Sterol 27-Hydroxylase Deficiency as a Cause of Neonatal Cholestasis: Report of 2 Cases and Review of the Literature
 Patryk Lipiński1,2*
Patryk Lipiński1,2*  Maja Klaudel-Dreszler2
Maja Klaudel-Dreszler2  Elzbieta Ciara3
Elzbieta Ciara3  Dorota Jurkiewicz3
Dorota Jurkiewicz3  Rafał Płoski4 Joanna Cielecka-Kuszyk5 Piotr Socha2
Rafał Płoski4 Joanna Cielecka-Kuszyk5 Piotr Socha2  Irena Jankowska2
Irena Jankowska2- 1Department of Pediatrics, Nutrition and Metabolic Diseases, The Children's Memorial Health Institute, Warsaw, Poland
- 2Department of Gastroenterology, Hepatology, Feeding Disorders and Pediatrics, The Children's Memorial Health Institute, Warsaw, Poland
- 3Department of Medical Genetics, The Children's Memorial Health Institute, Warsaw, Poland
- 4Department of Medical Genetics, Medical University of Warsaw, Warsaw, Poland
- 5Department of Pathology, The Children's Memorial Health Institute, Warsaw, Poland
Introduction: Inborn errors of primary bile acid (BA) synthesis are rare autosomal recessive disorders responsible for 1–2% of cases of neonatal cholestasis. Among them, cerebrotendinous xanthomatosis (CTX) is caused by mutations in the CYP27A1 gene resulting in the impairment of sterol 27-hydroxylase enzyme activity.
Patients and Methods: Here we present the study on two siblings with neonatal cholestasis diagnosed with sterol 27-hydroxylase deficiency. The clinical, biochemical, histological, and molecular presentation at the time of diagnosis and detailed follow-up were described. An extensive overview of the literature regarding patients with sterol 27-hydroxylase deficiency presenting with neonatal cholestasis was also provided.
Results: Patient 1 presented with cholestatic jaundice since 10 weeks of age and developed the end-stage liver disease requiring liver transplantation at 8 months of age but finally succumbed 3 years post-transplantation due to autoimmune hemolytic anemia and multiorgan failure development. Next-generation sequencing performed post mortem, revealed him to be homozygous for the known pathogenic splicing variant c.1184+1G>A in the CYP27A1 gene. Patient 2 (sibling) presented with cholestatic jaundice since the first day of life. Sanger sequencing of CYP27A1 revealed the same results. Chenodeoxycholic acid treatment was introduced just after diagnosis, at 4 months of age. Fourteen patients with sterol 27-hydroxylase deficiency presenting with neonatal cholestasis were reported in the literature, in most of them presenting as a self-limiting disease.
Conclusions: An early recognition and treatment initiation in CTX is essential.
Introduction
Inborn errors of primary bile acid (BA) synthesis are rare autosomal recessive disorders responsible for 1–2% of cases of neonatal cholestasis (1–3). The absence of pruritus, normal serum gamma-glutamyltranspeptidase (GGT) activity, and normal or low total serum BA concentration constitute the most characteristic features while the specific diagnosis is based on the mass spectrometry (MS) analysis of urinary BA confirmed by gene sequencing (1–4).
Cerebrotendinous xanthomatosis (CTX; # 213700) is caused by mutations in the CYP27A1 gene resulting in the impairment of sterol 27-hydroxylase enzyme (EC 1.14.15.15) activity with subsequent diminished cholic acid (CA) formation and no production of chenodeoxycholic acid (CDCA) (5–7). An increased deposition of cholesterol and cholestanol in tissues throughout the body, including the brain (white matter), lens, and tendons is observed (5). Clinical hallmarks include premature bilateral cataracts, intractable diarrhea, tendon xanthomas, and progressive neurologic dysfunction (5–7). Early diagnosis remains crucial regarding that early and long-term treatment of CDCA improves the neurological outcome and even could reverse the progression of the disease (8–10).
Here we present the study on two siblings with neonatal cholestasis diagnosed with sterol 27-hydroxylase deficiency based on molecular analysis, aiming to highlight the manifestation of disease as neonatal cholestasis, emphasize the diagnostic difficulties with depicting the role of genetic studies, and also report the favorable outcome of an oral CDCA therapy. A detailed overview of the literature regarding sterol 27-hydroxylase deficiency presenting with neonatal cholestasis was also provided.
