Trio-Based Whole-Exome Sequencing Identifies a De novo EFNB1 Mutation as a Genetic Cause in Female Infant With Brain Anomaly and Developmental Delay
 Ji Yoon Han
Ji Yoon Han Hyun Jeong Kim2
Hyun Jeong Kim2  In Goo Lee
In Goo Lee Joonhong Park
Joonhong Park- 1Department of Pediatrics, College of Medicine, Catholic University of Korea, Seoul, South Korea
- 2Department of Radiology, College of Medicine, The Catholic University of Korea, Seoul, South Korea
- 3Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, South Korea
- 4Department of Laboratory Medicine, College of Medicine, Catholic University of Korea, Seoul, South Korea
- 5Department of Laboratory Medicine, Jeonbuk National University Medical School and Hospital, Jeonju, South Korea
Background: Craniofrontonasal syndrome is a rare, X-linked disorder in which heterozygous females ironically reported the majority of patients and is caused by in the EFNB1 gene located at chromosome Xq13.1. Unlike previous reports, we present a female infant with a de novo EFNB1 missense mutation that was demonstrated in clinical diagnosis as global developmental delay (GDD) and brain anomaly without frontonasal dysplasia or other malformation.
Case Presentation: This study reports the genetic analysis of a 4-month-old female infant presenting brain anomaly and GDD. She was the only child of unrelated parents. Early developmental was characterized by delays in fine motor, achieving gross motor, language, and social–cognitive milestones. She could not control her head or hold objects until 4 months of age. Brain magnetic resonance imaging revealed schizencephaly and dysgenesis of corpus callosum. Trio-based whole-exome sequencing revealed a heterozygous c.943C>T (p.Pro315Ser) in the EFNB1. Sanger sequencing confirmed this heterozygous alteration occurring in a dominant de novo manner, as a consequence of phenotypic and genotypic wild type in both parents.
Conclusion: EFNB1 mutation is considered for a child with schizencephaly, and further study focusing on phenotyping is required to understand the possible contribution of environmental impact and genetic modifier in the expression of EFNB1.
Introduction
Craniosynostoses are malformations of the developing skull in which one or more of the cranial sutures of the skull bone fuse prematurely, thereby affecting brain development and skull shape (1). They have marked allelic and phenotypic heterogeneity and are classified into syndromic and nonsyndromic forms. Syndromic craniosynostoses have a typically monogenic etiology and are related with other malformations. Nonsyndromic craniosynostosis is an isolated finding and is categorized according to the suture(s) involved (2). Indeed, pathogenic variant is well described in six major genes such as EFNB1, ERF, FGFR2, FGFR3, TCF12, and TWIST1 causing the recurrent craniosynostosis syndromes (3): Apert, craniofrontonasal (CFN), Crouzon and Crouzon with acanthosis nigricans, Muenke, Pfeiffer, and Saethre-Chotzen syndrome. Among them, CFN syndrome (CFNS) (OMIM #304110) is an X-linked dominant disease caused by loss-of-function mutations in the ephrin B1 gene (EFNB1, OMIM *300035). The ephrin-Eph signaling pathway affects the cellular cytoskeleton, leading to cell repulsion primarily as well as to cell adhesion in some instances. This pathway plays a critical role in morphogenesis activating signaling pathways and allowing cell-to-cell communication over a short distance (4). Classically, CFNS is characterized by severe hypertelorism with a central nasal groove related to unilateral coronal craniosynostosis, other midline defects, dermatological manifestations, body asymmetry, and skeletal abnormalities (5). Most CFNS patients are females as heterozygous with respect to X-linked gene who show paradoxically a more severe symptom than hemizygous males, who have an uncertain clinical manifestation (6).
The current report demonstrates a female infant with CFNS caused by a previously unreported EFNB1 mutation that demonstrates the difference in clinical features including global developmental delay (GDD) and brain anomaly without frontonasal dysplasia or other malformations.
