Case Report: Liver Transplantation in Homozygous Familial Hypercholesterolemia (HoFH)—Long-Term Follow-Up of a Patient and Literature Review
 Matej Mlinaric1
Matej Mlinaric1  Nevenka Bratanic1 Vlasta Dragos2 Ajda Skarlovnik3
Nevenka Bratanic1 Vlasta Dragos2 Ajda Skarlovnik3  Matija Cevc3
Matija Cevc3  Tadej Battelino1,4
Tadej Battelino1,4  Urh Groselj1,4*
Urh Groselj1,4*- 1Department of Pediatric Endocrinology, Diabetes and Metabolic Diseases, University Children's Hospital, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 2Department of Dermatovenereology, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 3Department of Vascular Diseases, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 4Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
Homozygous familial hypercholesterolemia (HoFH) is a rare inherited metabolic disorder, frequently leading to an early cardiovascular death if not adequately treated. Since standard medications usually fail to reduce LDL-cholesterol (LDL-C) levels satisfactorily, LDL-apheresis is a mainstay of managing HoFH patients but, at the same time, very burdensome and suboptimally effective. Liver transplantation (LT) has been previously shown to be a promising alternative. We report on a 14 year-long follow-up after LT in a HoFH patient. At the age of 4, the patient was referred to our institution because of the gradually increasing number of xanthomas on the knees, elbows, buttocks, and later the homozygous mutation c.1754T>C (p.Ile585Thr) on the LDL-receptor gene was confirmed. Despite subsequent intensive treatment with the combination of diet, statins, bile acid sequestrant, probucol, and LDL-apheresis, the patient developed valvular aortic stenosis and aortic regurgitation by 12 years. At 16 years, the patient successfully underwent deceased-donor orthotopic LT. Nine years post-LT, we found total regression of the cutaneous xanthomas and atherosclerotic plaques and with normal endothelial function. Fourteen years post-LT, his clinical condition remained stable, but LDL-C levels have progressively risen. In addition, a systematic review of the literature and guidelines on the LT for HoFH patients was performed. Six of the 17 identified guidelines did not take LT as a treatment option in consideration at all. But still the majority of guidelines suggest LT as an exceptional therapeutic option or as the last resort option when all the other treatment options are inadequate or not tolerated. Most of the observed patients had some kind of cardiovascular disease before the LT. In 76% of LT, the cardiovascular burden did not progress after LT. According to our experience and in several other reported cases, the LDL-C levels are slowly increasing over time post LT. Most of the follow-up data were short termed; only a few case reports have followed patients for 10 or more years after LT. LT is a feasible therapeutic option for HoFH patients, reversing atherosclerotic changes uncontrollable by conservative therapy, thus importantly improving the HoFH patient's prognosis and quality of life.
Introduction
Familial hypercholesterolemia (FH), the most common autosomal dominant condition, is due to the defective LDL-receptor leading to a decreased clearance of LDL-cholesterol (LDL-C) from plasma. In consequence, there is 2–3-fold elevation in the levels of total cholesterol (TC) and LDL-C after birth (1). Although the heterozygous FH (HeFH) is common (1/200–1/500), homozygous FH (HoFH) is a rare disease, affecting only four to six people in a million (2). Patients with HoFH can develop cutaneous and tendon xanthomas, arcus cornealis, and progressive generalized atherosclerosis in their early childhood. If untreated, patients with HoFH develop vascular lesions and cardiovascular disease (CVD) before the second decade of life and die before the end of the third decade of life (3, 4). Slovenia is, according to the available data, the only country with implemented nationwide universal screening for FH in preschool children, detecting both HeFH and HoFH patients (5–8).
HoFH is very difficult to manage. The medical treatment combines several cholesterol-lowering drugs used in other hypercholesterolemias. Initially, statins with ezetimibe are introduced and, in responsive patients, also PCSK9 inhibitors. Frequently, they do not result in satisfactory reductions in either TC or LDL-C levels, especially in moderate and severe HoFH patients with the highest CVD risk (9, 10). For over 30 years, LDL-apheresis is used, becoming a mainstay in the management of HoFH. It is currently considered the only safe and effective treatment for HoFH (11, 12). If LDL-apheresis is not successful, liver transplantation (LT) can be an alternative, also considering that LT is shown to be a successful treatment option in other metabolic liver diseases (13).
We aimed to report on a 14 year-long follow-up after LT in a HoFH patient at our center. In addition, a systematic review of the literature on LT for HoFH patients was performed.
Methods
We collected all the available clinical information of a now 31 year-old male patient, who was followed at the Department of Endocrinology, Diabetes, and Metabolism of the University Children's Hospital Ljubljana, UMC Ljubljana, Slovenia from the age of 4. FH in the patient was detected incidentally by the dermatologist before implementing the Slovenian universal screening program (5–8). The medical records were collected and entered into the national FH registry database with informed consent from the patient. Genetic analysis was performed after obtaining informed consent as a part of a prospective study on clinical and genetic characteristics of FH patients approved by the National Medical Ethics Committee [the genetic analysis was explained previously in detail by Klančar et al. (6)].
For the systematic review, two approaches were applied. For the case reports on LT as a treatment for FH patients, the PubMed database was used. The following search terms were used: “liver transplantation” (AND) [“familial hypercholesterolemia” (OR) “homozygous hypercholesterolemia” (OR) “familial hyperlipidemia”]. We found 111 research articles. By reading all the abstracts and titles, we excluded 93 articles that did not meet the following conditions: (1) only articles in English and articles published after 1998 were used, but no limits were made on the country of research; (2) only articles that were fully accessible were included; (3) only the articles on humans and not on animals nor cell models were used; (4) only articles where the recipient of the liver transplant has had FH were used; and (5) only articles with follow-ups longer than 1 month were included. All the available articles that met the criteria were read in full-text form. In addition, other case reports were found through the articles' reference list. In the end, 23 articles were included. A systematic review of the clinical guidelines on LT was based on the search procedure used by Migliara et al. (14). In the end, we have found 17 guidelines.
