Renal Cell Carcinoma in von Hippel–Lindau Disease—From Tumor Genetics to Novel Therapeutic Strategies
 Emily Kim
Emily Kim Stefan Zschiedrich
Stefan Zschiedrich- 1Department of Radiation Oncology, Faculty of Medicine, Albert Ludwigs University of Freiburg, Freiburg, Germany
- 2German Cancer Consortium (DKTK), Partner Site Freiburg, Freiburg, Germany
- 3Renal Division, Department of Medicine IV, Faculty of Medicine, Albert Ludwigs University of Freiburg, Freiburg, Germany
von Hippel–Lindau (VHL) disease is an autosomal dominant syndrome caused by mutations in the VHL tumor-suppressor gene, leading to the dysregulation of many hypoxia-induced genes. Affected individuals are at increased risk of developing recurrent and bilateral kidney cysts and dysplastic lesions which may progress to clear cell renal cell carcinoma (ccRCC). Following the eponymous VHL gene inactivation, ccRCCs evolve through additional genetic alterations, resulting in both intratumor and intertumor heterogeneity. Genomic studies have identified frequent mutations in genes involved in epigenetic regulation and phosphoinositide 3-kinase–AKT–mechanistic target of rapamycin (mTOR) pathway activation. Currently, local therapeutic options include nephron-sparing surgery and alternative ablative procedures. For advanced metastatic disease, systemic treatment, including inhibition of vascular endothelial growth factor pathways and mTOR pathways, as well as immunotherapy are available. Multimodal therapy, targeting multiple signaling pathways and/or enhancing the immune response, is currently being investigated. A deeper understanding of the fundamental biology of ccRCC development and progression, as well as the development of novel and targeted therapies will be accelerated by new preclinical models, which will greatly inform the search for clinical biomarkers for diagnosis, prognosis, and response to treatment.
Introduction
von Hippel–Lindau (VHL) disease is an autosomal dominant, multiorgan visceral cysts and tumor syndrome. The disease name derives from the German ophthalmologist Eugen von Hippel who studied two cases of striking retinal angiomas and the Swedish pathologist Arvid Lindau who detected a connection between cerebellar hemangioblastomas, retinal angiomas, and other visceral tumors (1, 2). The first report of VHL dates from 1894, when Collins described vascular intraocular tumors in two siblings (3). VHL manifestations can be found in retinal hemangioblastoma, cerebellar and spinal hemangioblastoma, renal cysts and clear cell renal cell carcinoma (ccRCC), liver hemangioma, pancreatic cysts, pancreatic microcystic serous adenoma and pancreatic neuroendocrine tumors, pheochromocytoma (PCC), epididymal and broad ligament cystadenoma, and endolymphatic sac tumors. In 1964, Melmon and Rosen suggested clinical diagnostic criteria that are still valid today. VHL disease has been classified depending on the presence of PCC in VHL type 1 (without PCC) and type 2 (with PCC). VHL type 2 is further subclassified in type 2A (with PCC but without ccRCC), type 2B (with PCC and ccRCC), and type 2C (PCC only) (4–6). Practically, however, practitioners do not rely on this classification because families can move between clinical subtypes.
von Hippel–Lindau disease is caused by mutations in the VHL tumor-suppressor gene, located on chromosome 3p25-26. VHL was mapped to chromosome 3 in 1988 and cloned in 1993 (7, 8). The incidence of VHL disease is approximately 1:36,000 (9). Although VHL disease typically presents in early adulthood, manifestation of retinal angiomas, PCCs, and ccRCCs has been reported earlier; therefore, guidelines recommend starting surveillance programs for eye examination at 2–5 years and abdominal imaging at 6–10 years (10–12).
Most patients develop ccRCCs that arise from microscopic precursor lesions present in both kidneys. The number of nonmalignant cysts lined with clear cells in an average VHL kidney was estimated to be 1,100, and the number of clear cell renal neoplasms (solid and cystic) to be 600 (13). Optimally, early RCC can be detected, observed, and surgically removed before progression to metastatic disease (14). However, repeated surgery for multifocal bilateral disease is followed by increased risk of end-stage renal disease (ESRD) requiring renal transplantation or dialysis.
The Role of VHL
Whether hereditary or sporadic, ccRCC is characterized by mutations in the VHL tumor-suppressor gene on chromosome 3 (3p25-26) and a subsequent loss of heterozygosity. VHL, Elongin B (encoded by TCEB1), and Elongin C form a stable complex that targets hypoxia-inducible factor α (HIFα) subunits for proteolytic degradation under normoxic conditions (15). In the presence of hypoxia or in the absence of functional VHL tumor-suppressor protein, HIFα subunits HIF1α and HIF2α are stabilized, binding together with ARNT (HIF1β) to hypoxia-response elements to activate genes involved in angiogenesis, cell cycle, cell proliferation, glucose, and lipid metabolism, among others. Mutations of TCEB1 that abrogate binding of Elongin B to VHL can also increase HIFα expression in ccRCCs with intact VHL (16). Mutations in VHL and TCEB1 were mutually exclusive, supporting a permissive role for VHL complex degradation and HIFα stabilization in tumorigenesis.
Additional Gene Mutations
Approximately 15 years after VHL was identified as the genetic basis for VHL, further driver mutations for ccRCC were identified, summarized in Table 1. Exome sequencing of tumors identified additional inactivating mutations on chromosome 3 in tumor suppressors polybromo-1 (PBRM1), SET domain-containing 2 (SETD2), and BRCA1-associated protein-1 (BAP1), which are chromatin and histone regulators located at 3p21 (17–19). PBRM1 and BAP1 mutations are mutually exclusive, with BAP1 mutations correlating with higher grade disease (19, 20). Large-scale genomic sequencing showed that these three tumor-suppressor genes, located near VHL at 3p21, are the most frequently mutated genes in ccRCC after VHL (16, 21). Other significantly mutated genes included histone-modifiers KDM5C and KDM6A, previously implicated in ccRCC (17–19, 22), genes in the phosphoinositide 3-kinase (PI3K)/AKT pathway, such as mechanistic target of rapamycin (mTOR), PIK3CA, and PTEN, as well as TCEB1 (Elongin C) (16, 21). The tumor-suppressor gene TP53 is infrequently mutated, playing a lesser role than in many other solid tumors.