Case Presentation
A 4-month-old female infant was referred to the Department of Pediatric Neurology of Daejeon St. Mary's Hospital with a diagnosis of microcephaly and developmental delay. There was no family history of neurodevelopmental or genetic disorders, and she was the only child of Korean nonconsanguineous parents. The pregnancy had been uneventful. At age 4 months, the weight was 6.6 kg (25th percentile), length was 63 cm (50th percentile), and head circumstance was 35.5 cm (less than the third percentile). Her early development was characterized by delays in fine motor, achieving gross motor, language, and social–cognitive milestones. She could not control her head or hold objects until 4 months of age. Estimation using the Bayley Scale of Infant and Toddler Development, Third Edition, was performed at age 2 years and demonstrated marked GDDs (language cognitive, developmental, and motor ages: 8, 13, and 9–10 months, respectively). At age of 2 years, she was in the 20th percentile for weight (9.6 kg), the 50th percentile for height (81 cm), and first percentile for head circumference (43 cm). Physical examination demonstrated a well-nourished girl with microcephaly at age of 2 years and without CFN dysplasia (Figures 1A–D). Estimated orbital parameters at age of 2 years were all within the 10th percentile to 25th percentile (interpupillary distance 43 mm, inner canthal distance 24 mm, and outer canthal distance 71 mm). The palpebral fissures slightly sloped upward. Skull X-ray revealed no gross abnormality in cranial vault and no craniosynostosis (Figures 1E–H). Her ears, feet, and hands were normal in shape and size (Supplementary Figure 1). Skeletal surveillance including spine, both hands, and foot was done, and results were normal (Supplementary Figure 1). Deep tendon reflexes, muscle bulk, and muscle tone were normal. Brain magnetic resonance imaging revealed schizencephaly and dysgenesis of corpus callosum (Supplementary Figure 2). Her audiometry test result (auditory evoked potential and auditory brain stem response) was normal, and her response to sound seems to appropriate. The results of visual evoked potential and ophthalmologic tests were normal, but her visual perception was slow and did not seem accurate. Ophthalmological examinations of fundus and optic nerve were normal at ages 6 months and 2 years. Electroencephalography (EEG) observed continuous high-amplitude spikes and slow waves on both temporoparietal areas (Supplementary Figure 2). Despite abnormalities in EEG, she did not have a history of seizures. Pelvic sonogram screen reported no malformations. Echocardiogram showed no structural abnormalities. The laboratory tests such as growth hormone, thyroid function test, and metabolic workup were all within normal ranges.
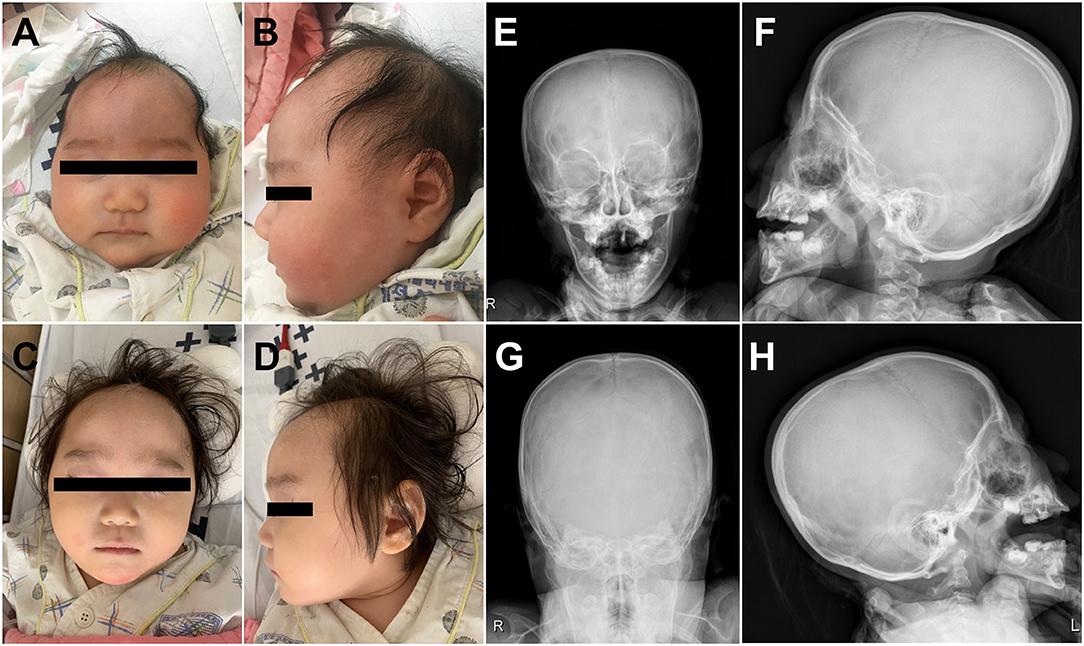
Figure 1. Clinical photographs of the proband showed microcephaly without craniofrontonasal dysmorphism at age of 4 months (A,B) and at age of 2 years (C,D). Skull X-ray of the proband revealed no craniosynostosis (E–H).
