 Lucia De Martino1†
Lucia De Martino1† Donatella Capalbo2†
Donatella Capalbo2† Nicola Improda1Paola Lorello1Carla Ungaro2
Nicola Improda1Paola Lorello1Carla Ungaro2 Raffaella Di Mase2Emilia Cirillo1
Raffaella Di Mase2Emilia Cirillo1 Claudio Pignata1
Claudio Pignata1 Mariacarolina Salerno1*
Mariacarolina Salerno1*
- 1Pediatric Section, Department of Translational Medical Sciences, Federico II University, Naples, Italy
- 2Department of Pediatrics, Federico II University, Naples, Italy
Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED), formerly known as autoimmune polyendocrine syndrome type 1, is a paradigm of a monogenic autoimmune disease caused by mutations of a gene, named autoimmune regulator (AIRE). AIRE acts as a transcription regulator that promotes immunological central tolerance by inducing the ectopic thymic expression of many tissue-specific antigens. Although the syndrome is a monogenic disease, it is characterized by a wide variability of the clinical expression with no significant correlation between genotype and phenotype. Indeed, many aspects regarding the exact role of AIRE and APECED pathogenesis still remain unraveled. In the last decades, several studies in APECED and in its mouse experimental counterpart have revealed new insights on how immune system learns self-tolerance. Moreover, novel interesting findings have extended our understanding of AIRE’s function and regulation thus improving our knowledge on the pathogenesis of APECED. In this review, we will summarize recent novelties on molecular mechanisms underlying the development of APECED and their clinical implications.
Introduction
Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED), formerly known as autoimmune polyendocrine syndrome type 1 (APS-1), is a rare disease caused by mutations of the autoimmune regulator (AIRE) which acts as a transcription regulator that promotes immunological central tolerance (1).
Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy represents a paradigm of genetically determined systemic autoimmunity. However, the great variability that characterizes APECED, irrespectively of AIRE genotype, implies that additional factors modulate the clinical expression of the disease.
Recent advances on how AIRE affects immunological tolerance and is linked to organ-specific autoimmunity have improved our understanding on the pathogenesis and the wide variability of clinical expression of APECED.
In this review, we will summarize new insights into AIRE genetics and functioning and its implications on APECED phenotype.
New Insights into Aire Function
Autoimmune regulator is known to exert a crucial role in central tolerance and negative selection of autoreactive T cells (1). The induction of central tolerance is an intricate process that occurs within the thymus where immature T lymphocytes are “committed” to become mature cells able to respond to a huge number of foreign antigens, but preventing autoimmune reactions. Medullary thymic epithelial cells (mTECs) have a primary role in the negative selection and, in this context, AIRE acts as a crucial transcriptional regulator. In mTECs, AIRE induces promiscuous gene expression (pGE) of tissue-specific antigens (TSAs), which are, then, presented to maturing T cells. Autoreactive T cells that recognize these TSAs with high affinity undergo negative selection through their apoptosis or, alternatively, regulatory T cells (Treg) are generated in order to prevent autoimmunity (2, 3).
Autoimmune regulator gene encodes a 545 amino acid protein with a molecular weight of 58 kDa (1). Starting from the amino terminus, AIRE is composed of a caspase recruitment domain (CARD)/homogeneously staining region (HSR), nuclear localization sequences (NLS), a SAND (Sp100, AIRE NucP41/75, and DEAF) domain, two planthomeodomain (PHD) zinc fingers, a proline-rich region (PRR), and four LXXLL motifs (where L stays for leucine) distributed among the domains (4). The CARD/HSR is involved in the process of AIRE homomultimerization and seems also to anchor AIRE to the chromatin (4, 5). The NLS has a stretch of basic amino acids at positions 131–133 important for nuclear import (4). The SAND domain does not have a distinct DNA-binding motif, but it is involved in promoting a protein–protein interaction with a transcriptional repressive complex (6). The two AIRE PHD fingers form a structural system for the recruitment of chromatin-related proteins and are engaged in AIRE transcriptional activity (7–9). LXXLL motif and PRR are implicated in promoting gene transcription (4).
Autoimmune regulator has a strict spatiotemporal regulation, being ubiquitously transcribed during the earliest stages of embryogenesis, and then restricted to thymic cells (mTECs and B cells) and extra thymic hematopoietic stem cells that may have a role in CD4 tolerization (10, 11).
At the transcriptional level, the expression of AIRE in mTECs and peripheral lymphoid organs is regulated by receptor activator of nuclear factor κB (RANK) signaling and therefore by nuclear factor κB (NF-κB)-induced transcription through an upstream conserved non-coding sequences (CNSs) of the Aire gene containing NF-κB-binding sites (12, 13). In addition, post-transcriptional mechanisms seem to modulate AIRE expression. A dioxygenase that catalyzes lysyl hydroxylation of splicing regulatory proteins (Jmjd6) is critical for AIRE expression. In fact, the intron 2 of Aire gene is not effectively spliced out in the absence of Jmjd6, resulting in marked reduction of mature Aire protein in mTECs and spontaneous development of multi-organ autoimmunity in mice (14).
The AIRE protein resides inside the nucleus, where it exhibits a speckled localization pattern (15). AIRE is a key regulator of TSA expression in mTECs and affects the transcription of thousands of TSA genes in a “stochastic” and “ordered” manner (16, 17). Indeed, a small percentage (1–3%) of the total number of mTECs expresses a particular TSA (18). Different sets of TSAs are regulated by AIRE within individual mTECs but whether a particular AIRE-regulated TSA is expressed in a given mTEC seems to be highly probabilistic (18, 19). Moreover, the “ordered” TSA expression refers to the increased likelihood that a particular set of TSA genes will be coexpressed in an individual mTEC (20) (Figure 1).