Case Description
A 4 year-old male patient was referred to our institution by a dermatologist because of the gradually increasing number of xanthomas on the knees, elbows, and buttocks that first appeared when he was 3 years old. He was born after an uneventful pregnancy and delivery as the second child of apparently non-consanguineous parents. The family history for premature CVD was negative. His early development was normal. Extremely high levels of TC (24.8 mmol/L; 928 mg/dl), LDL-C (21.6 mmol/L; 834 mg/dl), and triglycerides (TGC) (5.0 mmol/L; 443 mg/dl) and low levels of HDL-cholesterol (HDL-C) (0.3 mmol/L; 11.6 mg/dl) were measured at the first exam. Elevated levels of TC and LDL-C were also found in his parents, sister, and daughter (afterward) (Table 1). The clinical and biochemical picture suggested the diagnosis of HoFH, which was later confirmed genetically; a previously described homozygous mutation c.1754T>C (p.Ile585Thr) was found in the patient, while both his parents, his sister, and daughter were confirmed to be heterozygotes for the same mutation. Despite treatment with a highly restrictive diet, cholestyramine, statins, and LDL-apheresis (the exact timeline is described in Figure 1, Table 2), xanthomas extended progressively. At 7 years, increased IMT of the common carotid artery was measured with ultrasound. At 12 years, echocardiographically valvular aortic stenosis and aortic regurgitation were observed. Levels of LDL-C at the time of LDL-apheresis ranged from 2.4 to 11.6 mmol/L (92.8–449 mg/dl) in between treatments. Lp(a) levels ranged from 756 mg/L at the beginning to 135 mg/L after the last treatment. He refused to have apheresis for 3 years, but with psychological support, he continued.
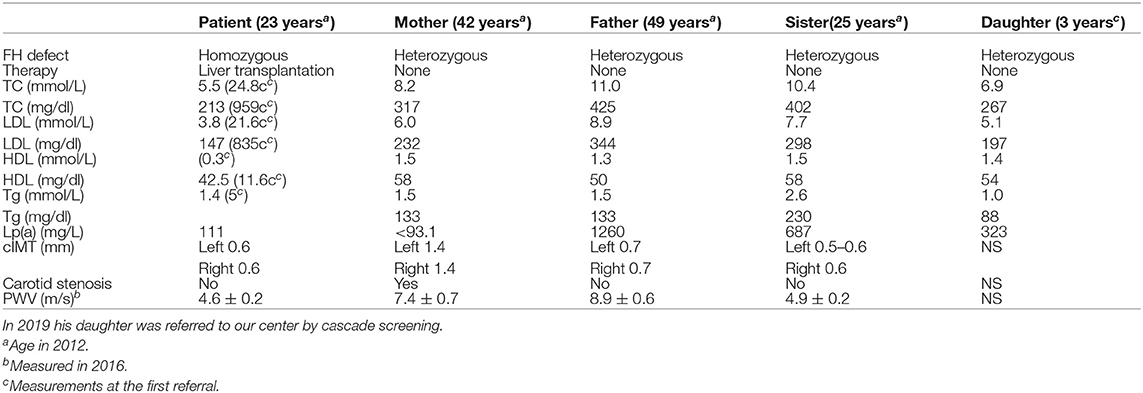
Table 1. Plasma levels of total, LDL, HDL cholesterol, and Lp(a) in patient, parents, and sister at the same time point (in 2012 and PWV values in 2016).
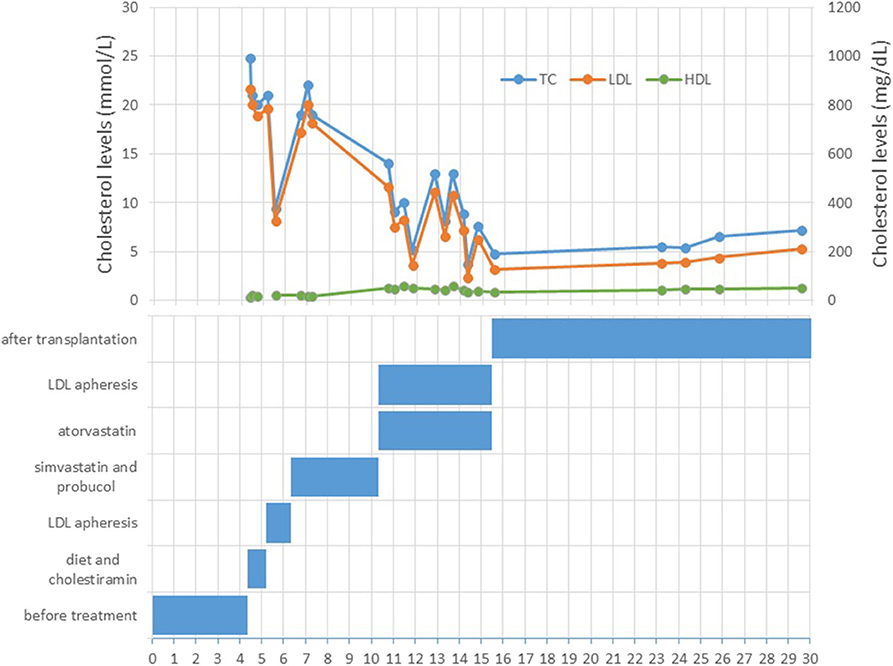
Figure 1. Overview of the cholesterol levels and therapy through the patient's lifetime.

Table 2. Timeline of clinical event.
Due to progressive worsening conditions (valvular aortic stenosis and aortic regurgitation), the patient underwent deceased-donor orthotopic LT when he was 16 years old. There were no intraoperative or postoperative complications. The patient's immunosuppression regimen first consisted of tacrolimus, methylprednisolone, and ursodeoxycholic acid, but the steroid was gradually tapered. Six months after the LT, the patient's xanthomas had regressed, and the TC levels have fallen to 5.2 mmol/L (201 mg/dl). A year and a half after the LT, the TC level was at the lowest value [4.4 mmol/L (170 mg/dl)]. Two years after the LT, an episode of acute rejection developed, which was successfully treated with steroids, and the TC level was measured at 5.1 mmol/L (197 mg/dl).
