starting from their own promoters. Only a small minority of miRNAs are located in protein-coding genomic regions (27, 28).
In their mature form, miRNAs are ~21-23 nucleotides in length and are mainly interspersed in non-coding regions of the human genome. However, mature miRNAs are not the final product of the transcription. Instead, they derive from a two-step cleavage process (29). After being transcribed by RNA polymerase II, primary miRNA transcripts (pri-miRNA) are cleaved into ∼60–80 nucleotides long RNAs by the microprocessor complex. This is essentially composed of the RNAase III Drosha and its cofactor DGCR8, a motif-specific binding protein. Drosha-cleaved RNA molecules are termed precursor miRNAs (pre-miRNAs). These typically maintain the hairpin structure presented by pri-miRNAs and are promptly exported to the cytoplasm, where the RNase type III Dicer further process them. Subsequently, two small single-stranded RNA molecules are produced, i.e., the mature miRNAs, originated from the 5′ (-5p form) and the 3′ (-3p form) arm of the pre-miRNA, respectively (29). One of these two miRNAs (termed guide strand) is loaded onto an Argonaute (AGO) protein to form the core unit of the miRNA-induced silencing complex (miRISC), while the other one (termed passenger strand) is degraded. This process is known as the canonical miRNA biogenesis pathway, by which most of the mature miRNAs are generated. However, some non-canonical biogenesis pathways also exist (29).
From the functional standpoint, miRNAs are important post-transcriptional regulators of gene expression exerting the role of translational repressors (30, 31). The basic requirement underlying such a modulatory role is a thermodynamically stable base pairing between a defined region of the miRNA sequence, referred to as the seed region, and one or multiple seed-complementary regions of an mRNA. By definition, the seed region includes nucleotides 2-8 at the 5’ terminus of miRNAs, while seed-complementary regions are usually assumed to be located within the 3’UTR (30, 31), although some exceptions to these rules have been reported in the literature (32–34). The vast majority of miRNA-mediated gene silencing is miRISC-dependent, occurs by non-perfect seed-mRNA base pairing, and requires the recruitment of additional cytoplasmic effectors, eventually causing translational repression and mRNA decay (30, 31).
MiRNAs are involved in a plethora of physiological functions and are essential for the regulation of gene expression during development, cell differentiation, and homeostasis maintenance (35–37). On the contrary, any dysfunction or alteration in their expression can lead to a wide range of pathological conditions, including cancer development (38). To date, it has been estimated that the human genome comprises ~1,900 annotated miRNA precursors giving rise to over 2,600 different mature miRNAs (39), which are thought to control the gene expression level of ~60% of human genes (40).
MiRNA expression is not homogeneous across human tissues, but is instead driven and modulated by several tissue-specific epigenetic mechanisms (41). In this regard, some conventional criteria have been proposed to classify miRNAs depending on differences in their tissue representativeness. For instance, in a wide-expression profiling study (42), miRNAs were classified as “tissue-specific” or “tissue-enriched”, depending on whether they were detected at ≥20-fold or <20-fold levels in the enriched tissue compared to the mean values for other tissues, respectively. More recently, the application of a Tissue Specificity Index (TSI) (43) on wide-transcriptome data has allowed the classification of miRNA genes into housekeepers, “intermediate” and tissue-specific (28, 44).
Myogenic miRNAs: An Overview
The striated muscle expresses its tissue-specific miRNAs, conventionally termed myomiRs. To date, the group of ascertained myomiRs includes miR-1-3p, -133a-3p, -133b, -206, miR-208a-3p, -208b-3p, and -499a-5p (Table 1) (45–48), with miR-208a-3p being cardiac muscle-specific while miR-206 being skeletal muscle-specific. Indeed, recent deep-sequencing analysis has let emerge further putative myomiRs (28, 44), although no meaningful information is available concerning their function in skeletal muscle physiology.
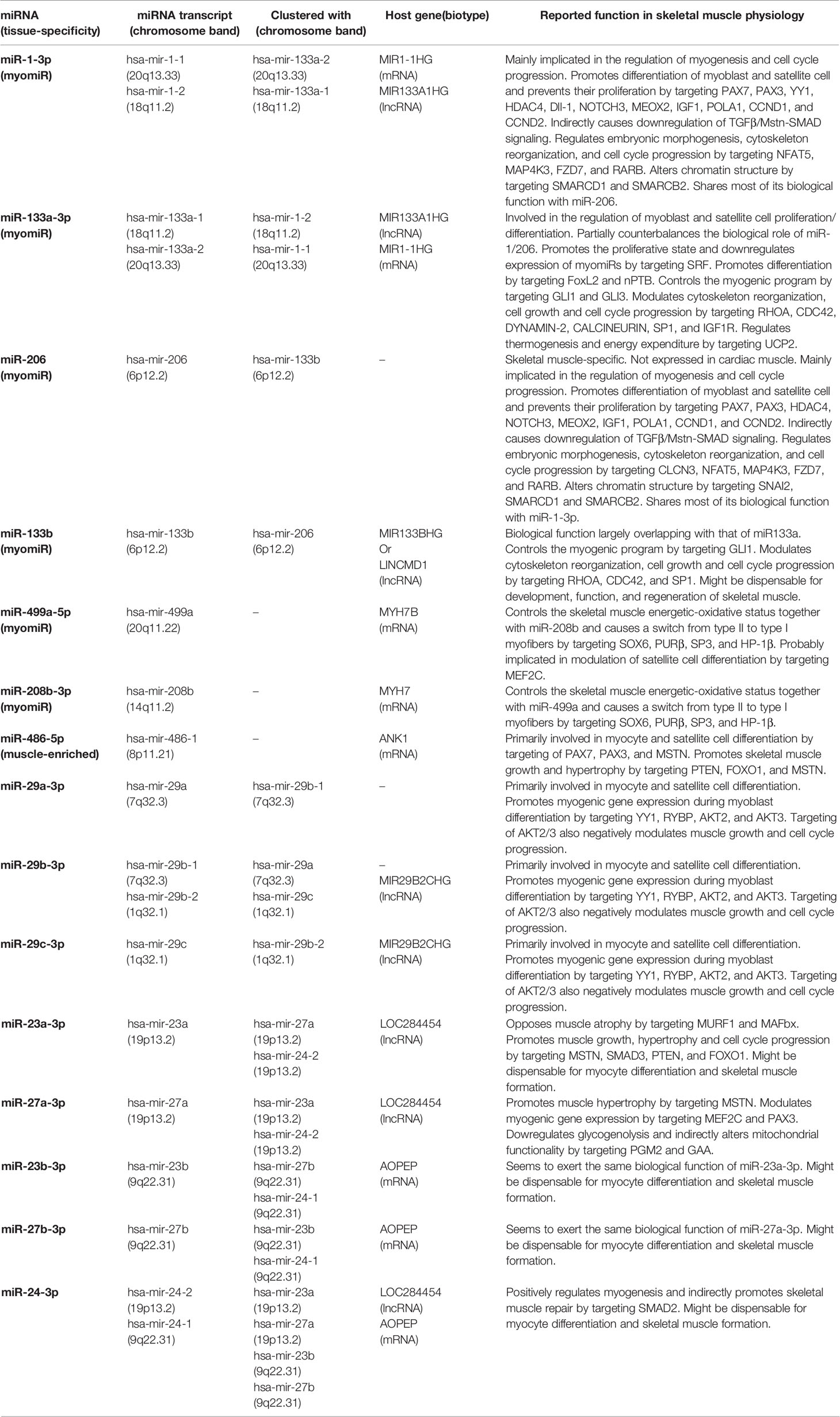
Table 1 List of ascertained myomiRs and most recognized miRNAs with myogenic functions, and their physiological role within the skeletal muscle.
MyomiR expression follows a distinct spatio-temporal pattern (49–51), aiming to properly regulate myogenesis, satellite cell differentiation, protein turnover, and muscle repair. Besides myomiRs, however, a subset of non-muscle-specific miRNAs also exert essential myogenic functions, including, for example, miRNAs of the miR-29 family (52), the miR-23a/b clusters (53), and miR-486-5p (48) (Table 1).
Overall, some of these miRNAs are clustered together and transcribed as bicistronic (46) or polycistronic (53) transcripts, whereas others are monocistronic and transcribed independently (47). Also, some of these miRNAs are intragenic, and their expression rate primarily depends on that of their host gene (47). In general, however, myomiR expression is mainly controlled by a set of muscle-specific transcription factors and cofactors, referred to as myogenic regulation factors (MRFs). Several MRFs are characterized by the basic helix-loop-helix (bHLH) motif and include myoblast determination protein (MyoD), myogenic factor 5 (Myf5), Myf6, and Myogenin. Others contain the MADS-box motif and include the myocyte enhancer factor-2 (Mef2) family of transcription factors (54, 55). Non-muscle-specific miRNAs with myogenic functions, instead, are often transcribed by a subset of non-myogenic transcription factors, albeit MRFs exert a significant influence on their expression.
Transcriptional Regulation of Myogenic miRNAs
The miR-1 (miR-1/miR-206) and miR-133 (miR-133a/miR-133b) families certainly represent the best-studied group of myomiRs. These consist of three bicistronic transcripts, specifically mir-1-1/mir-133a-2, mir-1-2/mir-133a-1, and mir-206/mir-133b, clustered into three different chromosome loci of the human genome and giving rise to four distinct mature miRNAs (46). Because of high sequence similarity, miR-1 shares a large fraction of targets with miR-206 and miR-133a with miR-133b. Intriguingly, these miRNAs are capable of regulating their expression through feedback loop mechanisms in several circumstances, as described hereafter. Mir-208b and mir-499a are instead monocistronic transcripts hosted in intronic regions of genes encoding for two isoforms of myosin heavy chain-β (β-MHC), i.e., myosin heavy chain 7 (MYH7) and MYH7B, respectively (47, 56).
MyoD, Mef2, and serum response factor (SRF) are notoriously responsible for transcription of myomiRs (55, 57, 58). Meanwhile, each of these MRFs is regulated by other factors. Concerning MyoD, it was demonstrated that its stability is under the control of the mammalian target of rapamycin complex-1 (mTORC1). In contrast, the blockage of mTOR kinase activity results in its degradation, with consequent underexpression of miR-1, -206, -499a, and several non-muscle-specific miRNAs (59). MyoD is also controlled by the paired box 7 (Pax7) transcription factor through a dual mechanism consisting of downregulation of MyoD expression and impairment of its transcriptional activity (60). The latter, in particular, involves the expression of two transcriptional targets of Pax7 and Pax3, namely inhibitor of DNA binding 2 (ID2) and ID3, which antagonize myogenic bHLH transcription factors (61, 62). Pax7/3-mediated control of MyoD is fundamental to allow the proliferation of skeletal muscle precursors and to maintain the status of quiescent satellite cells (61, 62). On the other hand, however, both Pax7 and Pax3are direct targets of miR-1/206 and miR-486, and their miRNA-mediated suppression occurs during the early phase of myogenic differentiation, when the expression of these miRNAs considerably increases (62–65). Also, miR-1, -133, -206, -29b-2, 29c, and Pax7 are under the control of Yin Yang1 (YY1), a ubiquitously expressed transcription factor positively regulated by the nuclear factor-kappaB (NF-κB) signaling (52, 66). Precisely, activation of the NF-κB signaling causes increased expression of YY1, which in turn causes the upregulation of Pax7 expression and the subsequent repression of skeletal myogenesis and satellite cell differentiation. At the same time, YY1 is a direct target of miR-1 and miRNAs of the miR-29 family (miR-29a/b/c), which suppress its expression during the early differentiation phase (52, 66).
Local chromatin remodeling undoubtedly exerts an essential role in the control of MRFs-mediated miRNA gene expression in skeletal muscles. For instance, it is known that the Snail DNA-binding proteins Snai1 and Snai2 act as transcriptional repressors of MYOD by recruiting histone deacetylase 1 and 2 (HDAC1/2) onto genes containing G/C-rich E box motifs, which are almost exclusively associated with differentiation. On the contrary, Snail-HDAC1/2 complexes are not recruited in MyoD-targeted genes containing A/T-rich E box motifs. Such an occurrence causes the blockage of MyoD-induced myogenic differentiation but does not prevent MyoD function in cell growth (67). In this context, miR-206 exerts an essential role in the switch from myoblast growth to myoblast differentiation by directly targeting SNAI2, while miR-30a inhibits expression of SNAI1. Therefore, it was noticed that the absence of such a regulatory loop would impede myogenic differentiation (67).
The regulation of Mef2 represents another important mechanism capable of modulating myogenic miRNA expression. Here, HDAC4 plays an essential role as a repressor of both Mef2 activity and myogenic miRNAs expression, and represents a point of convergence of several pathways. For example, the binding of HDAC4/5 and Mef2-interacting transcriptional repressor (MITR) to Mef2 hinder Mef2 transcriptional activity (68–70). However, upon increased concentrations of intracellular calcium ions (Ca2+), calmodulin-dependent protein kinase (CaMK) causes the dissociation of both these inhibitors from Mef2, rehabilitating its activity. Also, CaMK phosphorylates Mef2 cooperatively with p38, a member of the mitogen-activated protein (MAP) kinases, maximizing Mef2-mediated transcription (69, 70). The alpha subunit of the heterotrimeric G protein complex (Gαi2) also suppresses HDAC4 via an indirect mechanism involving the protein kinase C (PKC) signaling (71).
Importantly, HDAC4 is a direct target of several miRNAs with myogenic function, such as miR-1/206 and miRNAs of the mir-29 family (49, 72, 73). This generates further regulatory loops capable of determining the switch from proliferation to differentiation of myoblasts. One of such loops involves the control of Mef2/MyoD activity by transforming growth factor-beta (TGFβ) and myostatin (Mstn), two established negative regulators of MyoDand Mef2 and suppressors of myoblast differentiation (74–76). Interaction of TGFβ or Mstn with their cognate receptor induces phosphorylation of small mother against decapentaplegic 2 (SMAD2) and SMAD3 with consequent activation of the SMAD2/3/4 complex. The latter then translocates to the nucleus, where it functions as a transcription factor that inhibits the expression of miR-206 while favoring the upregulation of HDAC4 (73). Moreover, activated SMAD3 induces the recruitment of a nuclear complex composed of YY1, Ring1-YY1-binding protein (Rybp), and Polycomb-repressive complex (PRC), capable of negatively regulating the expression of several miRNAs with myogenic functions (77, 78). Conversely, under normal myogenic differentiation conditions, increased concentrations of both miR-206 and miR-29(a/b/c) prevent the induction of SMAD3 by an indirect mechanism and cause a decrease in basal levels of SMAD3 (73), eventually leading to the downregulation of HDAC4, YY1, Rybp, and PRC (77, 78). MiR-29 also targets the RYBP 3’UTR, leading to the rapid upregulation of genes involved in somitic myogenesis and differentiation (77).
