Advanced Understanding of Monogenic Inflammatory Bowel Disease
 Ryusuke Nambu1,2,3
Ryusuke Nambu1,2,3  Aleixo M. Muise1,2,4*
Aleixo M. Muise1,2,4*- 1Cell Biology Program, Research Institute, The Hospital for Sick Children, Toronto, ON, Canada
- 2SickKids Inflammatory Bowel Disease Center, The Hospital for Sick Children, Toronto, ON, Canada
- 3Department of Gastroenterology and Hepatology, Saitama Children's Medical Center, Saitama, Japan
- 4Department of Pediatrics, Institute of Medical Science and Biochemistry, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
Inflammatory bowel disease (IBD) is a group of chronic disorders that cause relapsing inflammation in the gastrointestinal tract and comprise three major subgroups of Crohn's disease (CD), ulcerative colitis (UC), and IBD-unclassified (IBDU). Recent advances in genomic technologies have furthered our understanding of IBD pathogenesis. It includes differentiation rare monogenic disorders exhibiting IBD and IBD-like inflammation (monogenic IBD) from patients with the common polygenic form of IBD. Several novel genes responsible for monogenic IBD have been elucidated, and the number of reports has increased due to advancements in molecular functional analysis. Identification of these pathogenic genetic mutations has helped in elucidating the details of the immune response associated with gastrointestinal inflammation and in providing individualized treatments for patients with severe IBD that is often unresponsive to conventional therapy. The majority of monogenic IBD studies have focused on young children diagnosed <6 years of age (very early-onset IBD); however, a recent study revealed high prevalence of monogenic IBD in older children aged >6 years of age as well. Meanwhile, although patients with monogenic IBD generally show co-morbidities and/or extraintestinal manifestation at the time of diagnosis, cases of IBD developing as the initial symptom with unremarkable prodromal symptoms have been reported. It is crucial that the physicians properly match genetic analytical data with clinical diagnosis and/or differential diagnosis. In this review, we summarize the essential clues that may physicians make a correct diagnosis of monogenic disease, including classification, prevalence and clinical phenotype based on available literatures.
Introduction
Inflammatory bowel disease (IBD) is a group of chronic disorders that cause relapsing inflammation in the gastrointestinal tract. They traditionally comprise three major subgroups: Crohn's disease (CD), ulcerative colitis (UC), and IBD-unclassified (IBDU) (1). Recent evidences indicates that IBD is more heterogeneous than has traditionally been recognized using these three major categories (2, 3). One of most important pieces of evidence is the existence of monogenic IBD, which is caused by single gene defects. Translational research involving monogenic IBD has proceeded rapidly over the past few years, and there have been several reports of novel single gene mutations causing monogenic IBD. Based on the literature, we summarize issues of fundamental importance to physicians, including understanding of the genetic basis of these conditions, prevalence and the clinical phenotypes in this review.
Genetics of IBD
Monogenic IBD With Mendelian Pattern
Recent advances in genomic technologies, such as next-generation sequencing, have made it possible to diagnose severe refractory IBD and IBD-like disease as rare monogenic disorders. IBD and IBD-like diseases caused by monogenic disorders transmitted according to Mendelian inheritance patterns have been described as “monogenic” IBD, in contrast to “polygenic,” or classical IBD. The majority of monogenic IBD cases occur in children diagnosed under 6 years of age. Glocker et al. first identified interleukin IL-10 receptor deficiencies in young infants with severe IBD and perianal disease (4). Since then, several novel genes responsible for monogenic IBD have been identified. The current number of monogenic IBD disorders continues to grow with genes classified into six categories according to the biologic mechanism including: defects in the epithelial barrier; T- and B-cell defects; hyperinflammatory and autoinflammatory disorders; phagocytic defects; immunoregulation, including IL-10 signaling defects; and other (5).
The recent advances in translational research into monogenic IBD has resulted in three major contributions to IBD pathogenesis and care including: (1) increased knowledge of pathogenesis of disease, (2) better phenotyping, (3) targeted therapy based on genetic variants (Figure 1A). First, it has revealed the role of novel protein-coding genes in the intestinal tract, their function in intestinal homeostasis, and several immunological pathways involved in the control of intestinal inflammation. Second, it has elucidated more detailed descriptions of the clinical phenotype and clinical course of monogenic IBD. Third, it has made it possible to provide appropriate treatment for those children with monogenic IBD refractory to standard treatment, and to avoid the necessity for surgery (6, 7). For example, hematopoietic stem cell transplantation (HSCT) for patients with an X-linked inhibitor of apoptosis protein (XIAP) deficiency, FOXP3 deficiency, and chronic granulomatous diseases (CGDs) can improve the prognoses of their IBD (8–10). HSCT has been shown to be ineffective for epithelial barrier dysfunctions, such as TTC7A deficiency and nuclear factor-kappa B essential modulator (NEMO) deficiency (11, 12). HSCT can correct immune defects, but it cannot change the expression of these protein on the intestinal epithelium which is outside the hematopoietic compartment. On the other hand, some monogenic disorders can be treated with medications not commonly used for IBD patients. For example, IL-1 receptor antagonist has been shown to have benefits for patients with mevalonate kinase deficiency (13). CTLA4-Ig immunoglobulin fusion protein (abatacept) is effective for patients with CTLA4 deficiency and lipopolysaccharide-responsive and beige-like anchor protein (LRBA) deficiency (14, 15). This is because LRBA deficiency results in very low CTLA4 expression, which explains the clinical overlap between LRBA- and CTLA4- deficiency. Granulocyte-colony stimulating factor for IBD with glycogen storage disease type 1b (GSD-1b) caused by a mutation in SLC37A4 and colchicine for Familial Mediterranean fever caused by a mutation in MEFV, are commonly used (16, 17).
