 Liya Ma1†
Liya Ma1† Bin Liang2†Huixian Hu3†Wenli Yang1†Shengyun Lin4
Bin Liang2†Huixian Hu3†Wenli Yang1†Shengyun Lin4 Lihong Cao5Kongfei Li6Yuemin Kuang7Lihong Shou8Weimei Jin9Jianping Lan10
Lihong Cao5Kongfei Li6Yuemin Kuang7Lihong Shou8Weimei Jin9Jianping Lan10 Xingnong Ye1,11Jing Le12
Xingnong Ye1,11Jing Le12 Huyi Lei13Jiaping Fu14Ying Lin15Wenhua Jiang16Zhiying Zheng4Songfu Jiang2Lijuan Fu17Chuanyong Su18XiuFeng Yin19
Huyi Lei13Jiaping Fu14Ying Lin15Wenhua Jiang16Zhiying Zheng4Songfu Jiang2Lijuan Fu17Chuanyong Su18XiuFeng Yin19 Lixia Liu20
Lixia Liu20 Jiayue Qin20
Jiayue Qin20 Jie Jin1Shenxian Qian21*Guifang Ouyang22*
Jie Jin1Shenxian Qian21*Guifang Ouyang22* Hongyan Tong1*
Hongyan Tong1*- 1Department of Hematology, The First Affiliated Hospital of Zhejiang University, Hangzhou, China
- 2Department of Hematology, The First Affiliated Hospital of Wenzhou University, Wenzhou, China
- 3Department of Hematology, Jinhua Central Hospital, Jinhua, China
- 4Department of Hematology, Zhejiang Provincial Hospital of Chinese Medicine, Hangzhou, China
- 5Department of Hematology, Shulan Hospital of Zhejiang Province, Hangzhou, China
- 6Department of Hematology, Ningbo Yinzhou People’s Hospital, Ningbo, China
- 7Department of Hematology, Jinhua People’s Hospital, Jinhua, China
- 8Department of Hematology, Huzhou Central Hospital, Huzhou, China
- 9Department of Hematology, Lishui People’s Hospital, Lishui, China
- 10Department of Hematology, Zhejiang Provincial People’s Hospital, Hangzhou, China
- 11Department of Hematology, The Fourth Affiliated Hospital of Zhejiang University, Yiwu, China
- 12Department of Hematology, Ningbo Lihuili Hospital, Ningbo, China
- 13Department of Hematology, The Affiliated Hospital of Shaoxing University of Arts and Sciences, Shaoxing, China
- 14Department of Hematology, Shaoxing People’s Hospital, Shaoxing, China
- 15Department of Hematology, The Second Affiliated Hospital of Wenzhou University, Wenzhou, China
- 16Department of Hematology, Taizhou First People’s Hospital, Taizhou, China
- 17Department of Hematology, Xinhua Hospital of Zhejiang Province, Hangzhou, China
- 18Department of Hematology, Tongde Hospital of Zhejiang Province, Hangzhou, China
- 19Department of Hematology, The Affiliated Shaoyifu Hospital of Zhejiang University, Hangzhou, China
- 20Department of Medical Affairs, Acornmed Biotechnology Co., Ltd., Tianjin, China
- 21Department of Hematology, Hangzhou First People’s Hospital, Hangzhou, China
- 22Department of Hematology, Ningbo First Hospital, Ningbo, China
The outcomes of myelodysplastic syndrome (MDS) patients with SF3B1 mutation, despite identified as a favorable prognostic biomarker, are variable. To comprehend the heterogeneity in clinical characteristics and outcomes, we reviewed 140 MDS patients with SF3B1 mutation in Zhejiang province of China. Seventy-three (52.1%) patients diagnosed as MDS with ring sideroblasts (MDS-RS) following the 2016 World Health Organization (WHO) classification and 118 (84.3%) patients belonged to lower risk following the revised International Prognostic Scoring System (IPSS-R). Although clonal hematopoiesis-associated mutations containing TET2, ASXL1 and DNMT3A were the most frequent co-mutant genes in these patients, RUNX1, EZH2, NF1 and KRAS/NRAS mutations had significant effects on overall survival (OS). Based on that we developed a risk scoring model as IPSS-R×0.4+RUNX1×1.1+EZH2×0.6+RAS×0.9+NF1×1.6. Patients were categorized into two subgroups: low-risk (L-R, score <= 1.4) group and high risk (H-R, score > 1.4) group. The 3-year OS for the L-R and H-R groups was 91.88% (95% CI, 83.27%-100%) and 38.14% (95% CI, 24.08%-60.40%), respectively (P<0.001). This proposed model distinctly outperformed the widely used IPSS-R. In summary, we constructed and validated a personalized prediction model of MDS patients with SF3B1 mutation that can better predict the survival of these patients.
Introduction
In recent years, the molecular landscape of myelodysplastic syndrome (MDS) has been elucidated with the application of next-generation sequencing (NGS) (1). More than half of MDS patients carried splicing factor gene mutations, which have been indicated to be the most frequent molecular abnormality in this disease (2). Among these mutations, splicing factor 3 subunit 1 (SF3B1) is the most commonly mutated one. SF3B1 locates at 2q33.1 with 25 exons and encodes a 1304 amino acid protein with a highly conserved nucleotide sequence, which is an important component of U2 snRNP (3). Base pairing between the U2 snRNP and the branch-point sequence is essential for pre-mRNA splicing (4). The SF3b/SF3a complex anchors the U2 snRNP to the pre-mRNA, and SF3B1 is a crucial component of the activated spliceosome that helps the branch-point adenosine in place for nucleophilic attack from the 5’ splice site (3). SF3B1 point mutations in MDS are limited to exons 14 through 16. The most common SF3B1 mutation is an A-to-G transition that results in a lysine to glutamic acid substitution at amino acid position 700 (K700E) (5). SF3B1 mutation alters U2 snRNP function by prompting alternative branch-point usage and induction of cryptic 3′ splice site selection, thereby forming aberrantly spliced mRNA transcripts subject to nonsense-mediated decay and downregulation of target transcripts and protein expression (6, 7).
About 20-28% of all MDS patients harbor SF3B1 mutation (5, 8, 9) and a much higher occurrence rate of mutations has been detected in MDS with ring sideroblasts (MDS-RS), such as 64~83% in MDS-RS with single lineage dysplasia (MDS-RS-SLD) and 57~76% in MDS-RS with multiple lineage dysplasia (MDS-RS-MLD) (5, 9–11). Importantly, recent study shows that SF3B1 mutation in MDS-RS can derive from the scarce hematopoietic stem cell compartment and is an initiating event in this disease (12). MDS patients with SF3B1 mutation have higher platelet counts and lower bone marrow blast percentage in comparison to MDS patients with wild-type SF3B1 (13). SF3B1 mutations appear more commonly in lower risk MDS patients and are independent predictive factors of favorable prognosis in MDS (8). SF3B1 mutations co-occur with mutations of genes involved in the regulation of DNA methylation, such as the methyltransferase DNMT3A and the methylcytosine dioxygenase TET2 (14–17).
The International Prognostic Scoring System (IPSS) and the revised IPSS (IPSS-R) are the most widely applied models in clinical practice and clinical trial evaluation in MDS (18). IPSS or IPSS-R based upon the peripheral blood counts, percentage of bone marrow blasts and presence of cytogenetic abnormalities. However, neither IPSS nor IPSS-R incorporates gene mutations.
