Overview of Cardiomyopathies in Childhood
 Anika Rath1
Anika Rath1  Robert Weintraub1,2,3*
Robert Weintraub1,2,3*- 1Department of Cardiology, Royal Children's Hospital, Melbourne, VIC, Australia
- 2Heart Research, Murdoch Children's Research Institute, Melbourne, VIC, Australia
- 3Department of Paediatrics, Melbourne University, Melbourne, VIC, Australia
Paediatric cardiomyopathies are a heterogenous group of rare disorders, characterised by mechanical and electrical abnormalities of the heart muscle. The overall annual incidence of childhood cardiomyopathies is estimated at about 1 per 100,000 children and is significantly higher during the first 2 years of life. Dilated cardiomyopathies account for approximately half of the cases. Hypertrophic cardiomyopathies form the second largest group, followed by the less common left ventricular non-compaction and restrictive phenotypes. Infectious, metabolic, genetic, and syndromic conditions account for the majority of cases. Congestive heart failure is the typical manifestation in children with dilated cardiomyopathy, whereas presenting symptoms are more variable in other phenotypes. The natural history is largely influenced by the type of cardiomyopathy and its underlying aetiology. Results from a national population-based study revealed 10-year transplant-free survival rates of 80, 62, and 48% for hypertrophic, dilated and left ventricular non-compaction cardiomyopathies, respectively. Long-term survival rates of children with a restrictive phenotype have largely been obscured by early listing for heart transplantation. In general, the majority of adverse events, including death and heart transplantation, occur during the first 2 years after the initial presentation. This review provides an overview of childhood cardiomyopathies with a focus on epidemiology, natural history, and outcomes.
Introduction
Paediatric cardiomyopathies form an uncommon and heterogenous group of disorders, which are characterised by structural, mechanical, and electrical abnormalities of the heart muscle (1, 2). Aetiologies are diverse, and include infections, toxin exposure, tachyarrhythmias, genetic mutations, and underlying metabolic or neuromuscular disorders (1–6). Large population registries together with national and multicentre studies have contributed considerably to the increasing knowledge on epidemiology and outcomes of childhood cardiomyopathies (5, 7–24). The overall annual incidence is estimated at about 1 per 100,000 children, with a significantly higher incidence during the first 2 years of life (7, 17, 18). Dilated and hypertrophic cardiomyopathies are the most common types, whereas left ventricular non-compaction and restrictive cardiomyopathy occur less frequently (7, 17). Arrhythmogenic ventricular cardiomyopathy is rarely diagnosed during childhood and will not be discussed in this article. The terms dilated, hypertrophic, restrictive, and non-compaction depict different phenotypes (see Figure 1) and thereby assist in grouping cardiomyopathies, however they do not describe specific disease entities. The European Society of Cardiology classifies cardiomyopathies according to their predominant phenotype, but does not recognise left ventricular non-compaction as a separate entity, whereas the American Heart Association classifies cardiomyopathies according to their aetiology, with left ventricular non-compaction considered to be a separate entity (25, 26). Some children may present with a mixed phenotype, and cardiomyopathy phenotypes may undulate or transition during the course of disease (1, 12, 13, 27). This review summarises the most common forms of paediatric cardiomyopathies, with a focus on epidemiology and natural history.
