Acute Cervical Longitudinally Extensive Transverse Myelitis in a Child With Lipopolysaccharide-Responsive-Beige-Like-Anchor-Protein (LRBA) Deficiency: A New Complication of a Rare Disease
 Matteo Chinello1*
Matteo Chinello1*  Margherita Mauro2
Margherita Mauro2  Gaetano Cantalupo3 Giacomo Talenti4 Sara Mariotto5 Rita Balter1
Gaetano Cantalupo3 Giacomo Talenti4 Sara Mariotto5 Rita Balter1  Massimiliano De Bortoli1
Massimiliano De Bortoli1  Virginia Vitale1 Ada Zaccaron1
Virginia Vitale1 Ada Zaccaron1  Elisa Bonetti1 Daniela Di Carlo6 Federica Barzaghi7
Elisa Bonetti1 Daniela Di Carlo6 Federica Barzaghi7  Simone Cesaro1
Simone Cesaro1- 1Pediatric Hematology Oncology, Azienda Ospedaliera Universitaria Integrata, Verona, Italy
- 2Pediatric Department, Santa Maria Degli Angeli Hospital, Pordenone, Italy
- 3Child Neuropsychiatry, University of Verona, Verona, Italy
- 4Department of Diagnostics and Pathology, Neuroradiology Unit, Verona University Hospital, Verona, Italy
- 5Neurology Unit, Department of Neurosciences, Biomedicine and Movement Sciences, University of Verona, Verona, Italy
- 6Pediatric Department, University of Verona, Verona, Italy
- 7Pediatric Immunohematology and Bone Marrow Transplantation Unit, San Raffaele Telethon Institute for Gene Therapy, Milan, Italy
Lipopolysaccharide responsive beige-like anchor protein (LRBA) deficiency is a primary immunodeficiency disorder (PID) that can cause a common variable immunodeficiency (CVID)-like disease. The typical features of the disease are autoimmunity, chronic diarrhea, and hypogammaglobulinemia. Neurological complications are also reported in patients affected by LRBA deficiency. We describe a 7-year old female with an acute cervical longitudinally extensive transverse myelitis (LETM) as a feature of LRBA deficiency. This is the first case of LETM associated with LRBA deficiency described in literature.
Introduction
Lipopolysaccharide responsive beige-like anchor protein (LRBA) deficiency is a primary immunodeficiency disorder (PID) described as a cause of common variable immunodeficiency (CVID)-like disease (1). Several genes responsible of different subgroups of CVID have been identified (2), but the majority of patients with CVID have an unknown genetic etiology. CVID is a diagnosis of exclusion and so it is not surprising that CVID has heterogeneous clinical and laboratory presentations (3). This disease can be caused by LRBA gene defects (1). LRBA is a member of BEACH-WD40 protein family and it is expressed in several tissues (1, 4). The LRBA gene is located on 4q31.3, contains 57 exons and encodes a protein containing 2851 amino acid residues (5). The LRBA protein is widely expressed in several cell types including hematopoietic, neural, gastrointestinal, and endocrine cells1 with an high expression especially in lymphocytes (1, 6). This intracellular protein regulates the lysosomal degradation of cytotoxic T lymphocytes antigen-4 (CTLA-4), an inhibitory checkpoint receptor on T cells (6). For this reason patients with LRBA deficiency present an increase in CTLA4 degradation with clinical signs similar to CTLA4 haploinsufficient individuals (7, 8). An increase expression of LRBA is also described in many cancers, suggesting that the protein promotes cell survival by inhibiting apoptosis (9). The clinical features in patients affected by LRBA deficiency are heterogeneous with age of presentation ranging from 2 months to 12 years. There is not a genotype-phenotype correlation (10). LRBA deficiency can present with a wide spectrum of clinical manifestations such as inflammatory bowel disease (IBD)-like enteropathy, splenomegaly, pneumonia, autoimmune disease (AID) like immune thrombocytopenia purpura (ITP) and autoimmune hemolytic anemia (AIHA), hypogammaglobulinemia, B-cell deficiency, reduction in numbers of CD4+ T cells and regulatory T cells and autophagy (1, 6, 10–13). LRBA deficiency is suspected on the basis of heterogeneous clinical manifestations and immunological dysregulation that can be found through blood tests. The diagnosis currently relies on gene sequencing approaches or on the detection of LRBA protein by flow cytometry (14). The conventional treatment options for this disease have included various immunosuppressive agents such as corticosteroids, sirolimus, abatacept (soluble CTLA4- immunoglobulin fusion protein) (6, 13, 15) or Hematopoietic Stem Cell Transplantation (HSCT) (16). Neurological complications in patient affected by LRBA deficiency are described (6, 10, 17) even if they are not one of the typical features of the disease. We describe a 7-year old female with an acute cervical longitudinally extensive transverse myelitis (LETM) as a feature of LRBA deficiency. This is the first case of LETM associated with LRBA deficiency described in literature.