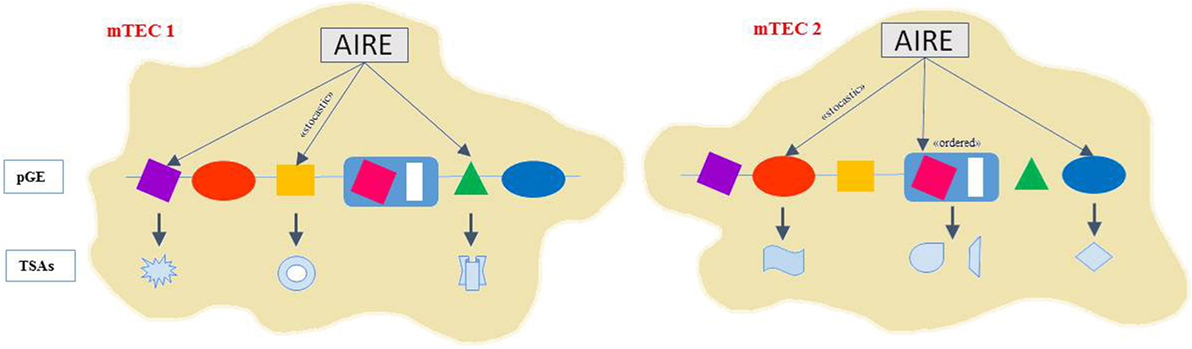
Figure 1. AIRE controls gene expression with ordered stochasticity. AIRE seems to regulate pGE in mTECs in an apparently stochastic manner. Thus, single mTECs would express TRAs of mixed tissue origin rather than emulating cell line age-affiliated patterns displaying the highest degree and diversity of pGE. Indeed, different sets of TSAs are expressed in mTECs but whether a particular AIRE-regulated TSA is expressed in a given mTEC seems to be highly probabilistic. The “ordered” TSA expression refers to the increased likelihood that a particular set of TSA genes will be coexpressed in an individual mTEC. Coexpressed gene loci tend to colocalize to the same nuclear subdomain and TSA subsets align along progressive differentiation stages within the mature mTEC subset.
Autoimmune regulator acts in a very unusual way among transcription regulators, as it has no clear DNA-binding motif but seems to recognize genes that possess silenced chromatin states (6, 8, 9, 16). AIRE does not directly initiate TSA gene transcription, but it promotes TSA expression through the release of stalled RNA polymerase, RNA elongation, and splicing of target TSAs (15, 21). Moreover, AIRE binds to several partners that have the potential for post-translational protein modification, including the modification of AIRE itself and that seem to be critical for its biological function (22–24).
Recent insights on AIRE’s regulation come from experimental studies which suggest that estrogen induces epigenetic changes in the AIRE gene, leading to reduced AIRE expression under a threshold that increases susceptibility to autoimmune diseases (25).
In summary, induction of pGE by AIRE is dependent on a complex regulatory mechanism which has only been partially unraveled so far.
In addition to the key role exerted on pGE, AIRE seems also to be critical for thymic generation of Treg cells during the perinatal period (3, 26). However, on this issue, further work is needed (3).
Moreover, recently a new hypothesis on Aire functioning in tolerance has been postulated. Aire may enforce immune tolerance by ensuring that autoreactive T cells differentiate into the Treg cell lineage; dysregulation of this process results in the diversion of Treg cell-biased clonotypes into pathogenic conventional T cells (27).
Furthermore, AIRE has several functions that are independent of its promotion of TSA expression in mTECs such as immunoregulatory functions in extrathymic AIRE-expressing cells and thymic B cells (15, 28). Moreover, AIRE enhances negative selection by regulating the repertoire of thymic dendritic cells and promoting apoptosis of mTECs (29, 30).
Finally, it has been postulated that AIRE regulates thymic maturation and architecture, probably through the expression of microRNAs (15, 31–34).
In summary, although our knowledge has increased in recent years, we still lack a coherent model incorporating and explaining all the intricacies of AIRE and its role in the regulation of immunological tolerance.
New Insights into Aire Mutations
In humans, AIRE, identified on chromosome 21q22.3 by positional cloning in 1997, consists of 14 exons spanning 11.9 kb of genomic DNA (15). Mutations in the AIRE gene result in the development of APECED, a rare autoimmune condition, but reported worldwide, with a higher prevalence in genetically isolated populations (1).
Nowadays, 101 APECED-causing mutations have been found throughout AIRE (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=AIRE). These mutations include nonsense/missense mutations, deletions, or insertions and often abolish AIRE transcriptional activity or its localization to nuclear bodies (15, 35).
Despite its monogenic nature, APECED is characterized by a wide variability of the clinical expression and no strong genotype–phenotype correlation has been found among several populations (1, 35). Noteworthy, this lack is exemplified by the significant intrafamilial differences even between siblings carrying the same mutation, suggesting that disease-modifying genes, environmental factors, and immune system dynamics may play a role in modulating clinical expression of the syndrome (36, 37).
Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy has been originally considered an autosomal–recessive disease, and most mutations were assumed to be inherited in an autosomal–recessive manner, except for one mutation in the SAND domain, p.G228W, which exerts a dominant inheritance pattern (38).
However, recent evidences highlight that also heterozygous mutations of the gene can be associated with increased susceptibility to autoimmune diseases or incomplete forms of APS-1 (39). Patients with atypical or incomplete manifestations of APECED or with other immune diseases carrying heterozygous mutations of AIRE have been also described (38, 40–49). Cervato et al. showed different AIRE mutations in heterozygous state in relatives of APECED patients with various degrees of autoimmune or non-autoimmune diseases, but none of which affected by one of the major components of APECED (50).