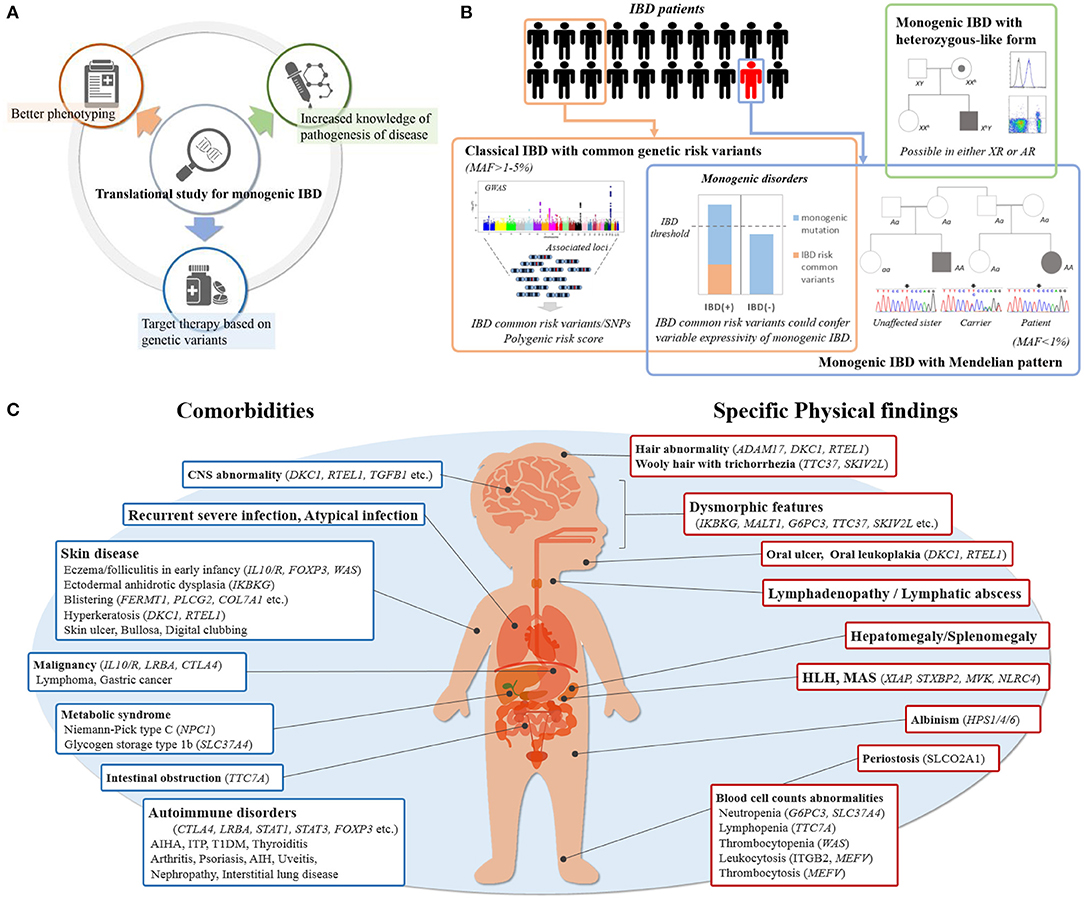
Figure 1. (A) Contribution by translational study for monogenic IBD. (B) Etiology of pediatric IBD with rare variants. Common genetic IBD risk variants are seen in some of patients, which show classical IBD. Pathogenic rare variants are seen in only a few of patients with IBD, which also has monogenic disorders in accordance with Mendelian pattern. Common genetic IBD risk variants may modulate the effects of pathogenic rare variants. The patients with only single-allele mutations causing monogenic disorders are rarely seen especially in using WES despite generally being X-linked or autosomal recessive. (C) Key indicators of monogenic IBD in clinical practice. Showing physical findings and comorbidities of which physicians should be aware at the initial physical examination and during follow-up.
Recent developments in scientific technology have led to the production of new drugs affecting the immunological pathways of intractable monogenic IBD. Using a phenotypic high-throughput drug screen, Jardine et al. showed that leflunomide could be used in TTC7A-deficient patient-derived colonoids. Clinical trial are currently underway (18).
Common Genetic Risk Variants for IBD
The impact of the genetics for classical IBD (polygenic IBD) is also significant and different from that of monogenic IBD. Genome-wide association studies (GWAS) have identified about 240 disease loci linked to classical adult-onset IBD and have substantially expanded our understanding of the genetic architecture and causative mechanisms of IBD (Figure 1B) (19, 20). Similarly, common IBD variants associated with pediatric-onset IBD have been investigated; most of these variants are shared with adult-onset IBD (21, 22). On the other hand, the common variants with risk in children with very-early onset (VEO)-IBD (those diagnosed at younger than 6 years of age) have not been cleared in detail.