The International Working Group for the Prognosis of MDS (IWG-PM) has suggested that SF3B1mut MDS as a distinctive entity which has a favorable prognosis with <1% peripheral blood or <5% BM blasts, absence of del (5q), inv (3), abnormal 3q26, monosomy 7, or complex karyotype (CK) and RUNX1 or EZH2 mutations (19). This classification was mainly established on a specific gene mutation, association with ring sideroblasts and favorable prognosis. However, MDS with SF3B1 mutation is a heterogeneous group since not all the patients had favorable survivals. The patients with excess blasts, poor cytogenetics and molecular genetic abnormalities had unfavorable survival (20).
In this study, we aimed to estimate the spectrum of SF3B1 mutation-harboring MDS patients in Zhejiang Province of China, to analyze their clinical and laboratory characteristics and molecular landscape, and to explore the prognostic impacts of co-mutations. Furthermore, we constructed a prognostic model involving IPSS-R and selected gene mutations of MDS patients with SF3B1 mutation to predict their outcomes.
Patients and Methods
Patients
We reviewed the diagnosed cases of MDS with SF3B1 mutation from January 1, 2011 through February 1, 2021 at the First Affiliated Hospital of Zhejiang University and other twenty-one hospitals in Zhejiang Province of China (Figure 1). Clinical, hematological, cytogenetic and molecular data were collected for all patients. Totally one hundred and forty patients were enrolled and classified according to 2016 WHO definition and classification of MDS (21). IPSS-R was used to evaluate the prognosis of each patient (18). This study was approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University consistent with Declaration of Helsinki.
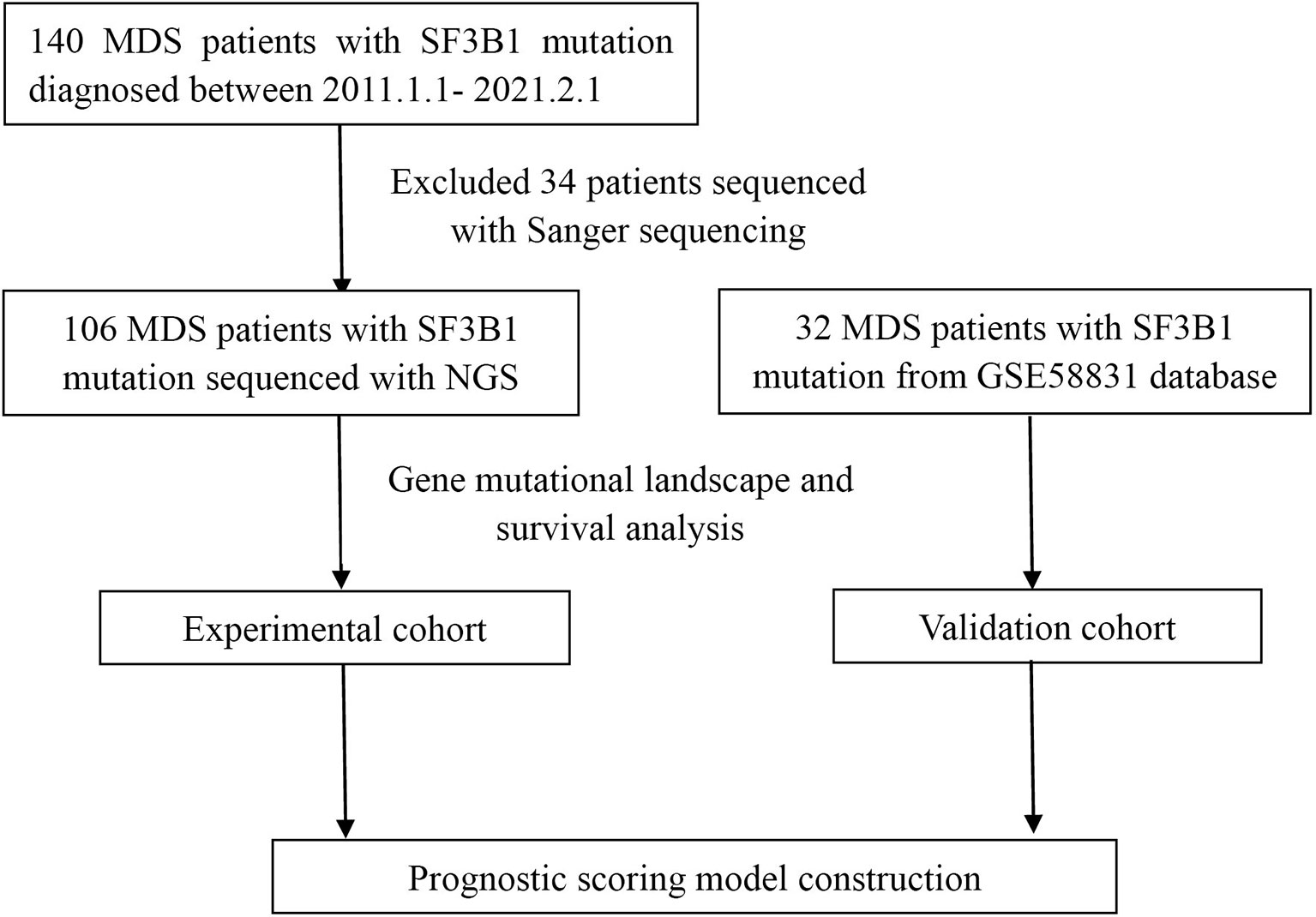
Figure 1 Flowchart of MDS patients with SF3B1 mutation.
Cytogenetic Analysis
Bone marrow (BM) aspirates were cultured for 24 or 48h without mitogens and metaphase cells were prepared for analysis. Chromosomal analysis was performed following standard protocols and the results were reported in accordance with International System for Human Cytogenetic Nomenclature (ISCN) 2016 (22). At least 20 metaphase cells were tested if available. On the basis of IPSS-R, cytogenetic risk was categorized into five groups: very good risk, good risk, intermediate risk, high risk and very high risk (18).
Gene Sequencing Analysis
Genomic DNA was extracted from mononuclear cells of BM samples at diagnosis of MDS. The Sanger sequencing was performed to detect gene mutations in 34 patients diagnosed from 2011 to 2014. NGS platforms covering 34~185 genes were performed to detect gene mutations in 106 patients diagnosed from 2015 to 2019 because NGS was widely applied since 2015 in Zhejiang Province. Multiplex libraries were sequenced using Illumina NovaSeq instrument. Burrows-Wheeler alignment (BWA, version 0.7.12) was used to align the trimmed reads. MarkDuplicates tool from Picard was performed to mark PCR duplicates. IndelRealigner and BaseRecalibrator from Genome Analysis Toolkit (GATK, version 3.8) were performed to realign and recalibrate the BWA data, respectively. Mutect2 was applied to call variants, including SNVs and InDels. ANNOVAR software was used for annotating all the variants including 1000G projects, COSMIC, PolyPhen and SIFT.