Eight years after the LT, the patient who was then 24 years old was doing well. The patient had been on maintaining tacrolimus therapy. His cutaneous xanthomas completely resolved. An echocardiogram showed complete regression of aortic stenosis, and his IMT was in the normal range (0.6 mm on both common carotid arteries). His TC and LDL-C levels were 5.4 and 3.9 mmol/L, respectively (209 and 151 mg/dl), and Lp(a) levels were 111 mg/L. His LDL-C levels were lower than the LDL-C levels measured on the same occasion in his parents and sister (Table 1). Twelve years after the LT, the PWV velocity was measured with ultrasound, and the values were in the normal range (4.6 ± 0.2 m/s) The RHI was 2.46.
Afterward, the patient was not responding to the invitations from our clinic for almost 5 years (at that time, he was only seeing his gastroenterologist) and returned for a follow-up visit only in the year 2018, almost 14 years after the LT. The total regression of xanthomas was evident at the last visit. At that point, he was almost 30 years old, employed, and had a child who was scheduled for a later visit at our lipid clinic and was confirmed to be heterozygote for the same mutation. His work was mostly sedentary, and he was not very physically active. In addition, he started smoking. He was still taking tacrolimus, but no antilipemic treatment was prescribed to him after the LT. His TC and LDL-C levels were 7.2 mmol/L (278 mg/dl) and 5.3 mmol/L (205 mg/dl), respectively. In addition, an elevated ApoB level was present (1.48 g/L), while his Lp(a) level was normal (145 mg/L). Furthermore, hsCRP was 2.11 mg/L. His carotid IMT level had increased to 0.90 mm on both carotid arteries (abnormal result for his age) (15). He was normotensive (97/67 mmHg), and his BMI was 18 kg/m2. Liver enzyme levels were in the normal range. The HbA1c level was 4.8%, and the fasting insulin level was 4.3 mE/L. The glomerular filtration rate was above 90 ml/min. At the last visit to our lipid clinic, the patient was recommended to start statin treatment (rosuvastatin, 10 mg) and was transitioned to the adult lipid clinic, where his first visit was scheduled in early 2019. At that visit, his TC and LDL-C levels were 7.36 mmol/L (284.6 mg/dl) and 5.9 mmol/L (228.15 mg/dl), respectively.
Discussion
HoFH frequently leads to early cardiovascular death if not adequately treated. However, the standard therapy with medications and LDL-apheresis at least in more severe cases often fail to address difficult clinical situation satisfactorily (16–18). The management of patients with HoFH represents a medical challenge despite the approval of new lipid-lowering agents (i.e., mipomersen, lomitapide, PCSK9 inhibitors) (19). An individualized approach in the management of the disease is of great importance (20). Diagnosis of FH and its early treatment is recommended in all guidelines (10, 21–23).
Lipid-lowering drug therapy is recommended for the treatment of HoFH in all age groups (24). LDL-apheresis has been used and progressively became a mainstay in the management of HoFH. In line with current guidelines, treatment should be started as soon as possible, ideally by age 5 and not later than 8 years. However, this and the frequency of treatment represent a compromise between access to centers, the severity of the disease, and the patient's choice and/or compliance (21, 25, 26). Currently, LDL-apheresis is recommended at weekly or biweekly intervals with concurrent administration of maximal doses of lipid-lowering agents in exceptional circumstances such as pregnancy; more frequent treatment without statins may be considered (4, 27, 28).
Despite the lack of randomized studies, there is clinical evidence that long-term lipoprotein apheresis can contribute to plaque regression and/or stabilization, slow coronary atherosclerosis progression, and improved prognosis (29). Thompson et al. have concluded in a 50 year follow-up study that improved treatment and treatment to lower TC levels have a better prognosis (30, 31). Combining apheresis with additional drug therapy to slow down the rapid rebound of LDL-C, which follows each procedure, and to keep the LDL-C level as low as possible for as long as possible is also essential (32, 33). A more recent study has shown that achieving a mean LDL-C level of 4.2 mmol/L (162.4 mg/dl) by weekly apheresis plus statin/ezetimibe therapy failed to prevent the progression of the aortic, coronary, and carotid disease (28, 34). The most recent statement on target levels for both HoFH and HeFH, which advocates lowering LDL-C to <3.5 mmol/L (135 mg/dl) in children and to <2.5 mmol/L (97 mg/dl) in adults, or even <1.8 mmol/L (70 mg/dl) in those at the highest risk can seldom be achieved in homozygotes with existing apheresis/drug therapy regimens (2, 10, 24, 35).
When our patient was 5 years old, biweekly LDL-apheresis first without and then with statins was used. Despite the early treatment initiation, he has developed valvular aortic stenosis. One week after LDL-apheresis, LDL-C levels were up to 10 mmol/L (386.7 mg/dl).
Therefore, LT was considered as a therapeutic option (19, 36), as it was also considered in 23 case reports and case series (19, 36–57), with a total of 90 patients with FH who have undergone LT. Data are presented in detail in Supplement Table 1. Of the 90 patients found, 47 were female. Most of them had HoFH (77.8%) (genetic or clinical diagnosis). Eight patients were just diagnosed with FH. Most of the patients had some kind of CVD before the LT, some having coronary stenting or dilatation (22 patients) or heart valve replacement (4 patients) before LT. Four patients also received a heart transplant in the same surgical procedure, and one also received a kidney transplant at the same time as LT. Although a successful therapeutic strategy (41, 42, 46, 47), there are obvious disadvantages, including the risk of post-transplantation surgical complications and mortality, the risk of acute or chronic organ rejection, and the need for life-long treatment with immunosuppressive therapy (58, 59). In most case studies (63.3%), the cardiovascular disease burden after LT was comparable to the one before LT. In 24.4%, however, the progression of CVD was observed, and in 12.62%, regression was observed (19, 36–57). The latter was also the case in our patient. Cardiovascular complications and disease progression were observed in 11 patients. Of them, six died because of heart failure after LT (19, 36, 38–40, 42, 45, 52). Another two patients died because of septicemia (19, 36). Interestingly, there were no deaths in the five patients with combined heart and liver transplantation (41, 43, 51, 53, 55). One patient lived for 20 years with a combined heart–liver transplant with an absence of CVD (41).