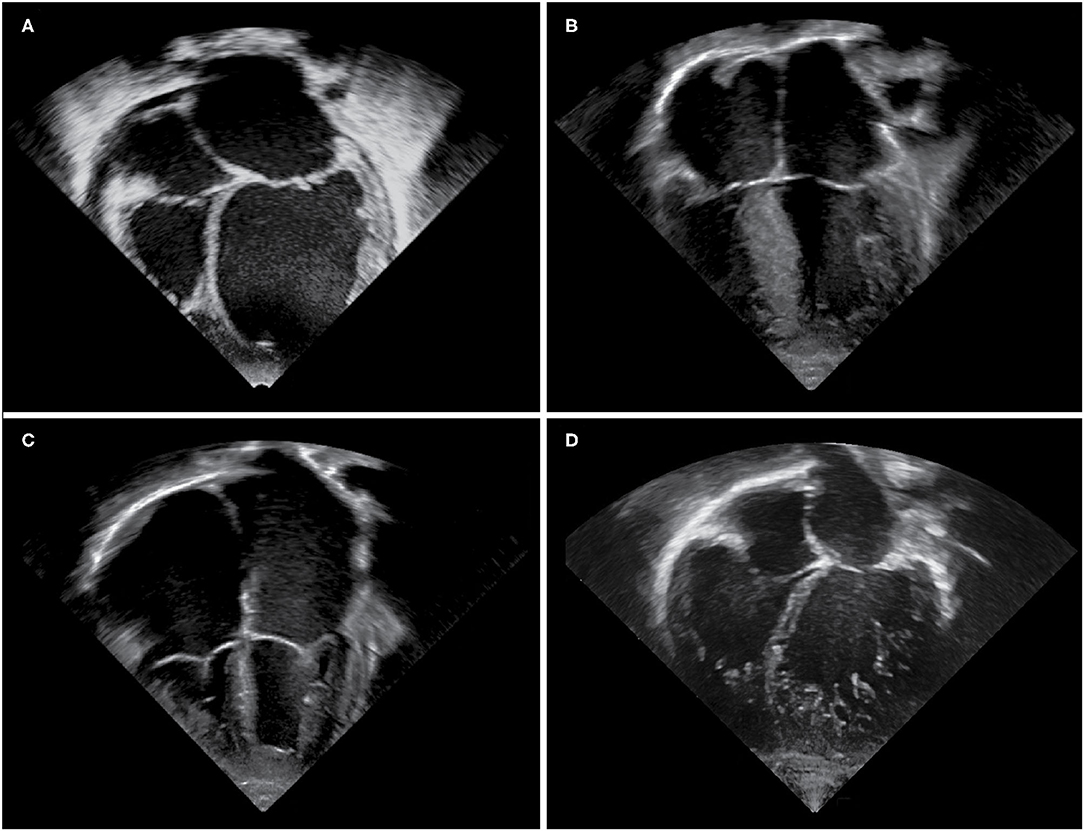
Figure 1. Echocardiographic images of cardiomyopathy phenotypes. Apical four chamber views demonstrating (A) a dilated left ventricle and left atrium in a DCM patient, (B) hypertrophy of the interventricular septum and left ventricular free wall in a HCM patient, (C) massively dilated atria and small right and left ventricular cavities in an RCM patient, (D) an extensively trabeculated myocardium with a compacted and non-compacted layer and deep intertrabecular recesses most prominent at the left ventricular apex and free wall in an LVNC patient.
Dilated Cardiomyopathy
Aetiologies
Dilated cardiomyopathy (DCM) is characterised by left ventricular dilation and systolic dysfunction. Important aetiologies in childhood include infections, toxic causes (including chemotherapy), genetic mutations, and other causes such as inborn errors of metabolism and neuromuscular disorders (1). Recent advances in genetic diagnostics, including the introduction of next-generation DNA sequencing technologies and extended cardiomyopathy gene panels, have increased the detection rate for pathogenic mutations in adult DCM patients to about 40% (28, 29). Sarcomere gene mutations are thought to be responsible for 35–40% of genetic cases (1). Evidence of myocarditis has been found in up to one third of children with DCM undergoing early endomyocardial biopsy (20, 21, 30). Individuals with variants in genes coding for cardiac structural proteins may be particularly susceptible to severe myocarditis (31, 32). However, despite significant diagnostic progress over the last decade, the aetiology of childhood DCM often remains unknown.
Epidemiology
DCM is the most common type of childhood cardiomyopathy. It comprised about half of all cases in the Paediatric Cardiomyopathy Registry (PCMR), a large multicentre North American study, as well as in the National Australian Childhood Cardiomyopathy Study (NACCS), a population-based cohort study which included children younger than 10 years of age at diagnosis. The overall incidence was 0.58–0.73 per 100,000 children and varied significantly by age (see Table 1). In both studies, the highest annual incidence was observed during the first year of life (see Table 1) (7, 17). A nationwide cardiomyopathy study from Finland, which included only idiopathic cardiomyopathy cases, demonstrated an overall DCM incidence of 0.34 per 100,000 children up to 20 years and an 11-fold higher incidence during the first year of life (see Table 1) (18). A large prospective study undertaken in the United Kingdom and Ireland assessed the incidence of paediatric new-onset heart failure due to heart muscle disease of known and unknown aetiologies. The authors reported an incidence of 0.76 symptomatic DCM cases per 100,000 children under 16 years (19). Lymphocytic myocarditis accounted for 22% of all cases in this study, and comprised 16–36% of DCM cases in the PCMR and NACCS (19–21). Endomyocardial biopsy is not universally performed in newly diagnosed cases of DCM and the presence of positive histological findings for lymphocytic myocarditis decreases rapidly within weeks after presentation, which may lead to an underestimation of inflammatory DCM in children (21, 33).
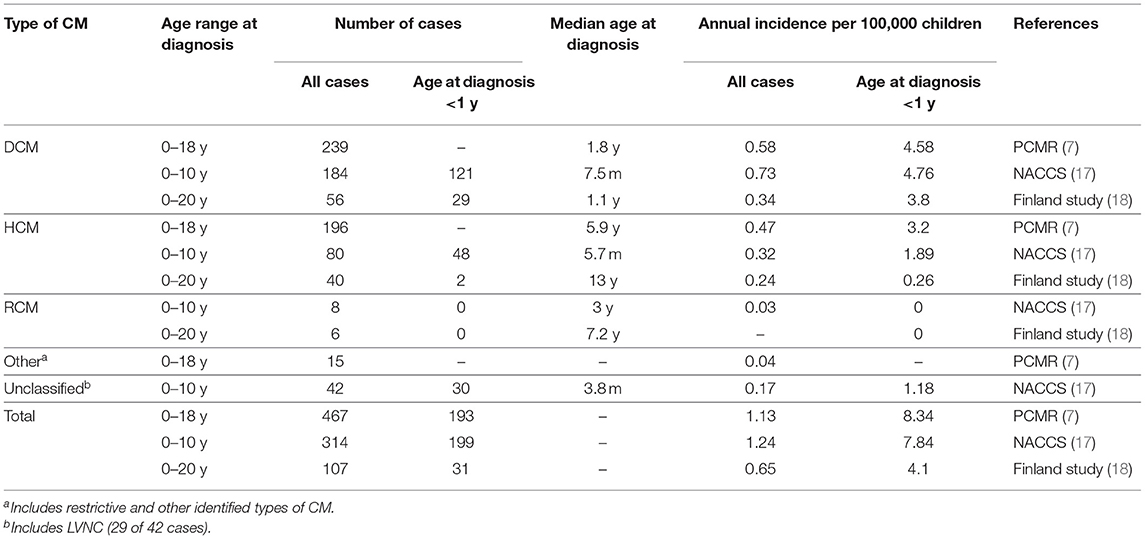
Table 1. Annual incidence and median age at diagnosis for each type of cardiomyopathy (CM).
