Two Novel Mutations (c.883-4_890del and c.1684C>G) of WDR62 Gene Associated With Autosomal Recessive Primary Microcephaly: A Case Report
 You Gyoung Yi
You Gyoung Yi Dong-Woo Lee3
Dong-Woo Lee3  Jaewon Kim
Jaewon Kim Dae-Hyun Jang
Dae-Hyun Jang- 1Department of Rehabilitation Medicine, National Traffic Injury Rehabilitation Hospital, Seoul, South Korea
- 2Department of Rehabilitation, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, South Korea
- 3Department of Rehabilitation Medicine, College of Medicine, The Catholic University of Korea, Seoul, South Korea
- 4Department of Laboratory Medicine, Green Cross Laboratories, Yongin-si, South Korea
- 5Green Cross Genome, Yongin-si, South Korea
Background: Autosomal recessive primary microcephaly (Microcephaly Primary Hereditary, MCPH) is a rare disorder, affecting 1 in 10,000 children in areas where consanguineous marriages are common. WDR62 gene mutations are the second most common cause of MCPH. Herein, we report a case of primary microcephaly caused by two novel WDR62 mutations, which is, to our knowledge, the first such case report in East Asia.
Case presentation: A 6-year-old girl visited our outpatient clinic as a result of microcephaly and delayed development. The patient was born at 36 weeks 4 days through cesarean section. Her birth weight was 1.8 kg (<1st percentile), and she was noted to have microcephaly (head circumference at birth was 28 cm, <−3SD). On examination, delayed speech development and microcephaly with an occipitofrontal head circumference of 43.5 cm (<−3SD) were noted. The patient's gross and fine motor development was normal. Her intelligence quotient was 43 (<0.1 percentile), the same as a 27-month-old child, and her social intelligence quotient was 76.92. Brain imaging revealed simplified gyral patterns of the cerebral cortex; however, laboratory findings, including organic acids, were normal. Multiplex ligation-dependent probe amplification technique for microdeletion syndrome and chromosomal microarray, showed no abnormality. Clinical exome sequencing test revealed two novel heterozygous variants in the WDR62 gene at two different sites: in the boundary of intron 7 and exon 8 (NM_001083961.1: c.883-4_890del) and in exon 13 (NM_001083961.1: c.1684C>G). The patient's parents were identified as heterozygous carriers for each variation.
Conclusion: We report on two novel heterozygous mutations in East Asia. Our data expand the understanding of WDR62 mutations.
Introduction
Microcephaly is a condition characterized by a head circumference measuring at least three standard deviations below the mean for a given population, gender, and age (1). Primary microcephaly is a congenital developmental defect in which the cerebral cortex may be thickened or disorganized and in which the gyral pattern may be normal or simplified (1–5). Autosomal recessive (AR) primary microcephaly (Microcephaly Primary Hereditary, MCPH) is a rare form of primary microcephaly, characterized by a marked reduction in brain size, and intellectual disability, which affects 1 in 30,000 children in Japan (6) and 1 in 10,000 children in areas where consanguineous marriages are common (1, 4).
MCPH has previously been described as a genetically heterogeneous disorder influenced by mutations in at least 20 genes including (MCPH1, WDR62, CDK5RAP2, KNL1, ASPM, CENPJ, STIL, CEP135, CEP152, ZNF335, PHC1, CDK6, CENPE, SASS6, MFSD2A, ANKLE2, CIT, AGMO, RTTN, and PGAP2) (2, 7). However, mutations in two particular genes are thought to be primarily responsible, with ASPM mutations being observed in over half of cases (1, 7, 8), and WDR62 mutations accounting for around 10% of MCPH cases (3, 5, 7, 8).
WDR62 is known to encode the WD repeat-containing protein 62, playing a significant role in neuronal progenitor cell proliferation and spindle formation (2, 5, 9, 10). Children with WDR62 mutations often present with intellectual disability alongside delayed development of speech and language skills and prominent microcephaly (2–5, 10, 11), whereas some also present with seizures (2, 3, 5, 11). Mutations in this gene are also reported to cause a range of other cortical malformations including polymicrogyria, pachygyria with cortical thickening, lissencephaly, schizencephaly, corpus callosum hypoplasia, simplified gyral patterns, cerebral hypoplasia, and band heterotopias (2–5, 11).