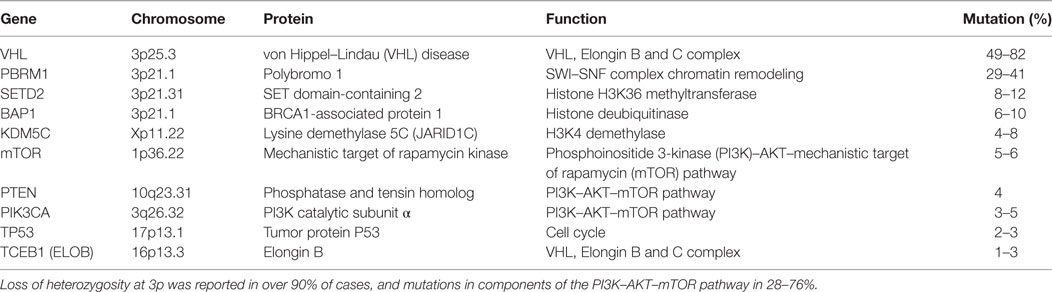
Tumor Heterogeneity
Clear cell renal cell carcinoma has a moderate somatic mutation frequency compared with other solid tumors and these mutations progress in a branched evolutionary manner (24, 25). The evolutionary history of sporadic ccRCC in 10 individuals was investigated by exome sequencing of multiregion samples from primary ccRCCs and metastases (26). Intratumor heterogeneity was present in all tumors, indicating that a single biopsy underestimates the genomic complexity of a tumor. Tumor phylogeny, similar to an evolutionary tree, showed that inactivation of VHL and loss of chromosome 3p were ubiquitous early truncal events. PBRM1 inactivation was a frequent mutation, occurring early as a truncal mutation in three tumors and as a later event in three others. Distinct subclones, spatially separated within a single tumor, contained mutations that appeared in a branched rather than linear manner. Subclonal driver mutations were similar to those identified by earlier studies, such as chromatin modifiers, regulators of mTORC1 pathway, and tumor-suppressor TP53. Parallel evolution was observed in certain genes, in which different evolutionary paths or branches in a tumor resulted in inactivation of the same gene by separate mechanisms. Recurrent mutations in PBRM1, SETD2, BAP1, and KDM5C suggest an evolutionary selection for epigenetic dysregulation in tumorigenesis. Branched subclonal mutations were highly variable and contained more C>T transitions than truncal mutations, potentially useful as prognostic and predictive biomarkers.
Genomic analysis of ccRCC has established the fundamental role for VHL inactivation and HIF dysregulation, the importance of chromatin regulation and histone modification, and the involvement of the mTORC1 pathway. The central role of chromosome remodeling in the development and the progression of ccRCC implicates epigenetic dysregulation as a permissive factor in tumorigenesis and a novel target for therapeutic agents and candidate biomarkers. However, the molecular mechanisms whereby epigenetic alterations result in transcriptional dysregulation is currently unclear. Mutations and copy number alterations were detected in mTOR (mammalian target of rapamycin), PIK3CA (PI3K catalytic subunit-α), TSC1, and PTEN (16, 21). Mutually exclusive gene alterations of the PI3K/Akt/mTOR pathway were detected in approximately 28% of tumors (21). Activation of the PI3K/Akt/mTOR pathway may underlie the efficacy of mTOR inhibitors, such as everolimus and temsirolimus.
In kidneys of individuals with VHL disease, VHL-deficient lesions with constitutive HIF activation were detectable by carbonic anhydrase IX staining, allowing the progression from single cells to ccRCC to be easily observed (27). HIF activation occurs extremely early in the disease. Most lesions are single cells, with very few multicellular dysplastic lesions and cystic lesions, showing that loss of VHL function alone is insufficient for ccRCC formation (27). Renal cysts are classified as benign, atypical, and malignant. HIF-1α is expressed in all VHL-deficient renal cells, whereas HIF-2α is highly expressed in renal tubular cysts and ccRCC (27).
Multiregion whole exome sequencing of four tumors from one individual with VHL disease delineated multifocal tumors of independent clonal origins (28). Each tumor exhibited a loss of chromosome 3p, each with a distinctly different breakpoint. Tumor evolution was more linear than branching compared with sporadic ccRCC, with markedly less intratumor heterogeneity. Convergent mTOR pathway activation was observed in all tumors through distinct gene mutations. The evolutionary history of 40 tumors from 6 individuals with VHL was examined by whole-genome sequencing (29). Tumors showed more genetic homogeneity than sporadically occurring tumors, which are generally removed at a later stage. However, the lack of overlapping sets of single-nucleotide variants as well as copy number variants between tumors indicated that ccRCCs evolved independently. A similar approach with different VHL subtypes could elucidate the effect of genetic background on the disease. For example, type 1 VHL disease (without PCC) is associated with truncation or exon deletion of germline VHL, whereas missense mutation is associated with type 2 disease (with PCC) (30). ccRCC occurs in Types 1 and 2B, which poorly downregulate HIF-1α but not 2A and 2C (6, 31).
Preclinical Models
Tumor xenografts using human ccRCC cell lines or tissue have been extensively used in mice to evaluate potential therapies. Likewise, injecting zebrafish with patient-derived xenografts and human cell lines is a rapid, low-cost preclinical model system of cancer (32, 33). Genetically engineered animal models of biallelic mutation of VHL alone in both mouse and zebrafish recapitulate features of early human disease, but not the formation of ccRCC. HIF activation appears to be necessary, but not sufficient for tumor formation in animal models of ccRCC.
Initial animal models were developed by genetically modifying levels of VHL and HIFα. Homozygous deletion of Vhl is embryonic lethal at 10.5–12.5 days in mice due to defective placental vasculogenesis; heterozygous mice fail to develop kidney tumors (34). Kidney-specific inactivation of Vhl is insufficient for ccRCC development, but results in multiple cysts with constitutive HIF-α expression and metabolic alterations marked by lipid and glycogen accumulation similar to early human disease (35). Transgenic overexpression of HIF-1α or HIF-2α resulted in simple cysts (36, 37). Likewise in zebrafish embryos, homozygous inactivation of VHL (vhl−/−) results in a kidney with enlarged proximal pronephric tubules, disorganized cilia, accumulated lipid and glycogen, cell proliferation, and apoptosis. This phenotype was rescued by a specific HIF2α inhibitor, showing that the zebrafish model system could be used to facilitate rapid screening of candidate drugs (38).