Presenting Features
The majority of children with DCM, particularly infants, present with varying severity of congestive cardiac failure, with symptoms ranging from feeding difficulties to cardiovascular collapse, and rarely sudden death. Older children with a positive family history and those with neuromuscular disorders or inborn errors of metabolism may be diagnosed as part of routine screening, prior to symptom onset. Lymphocytic myocarditis has a phenotype that may be indistinguishable from idiopathic DCM. A history of a prior viral illness is not always present and may be misleading (34). Data from the PCMR and NACCS showed that at the time of diagnosis, 71–90% of children with DCM had clinical evidence of congestive heart failure (20, 21). In the NACCS DCM cohort, 5% presented with sudden cardiac death, 2% with exercise intolerance or arrhythmia, and 3% for routine family screening. Intensive care unit admission was required in 45% and inotropic support was administered in 40% (21). Significant comorbidities associated with cardiomyopathy related intensive care unit admissions included renal failure, thromboembolic events and hepatic impairment (35).
Natural History
Outcomes for children with DCM are highly variable, ranging from complete recovery to death or requirement for transplantation. Beyond infancy, DCM is the most common indication for heart transplantation in children (36). Data from the PCMR and NACCS demonstrated an overall 5 year transplant-free survival of 54–65 % (see Table 2) (20, 21). Risk stratification helps to identify individuals who can be treated medically and those who require advanced heart failure therapy including listing for heart transplantation. Transplant-free survival analysis according to the underling aetiology from the PCMR is shown in Table 2 (20). Long-term outcome data from the NACCS revealed transplant-free survival of 56% after 20 years (22). The highest risk was observed early after diagnosis, with a 26% risk of death or transplantation within the first year and only 1% per year thereafter (22). Similar results were found in the UK study of children admitted with new-onset heart failure secondary to dilated cardiomyopathy. Event-free survival at 1 year was only 66 and 16% had undergone heart transplantation within the first year following diagnosis (19).
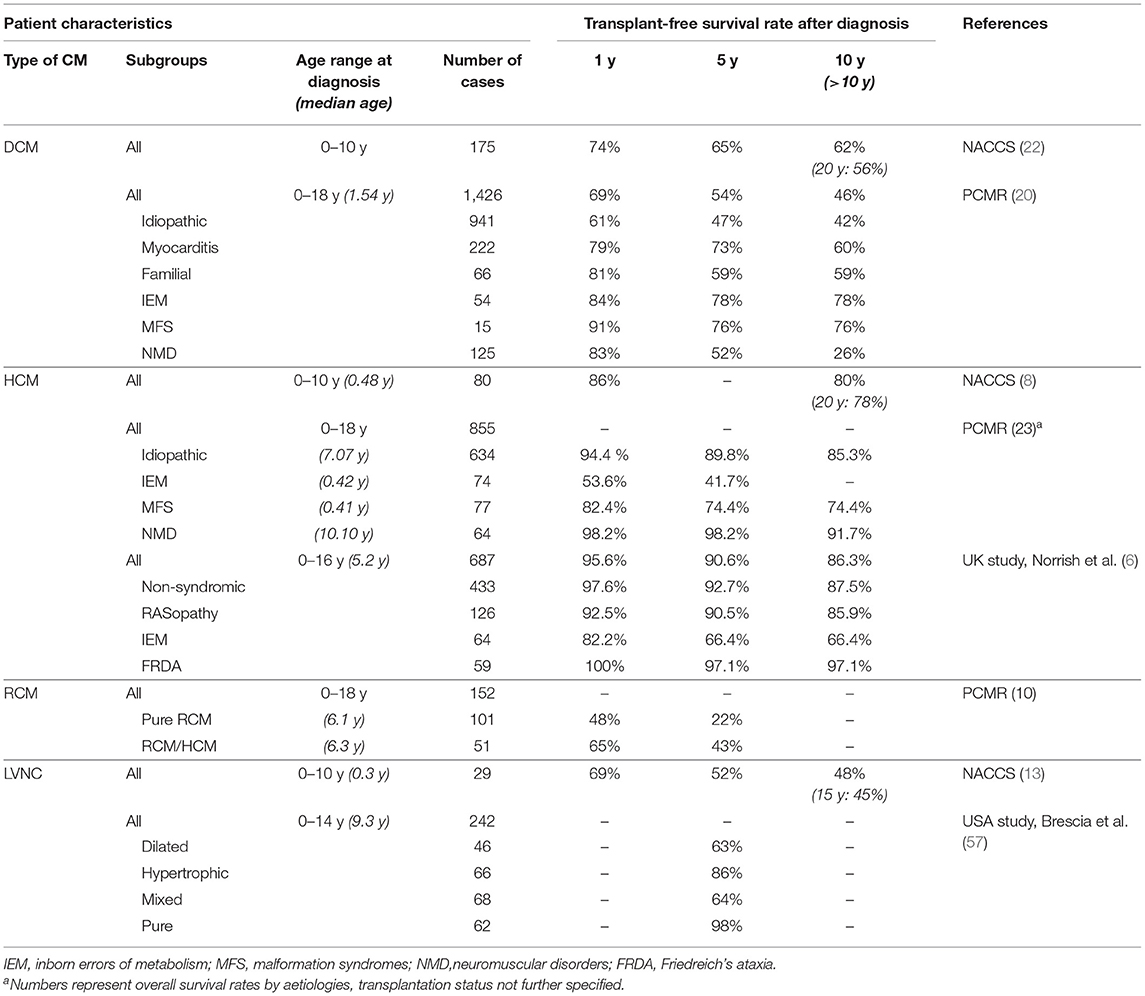
Table 2. Transplant-free survival for each type of cardiomyopathy (CM).
Other risk factors in children with DCM relate to age at diagnosis and the severity of cardiac dysfunction. Risk factors for death or transplantation in the NACCS comprised age below 4 weeks or above 5 years at presentation, familial dilated cardiomyopathy, a lower initial fractional shortening z-score and failure to improve fractional shortening z-score during follow-up (21, 22). Favourable outcomes including transplant-free survival and reverse remodelling were more frequent in children with lymphocytic myocarditis compared to other aetiologies, with echocardiographic normalisation of LV function found in 92% during follow-up (22). Although less frequent, reverse modelling has also been observed in children without proven or suspected myocarditis. Everitt