Corticosteroids, cyclosporine, tacrolimus, and sirolimus, the main treatments after transplantation, are all associated with elevated cholesterol levels (60). Cyclosporine interferes with the binding of LDL-C to LDL receptors and also interferes with bile acid synthesis acting on the enzyme 26 hydroxylases. Tacrolimus has similar but lesser effects on lipid metabolism, then cyclosporine (37). In addition, metabolic syndrome can develop after transplantation (19). In our patient, this was not the case. After successful LT, the cardiovascular complications regressed, endothelial dysfunction was not detected, and IMT was reversed.
In the gathered case reports and our case report interestingly in the short term, a decrease in TC levels after LT was seen, reaching the lowest level in the first years after LT. The main limitation of systematic review of the case reports is that most of the follow-up data were short termed; only a few case reports have followed patients for 10 or more years after LT. The longest report is 20 years (21, 22). Afterward, TC seems to increase steadily, in some patients, even despite taking cholesterol-lowering drugs (Figure 2) (51). Ibrahim et al. (41) report of an increase in TC levels from 3.4 mmol/L (132 mg/dl) to 6.3 mmol/L (244 mg/dl) 20 years after LT. An unhealthy lifestyle (excessive dietary intake of cholesterol and saturated fats), diabetes, obesity, proteinuria, age, genetic predisposition, and medications are known risk factors for elevated cholesterol levels in patients after LT for different defects (37). Some of them are also present in our patient.
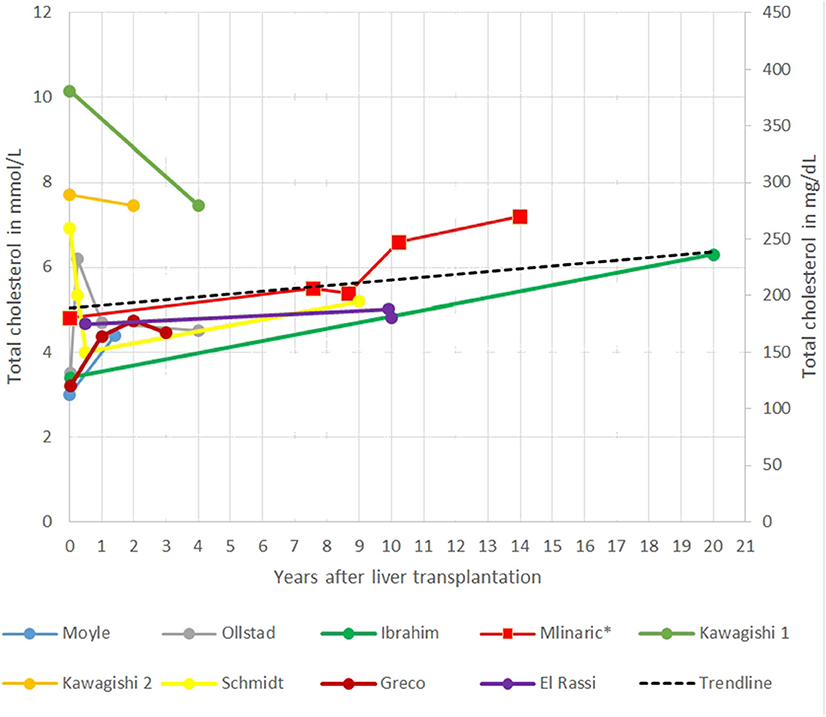
Figure 2. Representation of total cholesterol (TC) after liver transplantation (LT) in different case reports.
Out of the 16 guidelines (2, 4, 21, 22, 27, 61–71) proposed by professional societies and one by Raal et al. (10), 6 of the 17 did not take LT as a treatment option in consideration at all (2, 22, 64, 68, 69). In the guidelines that do mention LT, the majority suggests that it as an exceptional therapeutic option or as the last resort option when all the other treatment options are inadequate or not tolerated (4, 10, 22, 61–63, 65). The International FH foundation states that the LT decision should be in partnership with the patient and/or their relatives (4). In the article by Raal et al. (10), the LT is an acceptable option only for the treatment of HoFH patients that are unresponsive to conventional lipid-lowering therapy and possibly before the onset of significant CVD. However, if the CVD is discovered in preoperative cardiac investigations, the FH Australasia Network consensus group suggests that coronary artery bypass surgery and/or aortic valve replacement should also be considered (62). The same is proposed by the International FH group (4), also suggesting that a combined heart and LT should be considered according to the clinical context. EAS recommends that we should also have in mind the disadvantages of LT (i.e., the need for life-long immunosuppressive therapy, the paucity of donors, and possible surgical complications) (21).
LT is, therefore, especially indicated for patients with HoFH that do not otherwise respond to maximal medical therapy (4, 61–63, 65), but it is not a feasible option for all HoFH patients. However, LT has substantiated the development of other novel therapeutic approaches for patients with severe FH (e.g., liver-direct gene delivery, stem cell transplantation) (67).
The decision should be made with the patient and his relatives in an appropriate setting, and all the benefits or potential harms of the transplantation and of declining LT should be explained (22).
Conclusions
LT is a feasible therapeutic option, especially in patients with HoFH with progressive atherosclerotic disease that cannot be sufficiently controlled by medications and/or LDL-apheresis. In our HoFH patient, the long-term outcome after the LT was considered highly favorable, reversing the already severely progressed atherosclerosis before the procedure, despite the relatively early detection and intensive treatment with LDL-apheresis.