Statistical Analysis
The SPSS (version 25) and R (version 3.6.3) software were used to conduct statistical analysis. Mann-Whitney U test was applied for continuous variables and chi-square test was applied for categorical variables. Overall survival (OS) was calculated as the period from the day of diagnosis to the day of death regardless any cause or last contact. OS curves were constructed by the Kaplan-Meier method and the differences in survival curves were compared by the log-rank test. Cox proportional hazard regression analysis was used to examine different independent prognostic factors for OS. The least Absolute Shrinkage and Selector Operation (LASSO) Cox regression model was used for variable selection and predictive prognostic model construction. A two-tailed P < 0.05 was deemed as statistically significant.
Results
Clinical Characteristics of MDS Patients With SF3B1 Mutation
A total of 140 MDS patients from 22 hospitals in Zhejiang Province between January 2011 and February 2021 carried SF3B1 mutation. The clinical characteristics of the MDS patients were listed in Table 1. The patients contained 83 men and 57 women, with a median age of 66 (range, 26-95) years. The median percent of BM blasts was 1.5% (0-19%). According to 2016 WHO sub-classifications, 53 patients (37.9%) diagnosed as MDS-RS-SLD; 20 patients (14.3%) diagnosed as MDS-RS-MLD; 11 patients (7.9%) as MDS-SLD; 27 patients (19.3%) as MDS-MLD; 12 (8.6%) patients as MDS-EB1; 12 (8.6%) patients as MDS-EB2; 1 (0.7%) patient as del(5q) syndrome; and 4 (2.9%) patients as MDS-unclassifiable (MDS-U). According to the cytogenetic risk stratification, only one patient (0.7%) categorized to the very good group, 117 patients (83.6%) to the good group, 15 patients (10.7%) to the intermediate group, 6 patients (4.3%) to the poor group and one patient (0.7%) to the very poor group. Following the IPSS-R, 6 (4.3%) patients were very low risk; 67 (47.9%) patients were low risk, 45 (32.1%) patients were intermediate risk, 14 (10.0%) patients were high risk and 8 (5.7%) patients were very high risk. With respect to treatment, 69 (49.3%) patients received erythroid stimulating agents (ESA) alone or combined with testosterone undecanoate and retinoic acid, 47 (33.8%) received supportive care and 24 (17.2%) received hypomethylating agents (HMAs) alone or combined with chemotherapy. Only 9 patients (5.4%) transformed to Acute myeloid leukemia (AML) in the course of disease and 38 patients (27.1%) died during follow-up. With a median follow-up of 21.77 months (range, 11.33-52.77), the median time to AML progressions was 13.15 months (range, 4.77-47.7).

Table 1 Clinical and laboratory characteristics of 140 MDS patients with SF3B1 mutations.
Mutational Landscape of MDS Patients With SF3B1 Mutation
BM aspirates from 106 MDS patients with SF3B1 mutation underwent NGS analysis with 34~185 gene panels at the time of diagnosis. The median variant allele fraction (VAF) of SF3B1 mutations was 38.0% (range, 1.2% to 50.1%). The most frequent SF3B1 mutation site was K700E (n=84, 60.0%), followed by K666 (n=21, 15.0%), R625 (n=12, 8.6%), E622 (n=3, 2.1%), H662 (n=3, 2.1%) and others (n=17, 12.1%) (Figure 2). Ninety-eight mutant genes except SF3B1 were detected in the 106 patients, and the mutational landscape is described in Figure 2. Only 23 patients (21.7%) had SF3B1 mutation as the exclusive driver of MDS, while most patients (78.3%) had concomitant mutations. In order of decreasing frequency, commonly (> 5%) mutated genes included TET2 (33.0%), ASXL1 (23.6%), DNMT3A (16.0%), EZH2 (12.3%), RUNX1 (11.3%), KMT2D (11.3%), BCOR (8.5%), ATRX (7.5%), TP53 (7.5%), SETBP1 (6.6%), NF1 (5.7%) and ZRSR2 (5.7%). Furthermore, the genes were categorized by function, revealing that chromatin modifying genes (23.4%), DNA methylation related genes (18.0%), signaling pathway genes (15.2%), transcription factor genes (10.1%) and histone methylation (9.8%) were most common (Supplemental Figure 1). The gene association analysis was performed for mutated genes detected in more than five patients, showing interesting coexistence and mutual exclusion relationships (Figure 2). Significant associations were discovered in paired genes, including ZRSR2-TET2, EZH2-ASXL1, BCOR-RUNX1, RUNX1-EZH2, NF1-DNMT3A, JAK2-EZH2 and RUNX1-ASXL1, JAK2-KMT2D (P <0.001; P =0.001; P = 0.001; P = 0.018; P = 0.020; P = 0.022; P = 0.038; respectively) (Figure 2).
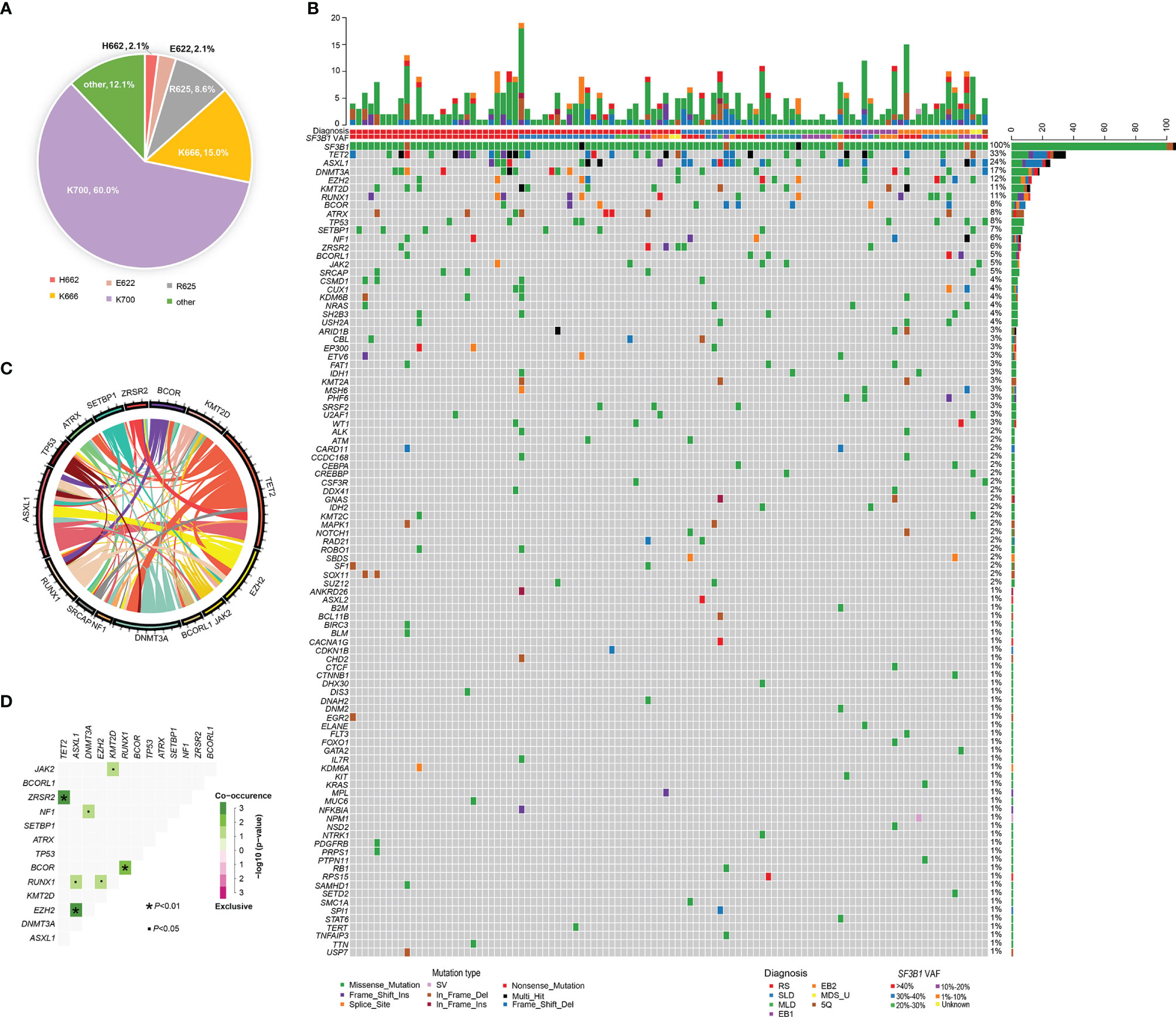
Figure 2 Genomic landscape of MDS patients with SF3B1 mutation. (A) Distribution and proportion of SF3B1 mutation sites in the 140 MDS patients. (B) Heatmap in 106 MDS patients with SF3B1 mutation. Each row represents mutated gene; each column represents a patient; the right side of the graph annotates the frequency and number of the mutated gene; the upper histogram shows the number of gene mutations per patient; different colors below the graph represent different mutation patterns. (C) Circos diagram shows gene association in 106 MDS patients with SF3B1 mutation, according to the relative frequency and pairwise co−occurrence of gene mutations on the basis of the mutated genes detected in ≥5 patients. (D) Diagram shows pairwise gene mutation correlations on the basis of the mutated genes detected in ≥5 patients, green color represents co-occurrence and pink color represents exclusivity.