et al. reported echocardiographic recovery of LV function within 2 years in 22% of cases with idiopathic DCM included in the PCMR. Younger age (<10 years) and less LV dilation at diagnosis were independent predictive factors of echocardiographic normalisation. Some patients developed recurrence of congestive cardiac failure following initial echocardiographic normalisation (37). Persistence of increased levels of N-terminal proBNP after initial stabilisation was found to be predictive for the risk of cardiac death in children with idiopathic DCM (38).
Pahl et al. reviewed risk factors for sudden cardiac death (SCD) in DCM from the PCMR and found a 5-year incidence of 3%. Younger age at diagnosis (<14.3 y), systolic LV dilation (LV end systolic dimension z-score >2.6) and posterior wall thinning were identified as risk factors for sudden death (39).
The overall survival for childhood DCM in North America has improved in the most recent era, despite rates of echocardiographic normalisation and cardiac transplantation that did not differ by comparison with a previous era (40). This may be due to improved resuscitation and/or the use of adult-based chronic heart failure therapies.
Hypertrophic Cardiomyopathy
Aetiologies
Hypertrophic Cardiomyopathy (HCM) is a condition characterised by left or biventricular hypertrophy in the absence of structural heart disease or increased ventricular afterload. Sarcomeric mutations represent the most common genetic cause in children and adults with a detection rate of about 60% in childhood HCM diagnosed beyond the first year of life, similar to that of adult HCM patients (41). By comparison with adult HCM, childhood HCM comprises a much more heterogenous group with diverse aetiologies and spectrum of disease (6). Genetic causes include inborn errors of metabolism, malformation syndromes (primarily RASopathies), neuromuscular disease as well as pathogenic mutations in genes coding for sarcomeric proteins. Typical causes within these four categories are Pompe disease, Noonan syndrome, Friedreich ataxia, and MYBPC3 or MYH7 mutations, respectively (1, 23).
Epidemiology
HCM forms the second commonest group of childhood cardiomyopathies, comprising 25–42% of all cases, respectively. The overall annual incidence is 0.24–0.47 per 100,000 children (see Table 1) (7, 17, 18). HCM caused by inborn errors of metabolism and malformation syndromes generally presents in infancy and contributes significantly to an early first peak in incidence. A second, smaller peak during adolescence and early adult life is largely due to sarcomeric protein mutations (1, 41). Data from the NACCS, which included children only aged 0–10 year at diagnosis and excluded those with metabolic and neuromuscular conditions, demonstrated a median age of 5.7 months at presentation. The NACCS and PCMR reported a marked decline in incidence between the first year and subsequent years of life (see Table 1). In the Finnish study, which also excluded patients with metabolic and neuromuscular aetiologies but included children up until 20 years at diagnosis, the median age at diagnosis was 13 years, and 39% of patients were over 15 years of age at presentation (see Table 1).