The Notch signaling represents another critical control point of myomiR expression as it prevents the activity of bHLH transcription factors and is determinant for the maintenance of quiescence in myoblasts (79–81). The binding of ligands with Notch receptors stimulates their proteolytical cleavage, causing the release of the Notch intracellular domain (ICD). The last shuttles into the nucleus, where it interacts with the recombination signal binding protein for immunoglobulin kappa J region (RBPJ). This event lastly leads to the formation of a Notch ICD-RBPJ transcriptional complex (80, 81), which antagonizes bHLH transcription factors (79). In this context, the Numb protein causes degradation of Notch1 in differentiating myoblasts and satellite cells (82, 83), allowing the bHLH-dependent transcription. In turn, Numb is negatively regulated by direct targeting of miR-146a, which thus turns the scale in favor of quiescence (84). MiR-1 can regulate the Notch signaling by inducing transcriptional repression of the Notch ligand Delta-like 1 (DLL-1) (85). Moreover, miR-1/206 modulate NOTCH3 levels during the later phase of differentiation by direct targeting of its 3’UTR, subsequently restoring Mef2 expression and p38 functionality (86, 87).
FoxO3, a non-myogenic transcription factor, was proved to be directly involved in the induction of miR-1/133a expression (88). FoxO3 is under the control of the insulin-like growth factor 1 (IGF1) – RAC serine/threonine-protein kinase Akt (Akt) signaling pathway, notoriously involved in muscle growth. The binding of IGF1 to its receptor (IGF1R) leads to activation of Akt, which in turn phosphorylates and inactivates FoxO3, causing a reduction in miR-1 expression. On the other hand, IGF1 is an established target of miR-1/206 (88, 89), and IGF1R transcript is directly targeted by miR-133a (90), giving rise to a reciprocal regulation between miR-1/206 and IGF1-Akt-FoxO3 signaling during myogenesis. MiR-29 also takes part in this regulatory feedback mechanism by directly suppressing the expression of AKT3/2 (91, 92).
Interestingly, in vitro experiments showed that sphingosine-1-phosphate (S1P) might cause a significant delay in the expression of miR-1, -206, and -486 by activation of sphingolipid signaling (93). However, no details were retrieved concerning the precise molecular mechanism.
Post-Transcriptional Regulation of Myogenic miRNAs
Regulation of myogenic miRNAs can also occur at the post-transcriptional level through mechanisms modulating either their processing or availability. For example, it was demonstrated that muscleblind-like splicing regulator 1 (MBNL1), a protein involved in alternative RNA splicing, regulates the processing of miR-1 specifically at the pre-miRNA level by binding to its loop region. This protects the pre-mir-1 loop region from post-transcriptional modifications (94). In contrast, nuclear sequestration of MBNL1 in individuals with myotonic dystrophy causes the replacement of MBNL1 with Lin28. The latter promotes the uridylation of the pre-miR-1 loop region, subsequently rendering pre-miR-1 resistance to Dicer cleavage (94). An analogous situation presumably occurs following MBNL1 suppression (95).
Bone morphogenetic proteins (BMPs), members of the TGFβ superfamily, regulate proliferation, differentiation, and apoptosis of various types of cells and organs, including skeletal muscles. For instance, BMPs are capable of preventing terminal differentiation of myogenic cells by inhibiting the transcription of the muscle-specific nuclear factors MyoD and Myogenin (96), thereby impacting on myogenic miRNA transcription. However, among BMPs, BMP2 was also shown to influence miRNA processing (97). Specifically, the interaction between BMP2 and its cognate receptor stimulates phosphorylation and activation of SMAD1. Phosphorylated SMAD1 interacts with SMAD4, forming the SMAD1/4 complex. The latter shuttles into the nucleus and impedes Drosha-mediated cleavage of miR-206 through an undefined mechanism. As a result, miR-206 is accumulated into the nucleus in the form of pri-mir-206, whereas concentrations of cytosolic miR-206 significantly decrease (97).
One further post-transcriptional regulatory mechanism reported to control the expression of myomiRs depends on KH-type splicing regulatory protein (KSRP), which was specifically demonstrated to regulate the processing of the miR-1 and miR-206 families of myomiRs in C2C12 cells (98).
Intriguingly, it was demonstrated that TAR DNA-binding protein 43 (TDP-43) physically associates with mature miR-1 and miR-206 in individuals with amyotrophic lateral sclerosis. This prevents their loading onto AGO proteins and thus decreases their availability, impeding them to exert their function as gene expression silencers (99).
Functions of Myogenic miRNAs in Skeletal Muscle Physiology
MyomiRs of the miR-1 and miR-133 families have a profound impact on skeletal muscle physiology, as they allow the fine-tuning of processes related to skeletal myogenesis and muscle regeneration (100–102). Meanwhile, several non-muscle-specific miRNAs, including miR-29 and -486, also cooperate in determining the switch between myoblast quiescence, proliferation, and differentiation (52, 62).
Besides targeting inhibitors of myogenic gene expression, microarray analysis demonstrated that miR-1 and -206 repress the expression of a small set of genes controlling muscle structure and function (103). Among the validated targets, the mesenchyme homeobox 2 (MEOX2) transcription factor has an established key role in somitogenesis (104). At the same time, chloride voltage-gated channel 3 (CLCN3) is involved in the regulation of cell volume and fibroblast-to-myofibroblast transition (105). MAP4K3, frizzled class receptor 7 (Fzd7), nuclear factor of activated T-cells 5 (NFAT5), and retinoic acid receptor beta (RARB) are non-muscle-specific components involved in embryonic morphogenesis, cytoskeletal changes, cell growth and differentiation (106–108). Smarcd1 and Smarcb2 are two well-known non-muscle-specific chromatin remodeling factors that might have a role in satellite cell differentiation and cytoskeletal reorganization (109).
MiR-1 and -206 also target a few cellular components involved in cell cycle progression. For instance, it was demonstrated that both these miRNAs directly target the POLA1 gene (50), encoding for the alpha 1catalytic subunit DNA polymerase, as well as two members of the cyclin family, i.e., cyclin D1 (CCND1) and CCND2 (110, 111), which function as positive regulators of the cyclin-dependent (CDK) kinases. MiR-206 might significantly impair the cell size in myogenic lineage through the inhibition of HDAC4 activity (112). In particular, an alteration of endogenous miR-206 expression associated with the hypertrophy and atrophy of muscles in mice. Nonetheless, manipulation of the miR-206/HDAC4 axis had no significant effect in post-natal muscle mass or adaptive responses.
MiR-133a takes part in myoblast differentiation as well, and it partially counterbalances the biological role of miR-1 (85). For instance, miR-133a directly targets the 3’UTR of SRF (49), required for skeletal muscle growth and maturation (113), thus causing its silencing and maintaining myoblasts and satellite cells in a proliferative state (49, 85). On the other hand, miR-133a targets the FOXL2 transcription factor (114) as well as the alternative splicing factor neuronal polypyrimidine tract-binding protein (nPTB) (115), promoting differentiation. Moreover, miR-133a was recently demonstrated to guide the myogenic program by exerting direct control over components of the Hedgehog pathway, including the GLI1 and GLI3 transcription factors. In contrast, miR-133a knockdown impaired myotome formation (116). Along with proliferation and differentiation, miR-133a also regulates the reorganization of the actin cytoskeleton and hypertrophy of striated muscles. This is achieved by the direct targeting of RAS homolog family member A (RHOA), Cell division cycle 42 (CDC42) (117), DYNAMIN-2 (118), and CALCINEURIN (119). Still, miR-133a is involved in the control of cell cycle – through transcriptional repression of Sp1, responsible for the expression of CCND1 (120) – thermogenesis and energy expenditure – by direct targeting of uncoupling protein 2 (UCP2), a mitochondrial anion carrier that affects myoblast differentiation (121).
Differently from miR-133a, very few pieces of evidence exist concerning the precise biological role of miR-133b in skeletal muscle physiology. However, studies from cancer research reveal that a large fraction of miR-133b targets overlaps with those of miR-133a, including FOXL2 (122), GLI1 (123), RHOA, CDC42 (124, 125), and SP1 (126), indicating overlapped biological functions. Nonetheless, knockout experiments performed on a murine model suggested that, indeed, the miR-206/133b cluster might be dispensable for development, function, and regeneration of skeletal muscle, probably because of compensation by miR-1/133a (127). Analyses performed on the soleus and plantaris muscles in mice showed that overload-induced hypertrophy resulted in decreased expression of miR-1 and miR-133a, and a parallel increase of muscle weight, hence suggesting that such miRNAs might play a regulatory role in mediating skeletal muscle response to functional overload (128).
In the context of skeletal muscles, expression of miR-208b and -499a is restricted to type I (slow-twitch) muscle fibers (47) and partly depends on the estrogen-related receptor-gamma and beta (ERRγ/β) (129). In contrast, peroxisome proliferator-activated receptor-alpha (PPARα) was shown to repress their expression (129). Accordingly, these two myomiRs determine a switch in myofiber gene program (from type II to type I) and control the skeletal muscle oxidative status by silencing the expression of a shared set of transcriptional repressors of β-MHC genes, consisting of SOX6, PURβ, SP3, and HP-1β (47, 129). In turn, activation of the type I myofiber gene program creates a positive regulatory loop via the expression of MYH7 and MYH7B, further reinforcing the slow-twitch muscle gene program. In addition, miR-499a might be implicated in the regulation of muscle differentiation by direct targeting of MEF2 isoform C (130).
A miRNA profiling performed on side population cells (a cell type that plays a crucial role in muscle regeneration after injury along with satellite cells) allowed the identification of a set of overexpressed molecules compared to main population cells (131). In particular, the overexpression of miR-128 in mice led to an impairment of cell proliferation and differentiation. Further analyses revealed that miR-128a mediated the maintenance of the quiescent state and regulation of cell differentiation through the modulation of genes involved in myogenesis, adipogenesis, and osteogenesis including Pax3, Runx1, and PPAR (131). The same authors later found that miR-128a also regulated genes involved in insulin signaling (such as Irs1) and that its inhibition mediated by TNF-α resulted myotube maturation and myofiber hypertrophy both in vivo and in vitro (132), paving the way for further investigations in human as miR-128 is conserved among species. MiR-486 is a potent regulator of the IGF1-Akt-mTORC1 pathway (48), which plays a major role in skeletal muscle growth (133). In particular, such regulation is achieved by direct targeting of phosphatase and tensin homolog (PTEN) and FOXO1, two negative modulators of the phosphoinositide-3-kinase (PI3K)-Akt signaling (48). In turn, the expression of miR-486 is negatively controlled by Mstn, which functions as an autocrine factor capable of inhibiting myogenesis (134). Accordingly, the overexpression of miR-486 was shown to induce striated muscle hypertrophy in mice knockdown for Mstn (134), while transfection of miR-486 mimic allowed the rescue of muscle mass in atrophic skeletal muscles (120). In a murine model of chronic kidney disease, characterized by increased muscle protein degradation mediated by E3 ligases, Atrogin-1/MAFbx, and MuRF-1, ectopic expression of miR-486 resulted in impaired skeletal muscle atrophy by the blockage of FoxO1 translation (135) (Figure 1).
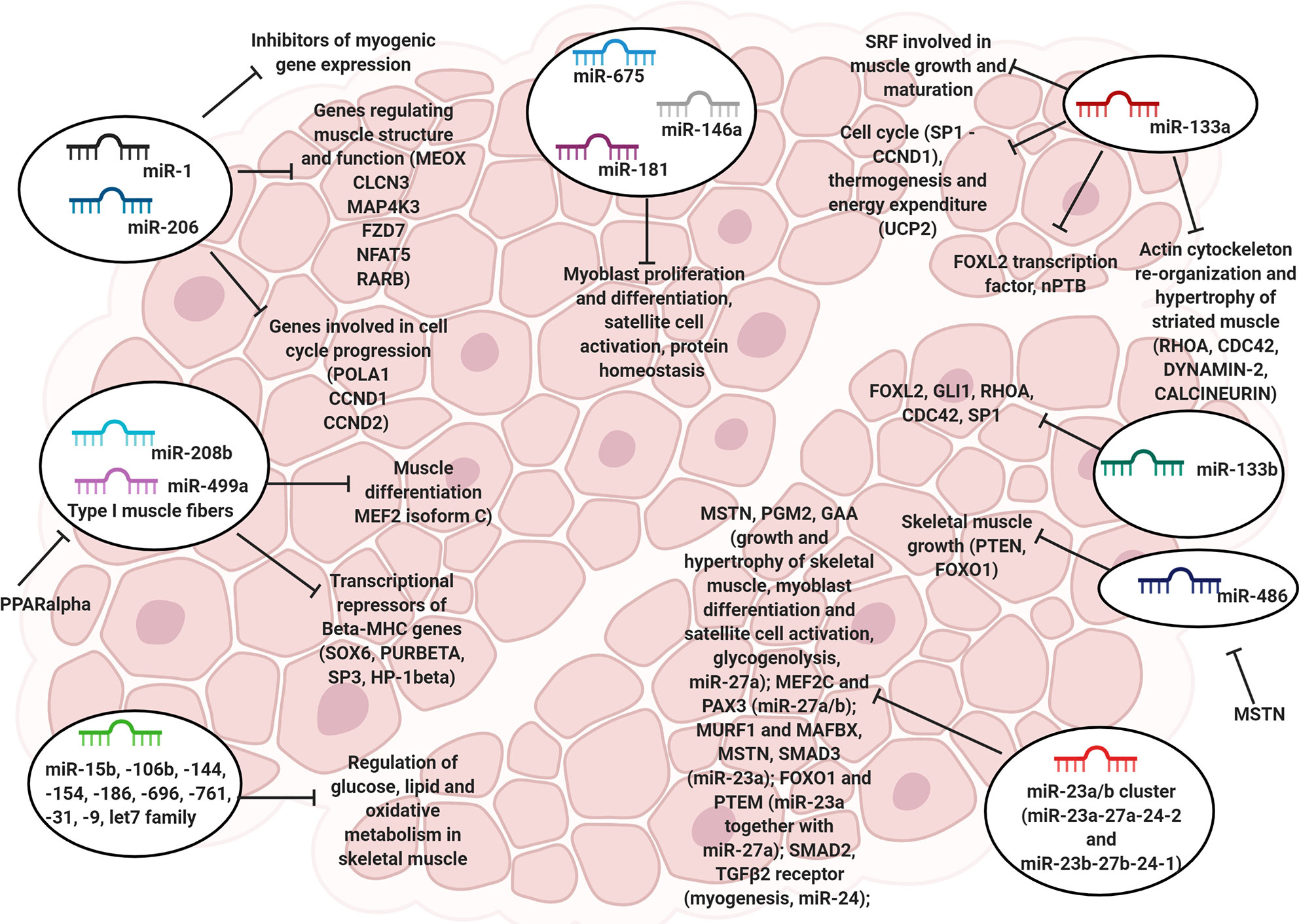
Figure 1 The role of myomiRs in skeletal muscle. Representative image showing the miRNAs involved in the regulation of skeletal muscle cell physiology, including proliferation, differentiation, satellite cell activation and cytoskeleton re-organization.