The identification of additional mutations underlying ccRCC has informed the development of genetically engineered mouse models that are more analogous to human disease. After VHL, the most frequently mutated genes in ccRCC were chromosome and histone regulators PBRM1, SETD2, and BAP1 (16, 21). Epigenetic genes were targeted in recent studies. Kidney-specific dual inactivation of Vhl and Bap1 or Pbrm1 using Pax8-Cre in mice recapitulated human ccRCC with cytoplasmic accumulation of glycogen and lipids (39). Bap1-deficient cystic tumors were high grade, whereas Pbrm1-deficient solid tumors showed a longer latency. Pbrm1-deficient tumors were converted from low to high grade by disruption of one Tsc1 allele, resulting in mTORC1 activation. Intriguingly, ccRCC appeared to arise from Bowman capsule cells rather than the proximal tubule based on gene inactivation using more specific Pax8-Cre drivers.
Kidney-specific inactivation of Vhl and Pbrm1 using Ksp-Cre model the histopathological and molecular features, and gradual onset of human ccRCC (40). Ksp-Cre is expressed both in renal tubular cells and the Bowman capsule. Bilateral, multifocal tumors were marked by the clear cytoplasm, high glycogen, and carbonic anhydrase IX staining similar to human ccRCC. A stepwise progression was observed from normal to cystic lesions over 6 months, developing into multifocal ccRCC at ~10 months. Loss of PBRM1 further amplified the activation of HIF1 (hetereodimer HIF-1α and HIF-1β) and STAT3 pathways caused by loss of Vhl. Activation of mTORC1 was implicated as the third event leading to ccRCC.
Inactivation of Vhl, Rb1, and Trp53 in mice induced precursor cysts and gradually developing ccRCC tumors (41). Mutations were observed in genes involved in the primary cilium, Kif3a and Kif3b. Transcriptional analysis showed a gene expression profile similar to that observed in human ccRCC, with upregulation of HIF-1α and HIF-2α, mTOR activity. Mouse tumors showed variable response to anti-angiogenic therapy, and partial response to acriflavine, which interferes with the dimerization of HIF-1α and HIF-2α.
Zebrafish with multiple mutations of VHL disease-related genes are another potential preclinical model for exploring the progression and metastasis of ccRCC with the advantage of live imaging.
Current Treatment Strategies
Active Surveillance
Clear cell renal cell carcinomas grow slowly and small tumors <3 cm are at low risk to metastasize in VHL (42, 43). Currently, active surveillance until a threshold size of 3–4 cm is recommended for surgical intervention (43–46), resulting in a recurrence-free survival rate of 76% at 5 years and 20% at 8 years (47).
Regular screening is advised to detect RCC at an early stage and small tumors are followed with serial imaging. To improve the quality of life and survival of these patients, a balance between two goals is paramount: preventing metastases and preserving renal function. The goal is to treat before the tumor metastasizes, but to minimize consequences of the treatment such as compromised renal function. Through better surveillance by regularly scheduled imaging, individuals are living longer; however, a longer lifespan increases the probability of developing multiple RCCs and other sequelae of the disease.
Treatment of Localized Disease
Surgery
Early RCC can be detected, observed, and surgically removed before progression to metastatic disease (14, 48). However, repeated surgery for multicentric bilateral disease is followed by increased risk of ESRD requiring renal replacement therapy. Nephron-sparing surgery at a tumor size of 3–4 cm is the current treatment standard, replacing radical nephrectomy which compromised renal function and resulted in early dialysis. However, repeated partial nephrectomy reduces renal function, eventually causing ESRD. Delaying the interval to kidney surgery without increasing the risk of metastases prolongs sufficient renal function and delays dialysis. Independently of VHL disease, chronic kidney disease is associated with increased risk of death, cardiovascular events, and hospitalization (49). Progression to ESRD not only impacts quality of life but further increases morbidity—the yearly mortality rate of patients receiving long-term dialysis is 15–20% (50).
Locally recurrent disease is not uncommon after both partial nephrectomy and ablative therapy. Therapy options include observation, initial or repeat ablation, initial or repeat partial nephrectomy, radical nephrectomy, or systemic therapy (51). Salvage operation has a high major complication rate approaching 20% (52).
As renal surveillance of VHL patients has shifted surgical treatment from the resection of large tumors to the management of multiple small asymptomatic tumors, nephron-sparing therapeutic options are increasingly used. These minimally invasive procedures include percutaneous radiofrequency ablation (RFA), microwave ablation (MWA), cryoablation, and stereotactic body radiotherapy (SBRT) which are compared with surgery in Table 2.
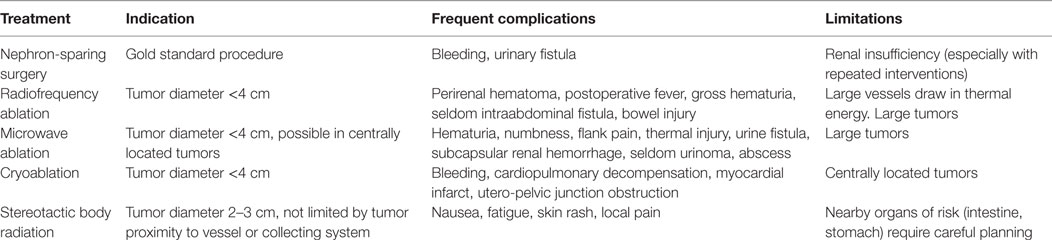
Table 2. Local treatment modalities for clear cell renal cell carcinoma, including indication, frequent complications and limitations of surgery, and ablation techniques.
Radiofrequency Ablation
Initially a treatment for non-surgical candidates, RFA has been used as a first-line procedure used to treat smaller and less numerous stage T1a lesions, and for renal salvage (46). Tumor necrosis is achieved with the heat of 50–100°C of radiofrequency energy transmitted by one or multiple needles in the tumor tissue. Placement of these needles can be achieved both laparoscopically or percutaneous (51).
Short-term local recurrence rates of RFA compare favorably with partial nephrectomy. A study of RFA treated T1 RCC reported a 13% retreatment rate for residual disease, a recurrence-free survival of 94%, and disease-free survival of 88% at 5 years (53). A study of T1a tumors treated with RFA versus partial nephrectomy reported a recurrence-free survival of 91.7 versus 94.6%, and an identical disease-free survival of 89% at 5 years (54). A recent study of 20 RCCs of 1–4 cm diameter in 9 VHL patients treated with RFA showed no recurrence and preserved kidney function with a median follow-up of 102 months (55). Zagoria and colleagues concluded that long-term tumor control could be achieved in lesions treated smaller than 4 cm diameter (56). Another group reported increased risk of residual tumor for lesions >3.5 cm diameter (57). Clearly, there is a decrease of disease-free survival with every centimeter increase of the lesion (56). Although RFA is performed in many centers, conclusions about long-term efficacy are limited by the relatively short follow-up interval and small study sizes.