Patients and Methods
The presentation at the time of diagnosis and detailed follow-up were described. An informed and written consent was obtained from the parents of patients. Ethical approval was obtained from the Children's Memorial Health Institute Bioethical Committee, Warsaw, Poland.
A retrospective chart review of patients' medical records concerning the biochemical (hemoglobin, platelets, serum transaminases, total and direct serum bilirubin, gamma-glutamyl transpeptidase, internal normalized ratio, serum bile acids), histopathological (microscopic examination of liver biopsy specimens) and molecular (CYP27A1 gene mutations) were collected.
The liver biopsy specimens, between 1.0 and 1.5 cm in length, were fixed in 4% formalin and stained by hematoxillin and eosin, PAS method (periodic acid + Schiff reagent) and PAS method after diastase digestion, Azan method and reticulin impregnation. To assess the histological activity of microscopical changes, the following categories of lesions were considered retrospectively: presence of inflammatory infiltrates in the portal spaces and lobules with or without piecemeal necrosis, degree of fibrosis, lobular disarray, rosette formations, proliferation of bile ducts and ductules with or without ductitis, lobular necrosis, hepatocyte giant cell transformation, steatosis and degenerative changes in the hepatocytes, canalicular bile plugs, cholestasis in the hepatocytes and in bile ducts, extramedullary hematopoesis.
Genomic DNA was extracted from peripheral blood samples of the patients. Next-generation sequencing (NGS) of targeted gene panel, created by The Children's Memorial Health Institute's for the simultaneous sequencing of 1,000 clinically relevant genes including 53 items related to cholestatic liver disorders was used. A detailed protocol have been published recently (11).
The nomenclature of molecular variants follows the Human Genome Variation Society guidelines (HGVS, http://varnomen.hgvs.org/) using human cDNA sequence of appropriate genes followed the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk).
The database PubMed (MEDLINE) was searched for relevant studies on September 30, 2020. The following keyword-based strategy was used: (“cerebrotendinous xanthomatosis” OR “CTX” OR “sterol 27-hydroxylase deficiency”) AND (“neonatal cholestasis”). All studies, letters, and abstracts that contained sufficient data were included. Available data regarding the first presentation, age at referral, biochemical and histological features, treatment and outcome, were extracted.
Patients' Presentation
Patient 1
The patient was the first child of non-consanguineous Polish parents born from an uneventful pregnancy at 39 weeks of gestation with a birth mass of 3,000 g. At the age of 10 weeks, he was referred to our hospital due to presence of jaundice. No history of hypo-/acholic stools was observed. Cholestasis with normal serum GGT accompanied by elevated serum transaminases and coagulopathy was diagnosed (Table 1). Normal liver and spleen volume were observed. HBV, HCV, CMV, EBV, HIV, and Toxoplasma gondii infections were excluded serologically; alpha-1-antitrypsin deficiency, cystic fibrosis, galactosemia were excluded as well. The liver biopsy performed at the age of 77 days revealed diffuse inflammatory infiltrates composed of lymphoid cells, situated in the portal tracts (Figure 1A). Abundant foci of extramedullary heamatopoiesis were found intraacinar in the sinusoids (Figure 1B). Multinucleated giant hepatocytes and hepatic rosettes with bilirubinostasis were found in all acini (Figure 1C). The patient was discharged home on ursodeoxycholic acid (UDCA) as well as fat-soluble vitamins treatment.