Presenting Features
The clinical status at presentation ranges from asymptomatic to symptoms of exercise intolerance, chest pain, palpitations, syncope, or cardiac arrest (1). Congestive heart failure or arrhythmic symptoms are found in 10–15% of cases at presentation (6, 24). Children with inborn errors of metabolism and malformation syndromes generally present earlier, and are more likely to have congestive heart failure at the time of diagnosis (23). Aborted sudden cardiac death or out of hospital arrest are an uncommon initial presentation in childhood HCM (6).
Natural History
The natural history and outcome of childhood HCM largely depend on the age at presentation and the underlying aetiology. The highest risk of mortality is seen in those diagnosed during the first year of life (24).
Overall survival free from death or transplantation was found to be about 90% at 5 years and 78% at 20 years from presentation (see Table 2) (6, 8, 24). The risk of mortality or transplantation was 14% during the first year after presentation, decreasing to 0.4% per subsequent year (8).
The worst outcomes are in infants with heart failure at the time of diagnosis and in older children with marked restrictive pathophysiology. Other risk factors include concentric left ventricular hypertrophy at diagnosis, Noonan syndrome, and increasing LV ventricular free wall thickness and worsening LV systolic function during follow-up (8). In the PCMR, children with non-syndromic HCM diagnosed before 1 year of age had a higher mortality, which reduced in those surviving infancy (23). Similarly, Noonan syndrome patients had a markedly reduced 1-year survival when diagnosed with congestive heart failure before 6 months of age (42). The highest 5 year survival rate was observed in HCM secondary to neuromuscular disease (see Table 2) (23).
While congestive heart failure accounts for the majority of early deaths in childhood HCM, the most common mode of death overall is SCD (6, 8). Arrhythmic events have been observed with a rate of 1.2 per 100 patient years in a large UK study, with more frequent occurrence in non-syndromic patients (6). The identification of specific paediatric risk factors for SCD is essential to guide implantable cardioverter defibrillator (ICD) insertion in a population that frequently experiences no cardiac symptoms in their daily life. A systematic review and meta-analysis of clinical risk factors for sudden cardiac death in childhood cardiomyopathy identified previous adverse cardiac events, non-sustained ventricular tachycardia, syncope, and extreme left ventricular hypertrophy as major factors (43). Norrish et al. recently described a novel risk prediction model for SCD in childhood HCM, with the objective of providing individualised risk estimates. Unexplained syncope, maximal left ventricular wall thickness, left atrial diameter, and non-sustained VT were found to have the strongest association with the composite outcome of SCD or an equivalent event (44). Miron et al. also described an SCD risk prediction model for paediatric HCM. This group used the above mentioned four risk factors as well as age at diagnosis for a clinical model and added the presence of a pathogenic gene variant for a combined clinical/genetic model (45). The relationship between left ventricular outflow obstruction and sudden death is complex, with some studies showing either a protective effect or an inverse relationship in children with the highest gradients (44–46). Arrhythmic events leading to sudden cardiac death continue to occur in adult cohorts at about 0.7% per year (47).
Restrictive Cardiomyopathy
Aetiologies
Restrictive cardiomyopathy (RCM) is a rare form of heart muscle disease defined by “normal or decreased volume of both ventricles associated with biatrial enlargement, normal left ventricular wall thickness and atrioventricular valves, impaired ventricular filling with restrictive physiology, and normal (or near normal) systolic function” (26). Nearly a quarter of patients with RCM have a family history of cardiomyopathy (10). An increasing number of genetic mutations in sarcomeric and non-sarcomeric proteins have been reported, providing evidence of overlap with other forms of cardiomyopathy (5, 48, 49). A significant subgroup of RCM cases has a mixed phenotype, most commonly combining characteristics of RCM and HCM (10). RCM has also been described in association with inborn errors of metabolism, infiltrative disease, and skeletal myopathy (1, 50).
Epidemiology
RCM is the rarest form of paediatric cardiomyopathy with an incidence of 0.03–0.04 per 100,000 children in Australia and the United States (see Table 1) (7, 17). RCM accounted for 2.5% of cases in the NACCS, and the PCMR reported 3% of pure RCM cases and an additional 1.5% of mixed RCM/HCM phenotype cases (7, 17). Age at diagnosis ranges from early infancy to late adulthood (1). Unlike other childhood cardiomyopathies, RCM becomes more frequent with increasing age. Only 10% of pure RCM cases in the PCMR were diagnosed during the first year of life (10).
Presenting Features
Early symptoms of RCM may be non-specific, including general fatigue and exercise intolerance. Clinical findings secondary to elevated systemic and pulmonary venous pressures include peripheral oedema, hepatomegaly, pulmonary oedema, and pulmonary hypertension (1). In the later stages of disease, patients may develop systolic dysfunction (1). Syncope is a non-specific but ominous presenting symptom, which may be caused by arrhythmias, coronary ischemia, or thromboembolic events (51).