Similarly to miR-29 and -486, the miR-23a/b clusters (miR-23a–27a–24-2 and miR-23b–27b–24-1) are also enriched in skeletal muscle (53, 136), exerting important regulatory roles. In particular, miR-27a assists both growth and hypertrophy of skeletal muscle by silencing MSTN expression and subsequently promoting myoblast differentiation and satellite cell activation (137, 138). Also, miR-27a targets phosphoglucomutase (PGM2) and acid α-glucosidase (GAA), two enzymes involved in glycogenolysis, and its combined action with miR-142 regulates glycogen and fatty acid metabolism, and alters mitochondrial functionality in both myoblasts and myofibers (139). Furthermore, miR-27a/b influences the myogenic gene expression program by targeting the 3’UTR of MEF2C (140) and PAX3 (141). MiR-23a attenuates muscle atrophy by directly targeting both muscle RING finger containing protein 1 (MURF1) and muscle atrophy F box protein (MAFBX) (136, 142), two E3 ubiquitin ligases of the ubiquitin-proteasome pathway that take part in the breakdown of short-lived and regulatory proteins (143). Moreover, miR-23a suppresses expression of both MSTN and SMAD3 and acts synergistically with miR-27a to downregulate FOXO1 and PTEN and upregulate PI3K-Akt signaling, opposing the loss of muscle mass and contractile function (144). MiR-24 has a role in myogenesis (145), which is partly exerted by direct targeting of SMAD2 (146). In the context of skeletal muscle injuries, miR-24 was shown to act concomitantly with miR-122, which targets the receptor II of TGFβ. Such an event would inhibit the TGFβ-SMAD signaling, thus favoring satellite cell activation and skeletal muscle repair (135, 146) (Figure 1). Nonetheless, loss-of-function experiments suggested that the miR-23a/b clusters would indeed be dispensable for myogenesis and skeletal muscle function, as mice knockout for these two clusters reported only subtle effects on skeletal muscle development and adaptation after exercise endurance (147).
Besides the aforementioned myogenic miRNAs, several other miRNAs are also known to exert either essential or dispensable roles in myoblast proliferation/differentiation, satellite cell activation, and protein homeostasis, including miR-675, miR-146a, and miR-181, respectively (148). Also, an extensive list of miRNAs involved in the control of glucose, lipids, and oxidative metabolism in skeletal muscle has been reported, including miR-15b, -106b, 144, -154, -186, -696, -761, -31, -9, and miRNAs of the let-7 family (135, 139, 149) (Figure 1).
Alteration of miRNA Expression in Skeletal Muscles During Cancer Cachexia
It is evident that, overall, miRNAs regulate almost all aspects of skeletal muscle physiology. Thus, any alteration in their expression or functionality is likely to result in impairments of relevant features, such as metabolic state, proteostasis, regenerative capabilities, and performance. This suggests the possibility for miRNAs to play a role as mediators and biomarkers of skeletal muscle loss in several debilitating diseases, including cancer cachexia. Accordingly, small RNA-seq analyses have been recently performed on skeletal muscle biopsies from cachectic tumor patients and mice, revealing a real correlation between changes in myogenic miRNA expression and cancer cachexia occurrence.MiRNA profiling on muscle biopsies from cachectic tumor patients
As yet, only two studies have been focused on the shift in myogenic miRNA expression in the context of human cancer cachexia (Table 2). In the former one, the myogenic miRNA expression profile from cachectic patients with pancreatic and colorectal cancers was compared with that from non-cachectic patients suffering from the same tumor types (150). The authors identified and validated eight differentially expressed miRNAs, i.e., let-7d-3p, miR-199a-3p, -1296-5p, -345-5p, -3184-3p, -423-5p, -423-3p, and -532-5p, between the two conditions. Precisely, these eight miRNAs were significantly upregulated in the muscles of cachectic patients and showed both predictive and prognostic value. The authors then identified 147 potential target genes in the cachectic condition, mainly related to myogenesis, inflammation, innate immune response, and signaling pathways involved in morphogenesis and development (150).
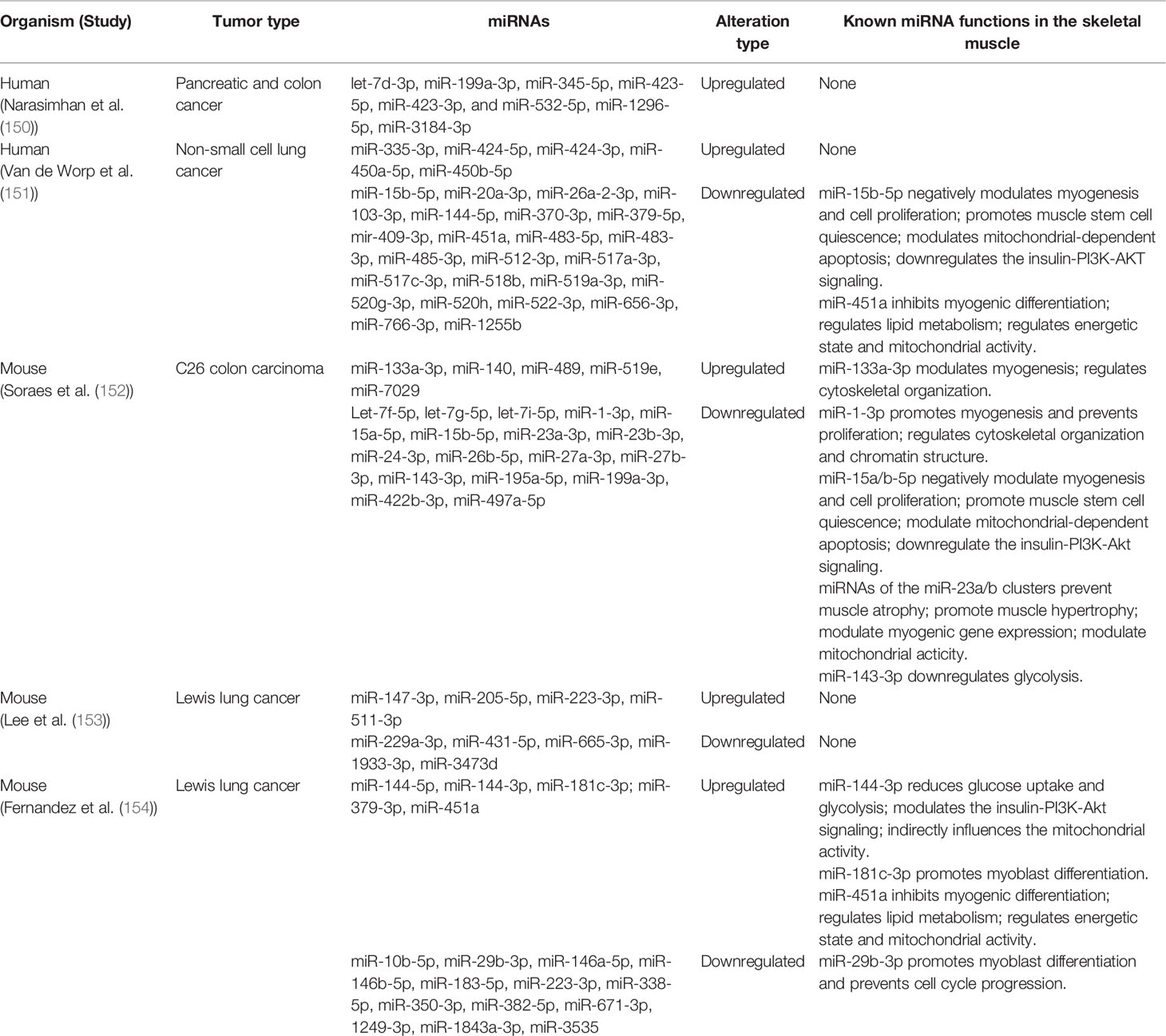
Table 2 List of significantly deregulated miRNAs in skeletal muscles of cachectic individuals suffering from tumors.
In the second study, the myogenic miRNA expression profile from cachectic patients with non-small cell lung cancer (NSCLC) was compared with that from matched healthy controls (151). Here, a signature of 28 differentially expressed miRNAs was identified, with 23 being down and five upregulated in cachectic patients. However, none of these overlapped with miRNAs identified in the earlier study, nor revealed any predictive or prognostic potential. Finally, 114 potential target genes functionally expressed in the skeletal muscle were identified, most of which were related either to muscle-specific degenerative or regenerative processes (151).
Results from Murine Models of Cancer Cachexia
A more extensive body of evidence has been reported in the case of murine models of cancer cachexia (Table 2). In a preliminary work, four distinct atrophic mice models – fasting, denervation, diabetes, and C26 colon carcinoma-induced cancer cachexia – were employed to frame the role of miRNAs in skeletal muscle loss (152). Noteworthy, the authors found that the miRNA signature was peculiar to each atrophic condition. Compared to the controls, muscle samples from cachectic mice presented 22 differentially expressed miRNAs, of which 17 were down and five upregulated. Of note, miRNAs of the miR-23a/b clusters, primarily involved in the prevention of protein catabolism, were among the most downregulated. MiR-143-3p, -199a-5p, -26b-5p, as well as miRNAs of the let-7 and miR-15a/b families, were also downregulated. These are involved in the modulation of insulin cascade, PI3K-Akt signaling, and glucose metabolism (149). Finally, miR-1-3p was significantly downregulated, whereas miR-133a-3p was upregulated (152).
The myogenic miRNA profiling of skeletal muscles from cachectic mice bearing Lewis lung carcinoma (LLC) revealed nine differentially expressed miRNAs compared to controls. Among these, miR-229a-3p, -431-5p, -665-3p, -1933-3p, and -3473d were downregulated, whereas miR-147-3p, -205-5p, -223-3p, and -511-3p were upregulated (153). The subsequent pathway analysis revealed that these miRNAs are likely to be involved in processes related to cell-to-cell communication, development and morphogenesis, cell cycle, and inflammatory disease, among others (153).
More recently, a myogenic miRNA profiling from skeletal muscles of cachectic LLC-bearing mice identified 18 differentially expressed miRNAs compared to the controls, with 13 being down and five upregulated (154). In particular, miR-144-3p and -451a, involved in the modulation of mitochondrial activity, energetic state, and glucose and lipid metabolism (149), were upregulated. MiR-181c, involved in myoblast differentiation (155), was upregulated as well. In contrast, miR-29b-3p was downregulated. By applying an integrated genome-wide approach combining miRNA-mRNA sequencing data, the authors identified 131 putative target genes, mostly involved in the extracellular matrix organization, cell migration, ion transport, and FoxO signaling (154).
Molecular Mechanisms Underlying Shifts in Myogenic miRNA Expression During Cancer Cachexia
As discussed above, dysregulation in myogenic miRNA expression usually represents the consequence of altered transcriptional mechanisms. In the context of cancer cachexia, some experimental studies suggest that the deregulation in MRFs expression or function is often the leading cause of altered miRNA expression in skeletal muscles. This would then contribute to maintain or even worsen the course of such metabolic syndrome.
One example regards tumor necrosis factor-alpha (TNFα) and TNF-weak inducer of apoptosis (TWEAK), two pro-inflammatory cytokines known to induce skeletal muscle wasting under conditions of experimental cachexia through indirect destabilization and suppression of MyoD (156, 157). This is achieved by the activation of MAPK/ERK/p38 and NF-κB signaling. p38 kinase induces activation of the CCAAT/enhancer-binding protein-beta (C/EBPβ) transcription factor, which is responsible for transcription of MAFBX and other genes involved in the ubiquitin-protease pathway (158). Activation of the NF-κB signaling, instead, leads to the overexpression of Pax7, which subsequently causes repression of MyoDand Myogenin transcriptional activity (159), as previously discussed. As a result, TNF exposition induces protein catabolism and simultaneous alteration of expression of several myogenic miRNAs, including miR-1/206, miR-451a, and -146a-5p (160, 161). The latter two miRNAs were shown to be involved in proteolytic degradation, inflammatory response, and extracellular matrix remodeling (160), while others, such as miR-361, -486, and -miR-98, were suggested to exert a role in myoblast fusion capacity (161). Similar mechanisms are assumed to occur following interaction between several other pro-inflammatory cytokines, such as interferons (INFs) and interleukins (ILs), and their cognate receptors on the myocyte membrane surface (162). Nonetheless, no concrete outcomes have been reached in this regard.
Expression of MyoD might eventually be inhibited by indirect action of heme oxygenase-1 (HO-1) following FoxO1-mediated HO-1 expression under conditions of muscle atrophy (163). Here, carbon monoxide derived from HO-1 enzymatic activity decreases nuclear translocation of C/EBP-gamma (C/EBPδ) and prevents its binding to promoters of myogenic genes, including MyoD (164). Moreover, expression of HO-1 is known to cause downregulation of DGCR8 and Lin28 (29), negatively impacting the pri-miRNA processing (164).
However, although these few studies focused their attention on MyoD transcriptional regulation, none of them detected any significant alteration in the expression of myomiRs in the context of cancer cachexia. Thus, it is more likely that such deregulation in miRNA expression involves ubiquitously expressed transcriptional factors.