Recently, Oftedal et al. reported a group of novel monoallelic and dominant-negative AIRE mutations clustered within the first PHD1 zinc finger domain in patients with various degrees of autoimmunity (39). The PHD1 domain is critical for AIRE’s transcription–transactivation activity and mutations in this domain seems to affect the structure and thus the function of the entire AIRE tetramer. However, the significance of these monoallelic mutations is still unclear since the same alterations were found in varying autoimmune phenotypes, ranging from milder phenotypes of late-onset APECED to autoimmune polyglandular syndrome type 2 (APS-2) and isolated organ-specific autoimmunity following incomplete inheritance. A possible explanation is that AIRE tetramers still have some residual activity sufficient to ensure partial self-tolerance. Moreover, PHD1 mutations scanned in a public databases revealed an estimated frequency of about 0.0008, which is in the range of several autoimmune conditions that affect about 1 in 1,000 people, thus suggesting that mutations in AIRE might be more widespread in patients with autoimmunity than previously thought (39).
Moreover, Sparks et al. identified additional dominant-negative AIRE mutations associated with the modulation of insulin gene expression in thymus which is essential to induce either insulin tolerance or the development of insulin autoimmunity and type 1 diabetes (51).
Apeced: From “Classical” to “Non-Classical” Phenotype
In the light of these new knowledges, the original classification of APECED as unique autosomal–recessive disease seems to be incomplete. Taking into account the huge spectrum of phenotypes related to AIRE mutations, Oftedal et al. interestingly proposed to differentiate APECED in two major forms: (1) “classical APECED,” characterized by recessive inheritance, presence of at least two of the three main components, and interferon (IFN) antibodies; and (2) “non-classical APECED,” characterized by dominant heterozygous mutations mainly in AIRE’s PHD1 zinc finger and a milder, less penetrant autoimmune phenotype (39) (Table 1).

Table 1. APECED in “classical” and “non-classical” forms.
Classical diagnosis of APECED has been originally defined by the presence of two of the three most common features: chronic mucocutaneous candidiasis (CMC), chronic hypoparathyroidism (CH), and Addison’s disease (AD) (52).
Neutralizing autoantibodies against type 1 IFN (especially IFN-ω and IFN-α) have been found to strictly correlate with AIRE deficiency, thus leading to consider these autoantibodies as a precocious diagnostic tool for APECED and an additional diagnostic criteria for the diagnosis of APECED (52, 53). However, IFN autoantibodies seem to be less prevalent in the “non-classical” form, probably reflecting some residual AIRE function (39).
Molecular analysis of Aire may help to confirm the clinical diagnosis, especially in those cases with an atypical presentation.
Both “classical” and “non-classical” phenotypes are characterized by a wide heterogeneity in the severity and in the number of components among affected subjects with a wide variability even between siblings with the same genotype (39, 54).
Chronic mucocutaneous candidiasis is the first sign to appear followed by CH, before the age of 10 years, and later by adrenal insufficiency. However, a precise chronological order is not always present (52).
In addition to the classic triad (CMC, CH, and AD), the phenotype of APECED includes several autoimmune manifestations, which in some cases may also precede the classical triad (52).
The spectrum of endocrinopathies associated with APECED includes hypergonadotropic hypogonadism, type 1 diabetes (T1D), autoimmune thyroid diseases (ATD), growth hormone (GH) deficiency, and other pituitary defects (52).
The appearance of ectodermal abnormalities is also quite common including dental enamel hypoplasia, pitted nail dystrophy, and alopecia. Keratopathy, vitiligo, calcifications of the tympanic membranes, and periodic maculopapular, morbilliform, or urticarial rash with fever (52) are also included in the clinical spectrum of APECED.
Furthermore, gastrointestinal autoimmunity in APECED may lead to autoimmune gastritis, autoimmune hepatitis (AIH), intestinal disorders with chronic diarrhea alternating with obstipation, and cholelitiasis (54).
Asplenia, tubulointerstitial nephritis, interstistial lung disease (ILD), vasculitis, Sjogren’s syndrome, cutaneous vasculitis, hemolytic anemia, scleroderma, metaphyseal dysplasia, and celiac disease have also been reported in APECED (55–58). Recently, a diagnosis of APECED was established by performing whole exome sequencing in a patient with increased renal echogenicity on renal ultrasound (59).
Muscle disease, with very similar clinical features of progressive limb-girdle myopathy, is a rare component of APECED (60).
To date, two patients with APECED have been affected by encephalitis leading to a severe and life-threatening condition (61, 62).
The “non-classical” form of APECED has been suggested to be characterized by variable autoimmune phenotypes, ranging from late-onset APECED to different combinations of autoimmune manifestations (APS-2), isolated organ-specific autoimmunity or autoantibodies, but no signs of autoimmune disease within individuals who harbor monoallelic AIRE mutations. In particular, families with vitamin B12 deficiency, pernicious anemia, and/or vitiligo at early age have been found to carry heterozygous PHD1 mutations, although the clinical phenotype has been expanded when larger materials were investigated. Indeed, organ-specific autoimmunity in the heterozygous cases seems to present in milder form and incomplete penetrance with respect to classical (39). These observations open a new window on the possibility that mutation carriers have a risk for developing some degree of APECED or other form of polyendocrinopathy.
However, more research is needed to determine the contributions of such AIRE variants to autoimmune susceptibility, especially in kindreds with a strong family history of autoimmunity.
In either “classical” or “non-classical” form of APECED, early diagnosis and regular surveillance, including periodic evaluation of hormonal and biochemical parameters, are essential to allow the prevention of severe and life-threatening events (i.e., hypocalcemia, adrenal crisis), even in the absence of clinical symptoms (63).
Old and New Autoantibodies
Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy features multi-organ autoimmunity and autoantibody responses against target molecules with restricted tissue expression profiles. Consequently, autoantibody markers have acquired a central role in research and clinical diagnosis of APECED, providing a tool for diagnosis, and as predicting factor for the clinical course of the disease.