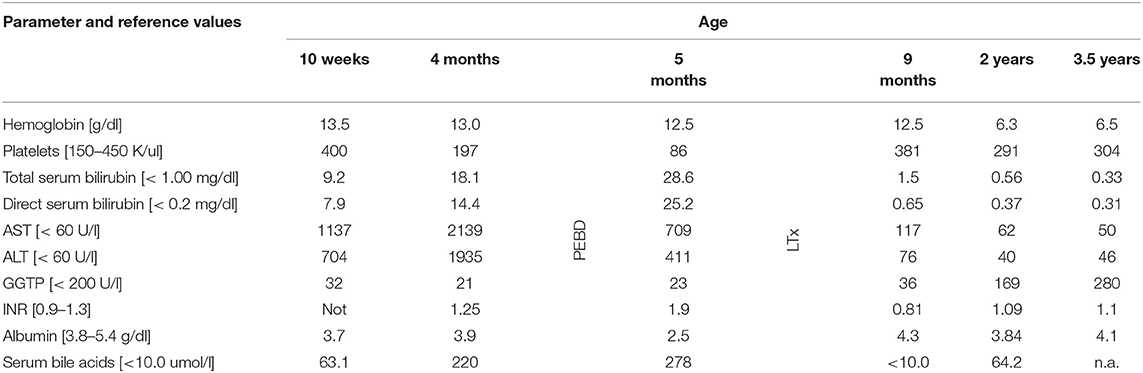
Table 1. Results of laboratory analyses of Patient 1.
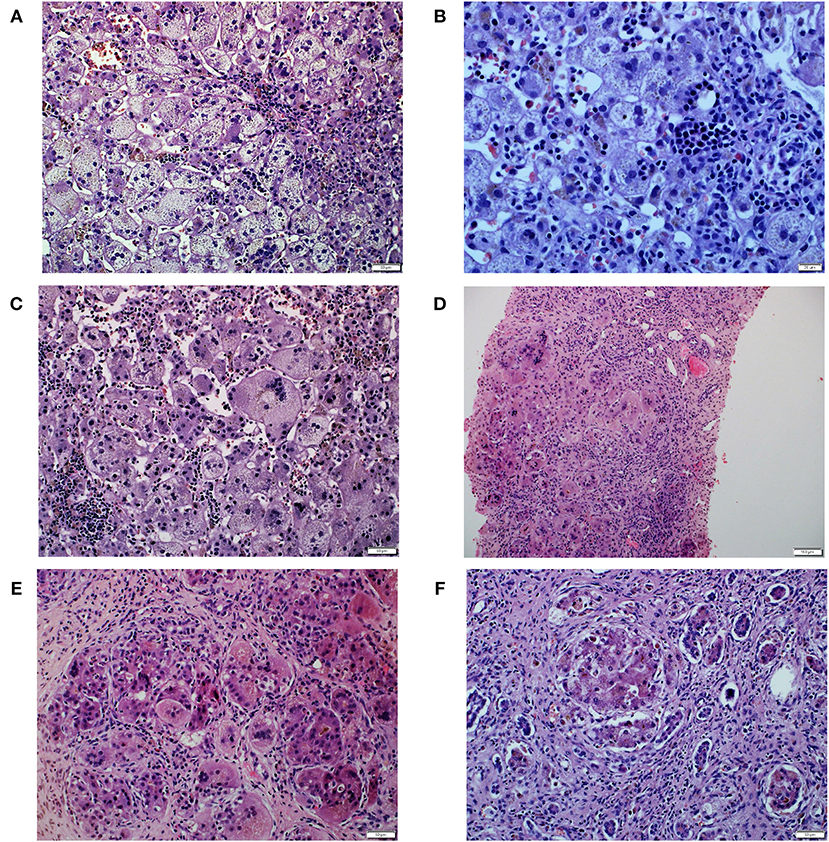
Figure 1. Histopathological liver features in Patient 1. (A) Diffuse inflammatory infiltrates composed of lymphoid cells, in the portal tracts. (B) Abundant foci of extramedullary heamatopoiesis in the sinusoids. (C) Multinucleated giant hepatocytes and hepatic rosettes with bilirubinostasis. (D) Multinucleated giant cells were inside cirrhotic nodules. (E) Intracellular cholestasis with non-specific inflammatory infiltrates composed of neutrophils and lymphocytes in the fibrous septa. (F) Late cirrhosis with only small remnants of liver cells surrounded by fibrous tissue.