Discrepancies Emerging From the Data Comparison
Despite the research efforts undertaken to elucidate the role of miRNAs in skeletal muscle loss during cancer cachexia, no conclusive results have been described yet. Downstream analyses of miRNA expression have indicated that deregulated miRNAs in cachectic muscles are usually involved in processes like cell cycle, myogenesis, inflammation, extracellular matrix organization, and myoblast fusion. However, very scarce or no overlap exists between the sets of differentially expressed miRNAs detected by the various authors. Such a low consensus might depend on multiple factors. For instance, different cancer types likely alter different molecular mechanisms, subsequently determining distinct molecular signatures and energetic states within the skeletal muscle (165, 166). In turn, miRNAs might be differentially expressed through feedback loop mechanisms in order to properly modulate fluctuations in myogenic gene expression and confer robustness in signaling outcomes for specific regulatory networks (167). Analytical bioinformatic pipelines and sampling times are also assumed to be highly influential in the determination of mRNA/miRNA signatures (154, 168).
Besides the mentioned explanations, the scarceness of experimental models capable to accurately recapitulate the molecular features of human cancer cachexia also represents a major limitation in this field of oncology (21, 26). Indeed, some studies have recently evidenced the relative inconsistency of the “traditional” xenograft models of cancer cachexia, as the molecular signatures found at the tissue level revealed sharply different patterns of gene expression compared to that obtained for human patients (169, 170). Thus, more reliable models should be considered for future studies. These include tamoxifen-induced genetically engineered mouse (GEM) (169), orthotopic models of patient-derived cancer cells (171), and syngeneic mouse models of GEM-derived tumors (172), which are currently being adopted as valuable and innovative alternatives (21).
Role of Tumor-Derived Circulating miRNAs in SM Wasting During Cancer Cachexia
Over the last decade, several studies have pointed out that cancer-secreted extracellular vesicles (EVs) profoundly contribute to the identification of new prognostic and diagnostic factors in cancer cachexia or even to the onset and progression of such syndrome. EVs are secreted by all cell types and consist of membrane-coated particles with different size: while the acronym “EVs” refers to a very heterogeneous group of secreted vesicles in general, such particles are classified as “exosomes” when their diameter ranges from 30 to 100 nm, “microvesicles” (MVs) when their size ranges from 50 to 1,000 nm, “oncosomes” (1 to 10 μm) and “apoptotic bodies” (100 nm to 5 μm) (173, 174). EVs harbor a wide array of different molecules, which most likely reflect the physiological status of the releasing cells: lipids, proteins, DNA fragments, and RNA molecules, including non-coding RNAs such as miRNAs, long non-coding RNAs (lncRNAs), rRNAs, and tRNAs (174). EVs can be isolated from different body fluids such as plasma, serum, milk, urine, saliva, spinal fluid (175). The percentage of miRNA content within EVs sharply differs from that of cytoplasmic miRNAs in donor cells, suggesting the existence of a controlled sorting mechanism by which miRNAs are selectively recruited and packaged into vesicles (176).
Several studies have demonstrated that miRNAs can also be found in body fluids not associated to EVs: in this scenario, miRNA molecules are associated to RNA-binding proteins, such as AGO2 (the effector member of the RISC complex), which protect them from degradation and maintain their stability in the extracellular environment. The mechanisms that mediate miRNA secretion through EVs rather than a vesicle-free system are still unknown, and this could occur through a highly regulated miRNA selection (177).
Although EVs were initially considered a system used by cells to get rid of unwanted molecules, several studies have instead demonstrated that secreted particles are a useful tool for the definition of specific non-invasive cancer-associated biomarkers. Examples are exosomal miR-30d-5p and let-7d-3p from plasma, which were found to be valuable diagnostic biomarkers for non-invasive screening of cervical cancer (178); upregulated miR-1290 in the serum of patients with high grade serous ovarian carcinoma allowed to discriminate these patients from those with other malignancy types (179); miR-25-3p and miR-92a-3p were identified as potential biomarkers for liposarcoma (180).
Besides their potential prognostic or diagnostic role, secreted miRNAs have a considerable impact on mediating cell-to-cell communication as they are biologically active molecules. In fact, once secreted by a donor cell, the circulating miRNAs can be adsorbed by recipient cells, where they efficiently target the 3’UTRs of mRNAs hence modulating the cell’s response to such stimulus. For example, it was demonstrated that the exosomal transfer of miRNAs in hepatocellular carcinoma (HCC) modulates carcinogenesis and promotes transformed cell growth (181). Here, the authors identified 11 secreted miRNAs, while TAK1, a member of the mitogen-activated protein kinase kinase kinase (MAP3K) family involved in homeostasis and tumorigenesis in HCC, was selected as a potential target. When Hep3B cells were incubated with Hep3B-derived exosomes, not only TAK1 protein expression was impaired in recipient cells, but also the activation of its associated pathway JNK1 as well as cell viability. Similarly, it showed that IL-4-activated macrophages secreted miRNAs through exosomes that targeted breast cancer cells (182). Among them, miR-223 promoted the invasion of recipient cells. In another study (183), it was demonstrated the endogenous transfer of exosomes between dendritic cells, whose cargo mirrored the level of cell maturation. In 2012 (184), our group demonstrated that miR-21 and miR-29a, secreted by lung cancer cells through EVs, were transferred to surrounding macrophages at the microenvironment level: here, miRNAs promoted tumor growth and spread by binding the TLR7/8 receptor and hence activating the NF-κB pathway. This study revealed, for the first time, a different mechanism used by secreted miRNAs than the post-transcriptional repression to promptly amplify a pro-tumoral response.
These findings opened the doors to new investigations aimed at understanding whether cancer-secreted miRNAs modulated lean mass wasting associated with cachexia (Figure 2). In 2014, we demonstrated that lung and pancreatic-secreted miR-21 and miR-29a promoted atrophy in recipient murine myoblasts through their binding to the TLR7/8 receptor and the activation of the JNK pathway (185). Such a process could be significantly inhibited by IMO-8503, a TLR7, 8 and 9 antagonist (186) (Figure 2A). Okugawa et al. (187) showed that an elevated level of circulating miR-203 in the serum of patients who had colorectal cancer was a risk factor for myopenia and that such miRNA promoted apoptosis and impaired cell proliferation by downregulating BIRC5 (Figure 2B). Conversely, in head and neck cancer patients, miR-130a levels in plasma negatively correlated with TNF-α concentration (188) and allowed to discriminate between cancer patients suffering from cachexia from patients mildly malnourished with high specificity, hence displaying potential clinical utility in the diagnosis of cachexia (Figure 2C). In rhabdomyosarcoma, several circulating miRNAs have been identified within patient sera: miR-1, miR-133a, miR-133b, and miR-206: in particular, miR-206 was able to discriminate between rhabdomyosarcoma and non-rhabdomyosarcoma tumors with a sensitivity of 1.0 and specificity of 0.913 (189, 190) (Figure 2D). Zhang et al. (144) reported that miR-23a and miR-27a, two miRNAs that regulated proteins involved in the atrophy process hence reducing muscle wasting, were involved in muscle-kidney cross talk as their expression levels were increased in both serum exosomes and kidneys. Their findings suggested that high levels of miR-23a and -27a could impair diabetes-induced loss of muscle mass and reduce renal fibrosis lesions (Figure 2E).
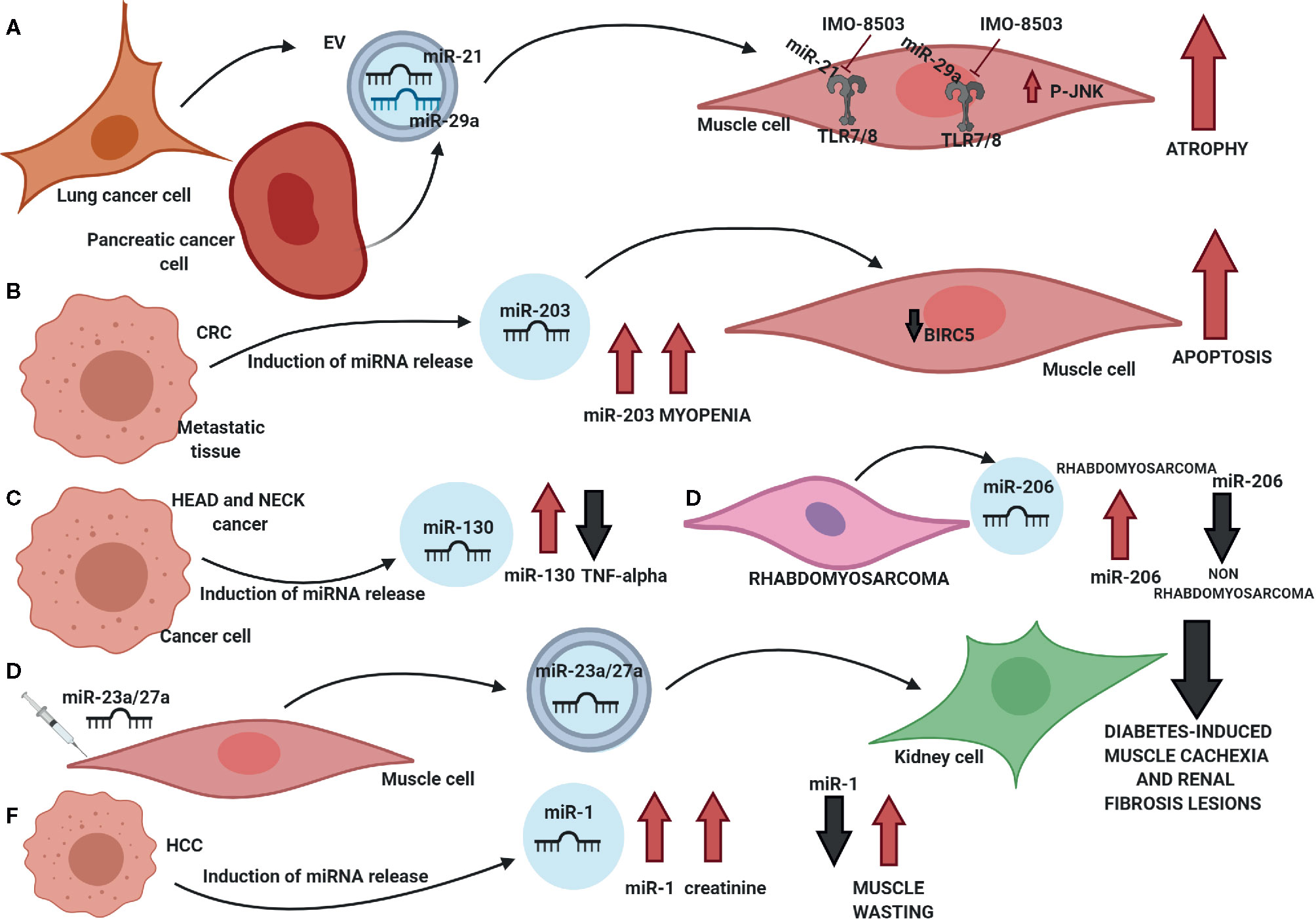
Figure 2 Involvement of circulating miRNAs in muscle wasting. (A) miR-21 and miR-29a are secreted through extracellular vesicles (EVs) by lung and pancreatic cancer cells: once internalized by myoblasts, both miRNAs bind the TLR7/8 receptor hence inducing the phosphorylation of JNK and triggering apoptosis. Such process can be inhibited by TLR7/8 inhibitor IMO-8503. (B) High levels of circulating miR-203 in colorectal cancer patients promote myopenia and induce apoptosis in muscle cells through the downregulation of BIRC5. (C) Circulating miR-130a concentration negatively correlates to TNF-α levels in the serum of head and neck cancer patients. (D) Circulating miR-206 can significantly discriminate between rhabdomyosarcoma and non-rhabdomyosarcoma patients. (E) miR-23a/27a mediate the cross-talk between muscle and kidney cells and impair both diabetes-related muscle atrophy and renal fibrosis lesions. (F) In hepatocellular carcinoma (HCC) patients, circulating high levels of miR-1 positively correlate with creatinine levels in the serum, while a low concentration of miR-1 is an indicator of muscle wasting.
On the other hand, Köberle et al. (191) evaluated the potential of serum miR-1 and miR-122 as prognostic biomarkers in patients suffering from hepatocellular carcinoma (HCC): by comparing the analyses performed on 195 sera of HCC patients and 54 patients with liver cirrhosis, the authors concluded that miR-1 significantly correlated with serum creatinine. Moreover, since miR-1 is known to be a regulator in muscle cells where it is also highly expressed, low levels of such miRNA in serum could be associated with muscle wasting and hence correlate with overall survival in advanced cancer (Figure 2F).
Taken together, these findings have shed light on the importance of tumor microenvironment and circulating miRNAs as relevant mediators of muscle wasting in cancer-associated cachexia.
LncRNAs and ADAR Enzymes as Possible Players in Skeletal Muscle Loss During Cancer Cachexia
Besides the “canonical” regulatory pathways described above, further epigenetic mechanisms are emerging that allow the fine-tuning of miRNA expression, availability, and function within the skeletal muscle. Long non-coding RNAs (lncRNAs) and adenosine deaminases acting on RNA (ADAR) enzymes might represent part of such mechanisms. Interestingly, some cues from the literature suggest their active involvement in skeletal muscle atrophy. However, no clear proves exist at present concerning their involvement in the context of cancer cachexia-induced muscle atrophy.
lncRNAs in the Skeletal Muscle
LncRNAs are 5’-capped transcripts, longer than 200 nucleotides, often spliced and polyadenylated at the 3’ terminus. LncRNAs are known to be involved in a wide range of cellular processes, such as chromatin remodeling, DNA repair, recruitment of transcriptional complexes, and enzyme activity (192). Also, lncRNAs seem to act as competitive endogenous RNAs (ceRNAs), or “sponge” RNAs, by competing with other transcripts for miRNA binding. This poses lncRNAs as relevant modulators of miRNAs availability (193).
Emerging studies have demonstrated that lncRNAs actively participate in the regulation of myogenic gene expression during skeletal myogenesis, myofiber regeneration, and muscle hypertrophy (194, 195). For instance, linc-RAM and SRA were demonstrated to foster myoblast differentiation by enhancing the formation of a transcriptional complex containing MyoD (MyoD-Baf60c-Brg1) (196) and the assembly of MyoD co-regulators (197), respectively. Linc-YY1 was found to promote myoblast differentiation and muscle regeneration by inducing the eviction of the YY1/PRC complex from the target loci and the inhibition of YY1 activity independently from PRC (198). Malat1 acts as a repressor of skeletal myogenesis and muscle repair by recruiting the Suv39h1 histone methyltransferase to MyoD-binding promoters (199). However, upon myocyte differentiation, miR-181a targets Malat1 and causes its degradation, with subsequent destabilization of the Suv39h1-repressive complex (199). Malat1 was also shown to act as a ceRNA by sponging miR-133a (200). In turn, this led toward increased expression of SRF and Mef2C, with consequent promotion of differentiation. Similar effects were reported for the miR-133b hosting transcript linc-MD1 (201). Lnc-mg sponges miR-125b, an inhibitor of IGF2 expression, whereas Gtl2 sponges miR‐135b-5p, an inhibitor of Mef2C. Thus, both these lncRNAs positively regulate muscle differentiation and hypertrophy (202, 203).