Case Report
A 7-year-old female affected by LRBA deficiency was referred to our hospital for fever, stiff neck, and right cervical lymphadenopathy, that were unresponsive to anti-inflammatory drugs and oral antibiotic therapy.
The patient was born to unrelated parents after a full-term gestation. The birth weight was 2,420 g. The girl had normal psychomotor development. From 6 months of age she began to present recurrent severe infections during neutropenia associated with diffuse lymphadenomegaly (inguinal, abscellar, laterocervical, and mesenteric) and a diagnosis of autoimmune neutropenia was made. At 18 months she was hospitalized for cholelithiasis and hepatomegaly with autoimmune hepatitis and initial signs of cirrhosis. At 2 years she developed chronic diarrhea, hypertriglyceridemia, diffuse lipodystrophy, and splenomegaly. Double negative T cells resulted high (2.6% of total lymphocytes, normal value NV < 1.7%) and FAS-mediated apoptosis was abnormal. Genetic tests for autoimmune lymphoproliferative syndrome (ALPS) were negative (FAS, FAS ligand and caspase 10). In the suspicion of (ALPS)-like phenotype disease a therapy with mycophenolate mofetil was started. After one year, for the worsening of the lipodystrophy, anakinra, and then canakinumab were administered with initial but temporary clinical improvement. For persistent hypogammaglobulinemia the patient needed monthly administrations of immunoglobulins. At 4.5 years she underwent splenectomy for splenic abscess and started prophylaxis with amoxicillin/clavulanate and antiplatelet therapy with aspirin. At 5 years a mutation of Insulin Receptor Substrate 1 (IRS-1) which could lead to insulin resistance, leptin deficiency, and steatohepatitis, was detected. Therefore, leptin was administered with reduction of liver dimension, improvement of triglycerides level and no longer needing of insulin. The patient started empirical immunosuppressive therapy with prednisone (max 1 mg/kg/day) and sirolimus due to poor response to previous therapy. After few months the LRBA deficiency was identified by next generation sequencing (NGS) demonstrating double heterozygosity for two LRBA mutations; one inherited from the father (non sense c.7681 C>T: p.Q2561X) and one from the mother (splice disruption, c.1359 + 1G>A). Only sirolimus therapy was ongoing at the time of admission to our department.
On admission, she was conscious alert and oriented. Her heart rate, respiratory rate, blood pressure and body temperature were within normal range. She presented severe cervical pain associated with left deviation of the neck. The low-grade fever (37.6–37.8°C) and the stiff neck appeared 10 days earlier, but the pain was initially moderate and the patient was treated with anti-inflammatory drugs and oral antibiotic therapy. She received the last administrations of immunoglobulins 2 days before hospitalization. On examination, a right laterocervical lymphadenomegaly was observed. After few hours she developed right upper limb hyposthenia and fecal incontinence. Blood exams showed an increase of white blood