Microwave Ablation
Electromagnetic microwaves, transmitted from one or multiple antennas placed in the tumor can create a thermal field that causes tumor tissue necrosis (58). The antennas can be placed percutaneously, laparoscopically, or less often in an open approach. The potential benefit of Microwave Ablation (MWA) compared with RFA is that intratumoral temperatures with MWA are less affected by the heat sink effect, since MWA is less dependent on the electrical conductivity of the tissue. Larger tumors can be ablated with MWA compared to RFA; therefore, a maximum diameter of 4 cm was recommended in most studies (59–62). Several authors suggest MWA can be performed in tumors close to renal sinus or collecting system—a clear advantage over the other ablation techniques (62, 63).
Cryoablation
Cryoablation either laparoscopic or percutaneous, is a minimally invasive procedure that freezes and destroys small tumors (64–67). The cryoprobe is cooled down to −185 to −195°C by a nitrogen-based liquid guided through the tip of the probe. Cryoablation is less precise than RFA as it requires three applicators and a tumor margin of 10 mm. Thus, RFA of a 2-cm mass ablates approximately 10 cm3 of normal tissue, whereas cryoablation treatment ablates 30 cm3 of normal tissue (68). The location of the tumor within the kidney plays a critical role in treatment success, as centrally located tumors more frequently fail effective tumor cell destruction (69–71). Treatment failure is also significantly associated with tumor size >4 cm (72).
Stereotactic Body Radiotherapy
Stereotactic body radiotherapy (SBRT) is a method of external beam radiotherapy that precisely targets tumors with high individual doses. Multiple 3D conformal beams or intensity-modulated RT ensure the delivery of highly conformal dose distribution with a steep gradient falloff at the tumor margin to minimize injury of surrounding normal tissue (73, 74). SBRT provides superior renal tumor control compared with conventional radiation therapy, both in vitro and in vivo (75–77). Despite concerns that radiation may accelerate mutational events, stereotactic treatment of VHL tumors (primary and metastatic RCC, as well as hemangioblastomas) showed no increase in tumor formation of surrounding tissue after doses of 30–40 Gy (78–81). Two reviews reported a local control rate of over 90% for large primary RCCs and up to 98% for small tumors (82, 83). The treatment is non-invasive, with a low toxicity and mild deterioration of renal function (84). Lesions close to collecting vessels are also amenable to therapy (85). Mild side effects included nausea, fatigue, skin rash, and local pain. Similar to other ablative therapies, assessment of the long-term efficacy is limited by the short follow-up interval and small study size.
In summary, because of the lack of comparing long-term studies, partial nephrectomy for tumors of 3–4 cm diameter is still the standard of care. However, minimal-invasive physically treatment with RFA, MWA, cryoablation, and SBRT carries certain treatment advantages. The choice of resection versus ablative treatment is dependent upon the tumor localization, treatment availability, and experience of the VHL center.
Treatment of Advanced ccRCC
Advances in imaging and localized therapy have greatly improved detection and survival rates for VHL patients with early ccRCC. Currently, metastatic ccRCC is difficult to cure despite the availability of multiple systemic therapies. Before 2005, systemic therapy consisted of cytokine therapy with interleukin-2 and interferon-alpha, which was marked by severe toxicity and low response rates. Drugs that target the VHL–HIF–vascular endothelial growth factor (VEGF) pathway such as VEGF receptor inhibitors (sunitinib, sorafenib, pazopanib, and axitinib) significantly improved outcome (86–89). Bevacizumab, an anti-VEGF monoclonal antibody, was the first recombinant human monoclonal antibody that showed clinical efficacy in advanced disease (90). Today, sunitinib and pazopanib are approved first-line tyrosine kinase inhibitors for metastatic ccRCC.
Mechanistic target of rapamycin inhibitors like everolimus and temsirolimus aim at the “mechanistic target of rapamycin” complex mTORC1 which controls fundamental cellular functions such as growth, proliferation, and apoptosis. Intravenous temsirolimus was approved in 2007 following a study showing improved progressive free survival (PFS) compared with interferon alone or combination therapy with interferon and temsirolimus (91). For oral everolimus, PFS was longer in the treated group versus placebo group (92).
In 2016, cabozantinib was approved as a second-line inhibitor of VEGF receptor and a broad range of type III receptor tyrosine kinases. Both PFS and median overal survival were longer for cabozantinib treated patients versus everolimus treatment (93, 94). Levantinib, an oral multityrosine kinase, showed a synergistic effect with everolimus in patients previously pretreated for advanced ccRCC (95, 96).
Checkpoint inhibitors target “programmed cell death 1” (PD1), PD1-ligand, and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) to activate T cell function. Nivolumab is a fully humanized IgG4 antibody against PD1, a negative regulator of T cell function, which improved survival in a subset of patients (97). Regardless of the targeted pathway, single agent treatment shows limited efficacy, with eventual treatment failure. Building on clinical experience favoring multiagent therapy (98), ongoing clinical trials are investigating combinations of kinase inhibitor treatments with immunotherapy, as well as combinations of immune modulators nivolumab and ipilimumab, an inhibitor of CTLA-4. A regression of metastases was observed after stereotactic radiotherapy in 4 of 28 renal cell carcinoma patients (99), suggesting that radiotherapy could enhance an immune response (98). Clinical trials testing the combination of radiotherapy with immunotherapy or targeted therapy are in progress (NCT02781506, NCT02019576, and NCT02978404).
Therapeutic options of advanced ccRCC have rapidly advanced over the past decade. Effective agents in advanced ccRCC have now been tested in earlier ccRCC stages and become appropriate first-line therapy drugs (100). Ideally, these drugs could serve as candidate perioperative agents with the potential to optimize postoperative outcome (101). In terms of precision medicine, the challenge now is to match a given patient with the optimal therapeutic agents with the help of robust molecular biomarkers.
Therapeutic Targets and Future Trials
Traditional chemotherapy and conventional radiotherapy, unspecifically directed against highly proliferative cells, is poorly effective in ccRCC. Multiple dysregulated signaling pathways have recently been identified and therapies that target these pathways have shown clinical efficacy in a subset of patients. ccRCCs are highly vascular and treated by currently approved anti-angiogenic agents. Downstream components of the VHL–HIF–VEGF pathway are modulated by receptor tyrosine kinase inhibitors (axitinib, cabozantinib, levantinib, pazopanib, sorafenib, and sunitinib), as well as anti-VEGF monoclonal antibodies (bevacizumab). The PI3K/Akt/mTOR pathway is targetable by mTOR inhibitors (everolimus, temsirolimus), which reduces the accumulation of HIF protein.