Replacing the liver where most of the LDL-C metabolism occurs represents a way of somatic gene therapy for HoFH, a frequently fatal inborn metabolic disorder. On the other hand, LT exposes the patient to considerable other clinical risks and burdens associated with the transplantation (e.g., the need for life-long immunosuppressive therapy, the paucity of donors, and possible surgical complications) (21). According to our experience and from the case reports, the LDL-C levels are slowly increasing years after the LT due to an unhealthy lifestyle, tacrolimus, or other therapy (37) and maybe because of LT organ regeneration with cells stemming from the patient. CVD and death of myocardial infarction are also reported in HoFH patients after LT (19, 39). LT might not represent the definite treatment of HoFH. Thus, after LT, the patients need to be further regularly followed at the lipid clinic (and also by the hepatologist), but at least in our patient, the LT vastly improved his quality of life (10, 19). The data indicate that early transplantation may be favorable and that actively preventing septicemia might be improving outcomes.
Finally, early detection programs and the clinical availability of novel therapeutic strategies are urgently needed to address the needs of all HoFH patients. Developing specific guidelines and international patient registries on LT for HoFH might be beneficial, following the example of the research group, who have made the International Registry on Lipoprotein Apheresis in Children with Homozygous Familial Hypercholesterolemia (72).
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MM collected the data and prepared the manuscript. NB and UG followed and treated the patient, collected the data, and prepared the first draft of the case report. VD made the first suspicion of the disease and referred him early for further evaluation and treatment and reviewed the manuscript. AS performed follow-up tests and reviewed the manuscript. MC, UG, and TB supervised the work and helped writing the manuscript. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Funding
This work was partly supported by the Slovenian National Research Agency, Bleiweisova cesta 30, SI-1000 Ljubljana, Slovenia (grants P3-0343, J3-4116, Jx3-6800 and J3-6798).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the patient and his family and all the other physicians and nurses involved in the management and treatment of the patient. We would like to acknowledged Gasper Klancar, PhD, and Katarina Trebusak Podkrajsek, PhD, for performed the genetic analysis in the family.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2020.567895/full#supplementary-material
Abbreviations
FH, familial hypercholesterolemia; HoFH, homozygous familial hypercholesterolemia; HeFH, heterozygous familial hypercholesterolemia; LDL-C, LDL-cholesterol; HDL-C, HDL-cholesterol; TC, total cholesterol; Tgc, triglycerides; LT, liver transplantation; CVD, cardiovascular disease; RHI, reactive hyperemia index; PWV, pulse wave velocity; IMT, intima-media thickness; BMI, body mass index.
References
1. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease, 8th Edn. New York, NY: McGraw-Hill (2001). p. 2863–2913.
2. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. (2020) 41:111–88. doi: 10.15829/1560-4071-2020-3826
3. Vallejo-Vaz AJ, Kondapally Seshasai SR, Cole D, Hovingh GK, Kastelein JJ, Mata P, et al. Familial hypercholesterolaemia: a global call to arms. Atherosclerosis. (2015) 243:257–9. doi: 10.1016/j.atherosclerosis.2015.09.021
4. Watts GF, Gidding S, Wierzbicki AS, Toth PP, Alonso R, Brown WV, et al. Integrated guidance on the care of familial hypercholesterolaemia from the international FH foundation: executive summary. J Atheroscler Thromb. (2014) 21:368–74. doi: 10.1016/j.ijcard.2013.11.025
5. Groselj U, Kovac J, Sustar U, Mlinaric M, Fras Z, Podkrajsek KT, et al. Universal screening for familial hypercholesterolemia in children: the Slovenian model and literature review. Atherosclerosis. (2018) 277:383–91. doi: 10.1016/j.atherosclerosis.2018.06.858
6. Klančar G, Grošelj U, Kovač J, Bratanič N, Bratina N, Trebušak Podkrajšek K, et al. Universal screening for familial hypercholesterolemia in children. J Am Coll Cardiol. (2015) 66:1250–7. doi: 10.1016/j.jacc.2015.07.017
7. Sedej K, Kotnik P, Avbelj Stefanija M, Grošelj U, Širca Campa A, Lusa L, et al. Decreased prevalence of hypercholesterolaemia and stabilization of obesity trends in 5-year-old children: possible effects of changed public health policies. Eur J Endocrinol. (2014) 170:293–300. doi: 10.1530/EJE-13-0566
8. Vallejo-Vaz AJ, De Marco M, Stevens CAT, Akram A, Freiberger T, Hovingh GK, EAS Familial Hypercholesterolaemia Studies Collaboration. Overview of the current status of familial hypercholesterolaemia care in over 60 countries - The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis. (2018) 277:234–55. doi: 10.1016/j.atherosclerosis.2018.08.051
9. Perez de Isla L, Alonso R, Watts GF, Mata N, Saltijeral Cerezo A, Muñiz O, et al. Attainment of LDL-cholesterol treatment goals in patients with familial hypercholesterolemia: 5-year safeheart registry follow-up. J Am Coll Cardiol. (2016) 67:1278–85. doi: 10.1016/j.jacc.2016.01.008