Chronic mucocutaneous candidiasis is a sign of the underlying immunodeficiency. Although the pathogenesis of CMC seems to be different from the all other autoimmune manifestations of the disease, an autoimmune pathogenesis for APECED-related CMC has also been proposed (64). APECED patients also develop high titer of neutralizing autoantibodies against IL-22, IL-17F, and IL-17A (64). Indeed, the neutralizing autoantibodies to Th17 cytokines or the impaired production of IL-22 and IL-17A seem to be associated with susceptibility to Candida infection (64).
The occurrence of endocrine manifestations is usually associated with a specific array of organ-specific autoantibodies. NATCH leucine-rich repeat protein 5 (NALP5) has been identified as the target for autoimmune attack in the parathyroid cells (65) in APECED. Autoantibodies specific for the steroidogenic enzymes (CYP21A2 and CYP17A1) and side chain cleavage enzyme (CYP11A1) are useful markers for the autoimmune destruction of the adrenal cortex even years before the clinical onset of the disease (5, 66). Autoantibodies to cytochrome CYP11A1 are associated with ovarian insufficiency (66). T1D is correlated with autoantibodies against insulin, IA-2 tyrosine phosphatase-like protein, and glutamic acid decarboxylase GAD65 (67).
Autoimmune hepatitis is characterized by the presence of autoantibodies specific for liver-expressed cytochromes CYP1A2 and CYP2A6 (67). Gastrointestinal symptoms have been associated with the presence of autoantibodies against tryptophan hydroxylase (TPH) (68). Enteroendocrine cells can also be the target of an autoimmune attack. Therefore, in some cases, the intestinal dysfunction might be viewed as an autoimmune endocrinopathy (69). Recently, circulating autoantibodies to Paneth cell-specific alpha 5 defensin and reduced numbers of Paneth cells in APECED patients have been reported and also associated with intestinal dysfunction (70).
Autoantibodies against TPH have been associated with alopecia, vitiligo, and enamel dysplasia and anti-SOX9/SOX10 antibodies with vitiligo (71, 72).
Autoantibodies directed against the potassium channel regulatory protein (KCNRG) and BPIFB1, found in epithelial cells of terminal bronchioles, have been suggested as a marker for pulmonary disease in APECED patients (64, 73).
The autoimmune nature of renal destruction has been confirmed by examining biopsy samples and by determining antiproximal tubular autoantibodies (74, 75). Furthermore, autoantibodies targeting kidney collecting ducts specific antigens [aquaporin 2 (AQP2) and two transcription factors regulating the aquaporin 2 promoter, namely homolog of the human homeobox B7 (HOXB7) and NF of activated T cells 5 (NFAT5)], have been recently identified in APECED patients affected with tubulointerstitial nephritis (76).
Recently, B cell response against a panel of over 9,000 human proteins has enabled to have a detailed profiling of known autoantigens and to identify novel immune targets in APECED. As for the lack of genotype–phenotype relationship, it has been shown that AIRE genotype did not appear to be an important determinant of autoantibody expression. Moreover, two novel gonadal autoantigens, melanoma antigen family B 2 (MAGEB2) and protein disulfide isomerase-like testis (PDILT), have been identified that potentially could contribute to infertility in male and female patients with APECED (77). Another mechanism proposed to explain subfertility in males with APECED is the presence of autoantibodies against the prostatic antigen transglutaminase 4 (TGM4), causing prostatitis, and possible abnormal sperm maturation (78).
Neutralizing autoantibodies specific for type I IFNs discovered in 2006 by Meager et al. (79) are hallmark of the “classical” APECED and are detectable in AIRE-deficient children as early as a few months of age, before the appearance of clinical symptoms or organ-specific autoantibodies (79). Autoantibodies against IFN seems to be less prevalent in “non-classical” APECED, probably reflecting some residual AIRE function (39).
In conclusion, we should take into account that the autoimmune response in APECED appears orders of magnitude more limited than could be expected. There is a great discrepancy between the number of AIRE-controlled genes (around 4,000) and the number of detected autoantigens in APECED (around 20). Several explanations must be considered. First, it could be that only a subset of self-antigens is able to activate autoimmune responses. Moreover, the peripheral tolerance mechanisms may provide additional filters for the development of autoimmunity. Finally, it has been observed that autoimmunity in APECED preferentially targets molecules with restricted tissue expression profiles.
New Insights into Aire Genetics and Functioning: Clinical Implications
The genetic basis of autoimmunity is a complex problem. The main lesson from recent evidence is that the mutations of AIRE can lead to various degrees of clinical autoimmunity, ranging from “classical” APECED to specific autoimmune conditions, which had not been previously mined for genetically determined conditions. Therefore, partial alterations of AIRE could play a role in common autoimmune disease; however, to measure the penetrance and the relative risk conferred by pathogenic AIRE mutations in its monoallelic variants, it will be necessary to sequence Aire in large cohorts of healthy individuals and autoimmune patients and to characterize experimentally in-depth all mutant alleles.
Furthermore, in the last decade, knowledge of AIRE’s function and regulation has been significantly expanded leading to the identification of several partners and regulators of AIRE. Taken together, these molecular insights open new perspectives in understanding the phenotypic variability related to AIRE mutations and might provide interesting targets for novel therapeutic approach.
Indeed, an unanimously accepted effective therapy for APECED is not currently available. The use of immunosuppressive treatment in this category of patients may lead to a transient immunodeficiency with the risk to worsen their CMC and seems not able to stop the progression of all APECED manifestations (80). Thus, the management is mainly based on the care of each individual component and is mainly characterized by substitutive treatments for hormone deficiencies and immunomodulators have been only used in selected severe phenotypes (80, 81).