Survival Analysis of MDS Patients With SF3B1 Mutation
With a median follow-up of 22.22 months (range, 0.87-141.57), the 3-year OS of 106 MDS patients with SF3B1 mutation was 68.30% (95%CI, 58.05-80.37%). The median number of co-mutant genes in these patients was 2 (0-17). The patients without ASXL1 mutation had better survival than the patients with ASXL1 mutation (79.60 months vs. 39.03 months, P=0.021) (Figure 3). Likewise, the patients without NF1 mutation had longer survival than the patients with NF1 mutation (48.76 months vs. 13.17 months, P=0.005) (Figure 3). RUNX1 mutation was significantly associated with shorter survival (21.03 months vs. 79.60 months, P=0.003) (Figure 3). In addition, KRAS/NRAS mutation remarkably correlated with shorter survival (11.33 months vs. 79.60 months, P=0.001) (Figure 3). Nevertheless, EZH2, TET2, DNMT3A, BCOR, KMT2D, ATRX, SETBP1, TP53 and IDH1/2 had no impact on OS (Figure 3 and Supplemental Figure 2). The patients with K700E had no survival advantage over the patients without K700E (52.77months vs. 39.03 months, P=0.174) (Figure 3). Furthermore, there was no difference in OS of patients with SF3B1 K700E mutation compared with SF3B1 K666N mutation (52.77 months vs. 29.63 months, P = 0.075) (Supplemental Figure 2).
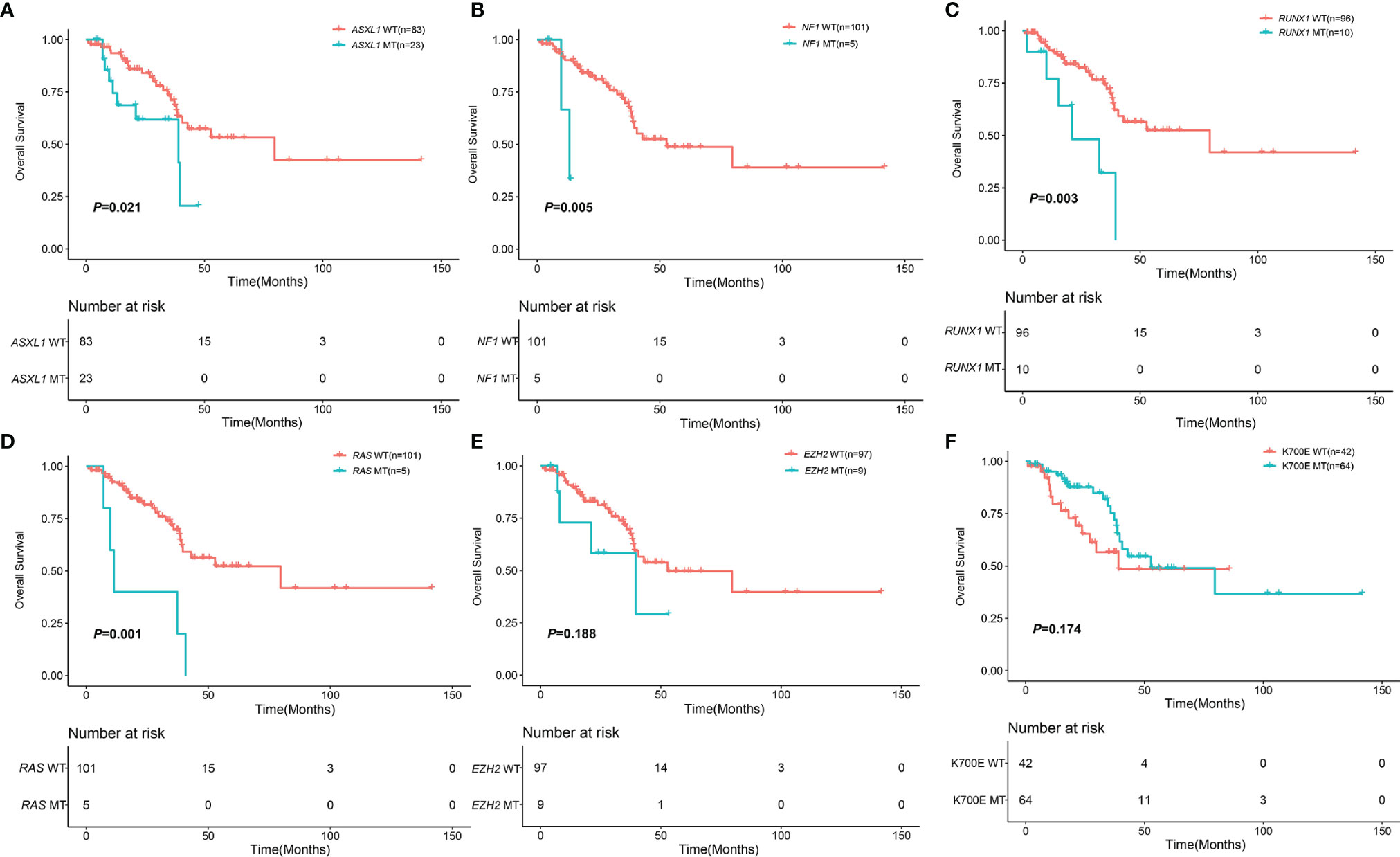
Figure 3 Impact of mutations on OS in 106 MDS patents with SF3B1 mutation. (A) Kaplan-Meier curves comparing the OS of patients with ASXL1 mutation (blue) compared with wild type (red) (39.03 months vs. 79.60 months, P = 0.021). (B) Kaplan-Meier curves comparing the OS of patients with NF1 mutation (blue) compared with wild type (red) (13.17 months vs. 52.77 months, P = 0.005). (C) Kaplan-Meier curves comparing the OS of patients with RUNX1 mutation (blue) compared with wild type (red) (21.03 months vs. 79.60 months, P = 0.003). (D) Kaplan-Meier curves comparing the OS of patients with KRAS/NRAS mutation (blue) compared with wild type (red) (11.33 months vs. 79.60 months, P = 0.001). (E) Kaplan-Meier curves comparing the OS of patients with EZH2 mutation (blue) compared with wild type (red) (39.57 months vs. 52.77 months, P = 0.188). (F) Kaplan-Meier curves comparing the OS of patients with SF3B1 K700E mutation (blue) compared with non SF3B1 K700E (red) (52.77 months vs. 39.03 months, P = 0.174).