10. Raal FJ, Hovingh GK, Catapano AL. Familial hypercholesterolemia treatments: guidelines and new therapies. Atherosclerosis. (2018) 277:483–92. doi: 10.1016/j.atherosclerosis.2018.06.859
11. Drouin-Chartier JP, Tremblay AJ, Bergeron J, Lamarche B, Couture P. High serum triglyceride concentrations in patients with homozygous familial hypercholesterolemia attenuate the efficacy of lipoprotein apheresis by dextran sulfate adsorption. Atherosclerosis. (2018) 270:26–32. doi: 10.1016/j.atherosclerosis.2018.01.005
12. Julius U. History of lipidology and lipoprotein apheresis. Atherosclerosis. (2017) 30:1–8. doi: 10.1016/j.atherosclerosissup.2017.05.034
13. Mc Kiernan PJ. Recent advances in liver transplantation for metabolic disease. J Inherit Metab Dis. (2017) 40:491–5. doi: 10.1007/s10545-017-0020-z
14. Migliara G, Baccolini V, Rosso A, D'Andrea E, Massimi A, Villari P, et al. Familial hypercholesterolemia: a systematic review of guidelines on genetic testing and patient management. Front Public Health. (2017) 5:252. doi: 10.3389/fpubh.2017.00252
15. Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J Hypertens. (2018) 36:1953–2014. doi: 10.1097/HJH.0000000000001940
16. Rajendran R, Srinivasa KH, Rangan K, Hegde M, Ahmed N. Supra-valvular aortic stenosis in a patient with homozygous familial hypercholesterolaemia. Eur Heart J Cardiovasc Imaging. (2013) 14:1023. doi: 10.1093/ehjci/jet072
17. Kolansky DM, Cuchel M, Clark BJ, Paridon S, McCrindle BW, Wiegers SE, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol. (2018) 102:1438–43. doi: 10.1016/j.amjcard.2008.07.035
18. Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. (2012) 223:262–8. doi: 10.1016/j.atherosclerosis.2012.02.019
19. Martinez M, Brodlie S, Griesemer A, Kato T, Harren P, Gordon B, et al. Effects of liver transplantation on lipids and cardiovascular disease in children with homozygous familial hypercholesterolemia. Am J Cardiol. (2016) 118:504–10. doi: 10.1016/j.amjcard.2016.05.042
20. Farnier M, Civeira F, Descamps O, FH expert working group. How to implement clinical guidelines to optimise familial hypercholesterolaemia diagnosis and treatment. Atheroscler Suppl. (2017) 26:25–35. doi: 10.1016/S1567-5688(17)30022-3
21. Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, et al. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Atherosclerosis. (2016) 253:281–344. doi: 10.1016/j.atherosclerosis.2016.08.018
22. National Institute for Health and Care Excellence. Familial Hypercholesterolaemia: Identification and Management. Guidance and Guidelines. National Institute for Health and Care Excellence. (2017). Available online at: https://www.nice.org.uk/guidance/cg71 (accessed May 18, 2020).
23. Daniels SR Gidding SS de Ferranti SD National Lipid Association Expert Panel on Familial Hypercholesterolemia. Pediatric aspects of familial hypercholesterolemias: recommendations from the national lipid association expert panel on familial hypercholesterolemia. J Clin Lipidol. (2011) 5:30–7. doi: 10.1016/j.jacl.2011.03.453
24. Hovingh GK, Goldberg AC, Moriarty PM. Managing the challenging homozygous familial hypercholesterolemia patient: academic insights and practical approaches for a severe dyslipidemia, a National Lipid Association Masters Summit. J Clin Lipidol. (2017) 11:602–16. doi: 10.1016/j.jacl.2017.03.008
25. Luirink IK, Hutten BA, Wiegman A. Optimizing treatment of familial hypercholesterolemia in children and adolescents. Curr Cardiol Rep. (2015) 17:629. doi: 10.1007/s11886-015-0629-1
26. Beliard S, Gallo A, Duchêne E, Carrié A, Bittar R, Chapman MJ, et al. Lipoprotein-apheresis in familial hypercholesterolemia: long-term patient compliance in a French cohort. Atherosclerosis. (2018) 277:66–71. doi: 10.1016/j.atherosclerosis.2018.08.007
27. Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, et al. The agenda for familial hypercholesterolemia. a scientific statement from the American Heart Association. Circulation. (2015) 132:2167–92. doi: 10.1161/CIR.0000000000000297
28. Stefanutti C, Julius U, Watts GF, Harada-Shiba M, Cossu M, Schettler VJ, et al. Toward an international consensus-Integrating lipoprotein apheresis and new lipid-lowering drugs. J Clin Lipidol. (2017) 11:858–71. doi: 10.1016/j.jacl.2017.04.114
29. Schuff-Werner P, Fenger S, Kohlschein P. Role of lipid apheresis in changing times. Clin Res Cardiol Suppl. (2012) 7:7–14. doi: 10.1007/s11789-012-0049-3
30. Thompson GR, Seed M, Naoumova RP, Neuwirth C, Walji S, Aitman TJ, et al. Improved cardiovascular outcomes following temporal advances in lipid-lowering therapy in a genetically-characterised cohort of familial hypercholesterolaemia homozygotes. Atherosclerosis. (2015) 243:328–33. doi: 10.1016/j.atherosclerosis.2015.09.029
31. Omer L, Hudson EA, Zheng S, Hoying JB, Shan Y, Boyd NL. CRISPR correction of a homozygous low-density lipoprotein receptor mutation in familial hypercholesterolemia induced pluripotent stem cells. Hepatol Commun. (2017) 1:886–98. doi: 10.1002/hep4.1110
32. Thompson GR. The evidence-base for the efficacy of lipoprotein apheresis in combating cardiovascular disease. Atheroscler Suppl. (2013) 14:67–70. doi: 10.1016/j.atherosclerosissup.2012.10.001
33. Stefanutti C, Julius U. Lipoprotein apheresis: state of the art and novelties. Atheroscler Suppl. (2013) 14:19–27. doi: 10.1016/j.atherosclerosissup.2012.10.021