While these GWAS studies have identified important pathways associated with IBD including autophagy, a clear understanding of the interactions of IBD risk loci and causal genes as most of these GWAS loci are in introns or intergenic regions. Additionally, the majority of common genetic risk polymorphisms detected by GWAS have small effect (odds ratio <1.5). Statistical analysis indicated that these variants could explain only 13% and 8% of the variance in disease heritability for CD and UC, respectively (23). In the future, meta-analysis research combined with transcriptome analysis, epigenetics, and GWAS data are expected to increase our understanding of these regions.
Expressivity of Monogenic IBD
Variable expressivity is defined as the degree of variation of the clinical phenotype in individuals with monogenic disorders. Expressivity is different from penetrance, a term referring to whether the patient has the clinical phenotype associated with a genetic modification (24). The expressivity of the IBD phenotype in monogenic IBD is often incomplete in both autosomal dominant and autosomal recessive disorders. For example, around 20–30% of patients with XIAP deficiency or with GSD-1b caused by SLC374A4 demonstrate an IBD-like phenotype (16, 25, 26) and 4–9% with Wiskott Aldrich Syndrome develop IBD (27, 28).
Incomplete expressivity is assumed to result from the effects of unlinked modifier genes, epigenetic changes, or environmental factors (24). Recently, common IBD risk variants have been reported to play a significant role in this expressivity (Figure 1B). Tronstad et al. reported GWAS meta-analysis in 22 patients with familial GUCY2C diarrhea syndrome (FGDS), caused by a mutation in GUCY2C. Of 22 FGDS patients, eight showed CD-like phenotype. Seven of the eight patients had either homozygous or heterozygous NOD2 variants, known to be IBD risk, compared with only two of 14 FGDS patients without IBD (29). Huang et al. showed CGD patients with IBD had on average a significantly greater number of variants associated with IBD risk than CGD patients without IBD (30). Thus, common genetic risk polymorphisms for IBD may modify the expressivity of monogenic IBD.
Monogenic IBD With Heterozygous-Like Form Inheritance
Monogenic IBD usually exhibits a Mendelian inheritance pattern, but some monogenic IBD disorders that are inherited as X-linked or autosomal recessive (Figure 1B) may have exceptions. For example, X-linked CGD may be observed in females CYBB female carrier as a result of X chromosome inactivation resulting in manifestation of the primary immunodeficiency (31). Also, Aguilar et al. reported two female cases of IBD patients with a heterozygous XIAP mutation (32).
Autosomal recessive forms of inheritance are different from X-linked inheritance. When using whole exome sequences (WES), we rarely find only single-allele mutations causing monogenic disorders, although the phenotypes may be compatible with monogenic disorders. There are potential explanations for this phenomenon: (1) the existence of copy-number variation or low frequency gene mosaicism, which may be missed by WES, (2) a second-hit allele may be located in an intron or a non-coding region, altering splice site recognition; or (3) unknown modifier genes may affect penetrance. Regardless of the explanation, an approach to determine whether a variant is disease causing, such as additional sequencing and functional analysis are necessary (Table 1A). For example, Wright et al. reported the case of a 4 years-old boy with IBD caused by a rare pathogenic heterozygote variant in the NCF4 gene (34). This variant was inherited from his healthy father. NCF4, which encodes p40-phox, and normally has an autosomal recessive form of inheritance, is classified as a genetic subgroup of CGD. NADPH oxidase activity in this patient's neutrophils was decreased, as were monocyte and phagocyte killing of bacteria. This patient underwent HSCT and improved without any conventional therapy for IBD. These results suggested that this heterozygote variant led to a defect in neutrophil function and indicated that any other factors are changing the penetrance or expressivity of this variant in this patient, such as may be explained by one of the three hypotheses listed above. Wright et al. suggested that this might be due to the presence of unlinked modifier genes in this report.
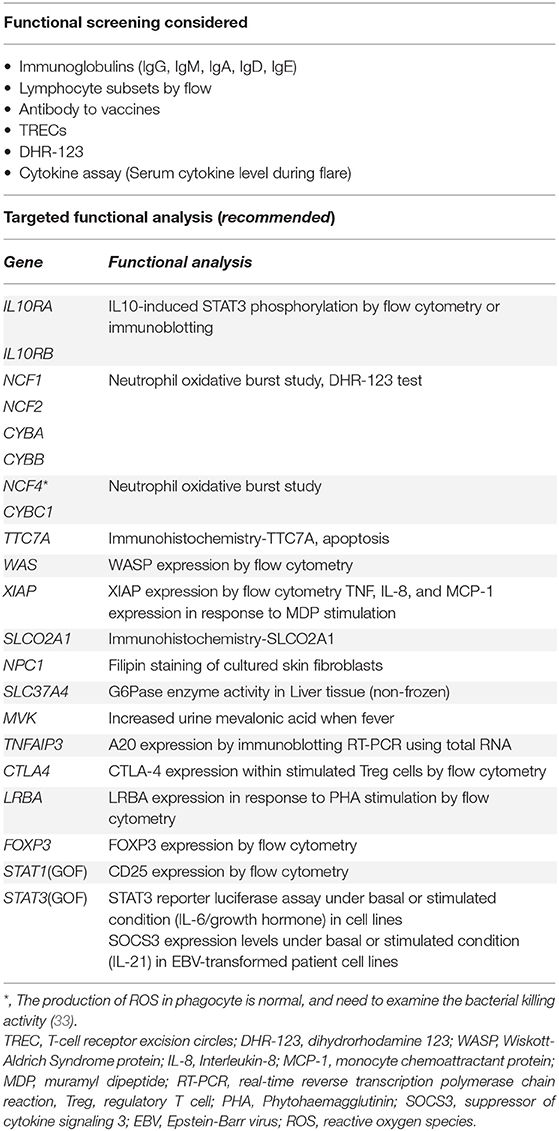
Table 1A. Functional analysis.