Although thymic transplantation has been proven useful in the treatment of differentiative thymic disorder (82), no data are available on this intervention on alterations of the thymic negative selection process. Thymic compartment can be targeted to modulate immune tolerance, for example, by enhancing AIRE expression, promoting deletion of self-reactive T cells and enhancing positive Treg cell selection, or inducing differentiation of TECs from pluripotent stem cells, offering new exciting possibility in therapeutic manipulation (15).
In conclusions, the new insights in the biology of AIRE and its control in immune tolerance offer exciting possibilities for the exploration of diagnostic and therapeutic strategies that would benefit APECED patients.
Author Contributions
All the authors contributed equally to this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Abramson J, Husebye ES. Autoimmune regulator and self-tolerance – molecular and clinical aspects. Immunol Rev (2016) 271(1):127–40. doi: 10.1111/imr.12419
2. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14(6):377–91. doi:10.1038/nri3667
3. Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science (2015) 348(6234):589–94. doi:10.1126/science.aaa7017
4. Perniola R, Musco G. The biophysical and biochemical properties of the autoimmune regulator (AIRE) protein. Biochim Biophys Acta (2014) 1842(2):326–37. doi:10.1016/j.bbadis.2013.11.020
5. Maslovskaja J, Saare M, Liiv I, Rebane A, Peterson P. Extended HSR/CARD domain mediates AIRE binding to DNA. Biochem Biophys Res Commun (2015) 468(4):913–20. doi:10.1016/j.bbrc.2015.11.056
6. Waterfield M, Khan IS, Cortez JT, Fan U, Metzger T, Greer A, et al. The transcriptional regulator Aire coopts the repressive ATF7ip-MBD1 complex for the induction of immunotolerance. Nat Immunol (2014) 15(3):258–65. doi:10.1038/ni.2820
7. Gaetani M, Matafora V, Saare M, Spiliotopoulos D, Mollica L, Quilici G, et al. AIRE-PHD fingers are structural hubs to maintain the integrity of chromatin-associated interactome. Nucleic Acids Res (2012) 40(22):11756–68. doi:10.1093/nar/gks933
8. Org T, Chignola F, Hetényi C, Gaetani M, Rebane A, Liiv I, et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep (2008) 9(4):370–6. doi:10.1038/sj.embor.2008.11
9. Koh AS, Kuo AJ, Park SY, Cheung P, Abramson J, Bua D, et al. Aire employs a histone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc Natl Acad Sci U S A (2008) 105(41):15878–83. doi:10.1073/pnas.0808470105
10. Poliani PL, Kisand K, Marrella V, Ravanini M, Notarangelo LD, Villa A, et al. Human peripheral lymphoid tissues contain autoimmune regulator-expressing dendritic cells. Am J Pathol (2010) 176(3):1104–12. doi:10.2353/ajpath.2010.090956
11. Gardner JM, Metzger TC, McMahon EJ, Au-Yeung BB, Krawisz AK, Lu W, et al. Extrathymic Aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4+ T cells. Immunity (2013) 39(3):560–72. doi:10.1016/j.immuni.2013.08.005
12. Haljasorg U, Bichele R, Saare M, Guha M, Maslovskaja J, Kõnd K, et al. A highly conserved NF-κB-responsive enhancer is critical for thymic expression of Aire in mice. Eur J Immunol (2015) 45(12):3246–56. doi:10.1002/eji.201545928
13. LaFlam TN, Seumois G, Miller CN, Lwin W, Fasano KJ, Waterfield M, et al. Identification of a novel cis-regulatory element essential for immune tolerance. J Exp Med (2015) 212(12):1993–2002. doi:10.1084/jem.20151069
14. Yanagihara T, Sanematsu F, Sato T, Uruno T, Duan X, Tomino T, et al. Intronic regulation of Aire expression by Jmjd6 for self-tolerance induction in the thymus. Nat Commun (2015) 6:8820. doi:10.1038/ncomms9820
15. Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat Rev Immunol (2016) 16(4):247–58. doi:10.1038/nri.2016.9
16. Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, et al. Population and single-cell genomics reveal the Aire dependency, relief from polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res (2014) 24(12):1918–31. doi:10.1101/gr.171645.113
17. Meredith M, Zemmour D, Mathis D, Benoist C. Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat Immunol (2015) 16(9):942–9. doi:10.1038/ni.3247
18. Derbinski J, Pinto S, Rösch S, Hexel K, Kyewski B. Promiscuous gene expression patterns in single medullary thymic epithelial cells argue for a stochastic mechanism. Proc Natl Acad Sci U S A (2008) 105(2):657–62. doi:10.1073/pnas.0707486105
19. Villaseñor J, Besse W, Benoist C, Mathis D. Ectopic expression of peripheral-tissue antigens in the thymic epithelium: probabilistic, monoallelic, misinitiated. Proc Natl Acad Sci U S A (2008) 105(41):15854–9. doi:10.1073/pnas.0808069105
20. Pinto S, Michel C, Schmidt-Glenewinkel H, Harder N, Rohr K, Wild S, et al. Overlapping gene coexpression patterns in human medullary thymic epithelial cells generate self-antigen diversity. Proc Natl Acad Sci U S A (2013) 110(37):E3497–505. doi:10.1073/pnas.1308311110
21. Giraud M, Yoshida H, Abramson J, Rahl PB, Young RA, Mathis D, et al. Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc Natl Acad Sci U S A (2012) 109(2):535–40. doi:10.1073/pnas.1119351109