Genomic profiling is a potential guide for treatment and prognosis. No predictive biomarkers have been validated for selecting treatment, but several candidate biomarkers for treatment response have been identified through retrospective patient studies using tumor DNA. For example, mutations in MTOR, TSC1, or TSC2 were associated with response to mTOR inhibitors, 21% in responders versus 11% in non-responders (102). But many responders (56%) had no mTOR pathway mutation. A positive response to first-line everolimus was associated with PBRM1 mutations, and a negative response with BAP1 mutations. In addition, KDM5 mutations were associated with better response with first-line sunitinib than with everolimus (103). These genomic biomarkers are currently being evaluated in prospective studies. Other potential biomarkers include RNA sequencing to detect structural rearrangement and transcription levels, microRNA sequencing, DNA methylation profiling, and metabolomics.
Given the inherent intratumor heterogeneity of ccRCC, therapy that targets truncal events involving VHL or chromosome 3p may be more effective than targeting subclonal pathways. VHL and TCEB1 are mutually exclusive mutations in ccRCC, resulting in VHL complex degradation and HIFα stabilization. HIF-2α, constitutively activated in ccRCC, is mainly expressed in renal, lung, hepatic, and endothelial cells. Although transcription factors are difficult to target, a novel HIF-2α inhibitor PT2399 was recently developed which prevents binding of HIF-2α to ARNT/HIF-1β to activate a HIF-responsive promoter. Treatment with a HIF-2α inhibitor was investigated by grafting human ccRCC cell lines or patient tumor cells into nude mice, and by inactivating vhl in a zebrafish model.
Specific HIF-2α inhibition resulted in tumor regression in a subset of ccRCC cell line xenografts in mouse models of primary and metastatic ccRCC, in 10 of 18 patient-derived RCC xenografts in mice, and a patient with extensively pretreated metastatic ccRCC, who remained progression free for 11 months. Sensitivity to PT2399 correlated with a higher level of HIF-2α expression, and the presence of p53 (104, 105). Clinical trials with the HIF-2α inhibitor PT2385 are currently in progress (NCT02293980 and NCT03108066). Given the diversity of response, predictive biomarkers, such as HIF-2α and p53, may be useful for effective, targeted treatment. In zebrafish, treatment of vhl−/− embryos with a specific HIF2α inhibitor rescued pronephric abnormalities similar to human precancerous disease (38).
Conclusion
Genomic sequencing has revolutionized the understanding of ccRCC by identifying multiple driver genes beyond VHL. Genetically engineered animal models to investigate combinations of VHL, epigenetic, and other genes provide a powerful preclinical model for elucidating the biology of ccRCC, developing novel combinatorial therapies, and identifying candidate biomarkers for clinical validation. In particular, uncovering the molecular basis driving tumor heterogeneity and the role of epigenetic genes will identify new pathways for intervention. Insights from these model systems will be clinically applicable to both hereditary and sporadic ccRCCs.
Surveillance and surgery remain standard of care in early ccRCC, while ablative therapies provide options for alternative treatment. For early ccRCC, current recommendations for intervention based solely on size could be better informed by prognostic tumor markers indicating a potential for aggressive growth, progression, and metastasis. Non-invasive biomarkers in blood or urine would be ideal for surveillance to avoid tissue biopsy. For advanced disease, a multiagent approach is supported by both clinical and preclinical observations, and ongoing clinical trials are currently in progress to evaluate treatment regimens and prognostic genomic biomarkers. In the future, genomic profiling is likely to be augmented by transcriptional and metabolomics analysis, as well as DNA methylation status. The efficacy of targeted therapy informed by tumor profiling may be limited by intratumor and intertumor heterogeneity. Immunotherapy has the potential to circumvent the high mutational load; clinical trials are in progress with single agent and multimodal therapy, targeting multiple signaling pathways or enhancing the immune response with stereotactic radiation.
Author Contributions
Both authors made substantial contribution to the work and approved the final version for publication.
Conflict of Interest Statement
The authors declare the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The article processing charge was funded by the German Research Foundation (DFG) and the University of Freiburg in the funding programme Open Access Publishing.
References
1. von Hippel E. Über eine sehr seltene Erkrankung der Netzhaut. Graefe Arch Opthalmol (1904) 59:83–106. doi:10.1007/BF01994821
2. Lindau A. Zur Frage der Angiomatosis Retinae und Ihrer Hirncomplikation. Acta Ophthalmol (1927) 4:193–226. doi:10.1111/j.1755-3768.1926.tb07786.x
4. Neumann HP, Wiestler OD. Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet (1991) 337:1052–4. doi:10.1016/0140-6736(91)91705-Y
5. Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat (1995) 5:66–75. doi:10.1002/humu.1380050109
6. Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG Jr. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet (2001) 10:1019–27. doi:10.1093/hmg/10.10.1019
7. Seizinger BR, Rouleau GA, Ozelius LJ, Lane AH, Farmer GE, Lamiell JM, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature (1988) 332:268–9. doi:10.1038/332268a0
8. Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science (1993) 260:1317–20. doi:10.1126/science.8493574
9. Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, Harris R, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet (1991) 28:443–7. doi:10.1136/jmg.28.7.443
10. Binderup ML, Bisgaard ML, Harbud V, Moller HU, Gimsing S, Friis-Hansen L, et al. Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition. Dan Med J (2013) 60:B4763.