At the age of 4 months he was hospitalized in the Hepatology Outpatient Clinic. Notably, raising parameters of cholestasis with high serum BA concentration as well as still highly elevated serum transaminases were noted (Table 1). The decision about partial external biliary diversion (PEBD) procedure was commenced. The liver biopsy done at the moment of PEBD revealed the presence of severe fibrosis with features of cirrhotic transformation. Multinucleated giant cells were seen inside cirrhotic nodules (Figure 1D). Intracellular cholestasis was more prominent as compared to the first biopsy and non-specific inflammatory infiltrates composed of neutrophils and lymphocytes were found in the fibrous septa (Figure 1E).
During control visit in the Hepatology Outpatient Clinic, 4 weeks after PEBD, the child presented with jaundice, huge ascites, and severe itching. Laboratory results revealed raising parameters of cholestasis with high serum BA concentration as well as coagulopathy and hypoalbuminemia (Table 1). The decision about qualification for liver transplantation (LTx) procedure was made.
The patient underwent AB0-compatible cadaveric liver transplantation (LTx) at 8 months of age. The examination of the explanted liver revealed features of late cirrhosis with only small remnants of liver cells surrounded by fibrous tissue (Figure 1F). An early post-LTx outcome was complicated with an acute rejection episode (1 month after LTx) and biliary complications (anastomotic strictures diagnosed 3 months after LTx). At the age of 2 years (1 year and 2 months after LTx) he developed a hemolytic anemia related to production of cold agglutinins (Table 1). Viral serologies significant for cold agglutinin syndrome were negative. No clinical evidence of post-transplant lymphoproliferative disorder was observed. Favorable resolution of the hemolytic anemia occurred following typical treatment with glucocorticosteroids.
The recurrence of hemolytic anemia refractory to steroids, plasmapheresis, and rituximab was observed at 3.5 years of age. The patient had finally died due to multiorgan failure development.
Patient 2
The patient was the younger sister of Patient 1, born from an uneventful second pregnancy at 40 weeks of gestation with a birth mass of 2,950 g. At the age of 9 days of life, she was referred to our hospital due to presence of jaundice observed since the first day of life. No history of hypo-/acholic stools was observed. Cholestasis with normal serum GGT accompanied by slightly elevated serum transaminases was diagnosed (Table 2). Normal liver and spleen volume were observed. The patient was discharged home on UDCA as well as fat-soluble vitamins treatment.
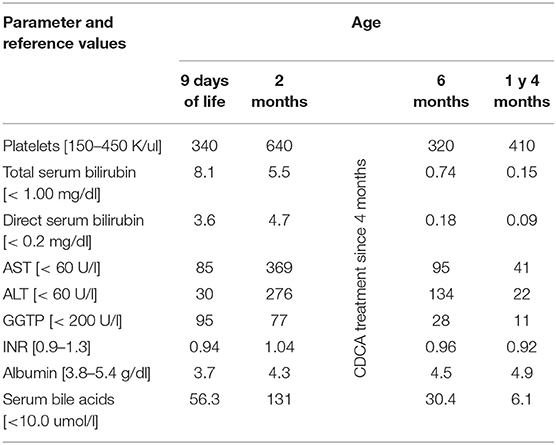
Table 2. Results of laboratory analyses of Patient 2.
At the age of 2 months she was hospitalized in the Hepatology Outpatient Clinic. Notably, raising parameters of cholestasis with high serum BA concentration as well as serum transaminases levels were noted (Table 2).
In the meanwhile, the results of next-generation sequencing study of her brother became available—the patient was found to be homozygous for the known pathogenic splicing variant c.1184+1G>A in the CYP27A1 gene (NM_000784.3). The parents were carriers of this variant. The Sanger sequencing of CYP27A1 in his sister revealed the same results; the diagnosis of CTX was established. The decision about chenodeoxycholic acid (CDCA) treatment was made and CDCA was finally introduced at the age of 4 months at a dosage of 5 mg/kg/day. After 1 year of treatment with CDCA maintained on a dosage of 5 mg/kg/day, the patient was in a good clinical condition presenting with normal results of liver function tests (Table 2). The therapy was well-tolerated by the patient.
Literature Review
Fourteen patients with sterol 27-hydroxylase deficiency presenting with neonatal cholestasis were reported, see Table 3 (11–19). To be noted that a detailed clinical presentation was not available in some of the reported cases (12, 14, 17, 19).