Trio-Based Whole Exome Sequencing
The study protocol was approved by the Institutional Review Board of the Catholic University of Korea. Written informed consent was collected from the parents on behalf of their children for the publication of any potentially identifiable images or data included in this article before blood sampling, and clinical data were achieved from the proband and her parents. The exomic DNA of the proband and her parents was enriched using the Agilent's SureSelect XT Human All Exon v5 (Agilent Technologies, Santa Clara, CA, USA), and paired-end sequencing was conducted on the Illumina HiSeq2500 (Illumina, San Diego, CA, USA). As a result, rare inherited or sporadic variants in each trio were identified by the mirTrios program based on the VCF files generated by GATK. Annotation of the identified variants with respect to the results of their mutation on reported genes (amino acid change, functional effect, etc.), mutation effect predicted by several in silico computational tools (MutationTaster, Polyphen2, SIFT, CADD-PHRED, etc.), and public genome databases (from 1,000 Genomes, ExAC, and gnomAD database) were estimated using the dbNSFP 2.4 and Variant Effect Predictor, which is a database constructed for annotation of presumptive nonsynonymous variants and functional prediction in the human genome.
Results
By estimating sequence quality along all sequences, average yield in target of 5,411 million reads was generated from the proband and her parents' samples. Mean depth (x) was 107, and percentage of bases above average 30x was achieved for the average target region of 91%. Trio-based whole-exome sequencing (WES) identified 21 rare variants as a candidate cause of schizencephaly and GDD (Table 1). Among them, only two rare variants of the EFNB1 and ABCC4 were inherited as a dominant de novo manner. Although two candidate missense variants were identified, there was no phenotypic description related to brain anomaly and developmental delay in OMIM database due to ABCC4 variant. Therefore, the ABCC4 variant seems to be less likely to be the pathogenic variant. Particularly, similar to the phenotype of the proband, a heterozygous c.943C>T of the EFNB1 gene causing a codon change of proline to serine at position 315 (NM_004429.4: c.943C>T, p.Pro315Ser; no rsID) had not been previously reported to be related to CFNS in the proband. Sanger sequencing was conducted in the proband and her parents to confirm that the mutation segregated with affected individuals and demonstrated this mutation as a dominant de novo state was present only in the proband but not in her parents showing phenotypic and genotypic wild type, respectively (Figure 2A). Allele frequency at this position has neither been reported in the gnomAD exome data or in 622 ethnically matched Korean, unrelated controls (http://coda.nih.go.kr/coda/KRGDB/index.jsp). This missense variant of the EFNB1 was predicted to be “deleterious,” “damaging,” or “disease causing” by computational in silico prediction (Supplementary Table 1). In addition, cross-species comparisons (phastCons and GERP) of protein sequences of EFNB1 protein demonstrated that this mutated region was conserved highly in vertebrates (phastCons 1 > cutoff of 0.8 and GERP 5.04 > cutoff of 4.4). An EFNB1 multiple sequence alignment of amino acids 300–331 was constructed using the following taxa showing phylogenetic diversity between fugu (Takifugu: SINFRUT00000172427) and human (Homo sapiens: HIT000037613) and using Evola (Figure 2B). As a result, amino acid sequence of the Pro315Ser residue is highly conserved across the species.