22. Incani F, Serra M, Meloni A, Cossu C, Saba L, Cabras T, et al. AIRE acetylation and deacetylation: effect on protein stability and transactivation activity. J Biomed Sci (2014) 21:85. doi:10.1186/s12929-014-0085-z
23. Chuprin A, Avin A, Goldfarb Y, Herzig Y, Levi B, Jacob A, et al. The deacetylase Sirt1 is an essential regulator of Aire-mediated induction of central immunological tolerance. Nat Immunol (2015) 16(7):737–45. doi:10.1038/ni.3194
24. Rattay K, Claude J, Rezavandy E, Matt S, Hofmann TG, Kyewski B, et al. Homeodomain-interacting protein kinase 2, a novel autoimmune regulator interaction partner, modulates promiscuous gene expression in medullary thymic epithelial cells. J Immunol (2015) 194(3):921–8. doi:10.4049/jimmunol.1402694
25. Dragin N, Bismuth J, Cizeron-Clairac G, Biferi MG, Berthault C, Serraf A, et al. Estrogen-mediated downregulation of AIRE influences sexual dimorphism in autoimmune diseases. J Clin Invest (2016) 126(4):1525–37. doi:10.1172/JCI81894
26. Leventhal DS, Gilmore DC, Berger JM, Nishi S, Lee V, Malchow S, et al. Dendritic cells coordinate the development and homeostasis of organ-specific regulatory T cells. Immunity (2016) 44(4):847–59. doi:10.1016/j.immuni.2016.01.025
27. Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA. Aire enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity (2016) 44(5):1102–13. doi:10.1016/j.immuni.2016.02.009
28. Yamano T, Nedjic J, Hinterberger M, Steinert M, Koser S, Pinto S, et al. Thymic B cells are licensed to present self antigens for central T cell tolerance induction. Immunity (2015) 42(6):1048–61. doi:10.1016/j.immuni.2015.05.013
29. Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, et al. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J Exp Med (2011) 208(2):383–94. doi:10.1084/jem.20102327
30. Gray D, Abramson J, Benoist C, Mathis D. Proliferative arrest and rapid turnover of thymic epithelial cells expressing Aire. J Exp Med (2007) 204(11):2521–8. doi:10.1084/jem.20070795
31. Matsumoto M, Nishikawa Y, Nishijima H, Morimoto J, Matsumoto M, Mouri Y. Which model better fits the role of aire in the establishment of self-tolerance: the transcription model or the maturation model? Front Immunol (2013) 22(4):210. doi:10.3389/fimmu.2013.00210
32. Macedo C, Oliveira EH, Almeida RS, Donate PB, Fornari TA, Pezzi N, et al. Aire-dependent peripheral tissue antigen mRNAs in mTEC cells feature networking refractoriness to microRNA interaction. Immunobiology (2015) 220(1):93–102. doi:10.1016/j.imbio.2014.08.015
33. Kisand K, Peterson P, Laan M. Lymphopenia-induced proliferation in aire-deficient mice helps to explain their autoimmunity and differences from human patients. Front Immunol (2014) 5:51. doi:10.3389/fimmu.2014.00051
34. Nishikawa Y, Nishijima H, Matsumoto M, Morimoto J, Hirota F, Takahashi S, et al. Temporal lineage tracing of Aire-expressing cells reveals a requirement for Aire in their maturation program. J Immunol (2014) 192(6):2585–92. doi:10.4049/jimmunol.1302786
35. Capalbo D, Mazza C, Giordano R, Improda N, Arvat E, Cervato S, et al. Molecular background and genotype-phenotype correlation in autoimmune-polyendocrinopathy-candidiasis-ectodermal-dystrophy patients from Campania and in their relatives. J Endocrinol Invest (2012) 35(2):169–73. doi:10.3275/7677
36. Capalbo D, De Martino L, Giardino G, Di Mase R, Di Donato I, Parenti G, et al. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy: insights into genotype-phenotype correlation. Int J Endocrinol (2012) 2012:353250. doi:10.1155/2012/353250
37. De Martino L, Capalbo D, Improda N, D’Elia F, Di Mase R, D’Assante R, et al. APECED: a paradigm of complex interactions between genetic background and susceptibility factors. Front Immunol (2013) 4:331. doi:10.3389/fimmu.2013.00331
38. Cetani F, Barbesino G, Borsari S, Pardi E, Cianferotti L, Pinchera A, et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectoderma dystrophy, acting in a dominant fashion and strongly consegregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab (2001) 86:4747–52. doi:10.1210/jc.86.10.4747
39. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity (2015) 42(6):1185–96. doi:10.1016/j.immuni.2015.04.021
40. Söderbergh A, Rorsman F, Halonen M, Ekwall O, Björses P, Kämpe O, et al. Autoantibodies against aromatic l-amino acid decarboxylase identifies a subgroup of patients with Addison’s disease. J Clin Endocrinol Metab (2000) 85(1):460–3. doi:10.1210/jc.85.1.460
41. Boe A, Knappskog M, Myhre A, Sørheim JI, Husebye ES. Mutational analysis of the autoimmune regulator (AIRE) gene in sporadic autoimmune Addison’s disease can reveal patients with unidentified autoimmune polyendocrine syndrome type 1. Eur J Endocrinol (2002) 146:519–22. doi:10.1530/eje.0.1460519
42. Buzi F, Badolato R, Mazza C, Giliani S, Notarangelo LD, Radetti G, et al. Autoimmune polyendogrinopathy-candidiasis-ectodermal dystrophy syndrome: time to review diagnostic criteria? J Clin Endocrinol Metab (2003) 88:3146–8. doi:10.1210/jc.2002-021495