Table 3. Review on reported patients with sterole 27-hydroxylase deficiency presenting with neonatal cholestasis.
Discussion
The paper presents a detailed clinical, biochemical, and histological phenotype of two patients (siblings) diagnosed with sterol 27-hydroxylase deficiency (cerebrotendinous xanthomatosis, CTX) manifested with neonatal cholestatic jaundice. The first patient, in fact, without adequate therapy progressed to liver failure and underwent LTx but finally died. CTX diagnosis was established post mortem. The second patient (sibling) was diagnosed with CTX at 4 months of age by next-generation sequencing (NGS) study. This fact raises the usefulness of NGS techniques in diagnostic approach to neonatal cholestasis.
The diagnosis of inborn errors of primary BA synthesis remains challenging, especially in the differential diagnosis of neonatal/infantile cholestasis. The absence of pruritus, normal serum GGT activity, and normal or low total serum BA concentration are the most characteristic features of inborn errors of BA synthesis (1, 2). In some patients (like Patient 1 and some reported patients) despite of lack of itching at first, a severe itching was observed in the course of disease. That, based on the clinical and biochemical features a diagnosis of progressive familial intrahepatic cholestasis (PFIC) could be established. PFIC patients generally present in the first few months of life with cholestatic jaundice and pruritus, high serum BA and transaminases, normal serum GGT levels (beyond PFIC type 3). Liver histology shows usually a marked intracellular cholestasis and an obvious giant-cell transformation. In our patient, the histopathological liver study was also miscellaneous (20). The massive gigantocellular transformation of hepatocytes as well as extramedullary hematopoiesis were not characteristic for PFIC. However, the giant-cell transformation of hepatocytes was the most common histopathological finding reported in patients with cholestatic liver disease in the course of CTX (Table 3). Thus, we recommend to consider CTX in a patient presenting with neonatal cholestasis, especially in the presence of giant cell hepatitis.
In the case of prolonged intrahepatic cholestasis with low levels of GGT, without other explanations, the analysis of bile alcohols by mass spectrometry is indicated. BA profiles of plasma and urine are useful in CTX diagnosis but depend on methodology, sample type and preparation, and experience of the laboratory performing the test. In our country these techniques are not routinely available (2). The introduction of NGS allowed us to diagnose CTX and other disorders which have not been recognized yet (21). Molecular biology constitutes an useful diagnostic tool, especially when mass spectrometry data may be inconclusive, such as during the early neonatal period, or if the patient had been treated with bile acid before analysis (13, 17).
There is an existing literature including diagnosis and some outcomes of infants with neonatal cholestasis with CTX. In 1995, Clayton et al. described the first infant with giant cell hepatitis in the course of CTX in whom the treatment with CDCA and CA led to a resolution of the hepatitis at the age of 12 years (12). The retrospective analysis of the past medical records of 50 Dutch patients with CTX, authored by Clayton et al., revealed that neonatal liver disease defined as prolonged jaundice with raised serum transaminases occurred much more often in CTX than in the general population (13). Up to now, neonatal cholestatic jaundice in the course of CTX has been reported in 14 patients, in most of them presenting as a self-limiting disease (11–19). However, a fatal neonatal cholestasis has been reported in 5 out of 12 patients (Table 3). In the largest cohort of 8 infants with CTX presenting with severe cholestasis, reported by Gong et al., 4 died from liver failure at the age of 8, 7, 9, and 5 months, respectively; in 3 other patients their jaundice resolved spontaneously. The authors proposed that CTX manifested as neonatal cholestasis could have a relatively adverse prognosis. One child had undergone LTx at 8 months of age; at the last follow-up (17 months) was presenting with a good outcome (17). Shen et al. reported the second child in the literature who underwent LTx (11). This patient did not require longer CDCA supplement therapy; LTx not only restored liver function but also corrected cholestanol metabolism. LTx could correct the metabolic defect but it is not the advisable therapeutic strategy for CTX, since there is an effective and safe therapy for this condition.