Even if only a single-allele mutation causing monogenic disorder is found, the clinicians should consider additional functional test if the patient's phenotype is consistent with that disorder.
Prevalence of Monogenic IBD in Pediatric IBD Patients
IBD Onset Before 6 Years of Age
In recent years, several cohort studies have been conducted, using WES and targeted genome panel sequence (TGPS) to determine the prevalence of monogenic IBD in children (Table 1B) (35–41). Among these previous studies, the prevalence of monogenic IBD in children with IBD has varied greatly. This may be due to differences in locality, differences in referral populations, and differences in the feature of the institution. Besides, the discrepancies in allele distribution among ethnic groups, and the cultural and social backgrounds of the regions, which could be influenced by factors such as consanguineous marriage might have an impact on the difference. For example; there have been two cohort studies conducted in tertiary facilities in China. In these studies, 24 of 27 cases and 5 of 9 cases with monogenic IBD had IL10R signaling defects. The percentage of colitis with IL10R signaling defects in Asia is higher than in Europe (35–41).
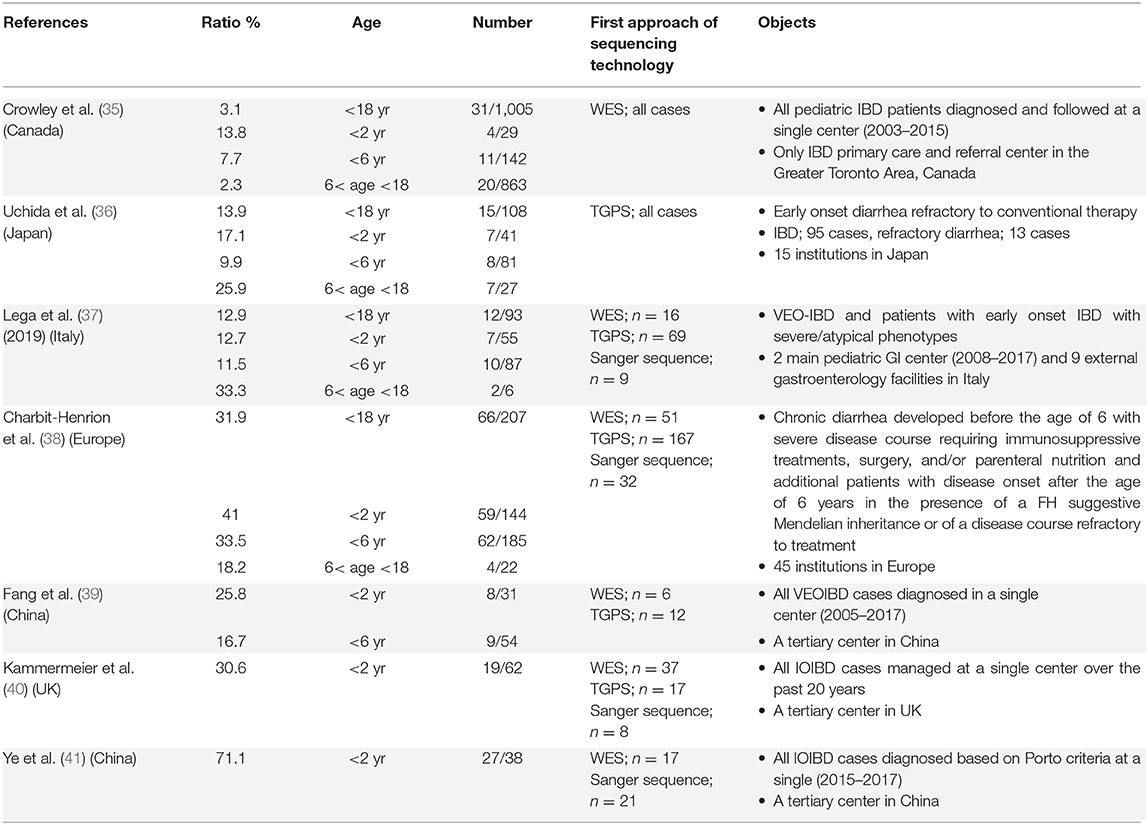
Table 1B. Cohort studies reporting the prevalence of monogenic IBD in pediatric IBD patients.
Recently, Crowley et al. reported data from a cohort of over 1,000 children with IBD at a single center in Canada (35). This center has the distinction of being the only IBD primary care and referral center in this area, and most children with IBD in this region are diagnosed and followed up at this center. All children with IBD from mild to severe underwent WES, and pathogenic variants of monogenic IBD were found to occur in 7.8% of children younger than 6 years of age and 13.8% of children younger than 2 years of age. This cohort includes a variety of racial groups, which may be representative of the real world. Based on these previous cohorts, monogenic IBD has been found to be about 1.5 times as prevalent in children younger than 2 years of age with IBD as in children younger than 6 years of age with IBD. The prevalence of monogenic IBD is at least 10–15% in children younger than 2 years of age with IBD and 7%-10% in children younger than 6 years of age with IBD, however, regional differences should be considered.
