Systemic Epstein–Barr Virus-Positive T/NK Lymphoproliferative Diseases With SH2D1A/XIAP Hypomorphic Gene Variants
 Masataka Ishimura1*
Masataka Ishimura1*  Katsuhide Eguchi1 Akira Shiraishi1 Motoshi Sonoda1 Yoshihiro Azuma2 Hiroyuki Yamamoto1 Ken-ichi Imadome3 Shouichi Ohga1
Katsuhide Eguchi1 Akira Shiraishi1 Motoshi Sonoda1 Yoshihiro Azuma2 Hiroyuki Yamamoto1 Ken-ichi Imadome3 Shouichi Ohga1- 1Department of Pediatrics, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan
- 2Department of Pediatrics, Yamaguchi University, Ube, Japan
- 3Division of Advanced Medicine for Virus Infections, National Research Institute for Child Health and Development, Tokyo, Japan
X-linked lymphoproliferative disease (XLP) is one of the X-linked primary immunodeficiency diseases (PIDs) with defective immune response to Epstein–Barr virus (EBV) infection. Chronic active EBV infection (CAEBV) and EBV-hemophagocytic lymphohistiocytosis (HLH) are recognized as systemic EBV-positive T-cell and natural killer (NK)-cell lymphoproliferative diseases (LPDs) arising from the clonal proliferations of EBV-infected T cells and NK cells. A high incidence of CAEBV in East Asia implies the unknown genetic predisposition. In patients with XLP, EBV-infected cells are generally B cells. No mutation of SH2D1A/XIAP genes has ever been identified in patients with systemic EBV-positive T-cell and NK-cell LPD. We report herewith a male case of NK-cell type CAEBV with SH2D1A hypomorphic mutation (c.7G > T, p.Ala3Ser), two male cases of CAEBV/EBV-HLH with XIAP hypomorphic variant (c.1045_1047delGAG, p.Glu349del), and another female case of CD4+CAEBV with the same XIAP variant. The female underwent bone marrow transplantation from an HLA-matched sister with the XIAP variant and obtained a complete donor chimerism and a cure of laryngeal LPD lesion, but then suffered from donor-derived CD4+ T cell EBV-LPD. These observations demonstrated that SH2D1A and XIAP genes are critical for the complete regulation of EBV-positive T/NK cell LPD. X-linked lymphoproliferative disease (XLP) is one of the X-linked primary immunodeficiency diseases (PIDs) reported to have a defective immune response to Epstein–Barr virus (EBV) infection. Mutations in SH2D1A and XIAP genes cause XLP. Systemic EBV-positive T-cell and natural killer (NK)-cell lymphoproliferative diseases (LPDs) consist of three major types: EBV-positive hemophagocytic lymphohistiocytosis (HLH), chronic active EBV infection (CAEBV), and EBV-positive T-cell/NK-cell lymphoma. CAEBV is recognized as a poor prognostic disease of EBV-associated T-cell and NK-cell LPD arising from the clonal proliferation of EBV-infected T cells (CD4+, CD8+, and TCRγδ+) and/or NK cells. The majority of cases with CAEBV were reported from East Asia and South America. In Caucasian patients with CAEBV disease, the target of infection is exclusively B cells. These imply a genetic predisposition to EBV-positive T/NK cell LPD according to ethnicity. In reported cases with XLP, EBV-infected cells are B cells. On the other hand, no mutation of SH2D1A/XIAP genes have been determined in patients with T/NK-cell-type (Asian type) CAEBV. We here describe, for the first time, four case series of CAEBV/EBV-HLH patients who carried the hypomorphic variants of XLP-related genes. These cases included a male patient with CAEBV carrying SH2D1A hypomorphic mutation (c.7G > T, p.Ala3Ser) and two male patients with CAEBV/EBV-HLH carrying the XIAP hypomorphic variant (c.1045_1047delGAG, p.Glu349del), along with another female patient with CAEBV carrying the same XIAP variant. The female case underwent bone marrow transplantation from a healthy HLA-matched sister having the same XIAP variant. Although a complete donor chimerism was achieved with the resolution of laryngeal LPD lesions, systemic donor-derived CD4+ T-cell EBV-LPD developed during the control phase of intractable graft- vs. -host-disease. These observations demonstrated that SH2D1A and XIAP genes are critical for the complete regulation of systemic EBV-positive T/NK-cell LPD.