Prognostic Scoring Model of MDS Patients With SF3B1 Mutations
To explore the prognostic factors of MDS patients with SF3B1 mutation, we regarded MDS patients with SF3B1 mutation in Zhejiang Province as the experimental cohort (n=106) and MDS patients with SF3B1 mutation from GSE58831 database (23) as the validation cohort (n=32). A comparison of the basic characteristics of the patients in the experimental cohort and the validation cohort was listed in Table 2.
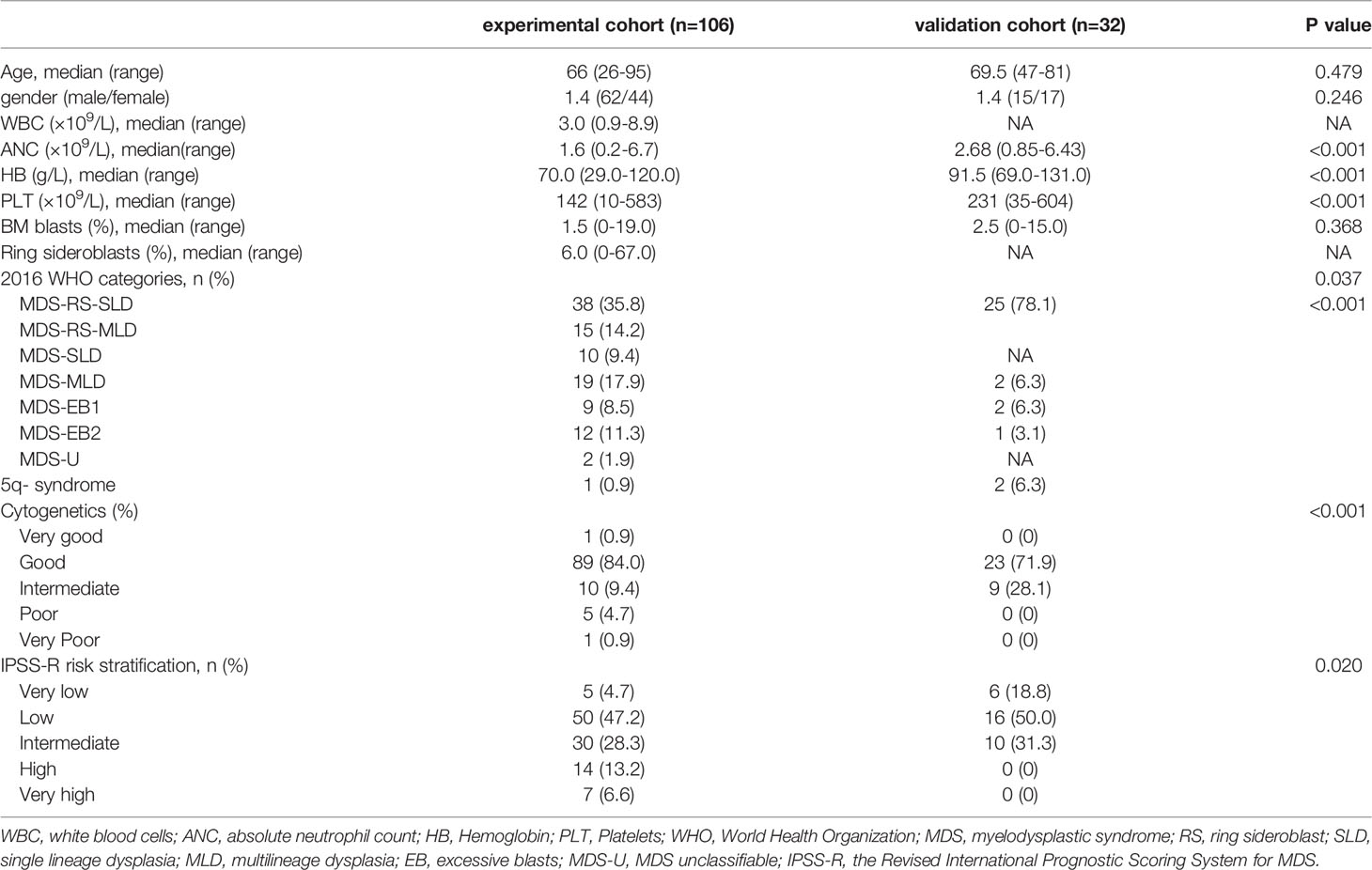
Table 2 Comparison of basic characteristics of patients in the experimental cohort and the verification cohort.
Prognostic factors with P < 0.2 in the univariate analysis were performed to develop the prognostic scoring system and the results of univariate analysis were listed in Supplemental Table 1. Five variables were incorporated in the novel scoring model using LASSO Cox regression model. The risk scoring model was constructed including the weighted coefficients of these variables: IPSS-R×0.4+RUNX1×1.1+EZH2×0.6+RAS×0.9+NF1×1.6 (IPSS-R scored as regular; RUNX1 mutation scored 1; EZH2 mutation scored 1; KRAS/NRAS mutation scored 1; NF1 mutation scored 1; 0 for other conditions). In the experimental cohort, 106 patients were classified into two subgroups on the basis of the risk score: low-risk (L-R, score <= 1.4, n = 60) and high risk (H-R, score > 1.4, n =46) groups. The 3-year OS for the L-R and H-R groups was 91.88% (95% CI, 83.27%-100%) and 38.14% (95% CI, 24.08%-60.40%), respectively (P<0.001) (Figure 4). In the validation cohort, the 3-year OS for the L-R and H-R groups was 88.54% (95% CI, 74.77%-100%) and 50.0% (95% CI, 18.77%-100%), respectively (P =0.052) (Figure 4). A prognostic nomogram that integrated all the five significantly independent variables from the LASSO Cox regression model was constructed (Figure 4). The nomogram was externally verified in the validation cohort. The predictive accuracy of the prognostic scoring model for OS in the experimental cohort evaluated with the C-index was 0.799 (95% CI, 0.764-0.834) which was higher than the C-index [0.765 (95% CI, 0.726-0.804)] of IPSS-R. Likewise, the C-index score [0.781 (95% CI, 0.715-0.847)] of the prognostic scoring model in the validation cohort was higher than the C-index [0.754 (95% CI, 0.687-0.821)] of IPSS-R. The calibration curves for predicting OS of patients after 3 years indicated an excellent conformity between the nomogram-predicted and actually observed values (Figures 5A–C).