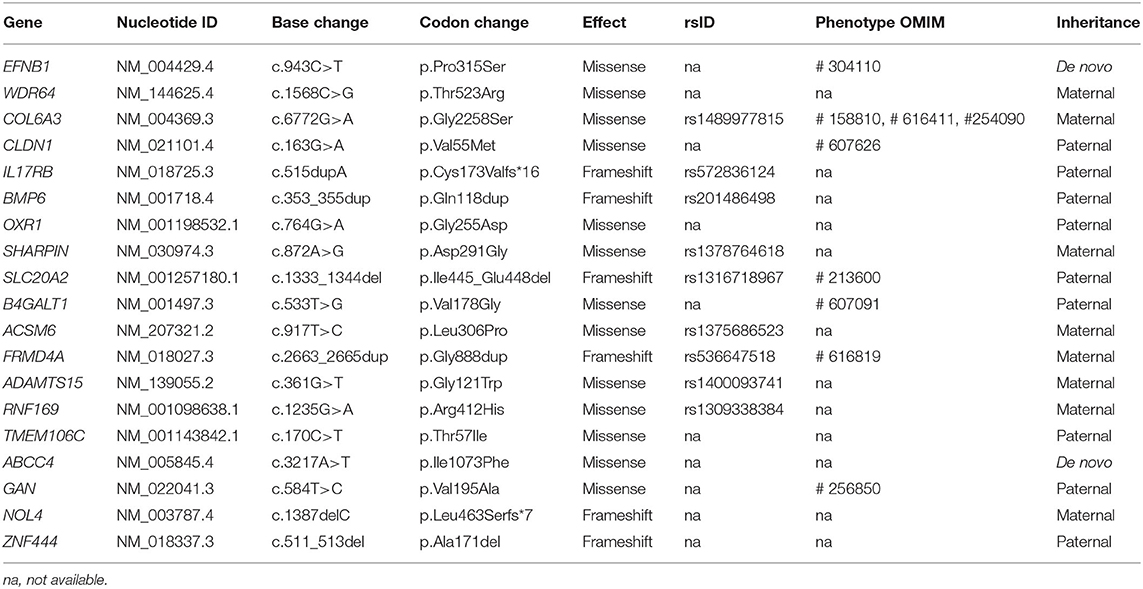
Table 1. Summary of 21 rare variants as a candidate cause of schizencephaly and global developmental delay identified by trio-based exome sequencing.
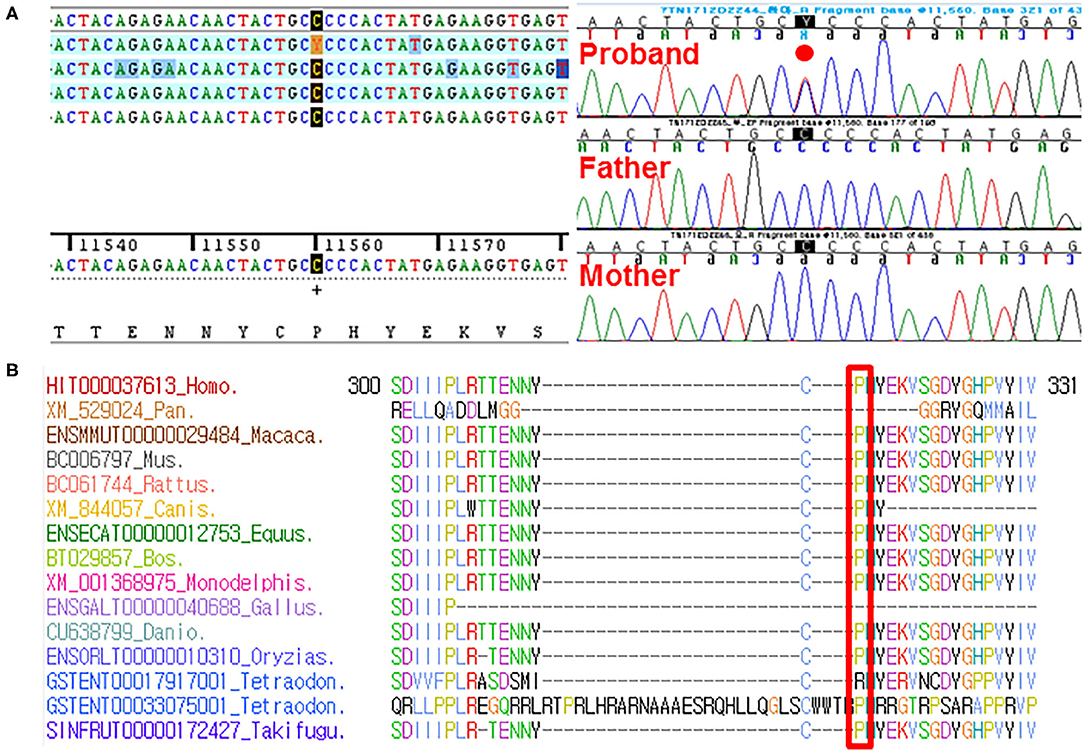
Figure 2. (A) Sanger sequencing confirmed this heterozygous c.943C>T (p.Pro315Ser) in the EFNB1 occurring in a dominant de novo manner, as a consequence of phenotypic and genotypic wild type in both parents. (B) Sequence alignment of the conserved cytoplasmic domain of the EFNB1 protein in multiple species. Protein sequence of the Pro315Ser residue is highly conserved across species compared. It is highlighted in the red box.