Introduction
Epstein–Barr virus (EBV) infects preferentially human B lymphocytes and epithelial cells. The majority of subjects are asymptomatic after a primary infection with EBV, and a part of them suffer from acute infectious mononucleosis (IM). EBV-positive T-cell and natural killer (NK)-cell lymphoproliferative diseases (LPDs) are classified into three major types: EBV-positive hemophagocytic lymphohistiocytosis (HLH), chronic active EBV infection (CAEBV), and EBV-positive T-cell/NK-cell lymphoma (1). CAEBV is a rare persistent active mononucleosis syndrome presenting with fever, liver dysfunction, cytopenias, and hepatoplenomegaly. The patients occasionally progress to the lethal course of HLH, LPD, and lymphoma. CAEBV is currently recognized as EBV-associated T/NK-cell LPD arising from the clonal proliferations of EBV-infected T cells (CD4+, CD8+, and TCRγδ+) and/or NK cells. The majority of cases were reported from East Asia and South America. The reported cases of chronic EBV disease in the United States were exclusively B-cell-type (Caucasian type) CAEBV (2). These may account for the genetic predisposition to T/NK-cell-type (Asian type) CAEBV (3). Recently, Okuno et al. (4) have reported that somatic mutations of infected cells and gene mutations of EBV were concurrently involved in the development of T/NK cell type (Asian type) CAEBV, which were considered to be similar to malignant lymphoma. Allogenic hematopoietic stem cell plantations (HSTs) are needed for curing of progressive CAEBV (5). On the other hand, there is little information on the prolonged survival of patients with indolent CAEBV.
X-linked lymphoproliferative disease (XLP) is one of the X-linked primary immunodeficiency diseases (PIDs) with defective immune response to EBV infection (6). The manifestations of XLP are typified by EBV-driven fatal IM and/or HLH, regenerative anemia, dysgammaglobulinemia, and B-cell lymphoma. In cases of XLP-related EBV-HLH, EBV-infected cells are predominantly B cells (7, 8). Currently, two causative genes have been determined for XLP1 and XLP2: SLAM-associated protein (SAP) deficiency due to SH2D1A gene mutation called XLP1 and XIAP (X-linked inhibitor of apoptosis) deficiency due to XIAP gene (previously termed BIRC4) mutation called XLP2. SAP is composed almost exclusively of an Src homology 2 (SH2) domain (9). SAP expressed in T cells, NK cells, and natural killer T (NKT) cells enhances immune response binding to signaling lymphocyte activation molecule (SLAM) families. In XLP1 patients, decreased cytotoxic activities in CD8+ T cells and NK cells are associated with the developing risk of HLH. In addition, the lack of invariant NKT (iNKT) cells leads to a nonexcludability of EBV-infected B cells and subsequent development of fatal IM or B-cell lymphoma. XIAP is a member of the inhibitor of apoptosis protein (IAP) family containing three baculovirus IAP repeat (BIR) domains and one really interesting new gene (RING) domain. XIAP regulates apoptotic cell death with the inhibition of procaspase 9 by BIR3 domain and the inhibition of caspase 3 and 7 by BIR2 domain (10). The RING domain ubiquitylates receptor-interacting serine/threonine-protein kinase 2 and recruits linear ubiquitin chain assembly complex (LUBAC) to nucleotide-binding oligomerization domain 2 (NOD2). LUBAC activity is required for efficient NF-κB activation and secretion of proinflammatory cytokines after NOD2 stimulation (11). Increased activation-induced cell death (AICD) in T cells and a decreased number of iNKT cells are found in XIAP-deficient patients (12). XLP2 patients with a mutation in the RING domain exhibit interference with ubiquitin ligase activity (11). The symptoms start usually in early childhood with recurrent infection, with HLH (frequently recurrent and of a more indolent course than seen in other primary HLH diseases) associated with chronic EBV disease, splenomegaly, and chronic colitis. In contrast to XLP1, B-cell lymphomas have not been reported in XLP2 patients (13).