Natural History
Whilst RCM has the worst outcomes of any childhood cardiomyopathy, the natural history has largely been obscured by early referral for transplantation. Long-term transplant-free survival data is therefore sparse. Because of its relentless and progressive nature, patients are at risk of sudden death, congestive heart failure, atrial and ventricular arrhythmias, conduction disorders and thromboembolism (1, 52). Transplantation free survival for children with pure RCM in the PCMR was 48 and 22% 1 and 5 years after diagnosis, respectively (10). The mixed phenotype group (RCM/HCM) demonstrated a 2-fold higher 5 year transplant-free survival (see Table 2). Overall freedom from death after 5 years was identical for both cohorts, indicating a preference for earlier transplantation in pure RCM patients (10). Russo et al. reviewed 21 cases of RCM in a single centre retrospective analysis, and reported transplantation free survival of 80.5 and 20% at 1 and 10 years, respectively (53). Anderson et al. analysed their institutional experience for children with RCM comparing a historical cohort of 9 cases, diagnosed between 1975 and 1993, with a contemporary cohort of 12 cases. Transplantation free survival over 5 years was 38% in both groups, however overall survival in the contemporary group was 80 vs. 38% in the historical group (54).
Due to the infrequence of childhood RCM and small study cohorts, assessment of risk factors for outcome has proven difficult, and results have been inconsistent. In the PCMR, heart failure symptoms and lower fractional shortening z-score at diagnosis were identified as independent risk factors for decreased transplant-free survival (10). Similarly, higher initial echocardiographic left atrial dimensions and a requirement for diuretics during follow-up have been associated with increased mortality (49). Anderson et al. observed an association of marked elevation of mitral valve Doppler E/e′ ratio on echocardiography with increased mortality (54). Rivenes et al. evaluated risks factors predictive of sudden death and cardiovascular collapse in 18 children with RCM from a single centre retrospective study. They reported an increased risk of ischemia-related complications and mortality in the entire patient group. The risk of sudden death was highest in girls with clinical signs suggestive of ischemia, in particular chest pain and syncope at presentation. The subgroup at risk of sudden death appeared well and had no clinical evidence of ongoing congestive heart failure (51). Walsh et al. observed that PR prolongation and a wider QRS complex on a baseline ECG were associated with an increased incidence of acute cardiac events, and they found a substantial risk for acute high-grade heart block in RCM patients (52). An elevated pulmonary vascular resistance is present in up to 40% of children with RCM and may impact on the timing for transplant referral (11). Serial cardiac catheterisation is often undertaken to detect this serious complication which may impact on transplant suitability (5, 11).
Left Ventricular Non-Compaction
Aetiologies
Left ventricular non-compaction (LVNC) is a heterogenous form of cardiomyopathy characterised by excessive trabeculation of the left or both ventricles with deep intertrabecular recesses, most frequently affecting the left ventricular apex. LVNC was classified as a separate cardiomyopathy by the American Heart Association in 2006 (26) however there is ongoing discussion about whether it is a distinct entity or a morphological phenotype (55). Arrest in normal endomyocardial morphogenesis with failure of trabecular compaction is thought to be causative especially in paediatric cases (3). A similar phenotype can manifest at any time in adult life secondary to conditions associated with an increased left ventricular preload (56). LVNC can be isolated or associated with other cardiomyopathy phenotypes, arrhythmias, or congenital heart disease. LVNC is commonly found in Barth syndrome, an X-linked recessive disorder caused by tafazzin gene mutations, and has also been reported in patients with inborn errors of metabolism, neuromuscular diseases, and genetic syndromes (1). Genetic testing detects variants in 30–45% of cases, with sarcomeric mutations found most frequently (3, 56).
Epidemiology
The rates of diagnosis of LVNC in children have increased during the last decades, which is thought to reflect increased awareness and improved imaging techniques rather than a rise in incidence (12). Data from the NACCS demonstrated an incidence of 0.11 per 100,000 children aged 0–10 years, and a 7-fold higher incidence in infants (see Table 1). LVNC was found in 9.2% of children diagnosed with cardiomyopathy under 10 years (17). In the PCMR, LVNC was present in 4.8% of cases. LVNC was found to be associated with dilated, hypertrophic and indeterminate phenotypes in 59, 11, and 8% of cases, respectively, and isolated LVNC occurred in the remaining 23% of cases. The median age at diagnosis was significantly higher in isolated LVNC (9.8 years) compared to cases with mixed phenotypes (0.4–0.6 years) (12).
Presenting Features
Patients with LVNC may be found on routine screening but may also present with thromboembolic events, arrhythmias, or congestive heart failure (3). The variability of presenting symptoms reflects the phenotypic diversity, and associated features of other types of cardiomyopathies contribute significantly to the clinical picture. In the largest cohort of paediatric LVNC patients, of which almost 40% were infants, 25% presented primarily with congestive heart failure, 17% with arrhythmias, 19% with a heart murmur, and 37% were asymptomatic (57). A positive family history of cardiomyopathy was present in 23% of all cases, however, only 25% of this subgroup had a family history of LVNC.