11. Verein_Vhl-Betroffener_Familien_E.V. (2017). Available: http://www.hippel-lindau.de/
12. VHLAlliance. (2017). Available: www.vhl.org
13. Walther MM, Lubensky IA, Venzon D, Zbar B, Linehan WM. Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: clinical implications. J Urol (1995) 154:2010–4; discussion 2014–5. doi:10.1097/00005392-199512000-00007
14. Jilg CA, Neumann HP, Glasker S, Schafer O, Ardelt PU, Schwardt M, et al. Growth kinetics in von Hippel-Lindau-associated renal cell carcinoma. Urol Int (2012) 88:71–8. doi:10.1159/000333348
15. Schoenfeld AR, Davidowitz EJ, Burk RD. Elongin BC complex prevents degradation of von Hippel-Lindau tumor suppressor gene products. Proc Natl Acad Sci U S A (2000) 97:8507–12. doi:10.1073/pnas.97.15.8507
16. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet (2013) 45:860–7. doi:10.1038/ng.2699
17. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature (2010) 463:360–3. doi:10.1038/nature08672
18. Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet (2011) 44:17–9. doi:10.1038/ng.1014
19. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature (2011) 469:539–42. doi:10.1038/nature09639
20. Pena-Llopis S, Vega-Rubin-De-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet (2012) 44:751–9. doi:10.1038/ng.2323
21. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature (2013) 499:43–9. doi:10.1038/nature12222
22. van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet (2009) 41:521–3. doi:10.1038/ng.349
23. Hakimi AA, Chen YB, Wren J, Gonen M, Abdel-Wahab O, Heguy A, et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur Urol (2013) 63:848–54. doi:10.1016/j.eururo.2012.09.005
24. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med (2012) 366:883–92. doi:10.1056/NEJMoa1113205
25. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature (2013) 499:214–8. doi:10.1038/nature12213
26. Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet (2014) 46:225–33. doi:10.1038/ng.2891
27. Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell (2002) 1:459–68. doi:10.1016/S1535-6108(02)00071-5
28. Fisher R, Horswell S, Rowan A, Salm MP, De Bruin EC, Gulati S, et al. Development of synchronous VHL syndrome tumors reveals contingencies and constraints to tumor evolution. Genome Biol (2014) 15:433. doi:10.1186/s13059-014-0433-z
29. Fei SS, Mitchell AD, Heskett MB, Vocke CD, Ricketts CJ, Peto M, et al. Patient-specific factors influence somatic variation patterns in von Hippel-Lindau disease renal tumours. Nat Commun (2016) 7:11588. doi:10.1038/ncomms11588
30. Crossey PA, Richards FM, Foster K, Green JS, Prowse A, Latif F, et al. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet (1994) 3:1303–8. doi:10.1093/hmg/3.8.1303
31. Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet (2001) 10:1029–38. doi:10.1093/hmg/10.10.1029
32. Stoletov K, Klemke R. Catch of the day: zebrafish as a human cancer model. Oncogene (2008) 27:4509–20. doi:10.1038/onc.2008.95
33. Konantz M, Balci TB, Hartwig UF, Dellaire G, Andre MC, Berman JN, et al. Zebrafish xenografts as a tool for in vivo studies on human cancer. Ann N Y Acad Sci (2012) 1266:124–37. doi:10.1111/j.1749-6632.2012.06575.x
34. Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A, et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci U S A (1997) 94:9102–7. doi:10.1073/pnas.94.17.9102
35. Rankin EB, Tomaszewski JE, Haase VH. Rend cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res (2006) 66:2576–83. doi:10.1158/0008-5472.CAN-05-3241
36. Fu L, Wang G, Shevchuk MM, Nanus DM, Gudas LJ. Generation of a mouse model of von Hippel-Lindau kidney disease leading to renal cancers by expression of a constitutively active mutant of HIF1alpha. Cancer Res (2011) 71:6848–56. doi:10.1158/0008-5472.CAN-11-1745
37. Fu L, Wang G, Shevchuk MM, Nanus DM, Gudas LJ. Activation of HIF2alpha in kidney proximal tubule cells causes abnormal glycogen deposition but not tumorigenesis. Cancer Res (2013) 73:2916–25. doi:10.1158/0008-5472.CAN-12-3983
38. Noonan HR, Metelo AM, Kamei CN, Peterson RT, Drummond IA, Iliopoulos O. Loss of vhl in the zebrafish pronephros recapitulates early stages of human clear cell renal cell carcinoma. Dis Model Mech (2016) 9:873–84. doi:10.1242/dmm.024380
39. Gu YF, Cohn S, Christie A, Mckenzie T, Wolff N, Do QN, et al. Modeling renal cell carcinoma in mice: Bap1 and Pbrm1 inactivation drive tumor grade. Cancer Discov (2017) 7:900–17. doi:10.1158/2159-8290.CD-17-0292
40. Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, Xie Y, et al. The SWI/SNF protein PBRM1 restrains VHL-loss-driven clear cell renal cell carcinoma. Cell Rep (2017) 18:2893–906. doi:10.1016/j.celrep.2017.02.074
41. Harlander S, Schonenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L, et al. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat Med (2017) 23:869–77. doi:10.1038/nm.4343
42. Duffey BG, Choyke PL, Glenn G, Grubb RL, Venzon D, Linehan WM, et al. The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J Urol (2004) 172:63–5. doi:10.1097/01.ju.0000132127.79974.3f
43. Jilg CA, Neumann HP, Glasker S, Schafer O, Leiber C, Ardelt PU, et al. Nephron sparing surgery in von Hippel-Lindau associated renal cell carcinoma; clinicopathological long-term follow-up. Fam Cancer (2012) 11:387–94. doi:10.1007/s10689-012-9525-7
44. Ploussard G, Droupy S, Ferlicot S, Ples R, Rocher L, Richard S, et al. Local recurrence after nephron-sparing surgery in von Hippel-Lindau disease. Urology (2007) 70:435–9. doi:10.1016/j.urology.2007.04.040
45. Matin SF, Ahrar K, Wood CG, Daniels M, Jonasch E. Patterns of intervention for renal lesions in von Hippel-Lindau disease. BJU Int (2008) 102:940–5. doi:10.1111/j.1464-410X.2008.07718.x
46. Joly D, Mejean A, Correas JM, Timsit MO, Verkarre V, Deveaux S, et al. Progress in nephron sparing therapy for renal cell carcinoma and von Hippel-Lindau disease. J Urol (2011) 185:2056–60. doi:10.1016/j.juro.2011.02.007