34. Græsdal A, Bogsrud MP, Holven KB, Nenseter MS, Narverud I, Langslet G, et al. Apheresis in homozygous familial hypercholesterolemia: the results of a follow-up of all Norwegian patients with homozygous familial hypercholesterolemia. J Clin Lipidol. (2012) 6:331–9. doi: 10.1016/j.jacl.2012.03.004
35. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Consensus Statement of the European Atherosclerosis Society. Eur Heart J. (2013) 34:3478–90. doi: 10.1093/eurheartj/eht273
36. Mansoorian M, Kazemi K, Nikeghbalian S, Shamsaeefar A, Mokhtari M, Dehghani SM, et al. Liver transplantation as a definitive treatment for familial hypercholesterolemia: a series of 36 cases. Pediatr Transplant. (2015) 19:605–11. doi: 10.1111/petr.12562
37. Akdur A, Kirnap M, Ayvazoglu Soy EH, Ozcay F, Moray G, Arslan G, et al. Unusual indications for a liver transplant: a single-center experience. Exp Clin Transplant. (2017) 15:128–32. doi: 10.6002/ect.mesot2016.P11
38. Alim A, Tokat Y, Erdogan Y, Gokkaya Z, Dayangac M, Yuzer Y, et al. Liver transplantation for homozygote familial hypercholesterolemia: the only curative treatment. Pediatr Transplant. (2016) 20:1060–4. doi: 10.1111/petr.12763
39. Greco M, Robinson JD, Eltayeb O, Benuck I. Progressive aortic stenosis in homozygous familial hypercholesterolemia after liver transplant. Pediatrics. (2016) 138:e20160740. doi: 10.1542/peds.2016-0740
40. El-Rassi I, Chehab G, Saliba Z, Alawe A, Jebara V. Fatal cardiac atherosclerosis in a child 10 years after liver transplantation: a case report and a review. J Clin Lipidol. (2011) 5:329–32. doi: 10.1016/j.jacl.2011.05.002
41. Ibrahim M, El-Hamamsy I, Barbir M, Yacoub MH. Translational lessons from a case of combined heart and liver transplantation for familial hypercholesterolemia 20 years post-operatively. J Cardiovasc Transl Res. (2012) 5:351–8. doi: 10.1007/s12265-011-9311-1
42. Palacio CH, Harring TR, Nguyen NT, Goss JA, O'Mahony CA. Homozygous familial hypercholesterolemia: case series and review of the literature. Case Rep Transplant. (2011) 2011:154908. doi: 10.1155/2011/154908
43. Alkofer BJ, Chiche L, Khayat A, Deshayes JP, Lepage A, Saloux E, et al. Liver transplant combined with heart transplant in severe heterozygous hypercholesterolemia: report of the first case and review of the literature. Transplant Proc. (2005) 37:2250–2. doi: 10.1016/j.transproceed.2005.03.037
44. Khalifeh M, Faraj W, Heaton N, Rela M, Sharara AI. Successful living-related liver transplantation for familial hypercholesterolemia in the Middle East. Transpl Int. (2005) 17:735–9. doi: 10.1007/s00147-004-0791-7
45. Shrotri M, Fernando BS, Sudhindran S, Delriviere L, Watson CJ, Gibbs P, et al. Long-term outcome of liver transplantation for familial hypercholesterolemia. Transplant Proc. (2003) 35:381–2. doi: 10.1016/S0041-1345(02)03910-6
46. Küçükkartallar T, Yankol Y, Kanmaz T, Topaloglu S, Acarli K, Kalayoglu M. Liver transplantation as a treatment option for three siblings with homozygous familial hypercholesterolemia. Pediatr Transplant. (2011) 15:281–4. doi: 10.1111/j.1399-3046.2010.01469.x
47. Maiorana A, Nobili V, Calandra S, Francalanci P, Bernabei S, El Hachem M, et al. Preemptive liver transplantation in a child with familial hypercholesterolemia. Pediatr Transplant. (2011) 15:25–9. doi: 10.1111/j.1399-3046.2010.01383.x
48. Kawagishi N, Satoh K, Akamatsu Y, Sekiguchi S, Ishigaki Y, Oikawa S, et al. Long-term outcome after living donor liver transplantation for two cases of homozygous familial hypercholesterolemia from a heterozygous donor. J Atheroscler Thromb. (2007) 14:94–8. doi: 10.5551/jat.14.94
49. Schmidt HH, Tietge UJ, Buettner J, Barg-Hock H, Offner G, Schweitzer S, et al. Liver transplantation in a subject with familial hypercholesterolemia carrying the homozygous p.W577R LDL-receptor gene mutation. Clin Transplant. (2008) 22:180–4. doi: 10.1111/j.1399-0012.2007.00764.x
50. López-Santamaria M, Migliazza L, Gamez M, Murcia J, Diaz-Gonzalez M, Camarena C, et al. Liver transplantation in patients with homozygotic familial hypercholesterolemia previously treated by end-to-side portocaval shunt and ileal bypass. J Pediatr Surg. (2000) 35:630–3. doi: 10.1053/jpsu.2000.0350630
51. Offstad J, Schrumpf E, Geiran O, Søreide O, Simonsen S. Plasma exchange and heart-liver transplantation in a patient with homozygous familial hypercholesterolemia. Clin Transplant. (2011) 15:432–6. doi: 10.1034/j.1399-0012.2001.150612.x
52. Popescu I, Simionescu M, Tulbure D, Sima A, Catana C, Niculescu L, et al. Homozygous familial hypercholesterolemia: specific indication for domino liver transplantation. Transplantation. (2003) 76:1345–50. doi: 10.1097/01.TP.0000093996.96158.44
53. van Heyningen C. Progressive vascular disease in homozygous familial hypercholesterolaemia despite apheresis and heart and liver transplantation. Br J Diabetes Vasc Dis. (2011) 11:270–2. doi: 10.1177/1474651411422652
54. Moyle M, Tate B. Homozygous familial hypercholesterolaemia presenting with cutaneous xanthomas: response to liver transplantation. Australas J Dermatol. (2004) 45:226–8. doi: 10.1111/j.1440-0960.2004.00103.x