We herein describe four novel cases of systemic EBV-related T/NK-LPD having the hypomorphic variants of SH2D1A/XIAP and discuss their association.
Methods
Genetic Analysis
Genomic DNA was extracted from peripheral blood and/or biopsied samples of the lesion obtained from patients according to the standard method, after informed consent was obtained from the individuals or parents. Mutation analysis of the genes responsible for familial HLH (PRF1, UNC13D, STX11, and STXBP2) and XLP or XLP-like (SH2D1A, XIAP, ITK, and CD27) was performed by a PCR-assisted DNA sequencing method with a capillary DNA sequencer (ABI3130/3730, Applied Biosystems, Foster City, CA, USA) in all cases, and the whole exome sequencing, using a short-read next-generation DNA sequencer (MiSeq, Illumina, San Diego, CA, USA) as described previously (14) in case 1 and case 4.
EBV Analysis
Analysis of EBV gene expression by real time-PCR was carried out as previously described (15). Briefly, peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation on a Lymphosepar I (Immuno-Biological Laboratories Co., Ltd., Gunma, Japan). Then, CD19+, CD4+, CD8+, and CD56+ cells were serially removed from PBMCs using the IMag Cell Separation System (BD Biosciences, Franklin Lakes, NJ, USA). DNA extraction was performed for each fraction, and quantification of EBV-DNA was performed with real-time quantitative PCR based on the TaqMan system (Applied Biosystems, Foster City, CA, USA).
Clinical Case Reports
Case 1: Male Patient With NK-Cell-Type CAEBV
An 8-years-old boy presented with high fever, photosensitivity, and hypersensitivity to mosquito bites and then received the diagnosis of NK-cell-type CAEBV. These manifestations have gradually relieved until 12 years of age. The comprehensive genetic analysis of peripheral blood-derived DNA revealed one reported pathological mutation of SH2D1A gene hemizygously (c. 7G > T, p.Ala3Ser) (16, 17). During the following 13 years, he has continued to have photosensitivity alone. Repeated laboratory tests have shown unremarkable titers of anti-EBV antibodies indicating past infection and low titer of EBV genome copies in peripheral blood (7.3 × 102/ml), with no any evidence of cytopenia, dysgammagulobulinemia, or elevation in soluble interleukin (IL)-2 receptor.
Case 2: Male Patient With NK/B-Cell-Type CAEBV
A 2-years-old boy had suffered from intermittent fever, diarrhea, and hypersensitivity to mosquito bites. An EBV genome load was high in CD19+ B cells (5.6 × 103 copies/μgDNA) and slightly positive levels in CD16+ NK cells (8.1 × 101 copies/μgDNA). The comprehensive genetic analysis of peripheral blood-derived DNA determined a reported hemizygous variant of XIAP gene (c.1045_1047delGAG, p.Glu349del) (7, 8). NK cell activity was 18 %lysis (reference range; 18–40). After the diagnosis of chronic EBV+B-LPD, four courses of anti-CD20 antibody (Rituxan®, Chugai Pharmaceutical Co., LTD., Tokyo, Japan) therapies led to a complete disappearance of the EBV genome in circulation and an improvement in hypersensitivity to mosquito bites. Six months after rituximab therapies, a reappearance of B cells in the peripheral blood without the detection of EBV genome indicated the eradication of EBV-B-LPD. However, EBV genome level was again positive (1.5 × 103 copies/μgDNA of whole peripheral blood) 10 months after rituximab therapy, but there were no symptoms or abnormal data, including immunoglobulin levels, in the follow-up screening tests.