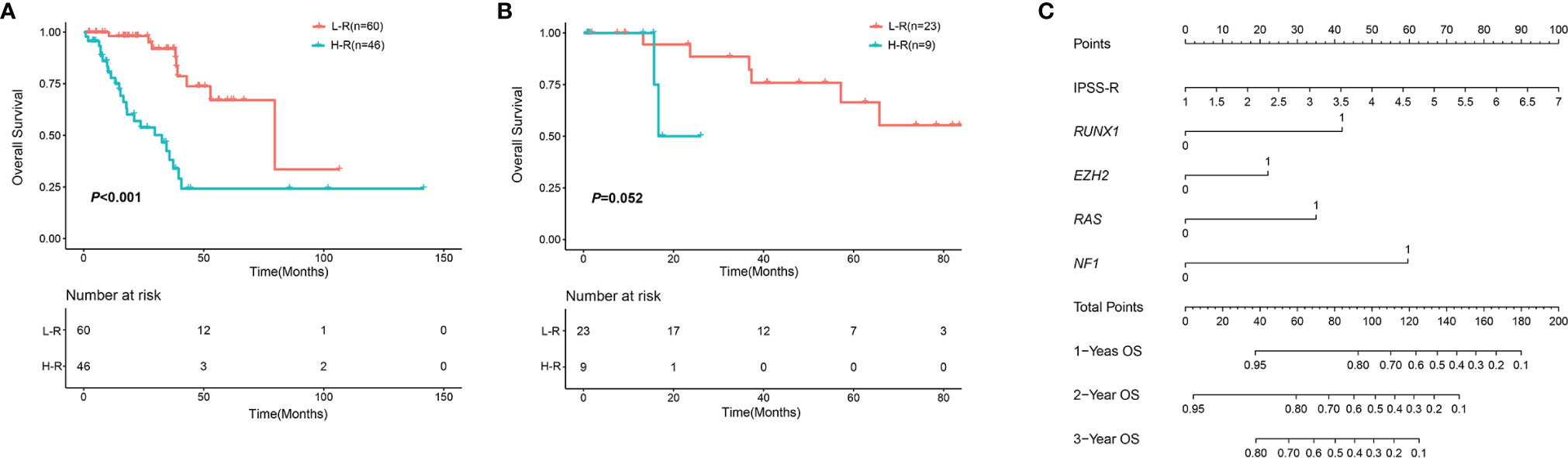
Figure 4 OS in different risk groups according to the novel scoring model. (A) OS in the experimental cohort. (B) OS in the validation cohort. (C) Nomogram for MDS patients with SF3B1 mutation. An individual’s value is located on each variable axis, and a line is drawn upward to determine the points received for each variable. Corresponding points for each variable: IPSS-R scored as regular; RUNX1 mutation scored 1; EZH2 mutation scored 1; KRAS/NRAS mutation scored 1; NF1 mutation scored 1; 0 for other conditions.
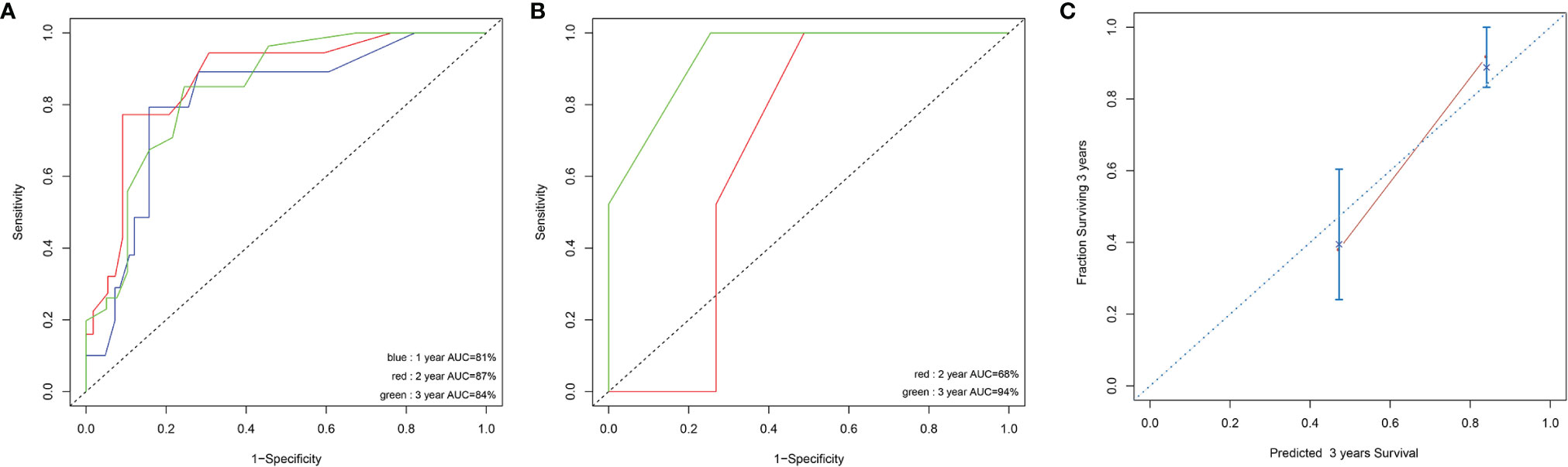
Figure 5 Discrimination ability with the use of the receiver operating characteristic curve in the experimental cohort (A) and validation cohort (B). Calibration curves for predicting OS of MDS patients at 3 years in experimental cohort (C). The sum of these points is located on the total point axis, and a line is drawn downward to the survival axis to determine the likelihood of 1,2, or 3-year OS.
Discussion
In this study, using a NGS platform we explored the mutation profile in MDS patients with SF3B1 mutations. We discovered that MDS patients with SF3B1 mutation had many coexisting gene mutations, and the interactions were very complicated. Meanwhile, we found RUNX1, EZH2, NF1 and KRAS/NRAS mutations had significant effects on prognosis. Based on these results, we proposed a scoring model combining both clinical features and gene mutations to predict outcomes in MDS patients with SF3B1 mutation. Our proposed model distinctly surpassed the widely used IPSS-R. Our study might help to investigate the risk stratification and prognostic prediction, make reasonable decision and select appropriate therapies in SF3B1 mutated MDS patients.
In accordance with previous reports (19, 24), more than half of MDS patients with SF3B1 mutation were diagnosed as MDS-RS. The majority were categorized into good karyotype risk group and lower risk group according to IPSS-R. K700E was the most common mutation site of SF3B1 in our study. But the patients with K700E had no survival advantage over the patients without K700E, which was inconsistent with the results from Rashmi KS, et al. showing that SF3B1 mutated MDS with K700E had a remarkably better OS in contrast to non-K700E mutations (25).
As for the co-mutant genes, clonal hematopoiesis-associated mutations including TET2, ASXL1 and DNMT3A were the most common co-mutant genes in the MDS patients with SF3B1 mutations. However, RUNX1, EZH2, NF1 and KRAS/NRAS mutations had significant effects on OS in our prognostic model, which coincided with the previous study (19) showing that RUNX1, EZH2 and NF1 mutations had significant effects on OS in SF3B1-mutant MDS patients within the IWG dataset.