Discussion
Craniosynostosis comprises a heterogeneous group of disorders correspondingly, in addition to monogenic causes, with reduced transduction of stretch forces from the growing brain due to poor intrinsic growth, intrauterine fetal head constraint, and polygenic background all likely to play substantial roles (7). Occasionally, extracranial manifestations include asymmetry of the pectoralis, breast, limbs, or thoracic skeleton; clinodactyly of the fifth finger, polydactylism, and diaphragmatic hernia are seen (8–12). Developmental delay or intellectual disability is not usually present (13–15). Our case showed severe GDD with schizencephaly and microcephaly, but no other skeletal anomaly or facial dysmorphism. However, mild to moderate intellectual disability or developmental delay has been reported in some patients, and it is somewhat related to uncorrected craniosynostosis. Neurocognitive development in CFNS is usually reported as normal in most published cases (16–18). It is not clear whether GDD represents primary features of CFNS or is secondary to the influence of premature fusion of sutures. Our patient had minor facial dysmorphisms such as hypertelorism, slightly broad nasal root, and palpebral fissures slightly sloped upward. However, compared to previous reports, our patient did not show definite facial dysmorphism and craniosynostosis. However, our patient shows severely retarded development at age of 2 years and schizencephaly, which is described as a new manifestation for the first time. In our case, trio-based WES results showed a heterozygous c.943C>T (p.Pro315Ser) in the EFNB1. This variant corresponds to the term “likely pathogenic” according to the American College of Medical Genetics guidelines: PS2, de novo in a patient with the disease and no family history; PS4, the prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls; PM5, novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. In previous study, prior to referral in the majority of cases, the number of EFNB1 variants was identified in individuals who had not been diagnosed clinically with CFNS (19). This finding supports that EFNB1 has a broader phenotypic spectrum, and a wider role in undiagnosed craniosynostosis, than previously recognized. The inquiry of whether mutations in other ephrins, in their receptors, or in membrane of the signaling cascade may lead to brain malformations and CFN dysplasia needs to be researched.
The mutation classifications in 89 cases included 3 whole-gene deletions, 3 intragenic deletions, 24 frameshifts, 13 nonsense mutations, 40 missense mutations, and 6 splice site mutations (5, 20–26). EFNB1 mutations have been dispersed across the gene, but mutations in exon 5, as found in our family, may be distributed infrequently compared with mutations in the first four exons of EFNB1. The most mutations were identified in exons 2 and 3 of EFNB1 gene encoding the extracellular ephrin domains (21). In our study, missense mutation c.943C>T was identified and results in the change of p.Pro315Ser located in exon 5 of EFNB1 gene coding the cytoplasmic domain (Figure 3). To date, most of the mutations in exon 5 have been deleterious mutations such as frameshift or nonsense mutations interfering with ephrin B1 reverse signaling that contribute to the CFNS phenotype. (19, 21, 23, 27). Unlike previous reports, this is the report missense EFNB1 mutation in exon 5 in atypical CFNS with brain anomaly but no CFN dysplasia. The effects of a missense mutation on molecular phenotype, function, and organism fitness can be extremely diverse. Alternatively, it may be mildly deleterious to compare splice site, frameshift, or nonsense mutations that can be interpreted easily as pathogenic in nature (28). Thus, the EFNB1 mutation type may have conferred a different phenotype in our case from those in previous studies. In addition, genotype and phenotype difference between ephrin (n = 62) and cytoplasmic (n = 7) domains was estimated; however, because of the small number of patients with mutation in cytoplasmic domain, it is difficult to fully verify the genotype–phenotype correlation (Supplementary Table 2).