43. Ferrera F, Rizzi M, Sprecacenere B, Balestra P, Sessarego M, Di Carlo A, et al. AIRE gene polymorphisms in systemic sclerosis associated with autoimmune thyroiditis. Clin Immunol (2007) 122:13–7. doi:10.1016/j.clim.2006.09.013
44. Lankisch TO, Mourier O, Sokal EM, Habes D, Lacaille F, Bridoux-Henno L, et al. AIRE gene analysis in children with autoimmune hepatitis type I or II. J Pediatr Gastroenterol Nutr (2009) 48(4):498–500. doi:10.1097/MPG.0b013e31818550de
45. Meyer G, Donner H, Herwig J, Böhles H, Usadel KH, Badenhoop K. Screening for an AIRE-1 mutation in patients with Addison’s disease, type 1 diabetes, Graves’ disease and Hashimoto’s thyroiditis as well as in APECED syndrome. Clin Endocrinol (Oxf) (2001) 54(3):335–8. doi:10.1046/j.1365-2265.2001.01230.x
46. Zhang J, Liu H, Liu Z, Liao Y, Guo L, Wang H, et al. A functional alternative splicing mutation in AIRE gene causes autoimmune polyendocrine syndrome type 1. PLoS One (2013) 8(1):e53981. doi:10.1371/journal.pone.0053981
47. Bellacchio E, Palma A, Corrente S, Di Girolamo F, Helen Kemp E, Di Matteo G, et al. The possible implication of the S250C variant of the autoimmune regulator protein in a patient with autoimmunity and immunodeficiency: in silico analysis suggests a molecular pathogenic mechanism for the variant. Gene (2014) 549(2):286–94. doi:10.1016/j.gene.2014.07.064
48. Tsai SL, Green J, Metherell LA, Curtis F, Fernandez B, Healey A, et al. Primary adrenocortical insufficiency case series: genetic etiologies more common than expected. Horm Res Paediatr (2016) 85(1):35–42. doi:10.1159/000441843
49. Cervato S, Morlin L, Albergoni MP, Masiero S, Greggio N, Meossi C, et al. AIRE gene mutations and autoantibodies to interferon omega in patients with chronic hypoparathyroidism without APECED. Clin Endocrinol (Oxf) (2010) 73:630–6. doi:10.1111/j.1365-2265.2010.03862.x
50. Cervato S, Mariniello B, Lazzarotto F, Morlin L, Zanchetta R, Radetti G, et al. Evaluation of the autoimmune regulator (AIRE) gene mutations in a cohort of Italian patients with autoimmune-polyendocrinopathy-candidiasis-ectodermal-dystrophy (APECED) and in their relatives. Clin Endocrinol (Oxf) (2009) 70(3):421–8. doi:10.1111/j.1365-2265.2008.03318.x
51. Sparks AE, Chen C, Breslin MB, Lan MS. Functional domains of autoimmune regulator, AIRE, modulate INS-VNTR transcription in human thymic epithelial cells. J Biol Chem (2016) 291(21):11313–22. doi:10.1074/jbc.M116.722488
52. Husebye ES, Perheentupa J, Rautemaa R, Kampe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J Intern Med (2009) 265:514–29. doi:10.1111/j.1365-2796.2009.02090.x
53. Meloni A, Furcas M, Cetani F, Marcocci C, Falorni A, Perniola R, et al. Autoantibodies against type I interferons as an additional diagnostic criterion for autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab (2008) 93:4389–97. doi:10.1210/jc.2008-0935
54. Capalbo D, Fusco A, Aloj G, Improda N, Vitiello L, Dianzani U, et al. High intrafamilial variability in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy: a case study. J Endocrinol Invest (2012) 35(1):77–81. doi:10.3275/8055
55. Friedman TC, Thomas PM, Fleisher TA, Feuillan P, Parker RI, Cassorla F, et al. Frequent occurrence of asplenism and cholelithiasis in patients with autoimmune polyglandular disease type I. Am J Med (1991) 91(6):625–30. doi:10.1016/0002-9343(91)90215-J
56. Orlova EM, Bukina AM, Kuznetsova ES, Kareva MA, Zakharova EU, Peterkova VA, et al. Autoimmune polyglandular syndrome type 1 in Russian patients: clinical variants and autoimmune regulator mutations. Horm Res Paediatr (2010) 73(6):449–57. doi:10.1159/000313585
57. Betterle C, Greggio NA, Volpato M. Clinical review 93: autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab (1998) 83(4):1049–55. doi:10.1210/jcem.83.4.4682
58. Improda N, Capalbo D, Cirillo E, Cerbone M, Esposito A, Pignata C, et al. Cutaneous vasculitis in patients with autoimmune polyendocrine syndrome type 1: report of a case and brief review of the literature. BMC Pediatr (2014) 14:272. doi:10.1186/1471-2431-14-272
59. Braun DA, Schueler M, Halbritter J, Gee HY, Porath JD, Lawson JA, et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int (2016) 89(2):468–75. doi:10.1038/ki.2015.317
60. Watanabe M, Ochi H, Arahata H, Matsuo T, Nagafuchi S, Ohyagi Y, et al. Myopathy in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Muscle Nerve (2012) 45(6):904–8. doi:10.1002/mus.23321
61. Capalbo D, Elefante A, Spagnuolo MI, Mazza C, Betterle C, Pignata C, et al. Posterior reversible encephalopathy syndrome in a child during an accelerated phase of a severe APECED phenotype due to an uncommon mutation of AIRE. Clin Endocrinol (Oxf) (2008) 69(3):511–3. doi:10.1111/j.1365-2265.2008.03206.x
62. Mazza C, Buzi F, Ortolani F, Vitali A, Notarangelo LD, Weber G, et al. Clinical heterogeneity and diagnostic delay of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome. Clin Immunol (2011) 139(1):6–11. doi:10.1016/j.clim.2010.12.021
63. Capalbo D, Improda N, Esposito A, De Martino L, Barbieri F, Betterle C, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy from the pediatric perspective. J Endocrinol Invest (2013) 36(10):903–12. doi:10.3275/8999
64. Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med (2010) 207:299–308. doi:10.1084/jem.20091669
65. Alimohammadi M, Dubois N, Skoldberg F, Hallgren A, Tardivel I, Hedstrand H, et al. Pulmonary autoimmunity as a feature of autoimmune polyendocrine syndrome type 1 and identification of KCNRG as a bronchial autoantigen. Proc Natl Acad Sci U S A (2009) 106:4396–401. doi:10.1073/pnas.0809986106
66. Soderbergh A, Myhre AG, Ekwall O, Gebre-Medhin G, Hedstrand H, Landgren E, et al. Prevalence and clinical associations of 10 defined autoantibodies in autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab (2004) 89:557–62. doi:10.1210/jc.2003-030279
67. Clemente MG, Meloni A, Obermayer-Straub P, Frau F, Manns MP, De Virgiliis S. Two cytochromes P450 are major hepatocellular autoantigens in autoimmune polyglandular syndrome type 1. Gastroenterology (1998) 114(2):324–8. doi:10.1016/S0016-5085(98)70484-6
68. Ekwall O, Hedstrand H, Grimelius L, Haavik J, Perheentupa J, Gustafsson J, et al. Identification of tryptophan hydroxylase as an intestinal autoantigen. Lancet (1998) 352(9124):279–83. doi:10.1016/S0140-6736(97)11050-9
69. Gianani R, Eisenbarth GS. Autoimmunity to gastrointestinal endocrine cells in autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab (2003) 88(4):1442–4. doi:10.1210/jc.2003-030247
70. Dobeš J, Neuwirth A, Dobešová M, Vobořil M, Balounová J, Ballek O, et al. Gastrointestinal autoimmunity associated with loss of central tolerance to enteric α-defensins. Gastroenterology (2015) 149(1):139–50. doi:10.1053/j.gastro.2015.05.009
71. Bratland E, Magitta NF, Boe Wolff AS, Ekern T, Knappskog PM, Kampe O, et al. Autoantibodies against aromatic amino acid hydroxylases in patients with autoimmune polyendocrine syndrome type 1 target multiple antigenic determinants and reveal regulatory regions crucial for enzymatic activity. Immunobiology (2013) 218:899–909. doi:10.1016/j.imbio.2012.10.006
72. Hedstrand H, Ekwall O, Olsson MJ, Landgren E, Kemp EH, Weetman AP, et al. The transcription factors SOX9 and SOX10 are vitiligo autoantigens in autoimmune polyendocrine syndrome type I. J Biol Chem (2001) 276:35390–5. doi:10.1074/jbc.M102391200
73. Shum AK, Alimohammadi M, Tan CL, Cheng MH, Metzger TC, Law CS, et al. BPIFB1 is a lung-specific autoantigen associated with interstitial lung disease. Sci Transl Med (2014) 5:206ra139. doi:10.1126/scitranslmed.3006998
74. Ulinski T, Perrin L, Morris M, Houang M, Cabrol S, Grapin C, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome with renal failure: impact of posttransplant immunosuppression on disease activity. J Clin Endocrinol Metab (2006) 91(1):192–5. doi:10.1210/jc.2005-1538
75. Al-Owain M, Kaya N, Al-Zaidan H, Bin Hussain I, Al-Manea H, Al-Hindi H, et al. Renal failure associated with APECED and terminal 4q deletion: evidence of autoimmune nephropathy. Clin Dev Immunol (2010) 2010:586342. doi:10.1155/2010/586342
76. Landegren N, Lindberg MP, Skov J, Hallgren Å, Eriksson D, Lisberg Toft-Bertelsen T, et al. Autoantibodies targeting a collecting duct specific water channel in tubulointerstitial nephritis. J Am Soc Nephrol (2016). doi:10.1681/ASN.2015101126
77. Landegren N, Sharon D, Freyhult E, Hallgren Å, Eriksson D, Edqvist PH, et al. Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine syndrome type 1. Sci Rep (2016) 6:20104. doi:10.1038/srep20104
78. Landegren N, Sharon D, Shum AK, Khan IS, Fasano KJ, Hallgren Å, et al. Transglutaminase 4 as a prostate autoantigen in male subfertility. Sci Transl Med (2015) 7(292):292ra101. doi:10.1126/scitranslmed.aaa9186
79. Meager A, Visvalingam K, Peterson P, Möll K, Murumägi A, Krohn K, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med (2006) 3:e289. doi:10.1371/journal.pmed.0030289
80. Kisand K, Peterson P. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J Clin Immunol (2015) 35(5):463–78. doi:10.1007/s10875-015-0176-y
81. Capalbo D, Giardino G, Martino LD, Palamaro L, Romano R, Gallo V, et al. Genetic basis of altered central tolerance and autoimmune diseases: a lesson from AIRE mutations. Int Rev Immunol (2012) 31(5):344–62. doi:10.3109/08830185.2012.697230
Keywords: APECED, autoimmune disease, diagnosis, AIRE, mutations
Citation: De Martino L, Capalbo D, Improda N, Lorello P, Ungaro C, Di Mase R, Cirillo E, Pignata C and Salerno M (2016) Novel Findings into AIRE Genetics and Functioning: Clinical Implications. Front. Pediatr. 4:86. doi: 10.3389/fped.2016.00086
Received: 14 June 2016; Accepted: 02 August 2016;
Published: 22 August 2016
Edited by:
Sergio Rosenzweig, National Institutes of Health, USAReviewed by:
Thomas Arthur Fleisher, National Institutes of Health, USAMichail Lionakis, National Institute of Allergy and Infectious Diseases, USA
Copyright: © 2016 De Martino, Capalbo, Improda, Lorello, Ungaro, Di Mase, Cirillo, Pignata and Salerno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariacarolina Salerno, c2FsZXJub0B1bmluYS5pdA==
† Lucia De Martino and Donatella Capalbo equally contributed to the manuscript.