Herein, we report a case of primary microcephaly with two novel mutations in the WDR62 gene. This is, to the best of our knowledge, the first such case report in East Asia. In this case, two novel heterozygous variants were found in the WDR62 gene at two different sites, reflecting cultural characteristics of South Korea, which prohibits consanguineous marriages and therefore results in a low incidence of rare AR diseases.
Case presentation
Clinical Presentation
A 6-year-old girl visited our outpatient clinic as a result of microcephaly and delayed development. Despite exhibiting delayed acquisition of language and cognitive milestones, her gross and fine motor functions were in correspondence with her age. The child was born by cesarean section at 36 weeks 4 days, with good Apgar scores. Her birth weight was 1.8 kg (<1st percentile), and microcephaly was detected (her head circumference at birth was 28 cm, <−3SD). Intrauterine growth restriction was detected on a prenatal ultrasonography test. She was capable of walking independently at the age of 14 months.
The patient had one 10-year-old brother who had a normal head circumference and showed no signs of developmental delay. Neither parent had any developmental abnormalities.
Upon examination, delayed speech development and microcephaly with an occipitofrontal head circumference of 43.5 cm (<−3SD, Figure 1) were noted. Her gross and fine motor development was found to be normal. She was capable of independent outdoor gait, running, jumping, hopping,walking up and down the stairs, touching the ground, and block building. However, she could perform only simple errands and had dysarthria. She found reading almost impossible, and she could not write at all. On the Preschool Receptive-Expressive Language Scale, the patient was determined to have the receptive and expressive language of a 41-month-old. On the Korean–Leiter International Performance Scale-Revised examination, the patient's Full-Scale Intelligence Quotient was 43 (<0.1 percentile), corresponding to the level of a 27-month-old child. Her social maturity scale score was 35.5, and her social intelligence quotient was 76.92.
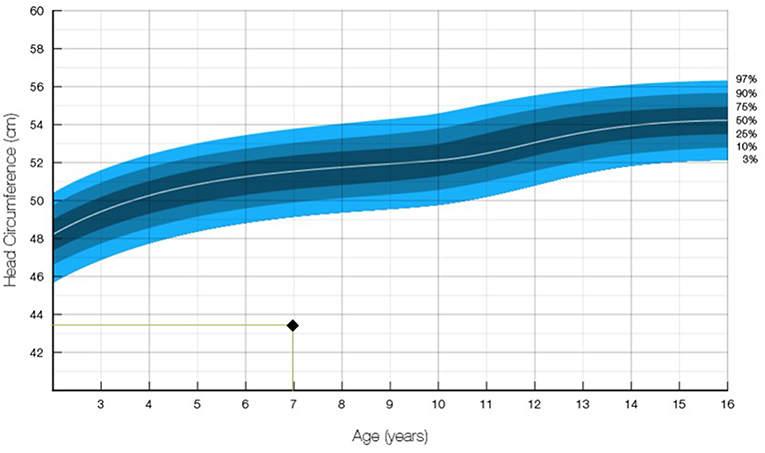
Figure 1. Occipitofrontal head circumference of the patient. The measurement of 43.5 cm at age 7 was in the 1.00 percentile and below −3 SD (12).
Brain magnetic resonance imaging revealed simplified gyral patterns of the cerebral cortex (Figure 2); however, laboratory tests, including organic acids, revealed no abnormal results.