In line with these findings, several recent studies have tried to define possible correlations between lncRNA alterations and muscular diseases by examining the expression profile of lncRNAs in various relevant myopathies, including dystrophies and atrophies (195, 204). However, only one study has yet reported a correlation between the expression level of some lncRNAs and cancer cachexia-induced muscle atrophy (204). Specifically, Gtl2 and IG-DMR, both involved in genomic imprinting and muscle development, were significantly decreased in skeletal muscles of cachectic C26-bearing mice. In contrast, none of the other examined long non-coding transcripts, like linc-MD1, linc-YY1, Malat1, or SRA, showed significant deregulations compared to the other conditions (204).
Lastly, it has been demonstrated that some lncRNAs are capable of stimulating the release of tumor-derived exosomes (205). This might represent an additional critical issue in cancer cachexia. For instance, the highly upregulated in liver cancer (HULC) lncRNA, a promoter of hepatocellular carcinoma progression, was recently shown to enhance the secretion of exosomes by human hepatoma cells (205). Such a mechanism was due to the ceRNA activity of HULC, which sponged miR-372-3p. In turn, miR-372-3p is an established suppressor of Rab11a, which is a key element in exosome formation.
ADAR1 and A-to-I RNA Editing in the Skeletal Muscle
ADAR1 and ADAR2 are conserved enzymes capable of converting adenosine (A) to inosine (I) by hydrolytic deamination. Their enzymatic function depends on their ability to interact with double-stranded RNA molecules (dsRNAs) through their dsRNA-binding domain (dsRBD) (206).
ADARs are involved in various biological processes, such as neuronal ion channel activity, increase of transcript diversity, immune response regulation, alternative splicing, RNA interference pathway, and miRNA biogenesis (206–208). The last, in particular, can be modulated by ADARs via editing-dependent or -independent mechanisms. In editing-dependent mechanisms, miRNA transcripts are co- or post-transcriptionally modified in their primary sequence at editing sites that prevent, or even enhance, the Drosha/Dicer-mediated cleavage (206). In editing-independent mechanisms, ADARs prevent miRNA processing by binding to their primary transcript via the dsRBD (209). Indeed, ADAR1 was also found to form a cytoplasmic complex with Dicer promoting pre-miRNA maturation (210). Similar findings were reported in the case of Drosha-mediated processing (211). Despite miRNA biogenesis, ADARs can also alter the function of mature miRNAs by editing their primary sequence, with particular regard to the seed region. Such an occurrence causes a change in the base-pairing ability of edited miRNAs, leading to a shift in their target repertoire (206, 212, 213).
ADARs have been demonstrated to exert essential roles for proper development. In fact, the deletion of ADAR genes caused embryonic lethality due to impaired organogenesis (ADAR1 KO), or post-natal death (ADAR2 KO) due to neuronal cell death (206). ADARs are not homogeneously expressed across tissues. Instead, they are subjected to spatio-temporal patterns of expression (214). In general, however, ADAR1 is ubiquitously expressed, while ADAR2 is highly expressed in the central nervous system, with much lower or null expression levels in other tissues (206).
A wide-transcriptome analysis revealed that ADAR1 is the sole responsible for RNA editing in the context of skeletal muscle. However, the A-to-I editing level in this tissue is significantly lower than in other tissues (214). ADAR1 was shown to be a regulatory constituent of myogenesis, and presumably of muscle repair. Specifically, ADAR1 contributes to the early phase of myogenesis by suppressing apoptotic processes in myoblasts, apparently in an independent manner from its catalytic activity (215). Indeed, more recent findings indicate that such a protective function might also involve the enzymatic action of ADAR1 (216, 217). In certain instances, RNA editing impedes melanoma differentiation-associated protein 5 (MDA5), a cytosolic sensor of viral RNA infection, from sensing endogenous dsRNAs as no-self, preventing the degradation of endogenous mRNAs (216, 218). Here, the p150 isoform of ADAR1 seems to exert an essential role, differently from the p110 isoform. However, both isoforms contribute to development (215, 216).
ADAR1’s editing activity is also indispensable for the regulation of expression of various genes associated with the myoblasts-myotubes transition and motility, hence influencing muscle development. This includes dynamin 1/2 (Dnm1/2) and annexin A4 (Anxa4), whose mRNAs are hyper-edited by nuclear ADAR1 and subsequently retained into the nucleus (215). A-to-I editing of the 3’UTR of Rho GTPase activating protein 26 (ARHGAP26) was instead shown to disrupt the binding sites for miR-30b-3p and miR-573, leading to increased levels of this protein (219). Moreover, a correlation analysis showed that ADAR1 also promotes MyoD and Myogenin expression, although the precise mechanism remained elusive (214).
Aminoacyl tRNA synthase complex-interacting multifunctional protein 2 (AIMP2) was identified as an important negative regulator of ADAR1 function and expression (214). AIMP2 presented significantly higher expression levels in the adult skeletal muscle compared with all other tissues, actively contributing to the low editing and expression levels of muscular ADAR1 (214). Interestingly, myomiR-1/206 were both demonstrated to base-pair with seed-complementary regions located in the 3’UTR of ADAR1, driving the temporal expression rate of ADAR1 across the skeletal myogenesis phases (215).
Some reports exist in the literature demonstrating that ADAR1 can moderately or highly edit several non-muscle specific miRNAs in skeletal muscles under physiological conditions (220–222). On the contrary, very low (<5%) (222) or no A-to-I editing was reported for myomiRs. However, the actual implications for miRNA biogenesis and targeting of such A-to-I editing events remain unknown, and no data have been reported about potential changes in miRNA editing levels during muscle atrophy.
Although no study has yet investigated the role of ADAR1 in skeletal muscles during cancer cachexia, one study demonstrated that ADAR1 is increasingly expressed and activated in skeletal muscles exposed to inflammatory stressors (223). These include TNFα, IFNγ, and TLR4, which are notorious mediators of muscle atrophy under conditions of experimental cancer cachexia (21). This event might depend on the significant decrease in myomiR expression detected under the same condition. Meanwhile, MyoD and Myogenin expression, as well as levels of active phosphorylated Akt, were shown to correlate positively with ADAR1 expression (223), in agreement with data reported by Tan et al. (214). Collectively, these data suggest a role for ADAR1 as a buffer of inflammatory stress in the skeletal muscle, by limiting muscle atrophy and promoting protein synthesis.
Conclusions
Despite the progress concerning the mechanisms underlying experimental cancer cachexia, this syndrome still represents a significant problem for the treatment of many tumor patients and remains mostly underdiagnosed at the clinical level. Unfortunately, the actual relevance of most of the conventional mediators of cancer cachexia has remained undefined or even controversial, impeding the establishment of effective therapeutic options. For such reason, cancer investigations are trying to identify novel mediators of cancer cachexia that are exploitable as both biomarkers of disease and targets of innovative therapies.
In recent times, miRNAs have emerged as essential regulators in skeletal muscle development and homeostasis. Accordingly, alterations in their expression rates were demonstrated to decrease muscle repair abilities and worsen muscle atrophy. In this context, several cues indicate that both myogenic and tumor-derived miRNAs might play a fundamental role in muscle wasting during cancer cachexia, hence representing potential biomarkers with predictive and prognostic value, as well as novel therapeutic targets. However, the findings are still preliminary and mainly based on experimental models of cancer cachexia.
MiRNA are, in turn, finely regulated by several epigenetic components, whose dysregulation alters their expression and function. One of such components is represented by lncRNAs, which directly influence the transcriptional activity of essential myogenic transcription factors like MyoD, Myogenin, and sponge myogenic miRNAs through their seed-complementary sequences. ADAR1 represents another essential epigenetic element, capable of modulating miRNA biogenesis and function either via editing-dependent or -independent mechanisms. However, albeit these mechanisms have been investigated in the skeletal muscle under physiological conditions, only a few cues exist concerning their influence on miRNAs during skeletal muscle atrophy.
Overall, these facts highlight the need to establish the real role of miRNAs in skeletal muscle cell death during cancer cachexia. This might lead to the identification of new reliable biomarkers of skeletal muscle wasting in cancer patients. Moreover, the precise role of miRNA modulators should also be studied to gain a better view of the complex network governing the myogenic gene expression throughout this debilitating syndrome.
Author Contributions
GM, GN, and FC wrote the manuscript. GM, GN, and FC contributed to the editing of the manuscript as needed while CC critically revised our work. All authors contributed to the article and approved the submitted version.
Acknowledgments
Figures of this manuscript were realized by using the software BioRender.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wolfe RR. The underappreciated role of muscle in health and disease. Am J Clin Nutr (2006) 84(3):475–82. doi: 10.1093/ajcn/84.3.475
2. Frontera WR, Ochala J. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int (2015) 96(3):183–95. doi: 10.1007/s00223-014-9915-y
3. Ortiz O, Russell M, Daley TL, Baumgartner RN, Waki M, Lichtman S, et al. Differences in skeletal muscle and bone mineral mass between black and white females and their relevance to estimates of body composition. Am J Clin Nutr (1992) 55(1):8–13. doi: 10.1093/ajcn/55.1.8
4. Thompson LV. Effects of age and training on skeletal muscle physiology and performance. Phys Ther (1994) 74(1):71–81. doi: 10.1093/ptj/74.1.71
5. Wang ZM, Gallagher D, Nelson ME, Matthews DE, Heymsfield SB. Total-body skeletal muscle mass: evaluation of 24-h urinary creatinine excretion by computerized axial tomography. Am J Clin Nutr (1996) 63(6):863–9. doi: 10.1093/ajcn/63.6.863
6. Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol (2012) 8(8):457–65. doi: 10.1038/nrendo.2012.49
7. Horton R. The neglected epidemic of chronic disease. Lancet (2005) 366(9496):1514. doi: 10.1016/S0140-6736(05)67454-5
8. Jackson MJ, McArdle A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J Physiol (2011) 589(Pt 9):2139–45. doi: 10.1113/jphysiol.2011.206623
9. Saitoh M, Ishida J, Doehner W, von Haehling S, Anker MS, Coats AJS, et al. Sarcopenia, cachexia, and muscle performance in heart failure: Review update 2016. Int J Cardiol (2017) 238:5–11. doi: 10.1016/j.ijcard.2017.03.155
10. Tényi Á, Cano I, Marabita F, Kiani N, Kalko SG, Barreiro E, et al. Network modules uncover mechanisms of skeletal muscle dysfunction in COPD patients. J Transl Med (2018) 16(1):34. doi: 10.1186/s12967-018-1405-y
11. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol (2011) 12(5):489–95. doi: 10.1016/S1470-2045(10)70218-7
12. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Primers (2018) 4:17105. doi: 10.1038/nrdp.2017.105
13. Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer (2002) 2(11):862–71. doi: 10.1038/nrc927
14. Argilés JM, Moore-Carrasco R, Fuster G, Busquets S, López-Soriano FJ. Cancer cachexia: the molecular mechanisms. Int J Biochem Cell Biol (2003) 35(4):405–9. doi: 10.1016/s1357-2725(02)00251-0
15. Anker MS, Holcomb R, Muscaritoli M, von Haehling S, Haverkamp W, Jatoi A, et al. Orphan disease status of cancer cachexia in the USA and in the European Union: a systematic review. J Cachexia Sarcopenia Muscle (2019) 10(1):22–34. doi: 10.1002/jcsm.12402
16. Freire PP, Fernandez GJ, de Moraes D, Cury SS, Dal Pai-Silva M, Dos Reis PP, et al. The expression landscape of cachexia-inducing factors in human cancers. J Cachexia Sarcopenia Muscle (2020) 11(4):947–61. doi: 10.1002/jcsm.12565
17. Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J (2007) 21(2):325–32. doi: 10.1096/fj.06-7227rev
18. Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer (2008) 8(11):887–99. doi: 10.1038/nrc2507
19. Tan BH, Ross JA, Kaasa S, Skorpen F, Fearon KC, Collaborative EPCR. Identification of possible genetic polymorphisms involved in cancer cachexia: a systematic review. J Genet (2011) 90(1):165–77. doi: 10.1007/s12041-011-0027-4
20. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol (2013) 10(2):90–9. doi: 10.1038/nrclinonc.2012.209
21. Marceca GP, Londhe P, Calore F. Management of Cancer Cachexia: Attempting to Develop New Pharmacological Agents for New Effective Therapeutic Options. Front Oncol (2020) 10:298. doi: 10.3389/fonc.2020.00298
22. Advani SM, Advani PG, VonVille HM, Jafri SH. Pharmacological management of cachexia in adult cancer patients: a systematic review of clinical trials. BMC Cancer (2018) 18(1):1174. doi: 10.1186/s12885-018-5080-4