The RUNX1 transcription factor is a pivotal regulator of embryogenesis and hematopoiesis in vertebrates (23). RUNX1 mutation is frequent in higher risk MDS such as MDS-MLD and MDS-EB. Furthermore, RUNX1 mutation is correlated with poor clinical outcomes, particularly higher probability and shorter period for progression to AML (26, 27). The MDS patients with RUNX1 mutation also have shorter OS (28, 29). Confirmed in our study, RUNX1 mutation is an independent predictive factor of poor survival in MDS patients with SF3B1 mutation (16, 30).
EZH2, located at chromosome 7q36, encodes for the catalytic subunit of the PRC2, which retains H3K27 methyltransferase activity. Inactivating mutation of EZH2 is also found in MDS, which resulted in down-regulation of its expression (31–33). Deletion of Ezh2 in mice leads to MDS/MPN-like diseases, thus confirming the role of EZH2 deficiency in disease development (31–34). In our study, although EZH2 mutation had no impact on OS in univariate analysis (P=0.188), it showed significance in the multivariate analysis. Consistent with our study, EZH2 mutation is also an independent predictive factor of poor survival in SF3B1 mutated MDS (19).
This study had some limitations. First, not all the samples from SF3B1 mutated patients were analyzed through NGS. Second, the number of patients in the verification cohort was relatively small. Therefore, a larger sample size study will be needed to verify our results.
In summary, we performed multi-gene sequencing and comprehensive prognostic analysis in MDS patients with SF3B1 mutation. Our study pointed out the SF3B1mutation profile, revealed a novel scoring model combining both genetic and clinical outcomes that could stratify patients into two subgroups with distinct clinical outcomes, which play an important role in improving accuracy of prediction.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: https://db.cngb.org/search/project/CNP0003053/.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University. The ethics committee waived the requirement of written informed consent for participation. Written informed consent was not obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Authors Contributions
LM designed the research study and draft the manuscript. WY, BL, HH, GO, SQ, SL, LC, KL, YK, LS, WMJ, JPL, XNY, JL, HL, JF, LF, WHJ, ZZ, YL, LF, CS and XFY collected the data. LL and JQ performed the statistical analysis and drew diagrams. WY and SJ completed the follow-up. JJ and HT reviewed and revised the paper. All authors read and approved the final manuscript.
Conflict of Interest
Authors LL and JQ are employed by Acornmed Biotechnology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.905490/full#supplementary-material
Supplementary Figure 1 | Categories of the co-mutant genes by function.
Supplementary Figure 2 | Impact of mutations on OS in 106 MDS patents with SF3B1 mutation based on different mutated genes or mutation sites, including (A) Kaplan-Meier curves comparing the OS of patients with TET2 mutation (blue) compared with wild type (red) (79.60 months vs. 40.67 months, P = 0.268). (B) Kaplan-Meier curves comparing the OS of patients with DNMT3A mutation (blue) compared with wild type (red) (42.93 months vs. 79.60 months, P = 0.253). (C) Kaplan-Meier curves comparing the OS of patients with BCOR mutation (blue) compared with wild type (red) (35.73 months vs. 52.77 months, P = 0.785). (D) Kaplan-Meier curves comparing the OS of patients with KMT2D mutation (blue) compared with wild type (red) (not reach vs. 42.93 months, P = 0.523). (E) Kaplan-Meier curves comparing the OS of patients with ATRX mutation (blue) compared with wild type (red) (not reach vs. 42.93 months, P = 0.535). (F) Kaplan-Meier curves comparing the OS of patients with SETBP1 mutation (blue) compared with wild type (red) (not reach vs. 42.93 months, P = 0.572). (G) Kaplan-Meier curves comparing the OS of patients with TP53 mutation (blue) compared with wild type (red) (not reach vs. 52.77 months, P = 0.971) (H) Kaplan-Meier curves comparing the OS of patients with IDH1/2 mutation (blue) compared with wild type (red) (29.63 months vs. 52.77 months, P = 0.530). (I) Kaplan-Meier curves comparing the OS of patients with SF3B1 K700E mutation (blue) compared with SF3B1 K666N mutation (red) (52.77 months vs. 29.63 months, P = 0.075).
Abbreviations
MDS, myelodysplastic syndrome; MDS-RS, MDS with ring sideroblasts; MDS-SLD, MDS with single lineage dysplasia; MDS- MLD, MDS with multiple lineage dysplasia; MDS-EB, MDS with excess blasts; MDS-U, MDS-unclassifiable; NGS, next-generation sequencing; SF3B1, splicing factor 3 subunit 1; RUNX1, Runt-related transcription factor 1; EZH2, Enhancer of zeste homolog 2; WHO, World Health Organization; IPSS-R, the revised International Prognostic Scoring System; IPSS, the International Prognostic Scoring System; IWG-PM, the International Working Group for the Prognosis of MDS; ISCN, International System for Human Cytogenetic Nomenclature; OS, Overall survival; LASSO, the least Absolute Shrinkage and Selector Operation; ESA, erythroid stimulating agents; HMAs, hypomethylating agents; AML, Acute myeloid leukemia; VAF, variant allele fraction.