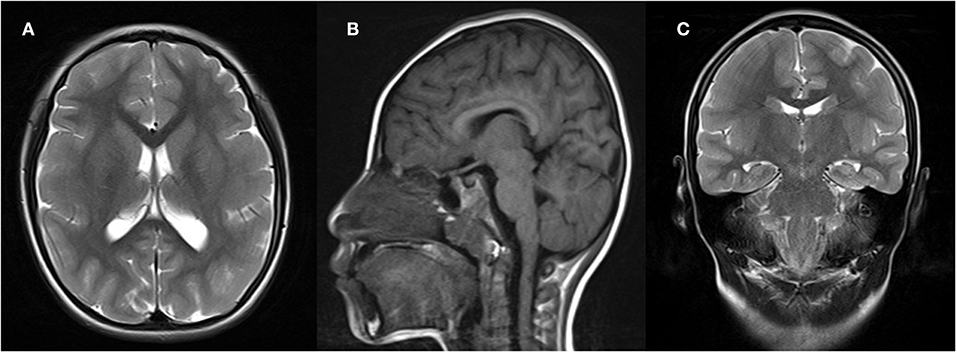
Figure 2. Brain magnetic resonance imaging (MRI) of the patient, showing microcephaly and simplified gyrus of the cerebral cortex. (A) Axial T1-weighted image; (B) sagittal T1-weighted image; (C) coronal T1-weighted image.
Cytogenetic and Molecular Analyses
Chromosomal study revealed a karyotype of 46, XX, inv (9) (p12q13), which had no clinical significance. Furthermore, multiplex ligation-dependent probe amplification (MLPA) technique for microdeletion syndrome showed no abnormality. MLPA analysis of patient was performed using the SALSA MLPA P064-C1 Reference Kit (MRC Holland, Amsterdam, The Netherlands). Chromosomal microarray (performed using an Affymetrix Cytoscan 750K array, CytoScan® 750K Cytogenetics Solution, Keppel Logistics Building, Singapore 629563, genome build: Hg19 method) was normal.
Genomic DNA was extracted from the peripheral blood of the child and her parents. Genomic DNA was enriched using the xGen Inherited Disease Panel (Integrated DNA Technologies, Inc., Coralville, Iowa, USA). Two compound heterozygous variants were observed in the WDR62 gene: a heterozygous deletion variant in the boundary of intron 7 and exon 8 (NM_001083961.1: c.883-4_890del) and a heterozygous missense variant in exon 13 (NM_001083961.1: c.1684C>G).
Sanger sequencing revealed that the parents were heterozygous carriers for each variant, occurring in trans configuration (Table 1). The patient's mother possessed mutations in the WDR62 gene boundary of intron 7 and exon 8 (NM_001083961.1: c.883-4_890del), as shown in Figure 3A, while her father possessed variant in the WDR62 gene at exon 13 (NM_001083961.1: c.1684C>G), as shown in Figure 3B.