cells (WBC) with low inflammatory markers (Table 1). Microbiological analysis were negative (blood cultures, urine culture, stool culture; Cytomegalovirus (CMV), Epstein Barr Virus (EBV), Adenovirus and Toxoplasma gondii serology; antistreptolysin antibodies, EBV, and CMV DNA copy number quantification by real-time polymerase chain reaction). Magnetic Resonance Imaging (MRI) was performed and showed extensive spinal cord T2- hyperintense lesion extending from the medulla oblongata to D3 level, with significant spinal cord swelling and marked contrast enhancement at C3–C5 level which was compatible with acute Longitudinally Extensive Transverse Myelitis (LETM) (Figure 1). Cerebrospinal fluid (CSF) analysis revealed an increase of white blood cells (100% lymphocytes), proteins and IgG levels with high CSF/serum albumin quotient and normal IgG-index (Table 2). CSF analysis for bacterial and viral infections yielded negative results (Escherichia coli, Haemophilus influenzae, Listeria monocytogenes, Neisseria meningitidis, Streptococcus agalatiae, Streptococcus pneumoniae, EBV, CMV, Herpes simplex Virus 1 (HSV1), Herpes simplex Virus 2 (HSV2), Human parechovirus, Varicella zoster virus (VZV), Enterovirus and Cryptococcus neoformans). Serum antibodies to myelin oligodendrocyte glycoprotein (MOG live cell-based assay) and aquaporin-4 (AQP4 fixed cell-based assay, Euroimmune commercial kit-4) were negative. Isoelectrofocusing revealed the presence of oligoclonal bands in both serum and CSF type 4 “mirror” pattern. An extensive autoimmune screening was negative [antinuclear antibodies (ANA), anti-double stranded DNA antibodies (anti-ds-DNA), anti-neutrophil cytoplasmic antibodies (ANCA, p-ANCA, c-ANCA, a-ANCA, anti PR3, anti myeloperoxidase antibodies MPO), anti-saccharomyces cerevisiae antibodies (ASCA), anti-smooth muscle antibody (ASMA), anti-liver-kidney microsome antibodies (LKM), anti-insulin antibody, TSH receptor antibodies (TRAb)]. The patient received intravenous dexamethasone at the dosage of 0.15 mg/kg for 27 days followed by a 9-week tapering dose of oral steroids which resulted in a rapid clinical improvement. Follow-up MRIs, performed after 20 and 55 days, respectively, (Figure 2), showed progressive improvement of spinal cord oedema and enhancement in parallel with a complete clinical recovery. The patient was then transferred to another institution for stem cell transplantation and the last follow-up MRI before transplant she was referred completely negative (image not available).
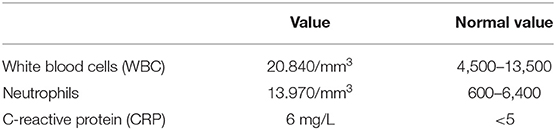
Table 1. Blood exams.
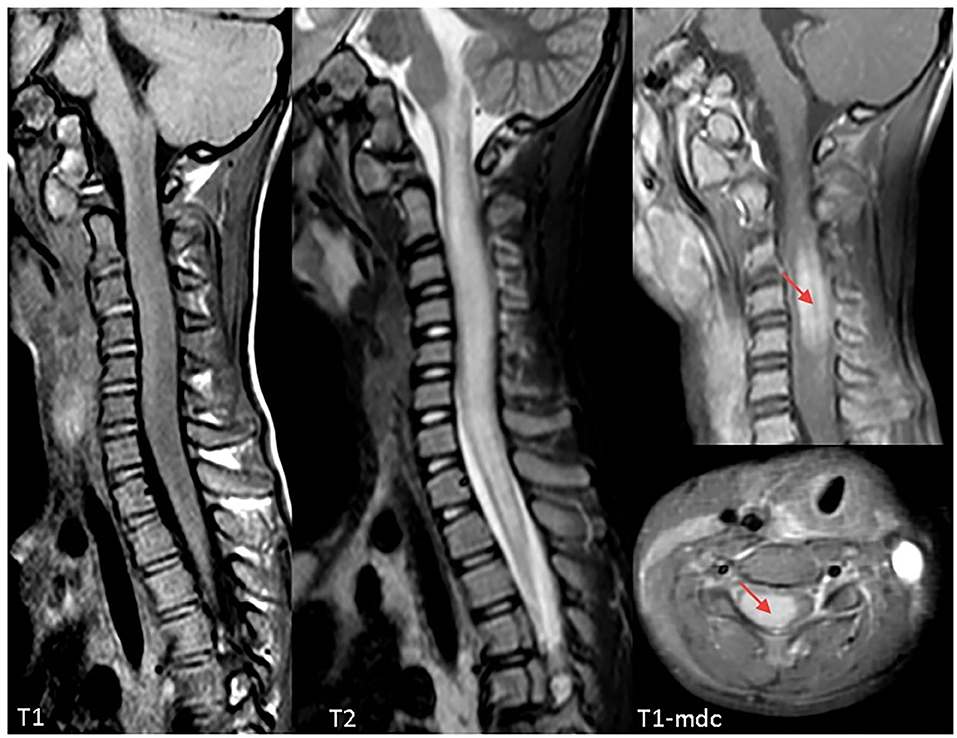
Figure 1. Cervical Magnetic Resonance Imaging (MRI) at clinical onset demonstrating a longitudinally extensive signal abnormality involving the cervical spinal cord, extending from the obex to D3. The cord appears markedly swollen, both gray and white matter are involved, and post-contrast sequences demonstrate marked cord enhancement between C3 and C5 (red arrows). Findings are compatible with acute longitudinally extensive transverse myelitis (LETM).