Early diagnosis remains crucial regarding that early and long-term treatment of CDCA improves the neurological outcome and even could reverse the progression of the disease (8–10). Our patient was started to treat with CDCA at 4 months at a dosage of 5 mg/kg/day. After 1 year of treatment with CDCA maintained on a dosage of 5 mg/kg/day, the patient was in a good clinical condition presenting with normal results of liver function tests. CDCA has been reported as hepatotoxic in one infant who started CDCA treatment at a dosage of 15 mg/kg/day but also as safe and effective in other pediatric patients (16). Degrassi et al. have recently reported a patient who started to be treated at 8 months of age with the initial dose of 10 mg/kg/day, increased until 15 mg/kg/day. The patient has been followed for 13 years and the therapy was well-tolerated (18).
The main limitation of our study is no biochemical data to confirm diagnosis of CTX and also follow-up of these patients. In our country it is not possible to perform the analysis of bile alcohols with tandem-mass spectrometry. The observation that CTX can present with neonatal cholestasis is also not novel thus an extensive review of the literature on pediatric cases of neonatal CTX cholestasis was provided, which may be helpful to stress the importance of an early diagnosis of this condition.
Conclusions
1. We recommend to consider CTX in a patient presenting with neonatal cholestasis of an unknown origin, especially in the presence of giant cell hepatitis.
2. An early recognition and treatment initiation in CTX is essential.
3. Panel-based next-generation sequencing could be a useful screening tool in patients with neonatal cholestais of an unknown origin, especially when other biochemical methods such us analysis of bile alcohols by mass spektrometry are not available.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by ethics committee of The Children's Memorial Health Institute in Warsaw. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
PL and IJ: project administration. IJ: supervision. PL, MK-D, EC, DJ, RP, JC-K, PS, and IJ: investigation and writing – review and editing. PL: writing – original draft. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by The Children's Memorial Health Institute intramural grant M28/17.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sundaram SS, Bove KE, Lovell MA, Sokol RJ. Mechanisms of disease: inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol. (2008) 5:456–68. doi: 10.1038/ncpgasthep1179
2. Heubi JE, Setchell KDR, Bove KE. Inborn errors of bile acid metabolism. Clin Liver Dis. (2018) 22:671–87. doi: 10.1016/j.cld.2018.06.006
3. Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. (2017) 64:154–68. doi: 10.1097/MPG.0000000000001334
4. Gonzales E, Matarazzo L, Franchi-Abella S, Dabadie A, Cohen J, Habes D, et al. Cholic acid for primary bile acid synthesis defects: a life-saving therapy allowing a favorable outcome in adulthood. Orphanet J Rare Dis. (2018) 13:190. doi: 10.1186/s13023-018-0920-5
5. Nie S, Chen G, Cao X, Zhang Y. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. (2014) 9:179. doi: 10.1186/s13023-014-0179-4
6. Salen G, Steiner RD. Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis(CTX). J Inherit Metab Dis. (2017) 40:771–81. doi: 10.1007/s10545-017-0093-8
7. Duell PB, Salen G, Eichler FS, DeBarber AE, Connor SL, Casaday L, et al. Diagnosis, treatment, and clinical outcomes in 43 cases with cerebrotendinous xanthomatosis. J Clin Lipidol. (2018) 12:1169–78. doi: 10.1016/j.jacl.2018.06.008
8. Mandia D, Chaussenot A, Besson G, Lamari F, Castelnovo G, Curot J, et al. Cholic acid as a treatment for cerebrotendinous xanthomatosis in adults. J Neurol. (2019) 266:2043–50. doi: 10.1007/s00415-019-09377-y