Table 1. WDR62 mutations of the patient and her parents.
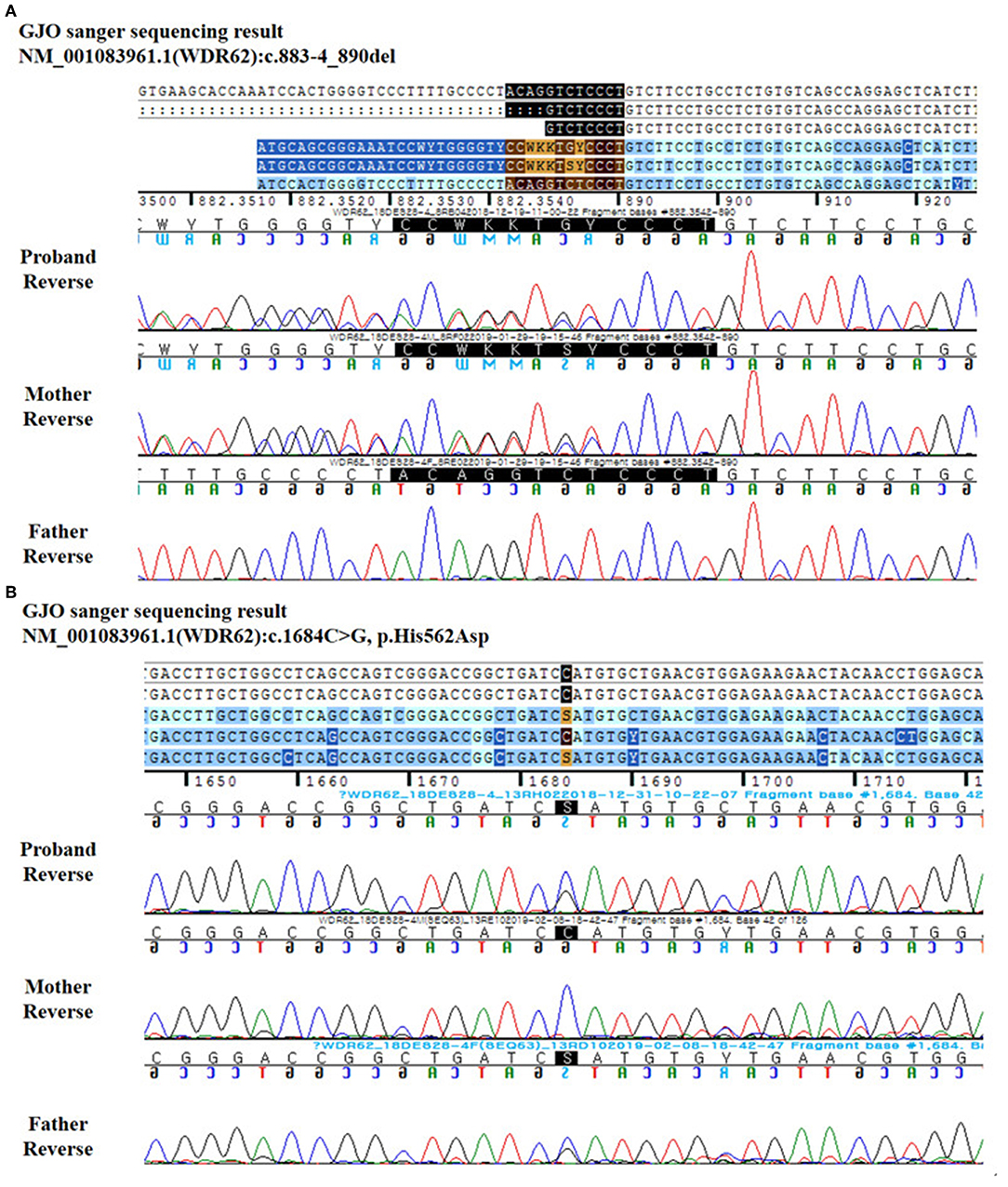
Figure 3. Reverse DNA sequence chromatography for the patient and her parents. (A) c.883-4_890del. (B) c.1684C>G.
Discussion
Based on clinical and molecular findings, the proband was diagnosed with AR primary microcephaly type 2. According to ClinVar, a public sequence database (https://www.ncbi.nlm.nih.gov/clinvar/), 36 pathogenic mutations have been identified in the WDR62 gene to date. WDR62 variants have previously been reported in people of Northern European descent and in Saudi Arabian, Indian, Mexican, Turkish, Iranian, Arabic, and Pakistani families. In a large consanguineous Saudi family, there were two affected siblings born to a consanguineous union in the family of heterozygous carriers of parents (2). In a non-consanguineous family of Northern European descent, compound heterozygous mutations in the WDR62 was reported as the cause of recurrent polymicrogyria in a sibling pair (13). Contrastingly, this is the first report of a WDR62 mutation in East Asian countries including China, Japan, and South Korea.
Consanguineous marriages are banned in South Korea, reducing the possibility of AR disease incidence. In the patient in this study, a WDR62 mutation phenotype might be expressed due to novel heterozygous mutations.
Functional Analysis of Two Novel Variants
We found compound heterozygous variants in the patient's WDR62, the deletion variant (c.883-4_890del) from the mother and the missense variant (c.1684C>G/p.His562Asp) from the father. Neither of these variants has previously been reported in control databases, such as the 1,000 Genomes Project, Exome Variant Server, Exome Aggregation Consortium, or the dbSNP Database.