Case 3: Male Patient With CD8+ T-Cell-Type EBV-HLH
A 1-year-old boy developed fever, skin eruptions, and hepatoplenomegaly with pancytopenia, hyperferritinemia (5,181 ng/ml), and elevated soluble IL-2 receptor (6,797 U/ml). Anti-EBV antibodies indicated a primary infection of EBV. High EBV loads in peripheral blood and CD8+ T cells of the patient (1 × 105 copies/ml and 1 × 106 copies/μgDNA, respectively) led to the diagnosis of EBV-HLH. NK-cell activity was 30 %lysis in normal (reference range; 18–40). Additional two courses of etoposide injection (100 mg/m2) were needed to control the relapsing HLH after the immunomodulation therapy using high-dose intravenous immunoglobulin, oral cyclosporine, and prednisolone. Circulating levels of EBV genome came to be undetectable after the immunochemotherapy. The comprehensive genetic analysis of peripheral blood-derived DNA determined a hemizygous variant of the XIAP gene (c.1045_1047delGAG, p.Glu349del). He is alive and well, without sequelae or dysgammaglobulinemia at 7 years of age. The numbers of CD19+IgD−CD27+ switched memory B cells and CD4+CD45RA−CXCR5+ follicular helper T cells were not decreased (data not shown).
Case 4: Female Patient With NK/CD4+ T-Cell-Type CAEBV
A 24-years-old woman was hospitalized because of dyspnea and hoarseness. The patient had received the diagnosis of NK-cell and CD4+ T-cell-type CAEBV because of recurrent fever and hypersensitivity to mosquito bites at age 14 years. Histopathological and molecular studies of the cutaneous lesion indicated clonal proliferation of EBV-infected cells. Thereafter, clinical resolution and declining levels of EBV load in circulation had allowed no treatment and observation. After admission, an urgent tracheostomy prevented airway obstruction by the laryngeal mass (Figure 1A). She was then transferred to our hospital for further management. Fluorodeoxyglucose-positron emission tomography (FDG-PET) showed increased levels of uptake in the stomach and terminal ileum as well as the laryngeal lesion (Figure 1B). Circulating EBV DNA was at undetectable levels. However, histopathological and molecular analysis of the laryngeal lesions demonstrated a proliferation of EBER-positive CD4+ cells and increased copy number of EBV-DNA (2–4 × 103 copies/μgDNA). The comprehensive genetic analysis of peripheral blood-derived DNA identified a heterozygous variant of XIAP gene (c.1045_1047delGAG, p.Glu349del) alone. A histocompatible sister aged 20 years carried the same XIAP variant. The anti-EBV antibody titers and undetectable EBV DNA in circulation verified a past infection of EBV in the healthy sister. The gene expression analysis indicated no skewing inactivation of X chromosome among DNA samples obtained from the bone marrow cells, PBMCs, and laryngeal tumor of the patient as well as PBMC of the sister (data not shown). After four courses of combined chemotherapies with cyclophosphamide, pirarubicin, vincristine, steroid, and etoposide (CHOP-VP), the patient underwent bone marrow transplantation from the sister. The laryngeal lesion disappeared after a compete donor chimerism was achieved (Figure 1C). However, systemic but not local proliferation of EBV-infected donor-derived CD4+ T cells (1 × 104 copies/ml of whole peripheral blood and 3 × 103 copies/μgDNA of CD4+T cells, respectively) developed 2 months posttransplantation (Figure 1D). Discontinuation of immunosuppresants and donor lymphocyte infusions effectively controlled the posttransplantation LPD, but she died of uncontrollable severe graft-vs.-host-disease with Candida sepsis.
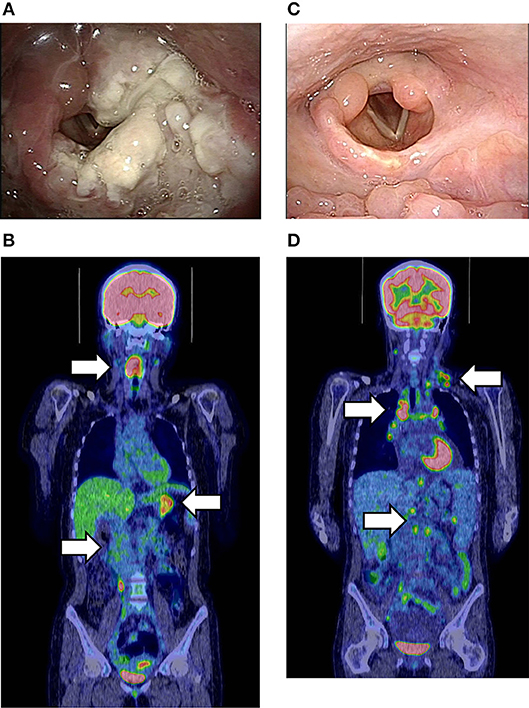
Figure 1. (A) Endoscopic findings of the laryngeal lesion of CD4+T-cell lymphoproliferative disease (LPD) lesion in case 4 prior to cancer chemotherapy. (B) Fluorodeoxyglucose-positron emission tomography (FDG-PET) at the onset of LPD. White arrows show the increased FDG uptake in the larynx, stomach, and terminal ileum. The maximum standardized uptake value (SUVmax) was 11.8. (C) Improvement of the laryngeal LPD lesion after bone marrow transplantation (BMT). (D) FDG-PET at the onset of donor-derived CD4+T-LPD after BMT. White arrows show the increased FDG uptake in multiple lymph nodes without laryngeal lesion. The SUVmax was 9.7.