IBD Onset at the Age 6 and Older
The majority of the studies on monogenic IBD have focused on children younger than 6 years of age. Recently, there have been several reports dealing with children older than 6 years of age. The aforementioned large cohort from Canada showed that 2.5% of pediatric IBD occurring between the ages of 6 and 18 years was monogenic IBD (35). Three other cohort studies have reported that the incidence of monogenic IBD in severe refractory IBD cases in individuals aged between 6 and 18 years was ~20–30%. Recent reports suggest that physicians should be aware of the presence of rare variants causing monogenic IBD in children diagnosed older than 6 years of age. IL10/R, TTC7A and IKBKG are known to cause severe colitis in early infancy (12, 42, 43), whereas XIAP deficiency, IBD with Hermansky–Pudlak Syndrome, and IBD caused by mutation in TNFAIP3 can occur in a wide range of ages from infancy to adulthood (32, 44–46). CGD-colitis with an autosomal recessive pattern of inheritance occurs more frequently in younger individuals (47). However, CYBC1 deficiency was recently identified occurring in CGD-colitis in adolescents aged on average 12.2 years (range 7-19 years) (48).
Identifying Monogenic IBD in Clinical Practice
The key indicators of monogenic IBD in clinical practice are age of onset, family history, and extraintestinal manifestations based on individual gene defects. Infant IBD (under 2 years of age at onset) and VEOIBD are more likely to be monogenic IBD, and aggressive genetic testing is recommended. Concerning family history, there are two issues to be aware of. The first is “family relationships” including consanguinity. The second is “family diseases” as it is not uncommon for disease presentation to differ among family members despite having the same gene defect (49). Therefore, it is important to ask for a detailed family history of autoimmune diseases of other organs and infectious diseases besides IBD.
In the following sections, we discuss comorbidities. Extraintestinal manifestation is a critical indicator of monogenic IBD. Physical findings and comorbidities of which physicians should be aware at the initial physical examination and during follow-up are shown in Figure 1C. Recurrent and atypical infections are commonly found in monogenic IBD, since a large proportion of underlying monogenic disorders are primary immune deficiencies. Specific skin lesions, lymphadenopathy, and hepatosplenomegaly on physical examination are also physical findings that should be raise suspicions of monogenic IBD. There are some monogenic disorders with characteristic complications that are relatively easy to differentiate. Severe perianal disease, folliculitis, and/or arthritis in early infancy suggest the presence of IL-10 signaling defects disorders (50). In cases of extraintestinal manifestation with autoimmune anemia, type 1 diabetes mellitus, autoimmune thrombocytopenia, autoimmune thyroiditis, interstitial pneumonia, or other multi-organ autoimmune disorders, IBD may be one of the symptoms of IPEX syndrome caused by FOXP3 deficiency or IPEX-like syndrome caused by mutations in CTLA4, LRBA, STAT1, STAT3, or CARMIL2 (14, 51–56). GSD-1b and Niemann pick C are monogenic metabolic disorders that could cause IBD (16, 57). In many cases, findings such as hypoglycemic symptoms and hepatomegaly point to underlying metabolic diseases in early infancy, and IBD occurs after a diagnosis of metabolic disorders.
XIAP Deficiency and Chronic Granulomatous Disease
We sometimes have difficulty diagnosing monogenic IBD because of the lack of severe complications. The most frequent disorders in this category are XIAP deficiency and CGD (47, 58, 59). These disorders should be considered in children younger than 6 years of age with IBD, and should be excluded using functional analysis described below (Table 1A) or targeted sequencing for patients older than 6 years of age with IBD refractory to conventional therapy or requiring surgical intestinal resection.
Regarding XIAP deficiency, flow cytometry to evaluate XIAP expression is valuable for diagnostic screening tests. However, there are patients with XIAP deficiency who may have a normal protein expression but lack function. Therefore, we need assays for evaluating the TNF production of monocytes stimulated by L18-MDP (muramyl dipeptides) (60). For the diagnosis of CGD, dihydrorhodamine 1,2,3 (DHR-123) is useful. Besides, neutrophil/monocyte respiratory burst tests should be considered, because patients with CGD caused by NCF4 with normal or mildly impaired DHR-123 (33).
Early diagnosis is beneficial for patients with both XIAP deficiency and CGD. XIAP can develop life-threatening hemophagocytic lymphohistiocytosis (HLH), therefore HSCT should be considered early before HLH occurs (8, 61). CGD patients have a high risk of infection, therefore anti-tumor necrosis factor (TNF) alpha is contraindicated as it may increase this risk (62).
Conclusions and Future Directions
In this paper, we have reviewed the essential features of monogenic IBD, as seen in clinical practice. The use of WES and TGPS has led to improved diagnosis of severe refractory IBD as monogenic IBD. In the future, more genetic mutations will be discovered, and the role of immunity in intestinal inflammation will be elucidated using techniques including transcriptomics, proteomics, and metabolomics. It is further expected that appropriate treatments for these monogenic disorders will be developed. Because monogenic IBD is very rare, the collaboration of specialized facilities around the world is required for the further development of individualized treatment strategies. The interNational Early Onset Pediatric IBD Cohort Study (NEOPICS) (http://www.neopics.org/) and (http://www.VEOIBD.org) keep working to identify the causes and to develop new treatments together with international pediatric gastroenterologists and scientists from academic centers around the world.