The c.883-4_890del variant involves splice acceptor site between intron 7 and exon 8, which might result in aberrant splicing, and exon 8 is not an alternatively splice exon among the transcript isoforms (PMID 20729831). This novel canonical splicing variant is suggested as the “likely pathogenic” variant, based on the American College of Medical Genetics and Genomics guidelines regarding the interpretation of sequence variations (PSV1+PM2) (14).
The c.1684C>G variant is located in conserved sequences across species (GERP++_RS 4.45 and phyloP20way_mammalian 0.852, Figure 4) and has been predicted to be deleterious by several in silico analysis tools [SIFT damaging (0.004), PolyPhen-2 deleterious (1), and MutationTaster (1)]. This novel missense variant is suggested as the possible pathogenic element (PM2 + PM3 + PP3 + PP4) (14). However, we were unable to perform some functional assay using in-vitro cell culture to further confirm the pathologic effects of those mutations.

Figure 4. Evolutionary conservation of the amino acid residues for mutant site. Multiple sequence alignment shows the amino acid site of a novel variant (c.1684C>G/p.His562Asp) is highly conserved among different species [Evola version 7.5 (www.h-invitational.jp/evola/)].
Clinical Features of WDR62 Mutations
In cases featuring WDR62 mutations, a broad range of clinical phenotypes have been observed by several independent research groups. These include microcephaly (2–5, 7–11, 15–21), intellectual disability (ID) (2–5, 10, 11, 19–22), speech delay (2, 3, 11, 16, 20), motor delay (3, 5, 16), spasticity (3, 7, 23), infantile spasm (3, 5), epilepsy (2–5, 7, 8, 11), behavior abnormalities (3, 4, 7, 11, 16), high-arched palate (3, 7, 16), dysmorphic face (3, 5, 11, 24), spastic quadriparesis (3, 7), micrognathia (3), dysconjugate gaze (25), and dysarthria (25). Brain imaging in these patients has variously revealed normal findings (26), simplified gyrus (2–5, 9, 10, 16, 20), pachygyria (2–5, 16), cortical thickening (2–5, 16), corpus callosum abnormalities (3–5, 7, 16), lissencephaly (2, 3, 11, 16, 20), schizencephaly (2–4, 16, 20), polymicrogyria (2–4, 16), and heterotopia (3, 4). In our patient, a simplified gyrus was observed alongside definite microcephaly, ID, and speech delay, which are all typical features of WDR62 mutation. However, the patient presented with no evidence of epilepsy, dysmorphic face, or spasticity.
Molecular and Neurodevelopmental Etiology of WDR62 Mutation
WDR62 is the second most frequently mutated gene in MCPH, accounting for ~10% of cases (8). This gene is located within the MCPH2 candidate junction of chromosome 19q13.12, with 32 functional exons and a genome size of 50230 bp (4). This gene encodes a protein with several WD40 domains, which mediate protein–protein interactions (5, 15, 20).
WDR62 functions to preserve centrosome and spindle pole integrity after bipolar spindle formation (2, 16). Loss of the WDR62 encoded protein leads to the dispersal of pericentriolar matrix components, including pericentrin, γ-tubulin, and CDK5 regulatory subunit-associated protein 2 (1, 7, 15, 20, 22, 24). These components interact with one another in metaphase centrosomes (1, 23, 24). In patients with heterozygous WDR62 mutations, lymphoblastoid cells displayed mitotic spindle defects and abnormal centrosome protein localization (8, 10, 24).
WDR62 exhibits remarkable cell cycle dependence (20). WDR62 is mainly associated with the nucleus during interphase, accumulating in the spindle pole, but not in the midbody in cytokinesis during mitosis (5, 8, 11, 16). Therefore, WDR62 mutations can result in various abnormal phenotypes of cerebral cortical development (20, 23).
Correlation Between Phenotypes and Genotypes in the Patient
The frameshift, missense, non-sense, and splice site mutations in the WDR62 are randomly distributed (5). Although missense mutations may cause a defect in neurogenesis resulting in primary microcephaly, it has been suggested that non-sense mutations may cause a more severe microcephaly phenotype by adding a cerebral cortex lamination defect (27). However, subsequent studies have not reported genotype–phenotype correlation. In the present study, splice site mutations in the WDR62 might be attributable to the pachygyria in addition to microcephaly, which is in line with previous studies (20, 28). Further cumulative data and molecular approaches are required to accurately identify genotype–phenotype correlations in WDR62.