References
1. Voso MT, Gurnari C. Have We Reached a Molecular Era in Myelodysplastic Syndromes? Hematol Am Soc Hematol Educ Program (2021) 2021(1):418–27. doi: 10.1182/hematology.2021000276
2. Cazzola M. Myelodysplastic Syndromes. N Engl J Med (2020) 383(14):1358–74. doi: 10.1056/NEJMra1904794
3. Isono K, Abe K, Tomaru Y, Okazaki Y, Hayashizaki Y, Koseki H. Molecular Cloning, Genetic Mapping, and Expression of the Mouse Sf3b1 (SAP155) Gene for the U2 snRNP Component of Spliceosome. Mamm Genome (2001) 12:192–8. doi: 10.1007/s003350010258
4. Gozani O, Potashkin J, Reed R. A Potential Role for U2AF-SAP 155 Interactions in Recruiting U2 snRNP to the Branch Site. Mol Cell Biol (1998) 18:4752–60. doi: 10.1128/MCB.18.8.4752
5. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 Mutation in Myelodysplasia With Ring Sideroblasts. N Engl J Med (2011) 365:1384–95. doi: 10.1056/NEJMoa1103283
6. Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-Associated SF3B1 Mutations Affect Alternative Splicing by Promoting Alternative Branchpoint Usage. Nat Commun (2016) 7:106–15. doi: 10.1038/ncomms10615
7. Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection Through Use of a Different Branch Point. Cell Rep (2015) 13:1033–45. doi: 10.1016/j.celrep.2015.09.053
8. Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, et al. Clinical Significance of SF3B1 Mutations in Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative Neoplasms. Blood (2011) 118:6239–46. doi: 10.1182/blood-2011-09-377275
9. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature (2011) 478:64–9. doi: 10.1038/nature10496
10. Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, et al. SF3B1 Mutations are Prevalent in Myelodysplastic Syndromes With Ring Sideroblasts But do Not Hold Independent Prognostic Value. Blood (2012) 119:569–72. doi: 10.1182/blood-2011-09-377994
11. Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, et al. SF3B1, a Splicing Factor is Frequently Mutated in Refractory Anemia With Ring Sideroblasts. Leukemia (2012) 26:542–5. doi: 10.1038/leu.2011.232
12. Mian SA, Rouault-Pierre K, Smith AE, Seidl T, Pizzitola I, Kizilors A, et al. SF3B1 Mutant MDS-Initiating Cells may Arise From the Haematopoietic Stem Cell Compartment. Nat Commun (2015) 6:10004. doi: 10.1038/ncomms10004
13. Malcovati L, Karimi M, Papaemmanuil E. Et Al, SF3B1 Mutation Identifies a Distinct Subset of Myelodysplastic Syndrome With Ring Sideroblasts. Blood (2015) 126:233–41. doi: 10.1182/blood-2015-03-633537
14. Mian SA, Smith AE, Kulasekararaj AG, Kizilors A, Mohamedali AM, Lea NC, et al. Spliceosome Mutations Exhibit Specific Associations With Epigenetic Modifiers and Proto-Oncogenes Mutated in Myelodysplastic Syndrome. Haematologica (2013) 98:1058–66. doi: 10.3324/haematol.2012.075325
15. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of Genetic Lesions in 944 Patients With Myelodysplastic Syndromes. Leukemia (2014) 28:241–7. doi: 10.1038/leu.2013.336
16. Papaemmanuil E, Gerstungm M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood (2013) 122:3616–27. doi: 10.1182/blood-2013-08-518886
17. Brück OE, Lallukka-Brück SE, Hohtari HR, Ianevski A, Ebeling FT, Kovanen PE, et al. Machine Learning of Bone Marrow Histopathology Identifies Genetic and Clinical Determinants in Patients With MDS. Blood Cancer Discovery (2021) 2(3):238–49. doi: 10.1158/2643-3230.BCD-20-0162
18. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood (2012) 120(12):2454–65. doi: 10.1182/blood-2012-03-420489
19. Malcovati L, Stevenson K, Papaemmanuil E, Neuberg D, Bejar R, Boultwood J, et al. SF3B1-Mutant MDS as a Distinct Disease Subtype: A Proposal From the International Working Group for the Prognosis of MDS. Blood (2020) 136:157–70. doi: 10.1182/blood.2020004850
20. Komrokji R, Volpe V, Chan O, Al Ali N, Swoboda D, Kuykendall A, et al. Validation of the International Working Group Proposal for SF3B1 Mutant Myelodysplastic Syndromes. Blood (2021) 138(11):989–92. doi: 10.1182/blood.2021010831
21. Cazzola M. Introduction to a Review Series: The 2016 Revision of the WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Blood (2016) 127(20):2361–4. doi: 10.1182/blood-2016-03-657379
22. McGowan-Jordan J, Simons A, Schmid M. An International System for Human Cytogenomic Nomenclature. Basel: Karger (2016).
23. Gerstung M, Pellagatti A, Malcovati L, Giagounidis A, Porta MG, Jädersten M, et al. Combining Gene Mutation With Gene Expression Data Improves Outcome Prediction in Myelodysplastic Syndromes. Nat Commun (2015) 6:5901. doi: 10.1038/ncomms6901
24. Ma L, Luo Y, Jiang L, Shen D, Li J, Xu W, et al. The Relation of SF3B1 Mutation and Intracellular Iron in Myelodysplastic Syndrome With Less Than 5% Bone Marrow Blasts. Leuk Lymphoma (2019) 60(5):1179–86. doi: 10.1080/10428194.2018.1520990
25. Kanagal-Shamanna R, Montalban-Bravo G, Sasaki K, Darbaniyan F, Jabbour E, Bueso-Ramos C, et al. Only SF3B1 Mutation Involving K700E (And Not Other Codons), Independently Predicts Overall Survival in Myelodysplastic Syndromes. Cancer (2021) 127(19):3552–65. doi: 10.1002/cncr.33745
26. Harada H, Harad Y. Recent Advances in Myelodysplastic Syndromes: Molecular Pathogenesis and its Implications for Targeted Therapies. Cancer Sci (2015) 106:329–36. doi: 10.1111/cas.12614
27. Tsai SC, Shih LY, Liang ST, Huang YJ, Kuo MC, Huang CF, et al. Biological Activities of RUNX1 Mutants Predict Secondary Acute Leukemia Transformation From Chronic Myelomonocytic Leukemia and Myelodysplastic Syndromes. Clin Cancer Res (2015) 21:3541–51. doi: 10.1158/1078-0432.CCR-14-2203
28. Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, et al. Validation of a Prognostic Model and the Impact of Mutations in Patients With Lower-Risk Myelodysplastic Syndromes. J Clin Oncol (2012) 30:3376–82. doi: 10.1200/JCO.2011.40.7379
29. Chen CY, Lin LI, Tang JL, Ko BS, Tsay W, Chou WC, et al. RUNX1 Gene Mutation in Primary Myelodysplastic Syndrome–the Mutation can be Detected Early at Diagnosis or Acquired During Disease Progression and is Associated With Poor Outcome. Br J Haematol (2007) 139:405–14. doi: 10.1111/j.1365-2141.2007.06811.x
30. Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell (2016) 30(3):404–17. doi: 10.1016/j.ccell.2016.08.006
31. Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, et al. Concurrent Loss of Ezh2 and Tet2 Cooperates in the Pathogenesis of Myelodysplastic Disorders. J Exp Med (2013) 210:2627–39. doi: 10.1084/jem.20131144
32. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating Mutations of the Histone Methyltransferase Gene EZH2 in Myeloid Disorders. Nat Genet (2010) 42:722–6. doi: 10.1038/ng.621
33. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tönnissen ER, et al. Somatic Mutations of the Histone Methyltransferase Gene EZH2 in Myelodysplastic Syndromes. Nat Genet (2010) 42:665–7. doi: 10.1038/ng.620
Keywords: Myelodysplastic syndrome (MDS), SF3B1 mutation, RUNX1, EZH2, Ras, prognostic scoring model
Citation: Ma L, Liang B, Hu H, Yang W, Lin S, Cao L, Li K, Kuang Y, Shou L, Jin W, Lan J, Ye X, Le J, Lei H, Fu J, Lin Y, Jiang W, Zheng Z, Jiang S, Fu L, Su C, Yin XF, Liu L, Qin J, Jin J, Qian S, Ouyang G and Tong H (2022) A Novel Prognostic Scoring Model for Myelodysplastic Syndrome Patients With SF3B1 Mutation. Front. Oncol. 12:905490. doi: 10.3389/fonc.2022.905490
Received: 27 March 2022; Accepted: 27 May 2022;
Published: 27 June 2022.
Edited by:
Cyrus Khandanpour, University Hospital Münster, GermanyReviewed by:
Leonor Arenillas, Hospital del Mar, SpainCarmelo Gurnari, Cleveland Clinic, United States
Copyright © 2022 Ma, Liang, Hu, Yang, Lin, Cao, Li, Kuang, Shou, Jin, Lan, Ye, Le, Lei, Fu, Lin, Jiang, Zheng, Jiang, Fu, Su, Yin, Liu, Qin, Jin, Qian, Ouyang and Tong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyan Tong, tonghongyan@zju.edu.cn; Guifang Ouyang, nbougf@163.com; Shenxian Qian, sxqian@hotmail.com
†These authors have contributed equally to this work and share first authorship