None of the four patients had a positive family history suggesting PID and/or chronic EBV disease. The clinical profile and treatment course of these patients are summarized in Table 1.
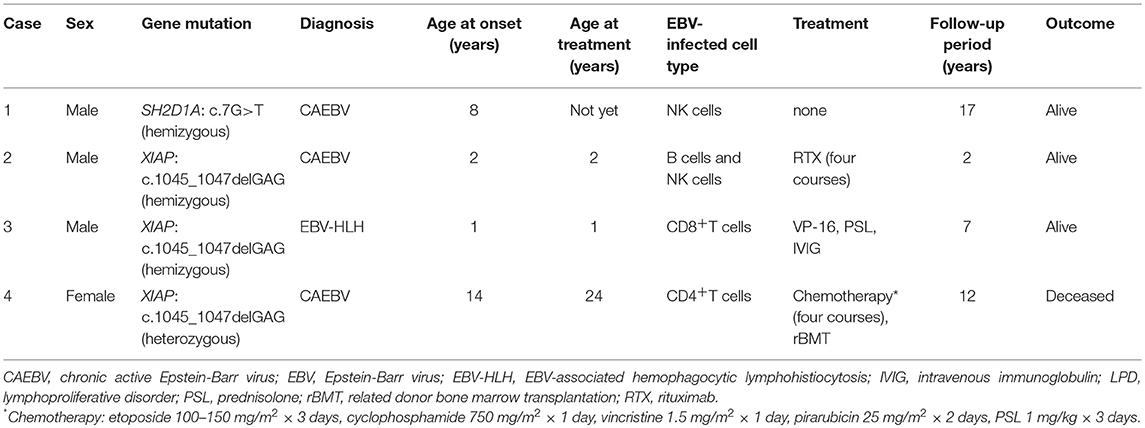
Table 1. Summary of the patients with SH2D1A/XIAP mutations who developed EBV-infected T/NK cell LPD.
Discussion
We report the first case series of CAEBV/EBV-HLH patients who carried the hypomorphic mutation of XLP-related genes. B cells are the major target of EBV infection in patients with XLP. No mutation in SH2D1A/XIAP genes has been reported in patients with CAEBV (4, 18). Patient 1 with SAP Ala3Ser and patients 2, 3, and 4 with XIAP Glu349del presented with the phenotype of NK+CAEBV, B+/NK+CAEBV, CD8+EBV-HLH, and NK+/CD4+CAEBV, respectively. EBV preferentially infects B cells via CD21 and also infects T cells or NK cells at a lower frequency during the acute phase of viremia (3). The absolute number of EBV-infected T/NK cells would be too small to have an advantage for proliferating in healthy individuals. On the other hand, SAP/XIAP variant carriers may have a modest ability to control the proliferation of EBV-infected T/NK cells.