Conclusion
We reported two novel heterozygous mutations in East Asia. Our data helped in the understanding of WDR62 mutations.
Data Availability Statement
The datasets generated for this study can be found in the Clinvar: SCV000924581, SCV000924580.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YY: acquisition of data, analysis and interpretation of data, writing, and critical revision of manuscript. S-ML, J-HJ, D-WL, and JK: acquisition of data, analysis and interpretation of data. D-HJ: study concept and design, acquisition of data, analysis and interpretation of data, study supervision, and critical revision of manuscript for intellectual content.
Funding
This research was supported by a Grant of Translational R&D Project through Clinical Research Laboratory, Incheon St. Mary's Hospital.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome? Trends Genet. (2009) 25:501–10. doi: 10.1016/j.tig.2009.09.011
2. Naseer MI, Rasool M, Sogaty S, Chaudhary RA, Mansour HM, Chaudhary AG, et al. A novel WDR62 mutation causes primary microcephaly in a large consanguineous Saudi family. Ann Saudi Med. (2017) 37:148–53. doi: 10.5144/0256-4947.2017.148
3. Nardello R, Fontana A, Antona V, Beninati A, Mangano GD, Stallone MC, et al. A novel mutation of WDR62 gene associated with severe phenotype including infantile spasm, microcephaly, and intellectual disability. Brain Dev. (2018) 40:58–64. doi: 10.1016/j.braindev.2017.07.003
4. Rupp V, Rauf S, Naveed I, Christian W, Mir A. A novel single base pair duplication in WDR62 causes primary microcephaly. BMC Med Genet. (2014) 15:1–6. doi: 10.1186/s12881-014-0107-4
5. Zombor M, Kalmár T, Nagy N, Berényi M, Telcs B, Maróti Z, et al. A novel WDR62 missense mutation in microcephaly with abnormal cortical architecture and review of the literature. J Appl Genet. (2019) 60:151–62. doi: 10.1007/s13353-019-00486-y
6. Komai T, Kishimoto K, Ozaki Y. Genetic study of microcephaly based on Japanese material. Am J Hum Genet. (1955) 7:51–65.
7. Zaqout S, Morris-Rosendahl D, Kaindl AM. Autosomal recessive primary microcephaly (MCPH): an update. Neuropediatrics. (2017) 48:135–42. doi: 10.1055/s-0037-1601448
8. Mahmood S, Ahmad W, Hassan MJ. Autosomal recessive primary microcephaly (MCPH): Clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J Rare Dis. (2011) 6:39. doi: 10.1186/1750-1172-6-39
9. Lim NR, Shohayeb B, Zaytseva O, Mitchell N, Millard SS, Ng DCH, et al. Glial-specific functions of microcephaly protein WDR62 and interaction with the mitotic kinase AURKA are essential for drosophila brain growth. Stem Cell Rep. (2017) 9:32–41. doi: 10.1016/j.stemcr.2017.05.015
10. Farag HG, Froehler S, Oexle K, Ravindran E, Schindler D, Staab T, et al. Abnormal centrosome and spindle morphology in a patient with autosomal recessive primary microcephaly type 2 due to compound heterozygous WDR62 gene mutation. Orphanet J Rare Dis. (2013) 8:178. doi: 10.1186/1750-1172-8-178
11. Poulton CJ, Schot R, Seufert K, Lequin MH, Accogli A, Annunzio GD, et al. Severe presentation of WDR62 mutation: Is there a role for modifying genetic factors? Am J Med Genet Part A. (2014) 164:2161–71. doi: 10.1002/ajmg.a.36611
12. Hall, Judith G. Handbook of Physical Measurements, 2nd ed. (2007) New York: Oxford University Press
13. Murdock DR, Clark GD, Bainbridge MN, Newsham I, Wu YQ, Muzny DM, et al. Whole-exome sequencing identifies compound heterozygous mutations in WDR62 in siblings with recurrent polymicrogyria. Am J Med Genet A. (2011) 155:2071–7. doi: 10.1002/ajmg.a.34165