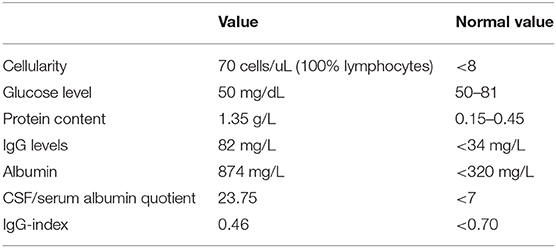
Table 2. Cerebrospinal fluid (CSF) analysis.
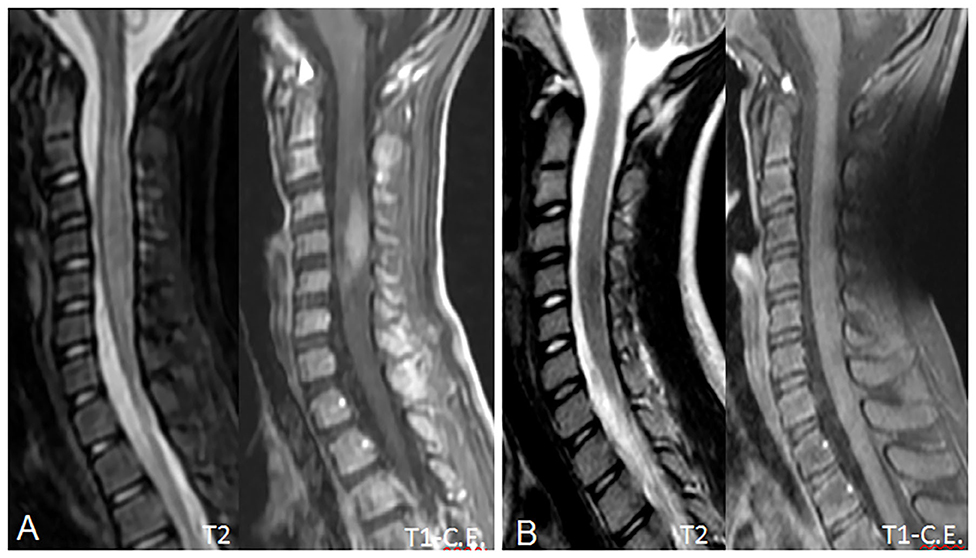
Figure 2. Follow-up MRI scans at 20 (A) and 55 (B) days, showing progressive reduction of the spinal cord swelling and contrast enhancement. In (B), contrast enhancement has completely regressed, due to blood-brain barrier restoration, and the spinal cord shows a near-normal appearance with no residual disease. No spinal cord atrophy is noted.
Discussion
Patients with LRBA deficiency typically present immunodeficiency, inflammatory bowel disease (IBD)-like enteropathy, and autoimmune disease (AID) (1, 11–13). Some studies report an imbalance in Th subsets, in particular in Th1-like Th17 and Treg cells and their corresponding cytokines in LRBA deficiency. This condition might be important in the immunopathogenesis of autoimmunity and enteropathy (18). According to some publications a defect of LRBA can be associated with an IPEX-like (19) or ALPS-like (20) clinical presentation thus similar to our patient's phenotype. Neurological complications of the disease have been described (6, 10, 17). Alkhairy et al. (10) reported that neurological features occur in 23% of patients. The authors, with the exception of myasthenia gravis which has an autoimmune etiology, describe patients with cerebral lesions and nervous tissue atrophy. They report two patients with cerebral granulomas, one with granuloma-like lesion with demyelination resulting in unilateral optic nerve atrophy, one with unilateral optic nerve atrophy, one with cerebral and cerebellar atrophy and another with parietal lobe lesion complicated by seizures (10). Tesi et al. described a patient with LRBA deficiency complicated with an acute disseminated encephalomyelitis (ADEM) who underwent hematopoietic stem cell transplant (17). ADEM has also been described in one CTLA-4 haploinsufficient patient (7) and in patient with CVID (21). To the best of our knowledge this is the first ever reported patient with LETM associated with LRBA deficiency. We believe that the high value of white blood count is a consequence of inflammation. The exclusion of infectious and neoplastic nature of the lesion, together with the good response to steroid treatment, allow us to consider it an inflammatory seronegative LETM. This condition is observed in a group of inflammatory autoimmune disorders, often in the context of neuromyelitis optica spectrum disorders (NMOSD) (22–25). These disorders are mostly related to the presence of antibodies against aquaporin (AQP4) or against myelin oligodendrocyte glycoprotein (MOG) (24–26). However, in some patients AQP4-Antibodies and MOG-Antibodies are lacking, thus these patients are usually labeled “double seronegative LETM” or “seronegative LETM” (26, 27). We consider LETM as one of the possible immune-mediated manifestations that compose the clinical spectrum of LRBA deficiency.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
MC: involvement in medical diagnosis and follow up of the patient, first writer of the manuscript. MM: involvement in medical diagnosis of the patient and she helped to write the manuscript. GC, GT, SM, RB, MD, VV, AZ, EB, and DD: involvement in diagnosis and management of the patient. FB involvement in genetic diagnosis. SC: involvement in diagnosis and management of the patient, supervision of the process of the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnote
1. ^GeneCards. http://www.genecards.org/cgi-bin/carddisp.pl?gene=LRBA