23. Yakovenko A, Cameron M, Trevino JG. Molecular therapeutic strategies targeting pancreatic cancer induced cachexia. World J Gastrointest Surg (2018) 10(9):95–106. doi: 10.4240/wjgs.v10.i9.95
24. Fearon KC. Cancer cachexia: developing multimodal therapy for a multidimensional problem. Eur J Cancer (2008) 44(8):1124–32. doi: 10.1016/j.ejca.2008.02.033
25. Solheim TS, Laird BJA, Balstad TR, Bye A, Stene G, Baracos V, et al. Cancer cachexia: rationale for the MENAC (Multimodal-Exercise, Nutrition and Anti-inflammatory medication for Cachexia) trial. BMJ Supp Palliat Care (2018) 8(3):258–65. doi: 10.1136/bmjspcare-2017-001440
26. Baracos VE. Bridging the gap: are animal models consistent with clinical cancer cachexia? Nat Rev Clin Oncol (2018) 15(4):197–8. doi: 10.1038/nrclinonc.2018.14
27. Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J (2007) 26(3):775–83. doi: 10.1038/sj.emboj.7601512
28. de Rie D, Abugessaisa I, Alam T, Arner E, Arner P, Ashoor H, et al. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat Biotechnol (2017) 35(9):872–8. doi: 10.1038/nbt.3947
29. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol (2014) 15(8):509–24. doi: 10.1038/nrm3838
30. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136(2):215–33. doi: 10.1016/j.cell.2009.01.002
31. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet (2015) 16(7):421–33. doi: 10.1038/nrg3965
32. Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci USA (2008) 105(39):14879–84. doi: 10.1073/pnas.0803230105
33. Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell (2010) 38(6):789–802. doi: 10.1016/j.molcel.2010.06.005
34. Moretti F, Thermann R, Hentze MW. Mechanism of translational regulation by miR-2 from sites in the 5’ untranslated region or the open reading frame. RNA (2010) 16(12):2493–502. doi: 10.1261/rna.2384610
35. Poliseno L, Tuccoli A, Mariani L, Evangelista M, Citti L, Woods K, et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood (2006) 108(9):3068–71. doi: 10.1182/blood-2006-01-012369
36. Li X, Jin P. Roles of small regulatory RNAs in determining neuronal identity. Nat Rev Neurosci (2010) 11(5):329–38. doi: 10.1038/nrn2739
37. Güller I, Russell AP. MicroRNAs in skeletal muscle: their role and regulation in development, disease and function. J Physiol (2010) 588(Pt 21):4075–87. doi: 10.1113/jphysiol.2010.194175
38. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet (2009) 10(10):704–14. doi: 10.1038/nrg2634
39. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res (2019) 47(D1):D155–62. doi: 10.1093/nar/gky1141
40. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell (2005) 120(1):15–20. doi: 10.1016/j.cell.2004.12.035
41. Bianchi M, Renzini A, Adamo S, Moresi V. Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation. Int J Mol Sci (2017) 18(4):840. doi: 10.3390/ijms18040840
42. Lee EJ, Baek M, Gusev Y, Brackett DJ, Nuovo GJ, Schmittgen TD. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA (2008) 14(1):35–42. doi: 10.1261/rna.804508
43. Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics (2005) 21(5):650–9. doi: 10.1093/bioinformatics/bti042
44. Ludwig N, Leidinger P, Becker K, Backes C, Fehlmann T, Pallasch C, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res (2016) 44(8):3865–77. doi: 10.1093/nar/gkw116
45. Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol (2004) 5(3):R13. doi: 10.1186/gb-2004-5-3-r13
46. McCarthy JJ. MicroRNA-206: the skeletal muscle-specific myomiR. Biochim Biophys Acta (2008) 1779(11):682–91. doi: 10.1016/j.bbagrm.2008.03.001
47. van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell (2009) 17(5):662–73. doi: 10.1016/j.devcel.2009.10.013
48. Small EM, O’Rourke JR, Moresi V, Sutherland LB, McAnally J, Gerard RD, et al. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc Natl Acad Sci USA (2010) 107(9):4218–23. doi: 10.1073/pnas.1000300107
49. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet (2006) 38(2):228–33. doi: 10.1038/ng1725
50. Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol (2006) 174(5):677–87. doi: 10.1083/jcb.200603008
51. Sweetman D, Goljanek K, Rathjen T, Oustanina S, Braun T, Dalmay T, et al. Specific requirements of MRFs for the expression of muscle specific microRNAs, miR-1, miR-206 and miR-133. Dev Biol (2008) 321(2):491–9. doi: 10.1016/j.ydbio.2008.06.019
52. Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell (2008) 14(5):369–81. doi: 10.1016/j.ccr.2008.10.006
53. Hernandez-Torres F, Aranega AE, Franco D. Identification of regulatory elements directing miR-23a-miR-27a-miR-24-2 transcriptional regulation in response to muscle hypertrophic stimuli. Biochim Biophys Acta (2014) 1839(9):885–97. doi: 10.1016/j.bbagrm.2014.07.009
54. Perry RL, Rudnick MA. Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci (2000) 5:D750–67. doi: 10.2741/perry
55. Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci USA (2006) 103(23):8721–6. doi: 10.1073/pnas.0602831103
56. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science (2007) 316(5824):575–9. doi: 10.1126/science.1139089
57. Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature (2005) 436(7048):214–20. doi: 10.1038/nature03817
58. Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, et al. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc Natl Acad Sci USA (2007) 104(52):20844–9. doi: 10.1073/pnas.0710558105
59. Sun Y, Ge Y, Drnevich J, Zhao Y, Band M, Chen J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J Cell Biol (2010) 189(7):1157–69. doi: 10.1083/jcb.200912093
60. Olguin HC, Olwin BB. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev Biol (2004) 275(2):375–88. doi: 10.1016/j.ydbio.2004.08.015
61. Kumar D, Shadrach JL, Wagers AJ, Lassar AB. Id3 is a direct transcriptional target of Pax7 in quiescent satellite cells. Mol Biol Cell (2009) 20(14):3170–7. doi: 10.1091/mbc.e08-12-1185
62. Dey BK, Gagan J, Dutta A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol (2011) 31(1):203–14. doi: 10.1128/MCB.01009-10
63. Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X, et al. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J Cell Biol (2010) 190(5):867–79. doi: 10.1083/jcb.200911036
64. Hirai H, Verma M, Watanabe S, Tastad C, Asakura Y, Asakura A. MyoD regulates apoptosis of myoblasts through microRNA-mediated down-regulation of Pax3. J Cell Biol (2010) 191(2):347–65. doi: 10.1083/jcb.201006025
65. Goljanek-Whysall K, Sweetman D, Abu-Elmagd M, Chapnik E, Dalmay T, Hornstein E, et al. MicroRNA regulation of the paired-box transcription factor Pax3 confers robustness to developmental timing of myogenesis. Proc Natl Acad Sci USA (2011) 108(29):11936–41. doi: 10.1073/pnas.1105362108
66. Lu L, Zhou L, Chen EZ, Sun K, Jiang P, Wang L, et al. A Novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PloS One (2012) 7(2):e27596. doi: 10.1371/journal.pone.0027596
67. Soleimani VD, Yin H, Jahani-Asl A, Ming H, Kockx CE, van Ijcken WF, et al. Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol Cell (2012) 47(3):457–68. doi: 10.1016/j.molcel.2012.05.046
68. Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J (1999) 18(18):5099–107. doi: 10.1093/emboj/18.18.5099
69. Youn HD, Grozinger CM, Liu JO. Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 4. J Biol Chem (2000) 275(29):22563–7. doi: 10.1074/jbc.C000304200
70. Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci U.S.A. (2000) 97(8):4070–5. doi: 10.1073/pnas.080064097
71. Minetti GC, Feige JN, Bombard F, Heier A, Morvan F, Nürnberg B, et al. Gαi2 signaling is required for skeletal muscle growth, regeneration, and satellite cell proliferation and differentiation. Mol Cell Biol (2014) 34(4):619–30. doi: 10.1128/MCB.00957-13
72. Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science (2009) 326(5959):1549–54. doi: 10.1126/science.1181046
73. Winbanks CE, Wang B, Beyer C, Koh P, White L, Kantharidis P, et al. TGF-beta regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J Biol Chem (2011) 286(16):13805–14. doi: 10.1074/jbc.M110.192625
74. Martin JF, Li L, Olson EN. Repression of myogenin function by TGF-beta 1 is targeted at the basic helix-loop-helix motif and is independent of E2A products. J Biol Chem (1992) 267(16):10956–60.
75. Liu D, Black BL, Derynck R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev (2001) 15(22):2950–66. doi: 10.1101/gad.925901
76. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem (2002) 277(51):49831–40. doi: 10.1074/jbc.M204291200
77. Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H. A novel target of microRNA-29, Ring1 and YY1-binding protein (Rybp), negatively regulates skeletal myogenesis. J Biol Chem (2012) 287(30):25255–65. doi: 10.1074/jbc.M112.357053
78. Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H. Inhibition of miR-29 by TGF-beta-Smad3 signaling through dual mechanisms promotes transdifferentiation of mouse myoblasts into myofibroblasts. PloS One (2012) 7(3):e33766. doi: 10.1371/journal.pone.0033766
79. Kopan R, Nye JS, Weintraub H. The intracellular domain of mouse Notch: a constitutively activated repressor of myogenesis directed at the basic helix-loop-helix region of MyoD. Development (1994) 120(9):2385–96.
80. Bjornson CR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells (2012) 30(2):232–42. doi: 10.1002/stem.773
81. Bröhl D, Vasyutina E, Czajkowski MT, Griger J, Rassek C, Rahn HP, et al. Colonization of the satellite cell niche by skeletal muscle progenitor cells depends on Notch signals. Dev Cell (2012) 23(3):469–81. doi: 10.1016/j.devcel.2012.07.014
82. Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell (2002) 3(3):397–409. doi: 10.1016/s1534-5807(02)00254-x
83. Beres BJ, George R, Lougher EJ, Barton M, Verrelli BC, McGlade CJ, et al. Numb regulates Notch1, but not Notch3, during myogenesis. Mech Dev (2011) 128(5-6):247–57. doi: 10.1016/j.mod.2011.02.002
84. Kuang W, Tan J, Duan Y, Duan J, Wang W, Jin F, et al. Cyclic stretch induced miR-146a upregulation delays C2C12 myogenic differentiation through inhibition of Numb. Biochem Biophys Res Commun (2009) 378(2):259–63. doi: 10.1016/j.bbrc.2008.11.041
85. Ivey KN, Muth A, Arnold J, King FW, Yeh RF, Fish JE, et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell (2008) 2(3):219–29. doi: 10.1016/j.stem.2008.01.016
86. Gagan J, Dey BK, Layer R, Yan Z, Dutta A. Notch3 and Mef2c proteins are mutually antagonistic via Mkp1 protein and miR-1/206 microRNAs in differentiating myoblasts. J Biol Chem (2012) 287(48):40360–70. doi: 10.1074/jbc.M112.378414
87. Zhang Z, Chen Y, Li B, Yao Y, Jiang A, Wei W, et al. Identification of a novel miR-206-Notch3 pathway regulating mouse myoblasts proliferation. Gene (2019) 695:57–64. doi: 10.1016/j.gene.2019.01.045
88. Elia L, Contu R, Quintavalle M, Varrone F, Chimenti C, Russo MA, et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation (2009) 120(23):2377–85. doi: 10.1161/CIRCULATIONAHA.109.879429
89. Shan ZX, Lin QX, Fu YH, Deng CY, Zhou ZL, Zhu JN, et al. Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochem Biophys Res Commun (2009) 381(4):597–601. doi: 10.1016/j.bbrc.2009.02.097
90. Huang MB, Xu H, Xie SJ, Zhou H, Qu LH. Insulin-like growth factor-1 receptor is regulated by microRNA-133 during skeletal myogenesis. PloS One (2011) 6(12):e29173. doi: 10.1371/journal.pone.0029173
91. Wei W, He HB, Zhang WY, Zhang HX, Bai JB, Liu HZ, et al. miR-29 targets Akt3 to reduce proliferation and facilitate differentiation of myoblasts in skeletal muscle development. Cell Death Dis (2013) 4:e668. doi: 10.1038/cddis.2013.184
92. Gong JN, Yu J, Lin HS, Zhang XH, Yin XL, Xiao Z, et al. The role, mechanism and potentially therapeutic application of microRNA-29 family in acute myeloid leukemia. Cell Death Diff (2014) 21(1):100–12. doi: 10.1038/cdd.2013.133
93. de la Garza-Rodea AS, Baldwin DM, Oskouian B, Place RF, Bandhuvula P, Kumar A, et al. Sphingosine phosphate lyase regulates myogenic differentiation via S1P receptor-mediated effects on myogenic microRNA expression. FASEB J (2014) 28(1):506–19. doi: 10.1096/fj.13-233155
94. Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol (2011) 18(7):840–5. doi: 10.1038/nsmb.2067
95. Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, et al. A muscleblind knockout model for myotonic dystrophy. Science (2003) 302(5652):1978–80. doi: 10.1126/science.1088583
96. Canalis E, Economides AN, Gazzerro E. Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr Rev (2003) 24(2):218–35. doi: 10.1210/er.2002-0023
97. Sato MM, Nashimoto M, Katagiri T, Yawaka Y, Tamura M. Bone morphogenetic protein-2 down-regulates miR-206 expression by blocking its maturation process. Biochem Biophys Res Commun (2009) 383(1):125–9. doi: 10.1016/j.bbrc.2009.03.142
98. Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, et al. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature (2009) 459(7249):1010–4. doi: 10.1038/nature08025
99. King IN, Yartseva V, Salas D, Kumar A, Heidersbach A, Ando DM, et al. The RNA-binding protein TDP-43 selectively disrupts microRNA-1/206 incorporation into the RNA-induced silencing complex. J Biol Chem (2014) 289(20):14263–71. doi: 10.1074/jbc.M114.561902
100. Yuasa K, Hagiwara Y, Ando M, Nakamura A, Takeda S, Hijikata T. MicroRNA-206 is highly expressed in newly formed muscle fibers: implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct Funct (2008) 33(2):163–9. doi: 10.1247/csf.08022
101. Greco S, De Simone M, Colussi C, Zaccagnini G, Fasanaro P, Pescatori M, et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J (2009) 23(10):3335–46. doi: 10.1096/fj.08-128579
102. Koutsoulidou A, Mastroyiannopoulos NP, Furling D, Uney JB, Phylactou LA. Expression of miR-1, miR-133a, miR-133b and miR-206 increases during development of human skeletal muscle. BMC Dev Biol (2011) 11:34. doi: 10.1186/1471-213X-11-34