Figure 3. Schematic diagram of the EFNB1 mutation spectrum in patients with CFNS. Below the putative EFNB1 protein structure mutation identified in the present study is highlighted in red, whereas those published previously are written in black.
High heterogeneity due to overlapping clinical features, as well as differences in expressivity and penetrance observed in craniosynostosis cases, often makes correct clinical diagnosis extremely difficult (2). Molecular genetic analysis is necessary to link as many phenotypes as possible with their underlying genetic cause, building the base for proper genotype–phenotype correlations, allowing for prediction of patient's clinical course and exact genetic counseling (14, 19, 29). The high apparent rate of rare actionable mutations detected in complex craniosynostosis without an obvious diagnosis may reflect developmental pleiotropy of signaling in the cranial sutures, with co-opting from more ancient uses in embryogenesis and multiple pathways, implicated at different stages of suture development (7, 19, 30, 31). As a result of the genetic diagnosis, potentially appropriate monitoring has been instigated.
In summary, EFNB1 mutation is considered for a child with schizencephaly, and further study focusing on phenotyping is required to understand the possible contribution of environmental impact and genetic modifier in the expression of EFNB1. Our case with atypical phenotype showed uncommon features. Most mutations were located in the ephrin domain (exons 2 and 3), and only some patients have mutation in cytoplasmic domain (exon 5). Mutation in the cytoplasmic domain (exon 5) may result in lesser extracranial abnormalities compared to mutation in the ephrin domain. Still, further analysis with more patients is necessary in order to establish the genotype–phenotype relations.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The study protocol was approved by the Institutional Review Board of the Catholic University of Korea. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JH made substantial contributions to interpretations of clinical medical record and was involved in drafting the manuscript. HK made substantial contributions to analysis radiologic findings. JJ contributed to the acquisition and interpretation of NGS data. JP made substantial contribution to analysis and interpretation of the data and was involved in drafting manuscript. IL was involved in revising critically for important intellectual content. All authors read and approved the manuscript for submission, contributed to the article, and approved the submitted version.
Funding
This research was supported by an alumni grant from the Department of Pediatrics, College of Medicine, The Catholic University of Korea in 2019. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1F1A1077316).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2020.00461/full#supplementary-material
Supplementary Figure 1. X-ray of the proband shows no gross abnormality in both feet (A, B) and both hands (C, D), and whole spine (E,F).
Supplementary Figure 2. Brain MRI performed at 2 years of age in the patient. T2-weighted axial (A) and coronal (B) MR images demonstrate bilateral CSF-filled brain defects extending from the lateral ventricle wall to pial surface and dysplastic gray matter lining the clefts (red arrows). (C) T1-weighted sagittal MR image shows focally thinned and dysgenetic corpus callosum (red arrow). (D) EEG shows continuous spikes and slow waves on both temporooccipital areas.
Supplementary Table 1. Results of predictive in-silico tools on missense variants identified by trio-based exome sequencing.
Supplementary Table 2. Comparison of clinical abnormalities according to EFNB1 mutation sites (5, 20–26).