References
1. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immunodeficiency and autoimmunity. Am J Hum Genet. (2012) 90:986–1001. doi: 10.1016/j.ajhg.2012.04.015
2. Abolhassani H, Sagvand BT, Shokuhfar T, Mirminachi B, Rezaei N, Aghamohammadi A. A review on guidelines for management and treatment of common variable immunodeficiency. Expert Rev Clin Immunol. (2013) 9:561–75. doi: 10.1586/eci.13.30
3. Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is Not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. (2011) 111:47–107. doi: 10.1016/B978-0-12-385991-4.00002-7
4. De Lozanne A. The role of BEACH proteins in Dictyostelium. Traffic. (2003) 4:6–12. doi: 10.1034/j.1600-0854.2003.40102.x
5. EnsemblGenomeBrowser. (2015). Available online at: http://www.ensembl.org/Homo_sapiens/Transcript/Sequence_Protein?db=core;g=ENSG00000198589;r=4:151185594–151936879;t=ENST00000510413 (accessed June 30, 2020).
6. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. (2015) 349:436–40. doi: 10.1126/science.aaa1663
7. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20:1410–6. doi: 10.1038/nm.3746
8. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. (2014) 345:1623–7. doi: 10.1126/science.1255904
9. Wang JW, Gamsby JJ, Highfill SL, Mora LB, Bloom GC, Yeatman TJ, et al. Deregulated expression of LRBA facilitates cancer cell growth. Oncogene. (2004) 23:4089–97. doi: 10.1038/sj.onc.1207567
10. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol. (2016) 36:33–45. doi: 10.1007/s10875-015-0224-7
11. Burns SO, Zenner HL, Plagnol V, Curtis J, Mok K, Eisenhut M, et al. LRBA gene deletion in a patient presenting with autoimmunity without hypogammaglobulinemia. J Allergy Clin Immunol. (2012) 130:1428–32. doi: 10.1016/j.jaci.2012.07.035
12. Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS, Aljebreen A, et al. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol. (2012) 130:481–8. doi: 10.1016/j.jaci.2012.05.043
13. Habibi S, Zaki-Dizaji M, Rafiemanesh H, Lo B, Jamee M, Gámez-Díaz L, et al. Clinical, immunologic, and molecular spectrum of patients with LPS-responsive beige-like anchor protein deficiency: a systematic review. J Allergy Clin Immunol Pract. (2019) 7:2379–86.e5. doi: 10.1016/j.jaip.2019.04.011
14. Gámez-Díaz L, Sigmund EC, Reiser V, Vach W, Jung S, Grimbacher B. Rapid flow cytometry-based test for the diagnosis of lipopolysaccharide responsive beige-like anchor (LRBA) deficiency. Front Immunol. (2018) 9:720. doi: 10.3389/fimmu.2018.00720
15. Azizi G, Abolhassani H, Mahdaviani SA, Chavoshzadeh Z, Eshghi P, Yazdani R, et al. Clinical, immunologic, molecular analyses and outcomes of iranian patients with LRBA deficiency: a longitudinal study. Pediatr Allergy Immunol. (2017) 28:478–84. doi: 10.1111/pai.12735
16. Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Inborn errors, clinical, and registry working parties of the european society for blood and marrow transplantation and the European society for Immunodeficiencies. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol. (2020) 145:1452–63. doi: 10.1016/j.jaci.2019.12.896
17. Tesi B, Priftakis P, Lindgren F, Chiang SC, Kartalis N, Löfstedt A, et al. Successful hematopoietic stem cell transplantation in a patient with LPS-responsive beige-like anchor (LRBA) gene mutation. J Clin Immunol. (2016) 36:480–9. doi: 10.1007/s10875-016-0289-y