103. Goljanek-Whysall K, Pais H, Rathjen T, Sweetman D, Dalmay T, Münsterberg A. Regulation of multiple target genes by miR-1 and miR-206 is pivotal for C2C12 myoblast differentiation. J Cell Sci (2012) 125(Pt 15):3590–600. doi: 10.1242/jcs.101758
104. Mankoo BS, Skuntz S, Harrigan I, Grigorieva E, Candia A, Wright CV, et al. The concerted action of Meox homeobox genes is required upstream of genetic pathways essential for the formation, patterning and differentiation of somites. Development (2003) 130(19):4655–64. doi: 10.1242/dev.00687
105. Yin Z, Tong Y, Zhu H, Watsky MA. ClC-3 is required for LPA-activated Cl- current activity and fibroblast-to-myofibroblast differentiation. Am J Physiol Cell Physiol (2008) 294(2):C535–42. doi: 10.1152/ajpcell.00291.2007
106. Habas R, Kato Y, He X. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell (2001) 107(7):843–54. doi: 10.1016/s0092-8674(01)00614-6
107. Kennedy KA, Porter T, Mehta V, Ryan SD, Price F, Peshdary V, et al. Retinoic acid enhances skeletal muscle progenitor formation and bypasses inhibition by bone morphogenetic protein 4 but not dominant negative beta-catenin. BMC Biol (2009) 7:67. doi: 10.1186/1741-7007-7-67
108. Zheng Y, Wang W, Liu B, Deng H, Uster E, Pan D. Identification of Happyhour/MAP4K as Alternative Hpo/Mst-like Kinases in the Hippo Kinase Cascade. Dev Cell (2015) 34(6):642–55. doi: 10.1016/j.devcel.2015.08.014
109. Saccone V, Consalvi S, Giordani L, Mozzetta C, Barozzi I, Sandoná M, et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev (2014) 28(8):841–57. doi: 10.1101/gad.234468.113
110. Li L, Sarver AL, Alamgir S, Subramanian S. Downregulation of microRNAs miR-1, -206 and -29 stabilizes PAX3 and CCND2 expression in rhabdomyosarcoma. Lab Invest (2012) 92(4):571–83. doi: 10.1038/labinvest.2012.10
111. Alteri A, De Vito F, Messina G, Pompili M, Calconi A, Visca P, et al. Cyclin D1 is a major target of miR-206 in cell differentiation and transformation. Cell Cycle (2013) 12(24):3781–90. doi: 10.4161/cc.26674
112. Winbanks CE, Beyer C, Hagg A, Qian H, Sepulveda PV, Gregorevic P. miR-206 represses hypertrophy of myogenic cells but not muscle fibers via inhibition of HDAC4. PloS One (2013) 8(9):e73589. doi: 10.1371/journal.pone.0073589
113. Li S, Czubryt MP, McAnally J, Bassel-Duby R, Richardson JA, Wiebel FF, et al. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc Natl Acad Sci U.S.A. (2005) 102(4):1082–7. doi: 10.1073/pnas.0409103102
114. Luo Y, Wu X, Ling Z, Yuan L, Cheng Y, Chen J, et al. microRNA133a targets Foxl2 and promotes differentiation of C2C12 into myogenic progenitor cells. DNA Cell Biol (2015) 34(1):29–36. doi: 10.1089/dna.2014.2522
115. Boutz PL, Chawla G, Stoilov P, Black DL. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev (2007) 21(1):71–84. doi: 10.1101/gad.1500707
116. Mok GF, Lozano-Velasco E, Maniou E, Viaut C, Moxon S, Wheeler G, et al. miR-133-mediated regulation of the Hedgehog pathway orchestrates embryo myogenesis. Development (2018) 145(12):dev159657. doi: 10.1242/dev.159657
117. Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med (2007) 13(5):613–8. doi: 10.1038/nm1582
118. Liu N, Bezprozvannaya S, Shelton JM, Frisard M II, Hulver MW, McMillan RP, et al. Mice lacking microRNA 133a develop dynamin 2–dependent centronuclear myopathy. J Clin Invest (2011) 121(8):3258–68. doi: 10.1172/JCI46267
119. Dong DL, Chen C, Huo R, Wang N, Li Z, Tu YJ, et al. Reciprocal repression between microRNA-133 and calcineurin regulates cardiac hypertrophy: a novel mechanism for progressive cardiac hypertrophy. Hypertension (2010) 55(4):946–52. doi: 10.1161/HYPERTENSIONAHA.109.139519
120. Zhang D, Li X, Chen C, Li Y, Zhao L, Jing Y, et al. Attenuation of p38-mediated miR-1/133 expression facilitates myoblast proliferation during the early stage of muscle regeneration. PloS One (2012) 7(7):e41478. doi: 10.1371/journal.pone.0041478
121. Chen X, Wang K, Chen J, Guo J, Yin Y, Cai X, et al. In vitro evidence suggests that miR-133a-mediated regulation of uncoupling protein 2 (UCP2) is an indispensable step in myogenic differentiation. J Biol Chem (2009) 284(8):5362–9. doi: 10.1074/jbc.M807523200
122. Dai A, Sun H, Fang T, Zhang Q, Wu S, Jiang Y, et al. MicroRNA-133b stimulates ovarian estradiol synthesis by targeting Foxl2. FEBS Lett (2013) 587(15):2474–82. doi: 10.1016/j.febslet.2013.06.023
123. Zhao Y, Huang J, Zhang L, Qu Y, Li J, Yu B, et al. MiR-133b is frequently decreased in gastric cancer and its overexpression reduces the metastatic potential of gastric cancer cells. BMC Cancer (2014) 14:34. doi: 10.1186/1471-2407-14-34
124. Qin W, Dong P, Ma C, Mitchelson K, Deng T, Zhang L, et al. MicroRNA-133b is a key promoter of cervical carcinoma development through the activation of the ERK and AKT1 pathways. Oncogene (2012) 31(36):4067–75. doi: 10.1038/onc.2011.561
125. Cheng Z, Liu F, Wang G, Li Y, Zhang H, Li F. miR-133 is a key negative regulator of CDC42-PAK pathway in gastric cancer. Cell Signal (2014) 26(12):2667–73. doi: 10.1016/j.cellsig.2014.08.012
126. Qiu T, Zhou X, Wang J, Du Y, Xu J, Huang Z, et al. MiR-145, miR-133a and miR-133b inhibit proliferation, migration, invasion and cell cycle progression via targeting transcription factor Sp1 in gastric cancer. FEBS Lett (2014) 588(7):1168–77. doi: 10.1016/j.febslet.2014.02.054
127. Boettger T, Wüst S, Nolte H, Braun T. The miR-206/133b cluster is dispensable for development, survival and regeneration of skeletal muscle. Skelet Muscle (2014) 4(1):23. doi: 10.1186/s13395-014-0023-5
128. McCarthy JJ, Esser KA. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol (2007) 102(1):306–13. doi: 10.1152/japplphysiol.00932.2006
129. Gan Z, Rumsey J, Hazen BC, Lai L, Leone TC, Vega RB, et al. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J Clin Invest (2013) 123(6):2564–75. doi: 10.1172/JCI67652
130. Xu Z, Han Y, Liu J, Jiang F, Hu H, Wang Y, et al. MiR-135b-5p and MiR-499a-3p Promote Cell Proliferation and Migration in Atherosclerosis by Directly Targeting MEF2C. Sci Rep (2015) 5:12276. doi: 10.1038/srep12276
131. Motohashi N, Alexander MS, Casar JC, Kunkel LM. Identification of a novel microRNA that regulates the proliferation and differentiation in muscle side population cells. Stem Cells Dev (2012) 21(16):3031–43. doi: 10.1089/scd.2011.0721
132. Motohashi N, Alexander MS, Shimizu-Motohashi Y, Myers JA, Kawahara G, Kunkel LM. Regulation of IRS1/Akt insulin signaling by microRNA-128a during myogenesis. J Cell Sci (2013) 126(Pt 12):2678–91. doi: 10.1242/jcs.119966
133. Schiaffino S, Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle (2011) 1(1):4. doi: 10.1186/2044-5040-1-4
134. Hitachi K, Nakatani M, Tsuchida K. Myostatin signaling regulates Akt activity via the regulation of miR-486 expression. Int J Biochem Cell Biol (2014) 47:93–103. doi: 10.1016/j.biocel.2013.12.003
135. Xu J, Li R, Workeneh B, Dong Y, Wang X, Hu Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int (2012) 82(4):401–11. doi: 10.1038/ki.2012.84
136. Wada S, Kato Y, Okutsu M, Miyaki S, Suzuki K, Yan Z, et al. Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy. J Biol Chem (2011) 286(44):38456–65. doi: 10.1074/jbc.M111.271270
137. Huang Z, Chen X, Yu B, He J, Chen D. MicroRNA-27a promotes myoblast proliferation by targeting myostatin. Biochem Biophys Res Commun (2012) 423(2):265–9. doi: 10.1016/j.bbrc.2012.05.106
138. McFarlane C, Vajjala A, Arigela H, Lokireddy S, Ge X, Bonala S, et al. Negative auto-regulation of myostatin expression is mediated by Smad3 and microRNA-27. PloS One (2014) 9(1):e87687. doi: 10.1371/journal.pone.0087687
139. Chemello F, Grespi F, Zulian A, Cancellara P, Hebert-Chatelain E, Martini P, et al. Transcriptomic Analysis of Single Isolated Myofibers Identifies miR-27a-3p and miR-142-3p as Regulators of Metabolism in Skeletal Muscle. Cell Rep (2019) 26(13):3784–97.e8. doi: 10.1016/j.celrep.2019.02.105
140. Chinchilla A, Lozano E, Daimi H, Esteban FJ, Crist C, Aranega AE, et al. MicroRNA profiling during mouse ventricular maturation: a role for miR-27 modulating Mef2c expression. Cardiovasc Res (2011) 89(1):98–108. doi: 10.1093/cvr/cvq264
141. Crist CG, Montarras D, Pallafacchina G, Rocancourt D, Cumano A, Conway SJ, et al. Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc Natl Acad Sci USA (2009) 106(32):13383–7. doi: 10.1073/pnas.0900210106
142. Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA (2009) 106(29):12103–8. doi: 10.1073/pnas.0811371106
143. Argilés JM, López-Soriano FJ. The ubiquitin-dependent proteolytic pathway in skeletal muscle: its role in pathological states. Trends Pharmacol Sci (1996) 17(6):223–6. doi: 10.1016/0165-6147(96)10021-3
144. Zhang A, Li M, Wang B, Klein JD, Price SR, Wang XH. miRNA-23a/27a attenuates muscle atrophy and renal fibrosis through muscle-kidney crosstalk. J Cachexia Sarcopenia Muscle (2018) 9(4):755–70. doi: 10.1002/jcsm.12296
145. Sun Q, Liu Q, Zheng Y, Cao X. Rapamycin suppresses TLR4-triggered IL-6 and PGE(2) production of colon cancer cells by inhibiting TLR4 expression and NF-kappaB activation. Mol Immunol (2008) 45(10):2929–36. doi: 10.1016/j.molimm.2008.01.025
146. Sun Y, Wang H, Li Y, Liu S, Chen J, Ying H. miR-24 and miR-122 Negatively Regulate the Transforming Growth Factor-β/Smad Signaling Pathway in Skeletal Muscle Fibrosis. Mol Ther Nucleic Acids (2018) 11:528–37. doi: 10.1016/j.omtn.2018.04.005
147. Lee M, Wada S, Oikawa S, Suzuki K, Ushida T, Akimoto T. Loss of microRNA-23-27-24 clusters in skeletal muscle is not influential in skeletal muscle development and exercise-induced muscle adaptation. Sci Rep (2019) 9(1):1092. doi: 10.1038/s41598-018-37765-3
148. Sharma M, Juvvuna PK, Kukreti H, McFarlane C. Mega roles of microRNAs in regulation of skeletal muscle health and disease. Front Physiol (2014) 5:239. doi: 10.3389/fphys.2014.00239
149. Massart J, Katayama M, Krook A. microManaging glucose and lipid metabolism in skeletal muscle: Role of microRNAs. Biochim Biophys Acta (2016) 1861(12 Pt B):2130–8. doi: 10.1016/j.bbalip.2016.05.006
150. Narasimhan A, Ghosh S, Stretch C, Greiner R, Bathe OF, Baracos V, et al. Small RNAome profiling from human skeletal muscle: novel miRNAs and their targets associated with cancer cachexia. J Cachexia Sarcopenia Muscle (2017) 8(3):405–16. doi: 10.1002/jcsm.12168
151. van de Worp WRPH, Schols AMWJ, Dingemans AC, Op den Kamp CMH, Degens JHRJ, Kelders MCJM, et al. Identification of microRNAs in skeletal muscle associated with lung cancer cachexia. J Cachexia Sarcopenia Muscle (2020) 11(2):452–63. doi: 10.1002/jcsm.12512
152. Soares RJ, Cagnin S, Chemello F, Silvestrin M, Musaro A, De Pitta C, et al. Involvement of microRNAs in the regulation of muscle wasting during catabolic conditions. J Biol Chem (2014) 289(32):21909–25. doi: 10.1074/jbc.M114.561845
153. Lee DE, Brown JL, Rosa-Caldwell ME, Blackwell TA, Perry RA, Brown LA, et al. Cancer cachexia-induced muscle atrophy: evidence for alterations in microRNAs important for muscle size. Physiol Genomics (2017) 49(5):253–60. doi: 10.1152/physiolgenomics.00006.2017
154. Fernandez GJ, Ferreira JH, Vechetti IJ, de Moraes LN, Cury SS, Freire PP, et al. MicroRNA-mRNA Co-sequencing Identifies Transcriptional and Post-transcriptional Regulatory Networks Underlying Muscle Wasting in Cancer Cachexia. Front Genet (2020) 11:541. doi: 10.3389/fgene.2020.00541
155. Naguibneva I, Ameyar-Zazoua M, Polesskaya A, Ait-Si-Ali S, Groisman R, Souidi M, et al. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat Cell Biol (2006) 8(3):278–84. doi: 10.1038/ncb1373
156. Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science (2000) 289(5488):2363–6. doi: 10.1126/science.289.5488.2363
157. Langen RC, Van Der Velden JL, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Tumor necrosis factor-alpha inhibits myogenic differentiation through MyoD protein destabilization. FASEB J (2004) 18(2):227–37. doi: 10.1096/fj.03-0251com
158. Zhang G, Jin B, Li YP. C/EBPβ mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J (2011) 30(20):4323–35. doi: 10.1038/emboj.2011.292
159. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest (2013) 123(11):4821–35. doi: 10.1172/JCI68523
160. Panguluri SK, Bhatnagar S, Kumar A, McCarthy JJ, Srivastava AK, Cooper NG, et al. Genomic profiling of messenger RNAs and microRNAs reveals potential mechanisms of TWEAK-induced skeletal muscle wasting in mice. PloS One (2010) 5(1):e8760. doi: 10.1371/journal.pone.0008760
161. Meyer SU, Thirion C, Polesskaya A, Bauersachs S, Kaiser S, Krause S, et al. TNF-α and IGF1 modify the microRNA signature in skeletal muscle cell differentiation. Cell Commun Signal (2015) 13:4. doi: 10.1186/s12964-015-0083-0
162. Georgantas RW, Streicher K, Greenberg SA, Greenlees LM, Zhu W, Brohawn PZ, et al. Inhibition of myogenic microRNAs 1, 133, and 206 by inflammatory cytokines links inflammation and muscle degeneration in adult inflammatory myopathies. Arthritis Rheumatol (2014) 66(4):1022–33. doi: 10.1002/art.38292
163. Kang J, Jeong MG, Oh S, Jang EJ, Kim HK, Hwang ES. A FoxO1-dependent, but NRF2-independent induction of heme oxygenase-1 during muscle atrophy. FEBS Lett (2014) 588(1):79–85. doi: 10.1016/j.febslet.2013.11.009
164. Kozakowska M, Ciesla M, Stefanska A, Skrzypek K, Was H, Jazwa A, et al. Heme oxygenase-1 inhibits myoblast differentiation by targeting myomirs. Antioxid Redox Signal (2012) 16(2):113–27. doi: 10.1089/ars.2011.3964
165. Fredrix EW, Soeters PB, Wouters EF, Deerenberg IM, von Meyenfeldt MF, Saris WH. Effect of different tumor types on resting energy expenditure. Cancer Res (1991) 51(22):6138–41.