Low levels of circulating EBV DNA continue to be detected in NK cells, but no symptoms other than photosensitivity have continued in patient 1 for 17 years. The SH2D1A gene variant (c.7G > T, p.Ala3Ser) is located at the N-terminal of the SAP protein, outside of the SH2 domain, with residual protein functions. This variant has no deleterious effect on the function depending on SH2 domain. A healthy elderly male carrier with c.7G > T SH2D1A variant indicates that the variant does not always develop XLP or lead to fatal outcomes for patients (17). However, one male patient with the same p.Ala3Ser variant demonstrated a marked reduction in SAP expression in CD8+ T cells (2.7%, reference range: 21.6–90.8%) and died at the age of 40 years with EBV infection and HLH (16). Another male patient with the hemizygous SH2D1A variant (c.7G > T) and heterozygous missense PRF1 mutation (c.127C > A, p.Leu43Met) reportedly suffered from severe HLH and required HST, and the other female with the heterozygous SH2D1A variant (c.7G > T) had a lethal HLH (19). The SH2D1A c.7G > T may be pathogenic in cases with late onset or indolent expression of disease.
The XIAP Glu349del (rs199683465) was reportedly detected in 3.5% of healthy Japanese people. The variant may be a single nucleotide polymorphism exerting the founder effect in Japanese people (20). Although the XIAP protein expression was normal in the Glu349del variant, the numbers of CD19+IgD−CD27+ switched memory B cells and CD4+CD45RA−CXCR5+ follicular helper T cells were decreased and immunoglobulin production was reduced in vitro. Immunoglobulin-related gene expression was also decreased in the variant carriers. On the other hand, they did not exhibit increased AICD because Glu349 is distant from the BIR2 or BIR3 domains. It remains unclear how the Glu349del mutation affects the NOD pathways (20).
Male patient 2, with persistent EBV infection in B/NK cells accompanied by hypersensitivity to mosquito bites, was diagnosed as having CAEBV. Rituximab therapy temporarily cleared EBV genome and all signs and symptoms. Although the EBV copy number was increased again, no symptoms recurred. Persistent EBV infection leads to the diagnosis of XLP2 without dysgammaglobulinemia or HLH. Patient 3 is the first Japanese individual who is a Glu349del carrier who developed EBV-HLH. No HLH developed in the three reported Japanese XLP2 patients with Glu349del (20). On the other hand, a 1-year-old Chinese male with XIAP Glu349del has been recently reported to present with HLH (21). Patient 3 suffered from severe CD8+EBV-HLH requiring chemotherapy, which did not recur after the first resolution. Unlike other XIAP mutations, the Glu349del variant would not lead to uncontrollable or relapsing HLH.
The major concern is the intractable course of patient 4 posttransplantation. How did a heterozygous XIAP variant affect the female patient? The XIAP variant is a genetic polymorphism found in 3.5% of healthy Japanese individuals. The healthy sister of patient 4 had experienced a primary infection of EBV. Therefore, heterozygous variant carriers are less likely to have acute fatal IM and/or chronic EBV diseases. X-linked recessive PID develops in females with X chromosome skewing (22, 23), but no skewing expression of XIAP was determined in the PBMC or LPD lesion of patient 4. EBV DNA levels did not increase in the peripheral blood even at the time of developing LPD after 10 years from the first CAEBV diagnosis. Additional factors such as somatic mutations and EBV genome mutations might contribute to the evolution of laryngeal tumor (4). Furthermore, as shown from her developing posttransplanted donor-derived EBV-related CD4+T-LPD, unknown host genetic predispositions other than XIAP or immunocompromised state of suppressing EBV-specific CTL activity might be involved in the onset of CAEBV (24). Somatic mutations of EBV-infected donor CD4+ T cells might also affect the developing posttransplantation EBV-T-LPD (4). Genetic backgrounds, including hypomorphic variants, may need to be considered in the selection of HST donors for the cure of systemic EBV-positive T/NK cell LPD.
Conclusions
We reported the first four case series of CAEBV/EBV-HLH with SH2D1A/XIAP gene variants. PID-related genetic predispositions to EBV infection should be considered for the treatment of EBV-T/NK cell LPD.
Ethics Statement
This study was carried out in accordance with the Declaration of Helsinki and the recommendations of the institutional review board of Kyushu University. The protocol was approved by the institutional review board of Kyushu University (#531-01). The subjects (or their parents) gave written informed consent about the study and the publication of this report.