55. Ahualli L, Stewart-Harris A, Bastianelli G, Radlovachki D, Bartolomé A, Trigo PL, et al. Combined cardiohepatic transplantation due to severe heterozygous familiar hypercholesteremia type II: first case in Argentina–a case report. Transplant Proc. (2007) 39:2449–53. doi: 10.1016/j.transproceed.2007.07.068
56. Gulsoy Kirnap N, Kirnap M, Bascil Tutuncu N, Moray G, Haberal M. The curative treatment of familial hypercholesterolemia: liver transplantation. Clin Transplant. (2019) 33:e13730. doi: 10.1111/ctr.13730
57. Cephus CE, Qureshi AM, Sexson Tejtel SK, Alam M, Moodie DS. Coronary artery disease in a child with homozygous familial hypercholesterolemia: regression after liver transplantation. J Clin Lipidol. (2019) 13:880–6. doi: 10.1016/j.jacl.2019.09.007
58. Malatack JJ. Liver transplantation as treatment for familial homozygous hypercholesterolemia: too early or too late. Pediatr Transplant. (2011) 15:123–5. doi: 10.1111/j.1399-3046.2010.01458.x
59. Al-Ashwal A, Alnouri F, Sabbour H, Al-Mahfouz A, Al-Sayed N, Razzaghy-Azar M, et al. Identification and treatment of patients with homozygous familial hypercholesterolaemia: information and recommendations from a middle east advisory panel. Curr Vasc Pharmacol. (2015) 13:759–70. doi: 10.2174/1570161113666150827125040
60. Agarwal A, Prasad GV. Post-transplant dyslipidemia: mechanisms, diagnosis and management. World J Transplant. (2016) 6:125–34. doi: 10.5500/wjt.v6.i1.125
61. Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the national lipid association expert panel on familial hypercholesterolemia. J Clin Lipidol. (2011) 5:1–8. doi: 10.1016/j.jacl.2011.04.003
62. Watts GF, Sullivan DR, Poplawski N, van Bockxmeer F, Hamilton-Craig I, Clifton PM, et al. Familial hypercholesterolaemia: a model of care for Australasia. Atheroscler Suppl. (2011) 12:221–63. doi: 10.1016/j.atherosclerosissup.2011.06.001
63. Descamps OS, Tenoutasse S, Stephenne X, Gies I, Beauloye V, Lebrethon MC, et al. Management of familial hypercholesterolemia in children and young adults: consensus paper developed by a panel of lipidologists, cardiologists, paediatricians, nutritionists, gastroenterologists, general practitioners and a patient organization. Atherosclerosis. (2011) 218:272–80. doi: 10.1016/j.atherosclerosis.2011.06.016
64. Village EG. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. (2011) 128:213–56. doi: 10.1542/peds.2009-2107C
65. France M, Rees A, Datta D, Thompson G, Capps N, Ferns G, et al. HEART UK statement on the management of homozygous familial hypercholesterolaemia in the United Kingdom. Atherosclerosis. (2016) 255:128–39. doi: 10.1016/j.atherosclerosis.2016.10.017
66. The Cardiac Society of Australia and New Zealand. Diagnosis and Management of Familial Hypercholesterolaemia – Position Statement. (2016). Available online at: https://www.csanz.edu.au/wp-content/uploads/2017/07/Familial-Hypercholesterolaemia_ratified_-25-Nov-2016.pdf (accessed May 18, 2020).
67. Santos RD, Gidding SS, Hegele RA, Cuchel MA, Barter PJ, Watts GF, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. (2016) 4:850–61. doi: 10.1016/S2213-8587(16)30041-9
68. Volpe M, Volpe R, Gallo G, Presta V, Tocci G, Folco E, et al. 2017 Position paper of the Italian society for cardiovascular prevention (SIPREC) for an updated clinical management of hypercholesterolemia and cardiovascular risk: executive document. High Blood Press Cardiovasc Prev. (2017) 24:313–29. doi: 10.1007/s40292-017-0211-6
69. Li YH, Ueng KC, Jeng JS, Charng MJ, Lin TH, Chien KL, et al. 2017 Taiwan lipid guidelines for high risk patients. J Formos Med Assoc. (2017) 116:217–48. doi: 10.1016/j.jfma.2016.11.013
70. Jellinger PS, Handelsman Y, Rosenblit PD, Bloomgarden ZT, Fonseca VA, Garber AJ, et al. American Association Of Clinical Endocrinologists and American College Of Endocrinology Guidelines for management of dyslipidemia and prevention of cardiovascular disease – executive summary. Endocr Pract. (2017) 23:479–97. doi: 10.4158/EP171764.APPGL
71. Harada-Shiba M, Ohta T, Ohtake A, Ogura M, Dobashi K, Nohara A, et al. Guidance for pediatric familial hypercholesterolemia 2017. J Atheroscler Thromb. (2018) 25:539–53. doi: 10.5551/jat.CR002
72. Luirink IK, Hutten BA, Greber-Platzer S, Kolovou GD, Dann EJ, de Ferranti SD, et al. Practice of lipoprotein apheresis and short-term efficacy in children with homozygous familial hypercholesterolemia: data from an international registry. Atherosclerosis. (2020) 299:24–31. doi: 10.1016/j.atherosclerosis.2020.01.031
Keywords: homozygous familial hypercholesterolemia, HoFH, fh, LDL-apheresis, liver transplantation, Slovenia, review
Citation: Mlinaric M, Bratanic N, Dragos V, Skarlovnik A, Cevc M, Battelino T and Groselj U (2020) Case Report: Liver Transplantation in Homozygous Familial Hypercholesterolemia (HoFH)—Long-Term Follow-Up of a Patient and Literature Review. Front. Pediatr. 8:567895. doi: 10.3389/fped.2020.567895
Received: 30 May 2020; Accepted: 28 August 2020;
Published: 09 October 2020.
Edited by:
Uma Ramaswami, Royal Free London NHS Foundation Trust, United KingdomReviewed by:
Nicolina Cristina Sorrentino, Telethon Institute of Genetics and Medicine (TIGEM), ItalyCharlotte L. Alston, Wellcome Trust Centre for Mitochondrial Research (WT), United Kingdom
Copyright © 2020 Mlinaric, Bratanic, Dragos, Skarlovnik, Cevc, Battelino and Groselj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Urh Groselj, urh.groselj@kclj.si