Author Contributions
RN conceived, designed, and drafted manuscript. AM drafted, edited, and revised manuscript. RN and AM approved final version of manuscript. All authors contributed to the article and approved the submitted version.
Funding
RN is supported by Uehara Memorial Foundation fellowship. AM is supported by the Leona M. and Harry B. Helmsley Charitable Trust, Canada Research Chair (Tier 1) in Pediatric IBD, CIHR Foundation Grant and NIDDK (RC2DK118640) Grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CD, Crohn disease; CGD, Chronic granulomatous diseases; FGDS, Familial GUCY2C diarrhea syndrome; GSD-1b, Glycogen storage disease type 1b; GWAS, Genome-wide association studies; HSCT, Hematopoietic stem cell transplantation; IBD, Inflammatory bowel disease; IBDU, Inflammatory bowel disease-unclassified; IPEX, Immune dysregulation, polyendocrinopathy, enteropathy, X-linked; LRBA, Lipopolysaccharide-responsive and beige-like anchor; NEMO, Nuclear factor-kappa B essential modulator; TGPS, Targeted genome panel sequencing; TNF, Tumor necrosis factor; UC, Ulcerative colitis; VEOIBD, Very early onset inflammatory bowel disease; WES, Whole exome sequences; XIAP, X-linked inhibitor of apoptosis protein.
References
1. Levine A, Koletzko S, Turner D, Escher JC, Cucchiara S, de Ridder L, et al. ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J Pediatr Gastroenterol Nutr. (2014) 58:795–806. doi: 10.1097/MPG.0000000000000239
2. Graham DB, Xavier RJ. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature. (2020) 578:527–39. doi: 10.1038/s41586-020-2025-2
3. Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, et al. Inherited determinants of crohn's disease and ulcerative colitis phenotypes: a genetic association study. Lancet. (2016) 387:156–67. doi: 10.1016/S0140-6736(15)00465-1
4. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. (2009) 361:2033–45. doi: 10.1056/NEJMoa0907206
5. Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. (2014) 147:990–1007.e3. doi: 10.1053/j.gastro.2014.07.023
6. Uhlig HH, Schwerd T. From genes to mechanisms: the expanding spectrum of monogenic disorders associated with inflammatory bowel disease. Inflamm Bowel Dis. (2016) 22:202–12. doi: 10.1097/MIB.0000000000000614
7. Denson LA, Curran M, McGovern DPB, Koltun WA, Duerr RH, Kim SC, et al. Challenges in IBD research: precision medicine. Inflamm Bowel Dis. (2019) 25:S31–9. doi: 10.1093/ibd/izz078
8. Ono S, Okano T, Hoshino A, Yanagimachi M, Hamamoto K, Nakazawa Y, et al. Hematopoietic stem cell transplantation for XIAP deficiency in Japan. J Clin Immunol. (2017) 37:85–91. doi: 10.1007/s10875-016-0348-4
9. Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J, et al. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. (2007) 109:383–5. doi: 10.1182/blood-2006-05-025072
10. Marsh RA, Leiding JW, Logan BR, Griffith LM, Arnold DE, Haddad E, et al. Chronic granulomatous disease-associated IBD resolves and does not adversely impact survival following allogeneic HCTJ. Clin Immunol. (2019) 39:653–67. doi: 10.1007/s10875-019-00659-8
11. Kammermeier J, Lucchini G, Pai SY, Worth A, Rampling D, Amrolia P, et al. Stem cell transplantation for tetratricopeptide repeat domain 7A deficiency: long-term follow-up. Blood. (2016) 128:1306–8. doi: 10.1182/blood-2016-01-696385
12. Miot C, Imai K, Imai C, Mancini AJ, Kucuk ZY, Kawai T, et al. Hematopoietic stem cell transplantation in 29 patients hemizygous for hypomorphic IKBKG/NEMO mutations. Blood. (2017) 130:1456–67. doi: 10.1182/blood-2017-03-771600
13. Levy M, Arion A, Berrebi D, Cuisset L, Jeanne-Pasquier C, Bader-Meunier B, et al. Severe early-onset colitis revealing mevalonate kinase deficiency. Pediatrics. (2013) 132:e779–83. doi: 10.1542/peds.2012-3344
14. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
15. Cagdas D, Halaçli SO, Tan Ç, Lo B, Çetinkaya PG, Esenboga S, et al. A spectrum of clinical findings from ALPS to CVID: several novel LRBA defects. J Clin Immunol. (2019) 39:726–38. doi: 10.1007/s10875-019-00677-6
16. Melis D, Fulceri R, Parenti G, Marcolongo P, Gatti R, Parini R, et al. Genotype/phenotype correlation in glycogen storage disease type 1b: a multicentre study and review of the literature. Eur J Pediatr. (2005) 164:501–8. doi: 10.1007/s00431-005-1657-4
17. Yildiz M, Adrovic A, Tasdemir E, Baba-Zada K, Aydin M, Koker O, et al. Evaluation of co-existing diseases in children with familial mediterranean fever. Rheumatol Int. (2020) 40:57–64. doi: 10.1007/s00296-019-04391-9
18. Jardine S, Anderson S, Babcock S, Leung G, Pan J, Dhingani N, et al. Drug screen identifies leflunomide for treatment of inflammatory bowel disease caused by TTC7A deficiency. Gastroenterology. (2020) 158:1000–15. doi: 10.1053/j.gastro.2019.11.019
19. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
20. de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. (2017) 49:256–61. doi: 10.1038/ng.3760
21. Shaw KA, Cutler DJ, Okou D, Dodd A, Aronow BJ, Haberman Y, et al. Genetic variants and pathways implicated in a pediatric inflammatory bowel disease cohort. Genes Immun. (2019) 20:131–42. doi: 10.1038/s41435-018-0015-2
22. Kugathasan S, Baldassano RN, Bradfield JP, Sleiman PM, Imielinski M, Guthery SL, et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat Genet. (2008) 40:1211–5. doi: 10.1038/ng.203
23. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
24. Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. (2013) 132:1077–130. doi: 10.1007/s00439-013-1331-2
25. Aguilar C, Latour S. X-linked inhibitor of apoptosis protein deficiency: more than an x-linked lymphoproliferative syndrome. J Clin Immunol. (2015) 35:331–8. doi: 10.1007/s10875-015-0141-9
26. Skakic A, Djordjevic M, Sarajlija A, Klaassen K, Tosic N, Kecman B, et al. Genetic characterization of GSD I in serbian population revealed unexpectedly high incidence of GSD Ib and 3 novel SLC37A4 variants. Clin Genet. (2018) 93:350–5. doi: 10.1111/cge.13093
27. Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, et al. Autoimmunity in wiskott-aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. (2003) 111:e622–7. doi: 10.1542/peds.111.5.e622
28. Imai K, Morio T, Zhu Y, Jin Y, Itoh S, Kajiwara M, et al. Clinical course of patients with WASP gene mutations. Blood. (2004) 103:456–64. doi: 10.1182/blood-2003-05-1480
29. Tronstad RR, Polushina T, Brattbakk HR, Stansberg C, von Volkmann HL, Hanevik K, et al. Genetic and transcriptional analysis of inflammatory bowel disease-associated pathways in patients with GUCY2C-linked familial diarrhea. Scand J Gastroenterol. (2018) 53:1264–73. doi: 10.1080/00365521.2018.1521867
30. Huang C, De Ravin SS, Paul AR, Heller T, Ho N, Wu Datta L, et al. Genetic risk for inflammatory bowel disease is a determinant of crohn's disease development in chronic granulomatous disease. Inflamm Bowel Dis. (2016) 22:2794–801. doi: 10.1097/MIB.0000000000000966
31. Leiding JW, Holland SM. Chronic Granulomatous Disease. GeneReviews. Seattle, WA: University of Washington (1993–2020)
32. Aguilar C, Lenoir C, Lambert N, Bègue B, Brousse N, Canioni D, et al. Characterization of crohn disease in x-linked inhibitor of apoptosis-deficient male patients and female symptomatic carriers. J Allergy Clin Immunol. (2014) 134:1131–41.e9. doi: 10.1016/j.jaci.2014.04.031
33. van de Geer A, Nieto-Patlán A, Kuhns DB, Tool AT, Arias AA, Bouaziz M, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest. (2018) 128:3957–75. doi: 10.1172/JCI97116
34. Wright M, Chandrakasan S, Okou DT, Yin H, Jurickova I, Denson LA, et al. Early onset granulomatous colitis associated with a mutation in NCF4 resolved with hematopoietic stem cell transplantation. J Pediatr. (2019) 210:220–5. doi: 10.1016/j.jpeds.2019.03.042
35. Crowley E, Warner N, Pan J, Khalouei S, Elkadri A, Fiedler K, et al. Prevalence and clinical features of inflammatory bowel diseases associated with monogenic variants, identified by whole-exome sequencing in 1000 children at a single center. Gastroenterology. (2020) 158:2208–20. doi: 10.1053/j.gastro.2020.02.023
36. Uchida T, Suzuki T, Kikuchi A, Kakuta F, Ishige T, Nakayama Y, et al. Comprehensive targeted sequencing identifies monogenic disorders in patients with early-onset refractory diarrhea. J Pediatr Gastroenterol Nutr. (2020) 71:333–9. doi: 10.1097/MPG.0000000000002796
37. Lega S, Pin A, Arrigo S, Cifaldi C, Girardelli M, Bianco AM, et al. Diagnostic approach to monogenic inflammatory bowel disease in clinical practice: a ten-year multicentric experience. Inflamm Bowel Dis. (2020) 26:720–7. doi: 10.1093/ibd/izz178
38. Charbit-Henrion F, Parlato M, Hanein S, Duclaux-Loras R, Nowak J, Begue B, et al. Diagnostic yield of next-generation sequencing in very early-onset inflammatory bowel diseases: a multicentre study. J Crohns Colitis. (2018) 12:1104–12. doi: 10.1093/ecco-jcc/jjy068
39. Fang YH, Luo YY, Yu JD, Lou JG, Chen J. Phenotypic and genotypic characterization of inflammatory bowel disease in children under six years of age in China. World J Gastroenterol. (2018) 24:1035–45. doi: 10.3748/wjg.v24.i9.1035
40. Kammermeier J, Dziubak R, Pescarin M, Drury S, Godwin H, Reeve K, et al. Phenotypic and genotypic characterisation of inflammatory bowel disease presenting before the age of 2 years. J Crohns Colitis. (2017) 11:60–9. doi: 10.1093/ecco-jcc/jjw118