Author Contributions
MI and SO were the principal investigators, taking primary responsibility for the paper. AS, KE, MS, and YA performed the clinical management with helpful discussion regarding the completion of the work. KI completed the EBV analysis. MS and HY completed the genetic analysis.
Funding
This work was supported in part by Practical Research Project for Rare/Intractable Diseases (15ek0109133) and Grant-in-Aids (15ek0109099h001) from the Japan Agency for Medical Research and Development (AMED) and the Research on Measures for Intractable Diseases Project and Health and Labor Sciences Research grants (Research on Intractable Diseases) from the Ministry of Health, Labor, and Welfare.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Koichi Ohshima in Kurume University (Kurume, Japan) for the pathological analysis of LPD, and Prof. Osamu Ohara and the staff in Kazusa DNA Research Institution (Chiba, Japan) for the genetic analysis of the patients.
Abbreviations
AICD, activation-induced cell death; BIR, baculovirus IAP repeat; BMT, bone marrow transplantation; CAEBV, chronic active Epstein–Barr virus infection; EBV, Epstein–Barr virus; FDG-PET, fluorodeoxyglucose-positron emission tomography; HLA, human leukocyte antigens; HST, hematopoietic stem cell transplantation; HLH, hemophagocytic lymphohistiocytosis; IL, interleukin; LPD, lymphoproliferative disease; LUBAC, recruits linear ubiquitin chain assembly complex; NK, natural killer; NOD2, nucleotide-binding oligomerization domain 2; PID, primary immunodeficiency disease; RING, really interesting new gene; SAP, SLAM-associated protein; SH2, Src homology 2; SNP, single nucleotide polymorphism; XIAP, X-linked inhibitor of apoptosis; XLP, X-linked lymphoproliferative disease.
References
1. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. (2016) 127:2375–90. doi: 10.1182/blood-2016-01-643569
2. Cohen JI, Jaffe ES, Dale JK, Pittaluga S, Heslop HE, Rooney CM, et al. Characterization and treatment of chronic active Epstein–Barr virus disease: a 28-year experience in the United States. Blood. (2011) 117:5835–49. doi: 10.1182/blood-2010-11-316745
3. Fujiwara S, Kimura H, Imadome K, Arai A, Kodama E, Morio T, et al. Current research on chronic active Epstein–Barr virus infection in Japan. Pediatr. Int. (2014) 56:159–66. doi: 10.1111/ped.12314
4. Okuno Y, Murata T, Sato Y, Muramatsu H, Ito Y, Watanabe T, et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. (2019) 4:404–13. doi: 10.1038/s41564-018-0334-0
5. Sawada A, Inoue M, Kawa K. How we treat chronic active Epstein-Barr virus infection. Int. J. Hematol. (2017) 105:406–18. doi: 10.1007/s12185-017-2192-6
6. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. doi: 10.1007/s10875-017-0465-8
7. Yang X, Kanegane H, Nishida N, Imamura T, Hamamoto K, Miyashita R, et al. Clinical and genetic characteristics of XIAP deficiency in Japan. J Clin Immunol. (2012) 32:411–20. doi: 10.1007/s10875-011-9638-z
8. Yang X, Wada T, Imadome K, Nishida N, Mukai T, Fujiwara M, et al. Characterization of Epstein–Barr virus (EBV)-infected cells in EBV-associated hemophagocytic lymphohistiocytosis in two patients with X-linked lymphoproliferative syndrome type 1 and type 2. Herpesviridae. (2012) 3:1. doi: 10.1186/2042-4280-3-1
9. Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. (1998) 395:462–9. doi: 10.1038/26683
10. Vaux DL, Silke J. IAPs, RINGs, and ubiquitylation. Nat Rev Mol Cell Biol. (2005) 6:287–97. doi: 10.1038/nrm1621
11. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. (2012) 46:746–58. doi: 10.1016/j.molcel.2012.04.014
12. Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. (2006) 444:110–4. doi: 10.1038/nature05257
13. Worth AJ, Houldcroft CJ, Booth C. Severe Epstein–Barr virus infection in primary immunodeficiency and the normal host. Br J Haematol. (2016) 175:559–76. doi: 10.1111/bjh.14339
14. Ono S, Nakayama M, Kanegane H, Hoshino A, Shimodera S, Shibata H, et al. Comprehensive molecular diagnosis of Epstein-Barr virus-associated lymphoproliferative diseases using next-generation sequencing. Int J Hematol. (2018) 108:319–28. doi: 10.1007/s12185-018-2475-6
15. Koizumi Y, Imadome K, Ota Y, Minamiguchi H, Kodama Y, Watanabe D, et al. Dual threat of Epstein–Barr virus:an autopsy case report of HIV-positive plasmablastic lymphoma complicating EBV-associated hemophagocytic lymphohistiocytosis. J Clin Immunol. (2018) 38:478–83. doi: 10.1007/s10875-018-0500-4
16. Zhao M, Kanegane H, Kobayashi C, Nakazawa Y, Ishii E, Kasai M, et al. Early and rapid detection of X-linked lymphoproliferative syndrome with SH2D1A mutations by flow cytometry. Cytometry B Clin Cytom. (2011) 80:8–13. doi: 10.1002/cyto.b.20552
17. Liu J, Tian W, Wang F, Teng W, Zhang Y, Tong C, et al. Maternal onset de novo SH2D1A mutation and lymphocytic choriomeningitis virus infection in a patient with X-linked lymphoproliferative disease type 1: a case report. Mol Med Rep. (2015) 11:3291–4. doi: 10.3892/mmr.2015.3173
18. Sumazaki R, Kanegane H, Osaki M, Fukushima T, Tsuchida M, Matsukura H, et al. SH2D1A mutations in Japanese males with severe Epstein–Barr virus-associated illnesses. Blood. (2001) 98:1268–70. doi: 10.1182/blood.V98.4.1268
19. Wang Y, Wang Z, Zhang J, Wei Q, Tang R, Qi J, et al. Genetic features of late onset primary hemophagocytic lymphohistiocytosis in adolescence or adulthood. PLoS ONE. (2014) 9:e107386. doi: 10.1371/journal.pone.0107386
20. Nishida N, Yang X, Takasaki I, Imai K, Kato K, Inoue Y, et al. Dysgammaglobulinemia associated with Glu349del, a hypomorphic XIAP mutation. J Investig Allergol Clin Immunol. (2015) 25:205–13.
21. Chen X, Wang F, Zhang Y, Teng W, Wang M, Nie D, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet. (2018) 94:200–12. doi: 10.1111/cge.13363
22. Takimoto T, Takada H, Ishimura M, Kirino M, Hata K, Ohara O, et al. Wiskott-Aldrich syndrome in a girl caused by heterozygous WASP mutation and extremely skewed X-chromosome inactivation: a novel association with maternal uniparental isodisomy 6. Neonatology. (2015) 107:185–90. doi: 10.1159/000370059
23. Takada H, Kanegane H, Nomura A, Yamamoto K, Ihara K, Takahashi Y, et al. Female agammaglobulinemia due to the Bruton tyrosine kinase deficiency caused by extremely skewed X-chromosome inactivation. Blood. (2004) 103:185–7. doi: 10.1182/blood-2003-06-1964
Keywords: Epstein–Barr virus, chronic active EBV infection, lymphoproliferative disease, hemophagocytic lymphohistiocytosis, SAP, XIAP, X-linked lymphoproliferative disease
Citation: Ishimura M, Eguchi K, Shiraishi A, Sonoda M, Azuma Y, Yamamoto H, Imadome K and Ohga S (2019) Systemic Epstein–Barr Virus-Positive T/NK Lymphoproliferative Diseases With SH2D1A/XIAP Hypomorphic Gene Variants. Front. Pediatr. 7:183. doi: 10.3389/fped.2019.00183
Received: 06 March 2019; Accepted: 23 April 2019;
Published: 21 May 2019.
Edited by:
Hiroshi Kimura, Nagoya University, JapanReviewed by:
Taizo Wada, Kanazawa University, JapanMichael Daniel Keller, Children's National Health System, United States
Copyright © 2019 Ishimura, Eguchi, Shiraishi, Sonoda, Azuma, Yamamoto, Imadome and Ohga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masataka Ishimura, ischii@pediatr.med.kyushu-u.ac.jp