47. Roupret M, Hopirtean V, Mejean A, Thiounn N, Dufour B, Chretien Y, et al. Nephron sparing surgery for renal cell carcinoma and von Hippel-Lindau’s disease: a single center experience. J Urol (2003) 170:1752–5. doi:10.1097/01.ju.0000092780.85876.de
48. Chittiboina P, Lonser RR. Von Hippel-Lindau disease. Handb Clin Neurol (2015) 132:139–56. doi:10.1016/B978-0-444-62702-5.00010-X
49. Go AS, Chertow GM, Fan D, Mcculloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med (2004) 351:1296–305. doi:10.1056/NEJMoa041031
50. Rocco MV, Frankenfield DL, Hopson SD, Mcclellan WM. Relationship between clinical performance measures and outcomes among patients receiving long-term hemodialysis. Ann Intern Med (2006) 145:512–9. doi:10.7326/0003-4819-145-7-200610030-00009
51. Ljungberg B, Bensalah K, Canfield S, Dabestani S, Hofmann F, Hora M, et al. EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol (2015) 67:913–24. doi:10.1016/j.eururo.2015.01.005
52. Johnson A, Sudarshan S, Liu J, Linehan WM, Pinto PA, Bratslavsky G. Feasibility and outcomes of repeat partial nephrectomy. J Urol (2008) 180:89–93; discussion 93. doi:10.1016/j.juro.2008.03.030
53. Psutka SP, Feldman AS, Mcdougal WS, Mcgovern FJ, Mueller P, Gervais DA. Long-term oncologic outcomes after radiofrequency ablation for T1 renal cell carcinoma. Eur Urol (2013) 63:486–92. doi:10.1016/j.eururo.2012.08.062
54. Olweny EO, Park SK, Tan YK, Best SL, Trimmer C, Cadeddu JA. Radiofrequency ablation versus partial nephrectomy in patients with solitary clinical T1a renal cell carcinoma: comparable oncologic outcomes at a minimum of 5 years of follow-up. Eur Urol (2012) 61:1156–61. doi:10.1016/j.eururo.2012.01.001
55. Allasia M, Soria F, Battaglia A, Gazzera C, Calandri M, Caprino MP, et al. Radiofrequency ablation for renal cancer in von Hippel-Lindau syndrome patients: a prospective cohort analysis. Clin Genitourin Cancer (2017). doi:10.1016/j.clgc.2017.07.027
56. Zagoria RJ, Pettus JA, Rogers M, Werle DM, Childs D, Leyendecker JR. Long-term outcomes after percutaneous radiofrequency ablation for renal cell carcinoma. Urology (2011) 77:1393–7. doi:10.1016/j.urology.2010.12.077
57. Iannuccilli JD, Dupuy DE, Beland MD, Machan JT, Golijanin DJ, Mayo-Smith WW. Effectiveness and safety of computed tomography-guided radiofrequency ablation of renal cancer: a 14-year single institution experience in 203 patients. Eur Radiol (2016) 26:1656–64. doi:10.1007/s00330-015-4006-7
58. Goldberg SN, Gazelle GS, Mueller PR. Thermal ablation therapy for focal malignancy: a unified approach to underlying principles, techniques, and diagnostic imaging guidance. AJR Am J Roentgenol (2000) 174:323–31. doi:10.2214/ajr.174.2.1740323
59. Li X, Liang P, Yu J, Yu XL, Liu FY, Cheng ZG, et al. Role of contrast-enhanced ultrasound in evaluating the efficiency of ultrasound guided percutaneous microwave ablation in patients with renal cell carcinoma. Radiol Oncol (2013) 47:398–404. doi:10.2478/raon-2013-0038
60. Moreland AJ, Ziemlewicz TJ, Best SL, Hinshaw JL, Lubner MG, Alexander ML, et al. High-powered microwave ablation of t1a renal cell carcinoma: safety and initial clinical evaluation. J Endourol (2014) 28:1046–52. doi:10.1089/end.2014.0190
61. Yu J, Zhang G, Liang P, Yu XL, Cheng ZG, Han ZY, et al. Midterm results of percutaneous microwave ablation under ultrasound guidance versus retroperitoneal laparoscopic radial nephrectomy for small renal cell carcinoma. Abdom Imaging (2015) 40:3248–56. doi:10.1007/s00261-015-0500-2
62. Gao Y, Liang P, Yu X, Yu J, Cheng Z, Han Z, et al. Microwave treatment of renal cell carcinoma adjacent to renal sinus. Eur J Radiol (2016) 85:2083–9. doi:10.1016/j.ejrad.2016.09.018
63. Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18-month follow-up. Urology (2011) 77:792–7. doi:10.1016/j.urology.2010.12.028
64. Uchida M, Imaide Y, Sugimoto K, Uehara H, Watanabe H. Percutaneous cryosurgery for renal tumours. Br J Urol (1995) 75:132–6; discussion 136–7. doi:10.1111/j.1464-410X.1995.tb07297.x
65. Ko YH, Choi H, Kang SG, Park HS, Lee JG, Kim JJ, et al. Efficacy of laparoscopic renal cryoablation as an alternative treatment for small renal mass in patients with poor operability: experience from the Korean single center. J Laparoendosc Adv Surg Tech A (2010) 20:339–45. doi:10.1089/lap.2009.0431
66. Tanagho YS, Bhayani SB, Kim EH, Figenshau RS. Renal cryoablation versus robot-assisted partial nephrectomy: Washington University long-term experience. J Endourol (2013) 27:1477–86. doi:10.1089/end.2013.0192
67. Georgiades CS, Rodriguez R. Efficacy and safety of percutaneous cryoablation for stage 1A/B renal cell carcinoma: results of a prospective, single-arm, 5-year study. Cardiovasc Intervent Radiol (2014) 37:1494–9. doi:10.1007/s00270-013-0831-8
68. Park BK, Kim CK, Park SY, Shen SH. Percutaneous radiofrequency ablation of renal cell carcinomas in patients with von Hippel Lindau disease: indications, techniques, complications, and outcomes. Acta Radiol (2013) 54:418–27. doi:10.1177/0284185113475441
69. Yoost TR, Clarke HS, Savage SJ. Laparoscopic cryoablation of renal masses: which lesions fail? Urology (2010) 75:311–4. doi:10.1016/j.urology.2009.09.038
70. Breen DJ, Bryant TJ, Abbas A, Shepherd B, Mcgill N, Anderson JA, et al. Percutaneous cryoablation of renal tumours: outcomes from 171 tumours in 147 patients. BJU Int (2013) 112:758–65. doi:10.1111/bju.12122
71. Yamanaka T, Yamakado K, Yamada T, Fujimori M, Takaki H, Nakatsuka A, et al. CT-guided percutaneous cryoablation in renal cell carcinoma: factors affecting local tumor control. J Vasc Interv Radiol (2015) 26:1147–53. doi:10.1016/j.jvir.2015.04.031
72. Kim HK, Pyun JH, Kim JY, Kim SB, Cho S, Kang SG, et al. Renal cryoablation of small renal masses: a Korea University experience. Korean J Urol (2015) 56:117–24. doi:10.4111/kju.2015.56.2.117
73. Lax I, Blomgren H, Naslund I, Svanstrom R. Stereotactic radiotherapy of malignancies in the abdomen. Methodological aspects. Acta Oncol (1994) 33:677–83. doi:10.3109/02841869409121782