18. Azizi G, Mirshafiey A, Abolhassani H, Yazdani R, Ghanavatinejad A, Noorbakhsh F, et al. The imbalance of circulating T helper subsets and regulatory T cells in patients with LRBA deficiency: correlation with disease severity. J Cell Physiol. (2018) 233:8767–777. doi: 10.1002/jcp.26772
19. Jamee M, Zaki-Dizaji M, Lo B, Abolhassani H, Aghamahdi F, Mosavian M, et al. Clinical, immunological, and genetic features in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like syndrome. J Allergy Clin Immunol Pract. (2020) 8:P2747–2760.E7. doi: 10.1016/j.jaip.2020.04.070
20. Revel-Vilk S, Fischer U, Keller B, Nabhani S, Gámez-Díaz L, Rensing-Ehl A, et al. Autoimmune lymphoproliferative syndrome-like disease in patients with LRBA mutation. Clin Immunol. (2015) 159:84–92. doi: 10.1016/j.clim.2015.04.007
21. Kondo M, Fukao T, Teramoto T, Kaneko H, Takahashi Y, Okamoto H, et al. A common variable immunodeficiency patient who developed acute disseminated encephalomyelitis followed by the lennox-gastaut syndrome. Pediatr Allergy Immunol. (2005) 16:357–60. doi: 10.1111/j.1399-3038.2005.00279.x
22. Hor JY, Asgari N, Nakashima I, Broadley SA, Isabel Leite M, Kissani N. et al. Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. Front Neurol. (2020) 11:501. doi: 10.3389/fneur.2020.00501
23. Sepúlveda M, Armangué T, Sola-Valls N, Arrambide G, Meca-Lallana JE, Oreja-Guevara C, et al. Neuromyelitis optica spectrum disorders: comparison according to the phenotype and serostatus. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e225. doi: 10.1212/nxi.0000000000000225
24. Lechner C, Baumann M, Hennes EM, Schanda K, Marquard K, Karenfort M, et al. Antibodies to MOG and AQP4 in children with neuromyelitis optica and limited forms of the disease. J Neurol Neurosurg Psychiatry. (2016) 87:897–905. doi: 10.1136/jnnp-2015-311743
25. Höftberger R, Sepulveda M, Armangue T, Blanco Y, Rostásy K, Cobo Calvo A, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler. (2015) 21:866–74. doi: 10.1177/1352458514555785
26. Maillart E, Durand-Dubief F, Louapre C, Audoin B, Bourre B, Derache N, et al. Outcome and risk of recurrence in a large cohort of idiopathic longitudinally extensive transverse myelitis without AQP4/MOG antibodies. J Neuroinflammation. (2020) 17:128. doi: 10.1186/s12974-020-01773-w
27. Álvaro C-C, María S, Raphael B-V, Anne R, David B, Sergio M-Y, et al. Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis: clinical and prognostic implications. Mult Scler J. (2016) 22:312–9. doi: 10.1177/1352458515591071
Keywords: lipopolysaccharide responsive beige-like anchor protein (LRBA), myelitis, acute cervical longitudinally extensive transverse myelitis, common variable immune deficiency (CVID), autommunity
Citation: Chinello M, Mauro M, Cantalupo G, Talenti G, Mariotto S, Balter R, De Bortoli M, Vitale V, Zaccaron A, Bonetti E, Di Carlo D, Barzaghi F and Cesaro S (2020) Acute Cervical Longitudinally Extensive Transverse Myelitis in a Child With Lipopolysaccharide-Responsive-Beige-Like-Anchor-Protein (LRBA) Deficiency: A New Complication of a Rare Disease. Front. Pediatr. 8:580963. doi: 10.3389/fped.2020.580963
Received: 07 July 2020; Accepted: 10 September 2020;
Published: 16 October 2020.
Edited by:
Michael Zemlin, Saarland University Hospital, GermanyReviewed by:
Gholamreza Azizi, Alborz University of Medical Sciences, IranCinzia Milito, Sapienza University of Rome, Italy
Copyright © 2020 Chinello, Mauro, Cantalupo, Talenti, Mariotto, Balter, De Bortoli, Vitale, Zaccaron, Bonetti, Di Carlo, Barzaghi and Cesaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matteo Chinello, matteo.chinello@aovr.veneto.it