References
1. Governale LS. Craniosynostosis. Pediatr Neurol. (2015) 53:394–401. doi: 10.1016/j.pediatrneurol.2015.07.006
2. Apostolopoulou D, Kaxira OS, Hatzaki A, Panagopoulos KP, Alexandrou K, Stratoudakis A, et al. Genetic analysis of syndromic and nonsyndromic patients with craniosynostosis identifies novel mutations in the TWIST1 and EFNB1 Genes. Cleft Palate Craniofac J. (2018) 55:1092–1102. doi: 10.1177/1055665618760412
3. Wilkie AO, Bochukova EG, Hansen RM, Taylor IB, Rannan-Eliya SV, Byren JC, et al. Clinical dividends from the molecular genetic diagnosis of craniosynostosis. Am J Med Genet A. (2007) 143a:1941–9. doi: 10.1002/ajmg.a.31905
4. Kania A, Klein R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol. (2016) 17:240–56. doi: 10.1038/nrm.2015.16
5. Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci U S A. (2004) 101:8652–7. doi: 10.1073/pnas.0402819101
6. Twigg SR, Babbs C, van den Elzen ME, Goriely A, Taylor S, McGowan SJ, et al. Cellular interference in craniofrontonasal syndrome: males mosaic for mutations in the X-linked EFNB1 gene are more severely affected than true hemizygotes. Hum Mol Genet. (2013) 22:1654–62. doi: 10.1093/hmg/ddt015
7. Twigg SR, Wilkie AO. A genetic-pathophysiological framework for craniosynostosis. Am J Hum Genet. (2015) 97:359–77. doi: 10.1016/j.ajhg.2015.07.006
8. Wieland I, Weidner C, Ciccone R, Lapi E, McDonald-McGinn D, Kress W, et al. Contiguous gene deletions involving EFNB1, OPHN1, PJA1 and EDA in patients with craniofrontonasal syndrome. Clin Genet. (2007) 72:506–16. doi: 10.1111/j.1399-0004.2007.00905.x
9. Srisupundit K, Brady PD, Devriendt K, Fryns JP, Cruz-Martinez R, Gratacos E, et al. Targeted array comparative genomic hybridisation (array CGH) identifies genomic imbalances associated with isolated congenital diaphragmatic hernia (CDH). Prenat Diagn. (2010) 30:1198–206. doi: 10.1002/pd.2651
10. Petit F, Andrieux J, Holder-Espinasse M, Bouquillon S, Pennaforte T, Storme L, et al. Xq12q13.1 microduplication encompassing the EFNB1 gene in a boy with congenital diaphragmatic hernia. Eur J Med Genet. (2011) 54:e525–7. doi: 10.1016/j.ejmg.2011.06.011
11. Evers C, Jungwirth MS, Morgenthaler J, Hinderhofer K, Maas B, Janssen JW, et al. Craniofrontonasal syndrome in a male due to chromosomal mosaicism involving EFNB1: further insights into a genetic paradox. Clin Genet. (2014) 85:347–53. doi: 10.1111/cge.12171
12. Acosta-Fernandez E, Zenteno JC, Chacon-Camacho OF, Pena-Padilla C, Bobadilla-Morales L, Corona-Rivera A. Extracranial midline defects in a patient with craniofrontonasal syndrome with a novel EFNB1 mutation. Am J Med Genet A. (2020) 182:1223–9. doi: 10.1002/ajmg.a.61506
13. Passos-Bueno MR, Sertie AL, Jehee FS, Fanganiello R, Yeh E. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front Oral Biol. (2008) 12:107–43. doi: 10.1159/000115035
14. Kutkowska-Kazmierczak A, Gos M, Obersztyn E. Craniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling. J Appl Genet. (2018) 59:133–47. doi: 10.1007/s13353-017-0423-4
15. Kilit C, Kilit TP. Report of a family with craniofrontonasal syndrome and Wolff-Parkinson-white syndrome: is it a new finding? Arq Bras Cardiol. (2019) 112:594–6. doi: 10.5935/abc.20190033
16. Goyal M, Pradhan G, Wieland I, Kapoor S. Craniofrontonasal syndrome: atrial septal defect with a novel EFNB1 gene mutation. Cleft Palate Craniofac J. (2015) 52:234–6. doi: 10.1597/13-354
17. Yoo H, Ko JM, Lim BC, Cheong HI. First Korean patients with craniofrontonasal syndrome confirmed by EFNB1 analysis. Ann Clin Lab Sci. (2016) 46:544–8