9. Stelten BML, Huidekoper HH, van de Warrenburg BPC, Brilstra EH, Hollak CEM, Haak HR, et al. Long-term treatment effect in cerebrotendinous xanthomatosis depends on age at treatment start. Neurology. (2019) 92:e83–95. doi: 10.1212/WNL.0000000000006731
10. Wong JC, Walsh K, Hayden D, Eichler FS. Natural history of neurological abnormalities in cerebrotendinousxanthomatosis. J Inherit Metab Dis. (2018) 41:647–56. doi: 10.1007/s10545-018-0152-9
11. Shen CH, Wang ZX. Liver transplantation due to cerebrotendinous xanthomatosis end-stage liver disease. World J Pediatr. (2018) 14:414–15. doi: 10.1007/s12519-018-0151-9
12. Clayton PT, Casteels M, Mieli-Vergani G, Lawson AM. Familial giant cell hepatitis with low bile acid concentrations and increased urinary excretion of specific bile alcohols: a new inborn error of bile acid synthesis? Pediatr Res. (1995) 37:424–31. doi: 10.1203/00006450-199504000-00007
13. Clayton PT, Verrips A, Sistermans E, Mann A, Mieli-Vergani G, Wevers R. Mutations in the sterol 27-hydroxylase gene (CYP27A) cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. J Inherit Metab Dis. (2002) 25:501–13. doi: 10.1023/A:1021211520034
14. von Bahr S, Björkhem I, Van't Hooft F, Alvelius G, Nemeth A, Sjövall J, et al. Sterol 27-hydroxylase gene associated with fatal cholestasis in infancy. J Pediatr Gastroenterol Nutr. (2005) 40:481–6. doi: 10.1097/01.MPG.0000150419.23031.2A
15. Pierre G, Setchell K, Blyth J, Preece MA, Chakrapani A, McKiernan P. Prospective treatment of cerebrotendinous xanthomatosis with cholic acid therapy. J Inherit Metab Dis. (2008) 31:241–45. doi: 10.1007/s10545-008-0815-z
16. Huidekoper HH, Vaz FM, Verrips A, Bosch AM. Hepatotoxicity due to chenodeoxycholic acid supplementation in an infant with cerebrotendinous xanthomatosis: implications for treatment. Eur J Pediatr. (2016) 175:143–6. doi: 10.1007/s00431-015-2584-7
17. Gong JY, Setchell KDR, Zhao J, Zhang W, Wolfe B, Lu Y, et al. Severe neonatal cholestasis in cerebrotendinous xanthomatosis: genetics, immunostaining, mass spectrometry. J Pediatr Gastroenterol Nutr. (2017) 65:561–8. doi: 10.1097/MPG.0000000000001730
18. Degrassi I, Amoruso C, Giordano G, Del Puppo M, Mignarri A, Dotti MT, et al. Case report: early treatment with chenodeoxycholic acid in cerebrotendinous xanthomatosis presenting as neonatal cholestasis. Front Pediatr. (2020) 8:382. doi: 10.3389/fped.2020.00382
19. Setchell KDR, O'Connell N, Russell DW, et al. A unique case of cerebrotendinous xanthomatosis presenting in infancy with cholestatic liver disease further highlights bile acid synthetic defects as an important category of metabolic liver disease (abstract). In: Van Berge Henegouwen GP, Keppler D, Leuschner U, Paumgartner G, Stiehl A, editors. Falk Symposium 120. XVI International Bile Acid Meeting. Biology of Bile Acids in Health and Disease. Den Haag (The Netherlands). Dordrecht: Kluwer (2001). p. 13–4.
20. Bull LN, Thompson RJ. Progressive familial intrahepatic cholestasis. Clin Liver Dis. (2018) 22:657–69. doi: 10.1016/j.cld.2018.06.003
Keywords: neonatal cholestasis, sterol 27-hydroxylase (CYP27A1), next-generation sequencing, chenodeoxycholic acid (CDCA), liver transplantation
Citation: Lipiński P, Klaudel-Dreszler M, Ciara E, Jurkiewicz D, Płoski R, Cielecka-Kuszyk J, Socha P and Jankowska I (2021) Sterol 27-Hydroxylase Deficiency as a Cause of Neonatal Cholestasis: Report of 2 Cases and Review of the Literature. Front. Pediatr. 8:616582. doi: 10.3389/fped.2020.616582
Received: 12 October 2020; Accepted: 14 December 2020;
Published: 13 January 2021.
Edited by:
Pietro Vajro, University of Salerno, ItalyReviewed by:
Hidde Huidekoper, Erasmus Medical Center, NetherlandsIrene Degrassi, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, Italy
Copyright © 2021 Lipiński, Klaudel-Dreszler, Ciara, Jurkiewicz, Płoski, Cielecka-Kuszyk, Socha and Jankowska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patryk Lipiński, p.lipinski@ipczd.pl