166. Patel HJ, Patel BM. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci (2017) 170:56–63. doi: 10.1016/j.lfs.2016.11.033
167. Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell (2012) 149(3):515–24. doi: 10.1016/j.cell.2012.04.005
168. Rahman M, Jackson LK, Johnson WE, Li DY, Bild AH, Piccolo SR. Alternative preprocessing of RNA-Sequencing data in The Cancer Genome Atlas leads to improved analysis results. Bioinformatics (2015) 31(22):3666–72. doi: 10.1093/bioinformatics/btv377
169. Talbert EE, Cuitiño MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, et al. Modeling Human Cancer-induced Cachexia. Cell Rep (2019) 28(6):1612–22.e4. doi: 10.1016/j.celrep.2019.07.016
170. Cernackova A, Mikova L, Horvathova L, Tillinger A, Mravec B. Cachexia induced by Yoshida ascites hepatoma in Wistar rats is not associated with inflammatory response in the spleen or brain. J Neuroimmunol (2019) 337:577068. doi: 10.1016/j.jneuroim.2019.577068
171. Delitto D, Pham K, Vlada AC, Sarosi GA, Thomas RM, Behrns KE, et al. Patient-derived xenograft models for pancreatic adenocarcinoma demonstrate retention of tumor morphology through incorporation of murine stromal elements. Am J Pathol (2015) 185(5):1297–303. doi: 10.1016/j.ajpath.2015.01.016
172. Michaelis KA, Norgard MA, Zhu X, Levasseur PR, Sivagnanam S, Liudahl SM, et al. Publisher Correction: The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat Commun (2019) 10(1):5257. doi: 10.1038/s41467-019-13151-z
173. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol (2018) 19(4):213–28. doi: 10.1038/nrm.2017.125
174. O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol (2020) 21:585–606. doi: 10.1038/s41580-020-0251-y
175. Cho YE, Song BJ, Akbar M, Baek MC. Extracellular vesicles as potential biomarkers for alcohol- and drug-induced liver injury and their therapeutic applications. Pharmacol Ther (2018) 187:180–94. doi: 10.1016/j.pharmthera.2018.03.009
176. Nolte-’t Hoen EN, Buermans HP, Waasdorp M, Stoorvogel W, Wauben MH, ‘t Hoen PA. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res (2012) 40(18):9272–85. doi: 10.1093/nar/gks658
177. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne) (2018) 9:402:402. doi: 10.3389/fendo.2018.00402
178. Zheng M, Hou L, Ma Y, Zhou L, Wang F, Cheng B, et al. Exosomal let-7d-3p and miR-30d-5p as diagnostic biomarkers for non-invasive screening of cervical cancer and its precursors. Mol Cancer (2019) 18(1):76. doi: 10.1186/s12943-019-0999-x
179. Kobayashi M, Sawada K, Nakamura K, Yoshimura A, Miyamoto M, Shimizu A, et al. Exosomal miR-1290 is a potential biomarker of high-grade serous ovarian carcinoma and can discriminate patients from those with malignancies of other histological types. J Ovarian Res (2018) 11(1):81. doi: 10.1186/s13048-018-0458-0
180. Casadei L, Calore F, Creighton CJ, Guescini M, Batte K, Iwenofu OH, et al. Exosome-Derived miR-25-3p and miR-92a-3p Stimulate Liposarcoma Progression. Cancer Res (2017) 77(14):3846–56. doi: 10.1158/0008-5472.CAN-16-2984
181. Kogure T, Lin WL, Yan IK, Braconi C, Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology (2011) 54(4):1237–48. doi: 10.1002/hep.24504
182. Yang M, Chen J, Su F, Yu B, Lin L, Liu Y, et al. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer (2011) 10:117. doi: 10.1186/1476-4598-10-117
183. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood (2012) 119(3):756–66. doi: 10.1182/blood-2011-02-338004
184. Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA (2012) 109(31):E2110–6. doi: 10.1073/pnas.1209414109
185. He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci USA (2014) 111(12):4525–9. doi: 10.1073/pnas.1402714111
186. Calore F, Londhe P, Fadda P, Nigita G, Casadei L, Marceca GP, et al. The TLR7/8/9 Antagonist IMO-8503 Inhibits Cancer-Induced Cachexia. Cancer Res (2018) 78(23):6680–90. doi: 10.1158/0008-5472.CAN-17-3878
187. Okugawa Y, Toiyama Y, Hur K, Yamamoto A, Yin C, Ide S, et al. Circulating miR-203 derived from metastatic tissues promotes myopenia in colorectal cancer patients. J Cachexia Sarcopenia Muscle (2019) 10(3):536–48. doi: 10.1002/jcsm.12403
188. Powrózek T, Mlak R, Brzozowska A, Mazurek M, Gołębiowski P, Małecka-Massalska T. miRNA-130a Significantly Improves Accuracy of SGA Nutritional Assessment Tool in Prediction of Malnutrition and Cachexia in Radiotherapy-Treated Head and Neck Cancer Patients. Cancers (Basel) (2018) 10(9):294. doi: 10.3390/cancers10090294
189. Miyachi M, Tsuchiya K, Yoshida H, Yagyu S, Kikuchi K, Misawa A, et al. Circulating muscle-specific microRNA, miR-206, as a potential diagnostic marker for rhabdomyosarcoma. Biochem Biophys Res Commun (2010) 400(1):89–93. doi: 10.1016/j.bbrc.2010.08.015
190. Siracusa J, Koulmann N, Banzet S. Circulating myomiRs: a new class of biomarkers to monitor skeletal muscle in physiology and medicine. J Cachexia Sarcopenia Muscle (2018) 9(1):20–7. doi: 10.1002/jcsm.12227
191. Köberle V, Kronenberger B, Pleli T, Trojan J, Imelmann E, Peveling-Oberhag J, et al. Serum microRNA-1 and microRNA-122 are prognostic markers in patients with hepatocellular carcinoma. Eur J Cancer (2013) 49(16):3442–9. doi: 10.1016/j.ejca.2013.06.002
192. St Laurent G, Wahlestedt C, Kapranov P. The Landscape of long noncoding RNA classification. Trends Genet (2015) 31(5):239–51. doi: 10.1016/j.tig.2015.03.007
193. Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet (2016) 17(5):272–83. doi: 10.1038/nrg.2016.20
194. Butchart LC, Fox A, Shavlakadze T, Grounds MD. The long and short of non-coding RNAs during post-natal growth and differentiation of skeletal muscles: Focus on lncRNA and miRNAs. Differentiation (2016) 92(5):237–48. doi: 10.1016/j.diff.2016.05.003
195. Li Y, Chen X, Sun H, Wang H. Long non-coding RNAs in the regulation of skeletal myogenesis and muscle diseases. Cancer Lett (2018) 417:58–64. doi: 10.1016/j.canlet.2017.12.015
196. Yu X, Zhang Y, Li T, Ma Z, Jia H, Chen Q, et al. Long non-coding RNA Linc-RAM enhances myogenic differentiation by interacting with MyoD. Nat Commun (2017) 8:14016. doi: 10.1038/ncomms14016
197. Caretti G, Schiltz RL, Dilworth FJ, Di Padova M, Zhao P, Ogryzko V, et al. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev Cell (2006) 11(4):547–60. doi: 10.1016/j.devcel.2006.08.003
198. Zhou L, Sun K, Zhao Y, Zhang S, Wang X, Li Y, et al. Linc-YY1 promotes myogenic differentiation and muscle regeneration through an interaction with the transcription factor YY1. Nat Commun (2015) 6:10026. doi: 10.1038/ncomms10026
199. Chen X, He L, Zhao Y, Li Y, Zhang S, Sun K, et al. regulates myogenic differentiation and muscle regeneration through modulating MyoD transcriptional activity. Cell Discovery (2017) 3:17002. doi: 10.1038/celldisc.2017.2
200. Han X, Yang F, Cao H, Liang Z. Malat1 regulates serum response factor through miR-133 as a competing endogenous RNA in myogenesis. FASEB J (2015) 29(7):3054–64. doi: 10.1096/fj.14-259952
201. Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell (2011) 147(2):358–69. doi: 10.1016/j.cell.2011.09.028
202. Zhu M, Liu J, Xiao J, Yang L, Cai M, Shen H, et al. Lnc-mg is a long non-coding RNA that promotes myogenesis. Nat Commun (2017) 8:14718. doi: 10.1038/ncomms14718
203. Liu M, Li B, Peng W, Ma Y, Huang Y, Lan X, et al. LncRNA-MEG3 promotes bovine myoblast differentiation by sponging miR-135. J Cell Physiol (2019) 234(10):18361–70. doi: 10.1002/jcp.28469
204. Hitachi K, Nakatani M, Funasaki S, Hijikata I, Maekawa M, Honda M, et al. Expression Levels of Long Non-Coding RNAs Change in Models of Altered Muscle Activity and Muscle Mass. Int J Mol Sci (2020) 21(5):1628. doi: 10.3390/ijms21051628
205. Cao SQ, Zheng H, Sun BC, Wang ZL, Liu T, Guo DH, et al. Long non-coding RNA highly up-regulated in liver cancer promotes exosome secretion. World J Gastroenterol (2019) 25(35):5283–99. doi: 10.3748/wjg.v25.i35.5283
206. Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol (2016) 17(2):83–96. doi: 10.1038/nrm.2015.4
207. Solomon O, Oren S, Safran M, Deshet-Unger N, Akiva P, Jacob-Hirsch J, et al. Global regulation of alternative splicing by adenosine deaminase acting on RNA (ADAR). RNA (2013) 19(5):591–604. doi: 10.1261/rna.038042.112
208. Eisenberg E, Levanon EY. A-to-I RNA editing - immune protector and transcriptome diversifier. Nat Rev Genet (2018) 19(8):473–90. doi: 10.1038/s41576-018-0006-1
209. Chen T, Xiang JF, Zhu S, Chen S, Yin QF, Zhang XO, et al. ADAR1 is required for differentiation and neural induction by regulating microRNA processing in a catalytically independent manner. Cell Res (2015) 25(4):459–76. doi: 10.1038/cr.2015.24
210. Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, et al. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell (2013) 153(3):575–89. doi: 10.1016/j.cell.2013.03.024
211. Bahn JH, Ahn J, Lin X, Zhang Q, Lee JH, Civelek M, et al. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat Commun (2015) 6:6355. doi: 10.1038/ncomms7355
212. Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science (2007) 315(5815):1137–40. doi: 10.1126/science.1138050
213. Nigita G, Acunzo M, Romano G, Veneziano D, Laganà A, Vitiello M, et al. microRNA editing in seed region aligns with cellular changes in hypoxic conditions. Nucleic Acids Res (2016) 44(13):6298–308. doi: 10.1093/nar/gkw532
214. Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, et al. Genome Browser Data Integration &Visualization—UCSC Genomics Institute: Dynamic landscape and regulation of RNA editing in mammals. Nature (2017) 550(7675):249–54. doi: 10.1038/nature24041
215. Hsieh CL, Liu H, Huang Y, Kang L, Chen HW, Chen YT, et al. ADAR1 deaminase contributes to scheduled skeletal myogenesis progression via stage-specific functions. Cell Death Diff (2014) 21(5):707–19. doi: 10.1038/cdd.2013.197
216. Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity (2015) 43(5):933–44. doi: 10.1016/j.immuni.2015.11.001
217. Walkley CR, Kile BT. Cell death following the loss of ADAR1 mediated A-to-I RNA editing is not effected by the intrinsic apoptosis pathway. Cell Death Dis (2019) 10(12):913. doi: 10.1038/s41419-019-2160-6
218. Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science (2015) 349(6252):1115–20. doi: 10.1126/science.aac7049
219. Wang Q, Hui H, Guo Z, Zhang W, Hu Y, He T, et al. ADAR1 regulates ARHGAP26 gene expression through RNA editing by disrupting miR-30b-3p and miR-573 binding. RNA (2013) 19(11):1525–36. doi: 10.1261/rna.041533.113
220. Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, et al. RNA editing of human microRNAs. Genome Biol (2006) 7(4):R27. doi: 10.1186/gb-2006-7-4-r27
221. Picardi E, Manzari C, Mastropasqua F, Aiello I, D’Erchia AM, Pesole G. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep (2015) 5:14941. doi: 10.1038/srep14941
222. Li L, Song Y, Shi X, Liu J, Xiong S, Chen W, et al. The landscape of miRNA editing in animals and its impact on miRNA biogenesis and targeting. Genome Res (2018) 28(1):132–43. doi: 10.1101/gr.224386.117
Keywords: cancer cachexia, skeletal muscle wasting, microRNAs, extracellular vesicles, long non-coding RNAs, ADAR
Citation: Marceca GP, Nigita G, Calore F and Croce CM (2020) MicroRNAs in Skeletal Muscle and Hints on Their Potential Role in Muscle Wasting During Cancer Cachexia. Front. Oncol. 10:607196. doi: 10.3389/fonc.2020.607196
Received: 16 September 2020; Accepted: 26 October 2020;
Published: 24 November 2020.
Edited by:
Daria Chudakova, The University of Auckland, New ZealandReviewed by:
Rodrigo Xavier Das Neves, National Cancer Institute, United StatesPaola Costelli, University of Turin, Italy
Copyright © 2020 Marceca, Nigita, Calore and Croce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federica Calore, federica.calore@osumc.edu; Gioacchino P. Marceca, gioacchinopaolo.marceca@unict.it
†These authors have contributed equally to this work