14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
15. Barbelanne M, Tsang WY. Molecular and cellular basis of autosomal recessive primary microcephaly. Biomed Res Int. (2014) 2014:547986 doi: 10.1155/2014/547986
16. Bhat V, Girimaji S, Mohan G, Arvinda H, Singhmar P, Duvvari M, et al. Mutations in WDR62, encoding a centrosomal and nuclear protein, in Indian primary microcephaly families with cortical malformations. Clin Genet. (2011) 80:532–40. doi: 10.1111/j.1399-0004.2011.01686.x
17. Cherkaoui Jaouad I, Zrhidri A, Jdioui W, Lyahyai J, Raymond L, Egéa G, et al. A novel non sense mutation in WDR62 causes autosomal recessive primary microcephaly: a case report. BMC Med Genet. (2018) 19:118. doi: 10.1186/s12881-018-0625-6
18. Pervaiz N, Abbasi AA. Molecular evolution of WDR62, a gene that regulates neocorticogenesis. Meta Gene. (2016) 9:1–9. doi: 10.1016/j.mgene.2016.02.005
19. Bacino CA, Arriola LA, Wiszniewska J, Bonnen PE. WDR62 missense mutation in a consanguineous family with primary microcephaly. Am J Med Genet Part A. (2012) 158:622–5. doi: 10.1002/ajmg.a.34417
20. Yu TW, Mochida GH, Tischfield DJ, Sgaier SK, Flores-Sarnat L, Sergi CM, et al. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat Genet. (2010) 42:1015–20. doi: 10.1038/ng.683
21. Von der Hagen M, Pivarcsi M, Liebe J, von Bernuth H, Didonato N, Hennermann JB, et al. Diagnostic approach to microcephaly in childhood: a two-center study and review of the literature. Dev Med Child Neurol. (2014) 56:732–41. doi: 10.1111/dmcn.12425
22. Passemard S, Titomanlio L, Elmaleh M, Afenjar A, Alessandri JL, Andria G, et al. Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology. (2009) 73:962–9. doi: 10.1212/WNL.0b013e3181b8799a
23. Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet. (2005) 76:717–28. doi: 10.1086/429930
24. Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, et al. Many roads lead to primary autosomal recessive microcephaly. Prog Neurobiol. (2010) 90:363–83. doi: 10.1016/j.pneurobio.2009.11.002
25. Bilgüvar K, Öztürk AK, Louvi A, Kwan KY, Choi M, Tatli B, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. (2010) 467:207–10. doi: 10.1038/nature09327
26. McDonell LM, Warman Chardon J, Schwartzentruber J, Foster D, Beaulieu CL, Majewski J, et al. The utility of exome sequencing for genetic diagnosis in a familial microcephaly epilepsy syndrome. BMC Neurol (2014) 14:22. doi: 10.1186/1471-2377-14-22
27. Nicholas AK, Khurshid M, Désir J, Carvalho OP, Cox JJ, Thornton G, et al. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet (2010) 42:1010–1014. doi: 10.1038/ng.682
Keywords: autosomal recessive primary microcephaly (MCPH), exome sequencing test, novel mutation, WDR62 gene mutation, neurodevelopment
Citation: Yi YG, Lee D-W, Kim J, Jang J-H, Lee S-M and Jang D-H (2019) Two Novel Mutations (c.883-4_890del and c.1684C>G) of WDR62 Gene Associated With Autosomal Recessive Primary Microcephaly: A Case Report. Front. Pediatr. 7:457. doi: 10.3389/fped.2019.00457
Received: 01 July 2019; Accepted: 21 October 2019;
Published: 07 November 2019.
Edited by:
Merlin G. Butler, University of Kansas Medical Center, United StatesCopyright © 2019 Yi, Lee, Kim, Jang, Lee and Jang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dae-Hyun Jang, dhjangmd@naver.com