18. Fischer M, Bansch PS, Unterecker S, Romanos M, Deckert J. Depression and hyperactivity in two patients with craniofrontonasal syndrome. Am J Med Genet A. (2016) 170:799–800. doi: 10.1002/ajmg.a.37482
19. Lee E, Le T, Zhu Y, Elakis G, Turner A, Lo W, et al. A craniosynostosis massively parallel sequencing panel study in 309 Australian and New Zealand patients: findings and recommendations. Genet Med. (2018) 20:1061–8. doi: 10.1038/gim.2017.214
20. Wieland I, Jakubiczka S, Muschke P, Cohen M, Thiele H, Gerlach KL, et al. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am J Hum Genet. (2004) 74:1209–15. doi: 10.1086/421532
21. Wieland I, Reardon W, Jakubiczka S, Franco B, Kress W, Vincent-Delorme C, et al. Twenty-six novel EFNB1 mutations in familial and sporadic craniofrontonasal syndrome (CFNS). Hum Mutat. (2005) 26:113–8. doi: 10.1002/humu.20193
22. Shotelersuk V, Siriwan P, Ausavarat S. A novel mutation in EFNB1, probably with a dominant negative effect, underlying craniofrontonasal syndrome. Cleft Palate Craniofac J. (2006) 43:152–4. doi: 10.1597/05-014.1
23. Twigg SR, Matsumoto K, Kidd AM, Goriely A, Taylor IB, Fisher RB, et al. The origin of EFNB1 mutations in craniofrontonasal syndrome: frequent somatic mosaicism and explanation of the paucity of carrier males. Am J Hum Genet. (2006) 78:999–1010. doi: 10.1086/504440
24. Vasudevan PC, Twigg SR, Mulliken JB, Cook JA, Quarrell OW, Wilkie AO. Expanding the phenotype of craniofrontonasal syndrome: two unrelated boys with EFNB1 mutations and congenital diaphragmatic hernia. Eur J Hum Genet. (2006) 14:884–7. doi: 10.1038/sj.ejhg.5201633
25. Torii C, Izumi K, Nakajima H, Takahashi T, Kosaki K. EFNB1 mutation at the ephrin ligand-receptor dimerization interface in a patient with craniofrontonasal syndrome. Congenit Anom (Kyoto). (2007) 47:49–52. doi: 10.1111/j.1741-4520.2006.00140.x
26. Hogue J, Shankar S, Perry H, Patel R, Vargervik K, Slavotinek A. A novel EFNB1 mutation (c.712delG) in a family with craniofrontonasal syndrome and diaphragmatic hernia. Am J Med Genet A. (2010) 152a:2574–7. doi: 10.1002/ajmg.a.33596
27. Wallis D, Lacbawan F, Jain M, Der Kaloustian VM, Steiner CE, Moeschler JB, et al. Additional EFNB1 mutations in craniofrontonasal syndrome. Am J Med Genet A. (2008) 146a:2008–12. doi: 10.1002/ajmg.a.32388
28. Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. (2007) 80:727–39. doi: 10.1086/513473
29. Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr. (2017) 29:622–8. doi: 10.1097/MOP.0000000000000542
30. Miller KA, Twigg SR, McGowan SJ, Phipps JM, Fenwick AL, Johnson D, et al. Diagnostic value of exome and whole genome sequencing in craniosynostosis. J Med Genet. (2017) 54:260–8. doi: 10.1136/jmedgenet-2016-104215
Keywords: trio exome sequencing, EFNB1 mutation, schizencephaly, global developmental delay, craniofrontonasal dysplasia
Citation: Han JY, Kim HJ, Jang JH, Lee IG and Park J (2020) Trio-Based Whole-Exome Sequencing Identifies a De novo EFNB1 Mutation as a Genetic Cause in Female Infant With Brain Anomaly and Developmental Delay. Front. Pediatr. 8:461. doi: 10.3389/fped.2020.00461
Received: 07 May 2020; Accepted: 30 June 2020;
Published: 01 September 2020.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
Charlotte L. Alston, Wellcome Trust Centre for Mitochondrial Research (WT), United KingdomBixia Zheng, Nanjing Children's Hospital, China
Copyright © 2020 Han, Kim, Jang, Lee and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joonhong Park, miziro@catholic.ac.kr