41. Ye Z, Zhou Y, Huang Y, Wang Y, Lu J, Tang Z, et al. Phenotype and management of infantile-onset inflammatory bowel disease: experience from a tertiary care center in China. Inflamm Bowel Dis. (2017) 23:2154–64. doi: 10.1097/MIB.0000000000001269
42. Zheng C, Huang Y, Hu W, Shi J, Ye Z, Qian X, et al. Phenotypic characterization of very early-onset inflammatory bowel disease with interleukin-10 signaling deficiency: based on a large cohort study. Inflamm Bowel Dis. (2019) 25:756–66. doi: 10.1093/ibd/izy289
43. Jardine S, Dhingani N, Muise AM. TTC7A: steward of intestinal health. Cell Mol Gastroenterol Hepatol. (2019) 7:555–70. doi: 10.1016/j.jcmgh.2018.12.001
44. Hussain N, Quezado M, Huizing M, Geho D, White JG, Gahl W, et al. Intestinal disease in hermansky-pudlak syndrome: occurrence of colitis and relation to genotype. Clin Gastroenterol Hepatol. (2006) 4:73–80. doi: 10.1016/s1542-3565(05)00858-x
45. Zheng C, Huang Y, Ye Z, Wang Y, Tang Z, Lu J, et al. Infantile onset intractable inflammatory bowel disease due to novel heterozygous mutations in TNFAIP3 (A20). Inflamm Bowel Dis. (2018) 24:2613–20. doi: 10.1093/ibd/izy165
46. Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis. (2018) 77:728–35. doi: 10.1136/annrheumdis-2017-212403
47. Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. (2008) 152:211–8. doi: 10.1111/j.1365-2249.2008.03644.x
48. Arnadottir GA, Norddahl GL, Gudmundsdottir S, Agustsdottir AB, Sigurdsson S, Jensson BO, et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat Commun. (2018) 9:4447. doi: 10.1038/s41467-018-06964-x
49. Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. (2020) 583:90–5. doi: 10.1038/s41586-020-2265-1
50. Huang Z, Peng K, Li X, Zhao R, You J, Cheng X, et al. Mutations in interleukin-10 receptor and clinical phenotypes in patients with very early onset inflammatory bowel disease: a Chinese VEO-IBD collaboration group survey. Inflamm Bowel Dis. (2017) 23:578–90. doi: 10.1097/MIB.0000000000001058
51. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front Immunol. (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
52. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20:1410–16. doi: 10.1038/nm.3746
53. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. (2012) 90:986–1001. doi: 10.1016/j.ajhg.2012.04.015
54. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
55. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. (2015) 125:591–9. doi: 10.1182/blood-2014-09-602763
56. Magg T, Shcherbina A, Arslan D, Desai MM, Wall S, Mitsialis V, et al. CARMIL2 deficiency presenting as very early onset inflammatory bowel disease. Inflamm Bowel Dis. (2019) 25:1788–95. doi: 10.1093/ibd/izz103
57. Schwerd T, Pandey S, Yang HT, Bagola K, Jameson E, Jung J, et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in niemann-pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn's disease. Gut. (2017) 66:1060–73. doi: 10.1136/gutjnl-2015-310382
58. Quaranta M, Wilson R, Gonçalves Serra E, Pandey S, Schwerd T, et al. Consequences of identifying XIAP deficiency in an adult patient with inflammatory bowel disease. Gastroenterology. (2018) 155:231–4. doi: 10.1053/j.gastro.2018.03.069
59. Colin de Verdière S, Noel E, Lozano C, Catherinot E, Martin M, Rivaud E, et al. Respiratory complications lead to the diagnosis of chronic granulomatous disease in two adult patients. J Clin Immunol. (2017) 37:113–6. doi: 10.1007/s10875-017-0370-1
60. Ammann S, Elling R, Gyrd-Hansen M, Dückers G, Bredius R, Burns SO, et al. A new functional assay for the diagnosis of X-linked inhibitor of apoptosis (XIAP) deficiency. Clin Exp Immunol. (2014) 176:394–400. doi: 10.1111/cei.12306
61. Lekbua A, Ouahed J, O'Connell AE, Kahn SA, Goldsmith JD, Imamura T, et al. Risk-factors associated with poor outcomes in VEO-IBD secondary to XIAP deficiency: a case report and literature review. J Pediatr Gastroenterol Nutr. (2019) 69:e13–8. doi: 10.1097/MPG.0000000000002297
Keywords: monogenic IBD, VEOIBD, inflammatory bowel disease, whole exome sequences, pediatric, genetics
Citation: Nambu R and Muise AM (2021) Advanced Understanding of Monogenic Inflammatory Bowel Disease. Front. Pediatr. 8:618918. doi: 10.3389/fped.2020.618918
Received: 19 October 2020; Accepted: 31 December 2020;
Published: 22 January 2021.
Edited by:
Wael El Matary, University of Manitoba, CanadaReviewed by:
Séamus Hussey, National Children's Research Centre (NCRC), IrelandChristiane Sokollik, University Children's Hospital Bern, Switzerland
Copyright © 2021 Nambu and Muise. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleixo M. Muise, aleixo.muise@sickkids.ca