74. Lo SS, Fakiris AJ, Chang EL, Mayr NA, Wang JZ, Papiez L, et al. Stereotactic body radiation therapy: a novel treatment modality. Nat Rev Clin Oncol (2010) 7:44–54. doi:10.1038/nrclinonc.2009.188
75. Onufrey V, Mohiuddin M. Radiation therapy in the treatment of metastatic renal cell carcinoma. Int J Radiat Oncol Biol Phys (1985) 11:2007–9. doi:10.1016/0360-3016(85)90285-8
76. Deschavanne PJ, Fertil B. A review of human cell radiosensitivity in vitro. Int J Radiat Oncol Biol Phys (1996) 34:251–66. doi:10.1016/0360-3016(95)02029-2
77. DiBiase SJ, Valicenti RK, Schultz D, Xie Y, Gomella LG, Corn BW. Palliative irradiation for focally symptomatic metastatic renal cell carcinoma: support for dose escalation based on a biological model. J Urol (1997) 158:746–9. doi:10.1097/00005392-199709000-00013
78. Beitler JJ, Makara D, Silverman P, Lederman G. Definitive, high-dose-per-fraction, conformal, stereotactic external radiation for renal cell carcinoma. Am J Clin Oncol (2004) 27:646–8. doi:10.1097/01.coc.0000145289.57705.07
79. Wersall PJ, Blomgren H, Lax I, Kalkner KM, Linder C, Lundell G, et al. Extracranial stereotactic radiotherapy for primary and metastatic renal cell carcinoma. Radiother Oncol (2005) 77:88–95. doi:10.1016/j.radonc.2005.03.022
80. Svedman C, Karlsson K, Rutkowska E, Sandstrom P, Blomgren H, Lax I, et al. Stereotactic body radiotherapy of primary and metastatic renal lesions for patients with only one functioning kidney. Acta Oncol (2008) 47:1578–83. doi:10.1080/02841860802123196
81. Asthagiri AR, Mehta GU, Zach L, Li X, Butman JA, Camphausen KA, et al. Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel-Lindau disease. Neuro Oncol (2010) 12:80–6. doi:10.1093/neuonc/nop018
82. Siva S, Pham D, Gill S, Corcoran NM, Foroudi F. A systematic review of stereotactic radiotherapy ablation for primary renal cell carcinoma. BJU Int (2012) 110:E737–43. doi:10.1111/j.1464-410X.2012.11550.x
83. De Meerleer G, Khoo V, Escudier B, Joniau S, Bossi A, Ost P, et al. Radiotherapy for renal-cell carcinoma. Lancet Oncol (2014) 15:e170–7. doi:10.1016/S1470-2045(13)70569-2
84. Siva S, Jackson P, Kron T, Bressel M, Lau E, Hofman M, et al. Impact of stereotactic radiotherapy on kidney function in primary renal cell carcinoma: establishing a dose-response relationship. Radiother Oncol (2016) 118:540–6. doi:10.1016/j.radonc.2016.01.027
85. Staehler M, Bader M, Schlenker B, Casuscelli J, Karl A, Roosen A, et al. Single fraction radiosurgery for the treatment of renal tumors. J Urol (2015) 193:771–5. doi:10.1016/j.juro.2014.08.044
86. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med (2007) 356:125–34. doi:10.1056/NEJMoa060655
87. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med (2007) 356:115–24. doi:10.1056/NEJMoa065044
88. Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol (2010) 28:1061–8. doi:10.1200/JCO.2009.23.9764
89. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet (2011) 378:1931–9. doi:10.1016/S0140-6736(11)61613-9
90. Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med (2003) 349:427–34. doi:10.1056/NEJMoa021491
91. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor, et al. Temsirolimus, Interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med (2007) 356:2271–81. doi:10.1056/NEJMoa066838
92. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer (2010) 116:4256–65. doi:10.1002/cncr.25219
93. Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373:1814–23. doi:10.1056/NEJMoa1510016
94. Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol (2016) 17:917–27. doi:10.1016/S1470-2045(16)30107-3
95. Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol (2015) 16:1473–82. doi:10.1016/S1470-2045(15)00290-9
96. Motzer RJ, Hutson TE, Ren M, Dutcus C, Larkin J. Independent assessment of lenvatinib plus everolimus in patients with metastatic renal cell carcinoma. Lancet Oncol (2016) 17:e4–5. doi:10.1016/S1470-2045(15)00543-4
97. Motzer RJ, Escudier B, Mcdermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373:1803–13. doi:10.1056/NEJMoa1510665
98. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520:373–7. doi:10.1038/nature14292
99. Wersall PJ, Blomgren H, Pisa P, Lax I, Kalkner KM, Svedman C. Regression of non-irradiated metastases after extracranial stereotactic radiotherapy in metastatic renal cell carcinoma. Acta Oncol (2006) 45:493–7. doi:10.1080/02841860600604611
100. Choueiri TK, Halabi S, Sanford BL, Hahn O, Michaelson MD, Walsh MK, et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: the alliance A031203 CABOSUN trial. J Clin Oncol (2017) 35:591–7. doi:10.1200/JCO.2016.70.7398
101. Ravaud A, Motzer RJ, Pandha HS, George DJ, Pantuck AJ, Patel A, et al. Adjuvant sunitinib in high-risk renal-cell carcinoma after nephrectomy. N Engl J Med (2016) 375:2246–54. doi:10.1056/NEJMoa1611406
102. Kwiatkowski DJ, Choueiri TK, Fay AP, Rini BI, Thorner AR, De Velasco G, et al. Mutations in TSC1, TSC2, and mTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res (2016) 22:2445–52. doi:10.1158/1078-0432.CCR-15-2631
103. Hsieh JJ, Chen D, Wang PI, Marker M, Redzematovic A, Chen YB, et al. Genomic biomarkers of a randomized trial comparing first-line everolimus and sunitinib in patients with metastatic renal cell carcinoma. Eur Urol (2017) 71:405–14. doi:10.1016/j.eururo.2016.10.007
104. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature (2016) 539:112–7. doi:10.1038/nature19796
Keywords: von Hippel–Lindau disease, renal cell carcinoma, cancer genetics, predictive biomarkers, preclinical models, new therapeutic targets
Citation: Kim E and Zschiedrich S (2018) Renal Cell Carcinoma in von Hippel–Lindau Disease—From Tumor Genetics to Novel Therapeutic Strategies. Front. Pediatr. 6:16. doi: 10.3389/fped.2018.00016
Received: 20 October 2017; Accepted: 16 January 2018;
Published: 09 February 2018
Edited by:
Max Christoph Liebau, Universitätsklinikum Köln, GermanyCopyright: © 2018 Kim and Zschiedrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Zschiedrich, stefan.zschiedrich@uniklinik-freiburg.de