Natural History
The outcome of paediatric LVNC is highly variable and depends on the underlying pathophysiology (1). Brescia et al. reviewed the risk of mortality and sudden death in the largest published cohort of paediatric LVNC patients. They found a strong relationship between the identified phenotype and the risk of death or transplantation. Five-year transplant-free survival rate was excellent for the normal-dimension phenotype, intermediate for the hypertrophic phenotype and worst for the dilated and mixed phenotypes (see Table 2). The greatest risk factors for death or transplantation were the presence of systolic dysfunction and/ or arrhythmias. Sudden cardiac death occurred in 6.2% of cases over a 19-year study period, with systolic dysfunction present in 95% and documented arrythmia in 60% of these patients. Early presentation during the first year of life was an additional independent risk factor, with a 25% risk of death or transplantation in infantile LVNC (57). Jefferies et al. similarly observed the worst outcome for patients with LVNC and the dilated or indeterminate phenotype in the PCMR. The risk of death was highest in the first year after diagnosis (12). Shi et al. reviewed long-term outcomes of children with LVNC from the NACCS. This cohort included mainly young and severely affected infants, with a median age of 0.3 years at diagnosis and congestive heart failure present in 83% at the time of diagnosis. Freedom from death and transplantation was 45% at 15 years after diagnosis (see Table 1). Propensity score matching suggested a 2-fold higher risk of death and transplantation for patients with LVNC and a dilated phenotype compared to children with DCM from the same registry (13). Children with an isolated LVNC phenotype and without observable cardiac dysfunction have been found to have a favourable outcome (12). However, progression to an associated cardiomyopathy phenotype with a risk of mortality has been observed in a small proportion of cases, therefore ongoing surveillance is recommended (12).
Discussion
Over the last 2 decades, registries and national studies have provided important data on epidemiology and outcomes of childhood cardiomyopathies. The reported overall annual incidence was between 0.65 and 1.24 per 100,000 children (7, 17, 18). Consistently throughout these epidemiological studies, the annual incidence of cardiomyopathy during the first year of life was 6–7 times higher than the above-mentioned average incidences (7, 17, 18). This peak incidence during infancy was found in all types of cardiomyopathy except for RCM which typically presents later in childhood (7, 17, 18). PCMR data demonstrated a small second peak during adolescence which was related to HCM and cardiomyopathies secondary to neuromuscular diseases (7).
LVNC cardiomyopathy has increasingly gained attention over the last two decades and there is ongoing discussion with regards to its diagnostic criteria (25, 26). Increasing rates of diagnosis reflect an increasing awareness of this entity compared to prior eras (12).
While there has been a dramatic improvement in survival of congenital heart disease over the last decades, the overall outcomes for childhood cardiomyopathies remain unfavourable (58, 59). Recovery from inflammatory cardiomyopathies or successful rhythm control in arrhythmogenic heart failure represent exemptions. Childhood cardiomyopathies comprise the most common indication for cardiac transplantation beyond the first year of age (36). The highest risk of death or cardiac transplantation occurs during the first year after diagnosis (19, 22). Knowledge of the natural history and risk factors for adverse outcomes, gained from registries and large multicentre studies, assists in risk stratification and case selection for advanced heart failure therapies, and for ICD insertion for prevention of sudden cardiac death.
Conclusion
Paediatric cardiomyopathies, although rare, carry a substantial burden of disease due to the risk of morbidity and mortality and a lack of curative therapy. Established registries have provided valuable insights into the natural history and risk factors, assisting in decision-making on sudden cardiac death prevention and cardiac transplantation.
Author Contributions
AR drafted the manuscript and designed figures and tables. RW supervised the project and contributed substantially to the final version of the manuscript. Both authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, et al. Pediatric cardiomyopathies. Circ Res. (2017) 121:855–73. doi: 10.1161/CIRCRESAHA.116.309386
2. Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the american heart association. Circulation. (2019) 140:9–68. doi: 10.1161/CIR.0000000000000682
3. Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet. (2015) 386:813–25. doi: 10.1016/S0140-6736(14)61282-4
4. Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. (2017) 390:400–14. doi: 10.1016/S0140-6736(16)31713-5
5. Denfield SW, Webber SA. Restrictive cardiomyopathy in childhood. Heart Fail Clin. (2010) 6:445–52. doi: 10.1016/j.hfc.2010.05.005
6. Norrish G, Field E, McLeod K, Ilina M, Stuart G, Bhole V, et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: a retrospective study in United Kingdom. Eur Heart J. (2019) 40:986–93. doi: 10.1093/eurheartj/ehy798
7. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. (2003) 348:1647–55. doi: 10.1056/NEJMoa021715
8. Alexander PMA, Nugent AW, Daubeney PEF, Lee KJ, Sleeper LA, Schuster T, et al. Long-term outcomes of hypertrophic cardiomyopathy diagnosed during childhood: results from a national population-based study. Circulation. (2018) 138:29–36. doi: 10.1161/CIRCULATIONAHA.117.028895
9. Maurizi N, Passantino S, Spaziani G, Girolami F, Arretini A, Targetti M, et al. Long-term outcomes of pediatric-onset hypertrophic cardiomyopathy and age-specific risk factors for lethal arrhythmic events. JAMA Cardiol. (2018) 3:520–5. doi: 10.1001/jamacardio.2018.0789
10. Webber SA, Lipshultz SE, Sleeper LA, Lu M, Wilkinson JD, Addonizio LJ, et al. Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: a report from the pediatric cardiomyopathy registry. Circulation. (2012) 126:1237–44. doi: 10.1161/CIRCULATIONAHA.112.104638
11. Weller RJ, Weintraub R, Addonizio LJ, Chrisant MRK, Gersony WM, Hsu DT. Outcome of idiopathic restrictive cardiomyopathy in children. Am J Cardiol. (2002) 90:501–6. doi: 10.1016/S0002-9149(02)02522-5
12. Jefferies JL, Wilkinson JD, Sleeper LA, Colan SD, Lu M, Pahl E, et al. Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: results from the pediatric cardiomyopathy registry. J Card Fail. (2015) 21:877–84. doi: 10.1016/j.cardfail.2015.06.381
13. Shi WY, Betancur MM, Nugent AW, Cheung M, Colan S, Turner C, et al. Long-term outcomes of childhood left ventricular noncompaction cardiomyopathy: Results from a national population-based study. Circulation. (2018) 138:367–76. doi: 10.1161/CIRCULATIONAHA.117.032262
14. Wilkinson JD, Sleeper LA, Alvarez JA, Bublik N, Lipshultz SE. The pediatric cardiomyopathy registry: 1995-2007. Prog Pediatr Cardiol. (2008) 25:31–6. doi: 10.1016/j.ppedcard.2007.11.006
15. Wilkinson JD, Landy DC, Colan SD, Towbin JA, Sleeper LA, Orav EJ, et al. The pediatric cardiomyopathy registry and heart failure: key results from the first 15 years. Heart Fail Clin. (2010) 6:401–13. doi: 10.1016/j.hfc.2010.05.002
16. Puggia I, Merlo M, Barbati G, Rowland TJ, Stolfo D, Gigli M, et al. Natural history of dilated cardiomyopathy in children. J Am Heart Assoc. (2016) 5:1–10. doi: 10.1161/JAHA.116.003450
17. Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. (2003) 348:1639–46. doi: 10.1056/NEJMoa021737
18. Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, et al. Epidemiology of idiopathic cardiomyopathies in children and adolescents: a nationwide study in Finland. Am J Epidemiol. (1997) 146:385–93. doi: 10.1093/oxfordjournals.aje.a009291
19. Andrews RE, Fenton MJ, Ridout DA, Burch M. New-onset heart failure due to heart muscle disease in childhood: a prospective study in the United Kingdom and Ireland. Circulation. (2008) 117:79–84. doi: 10.1161/CIRCULATIONAHA.106.671735
20. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, et al. Incidence, causes, and outcome of dilated cardiomyopathy in children. JAMA Cardiol. (2006) 296:1867–76. doi: 10.1001/jama.296.15.1867
21. Daubeney PEF, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, et al. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation. (2006) 114:2671–8. doi: 10.1161/CIRCULATIONAHA.106.635128
22. Alexander PMA, Daubeney PEF, Nugent AW, Lee KJ, Turner C, Colan SD, et al. Long-term outcomes of dilated cardiomyopathy diagnosed during childhood: results from a national population-based study of childhood cardiomyopathy. Circulation. (2013) 128:2039–46. doi: 10.1161/CIRCULATIONAHA.113.002767
23. Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the pediatric cardiomyopathy registry. Circulation. (2007) 115:773–81. doi: 10.1161/CIRCULATIONAHA.106.621185
24. Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Colan SD, Cheung M, et al. Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation. (2005) 112:1332–8. doi: 10.1161/CIRCULATIONAHA.104.530303
25. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. (2008) 29:270–6. doi: 10.1093/eurheartj/ehm342
26. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an american heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and function. Circulation. (2006) 113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287
27. Konta L, Franklin RCG, Kaski JP. Nomenclature and systems of classification for cardiomyopathy in children. Cardiol Young. (2015) 25:31–42. doi: 10.1017/S1047951115001201
28. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. (2017) 121:731–48. doi: 10.1161/CIRCRESAHA.116.309396
29. Bondue A, Arbustini E, Bianco A, Ciccarelli M, Dawson D, De Rosa M, et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: scientific update from the working group of myocardial function of the european society of cardiology. Cardiovasc Res. (2018) 114:1287–303. doi: 10.1093/cvr/cvy122
30. Nugent AW, Davis AM, Kleinert S, Wilkinson JL, Weintraub RG. Clinical, electrocardiographic, and histologic correlations in children with dilated cardiomyopathy. J Heart Lung Transplant. (2001) 20:1152–7. doi: 10.1016/S1053-2498(01)00334-5
31. Belkaya S, Kontorovich AR, Byun M, Mulero-Navarro S, Bajolle F, Cobat A, et al. Autosomal recessive cardiomyopathy presenting as acute myocarditis. J Am Coll Cardiol. (2017) 69:1653–65. doi: 10.1016/j.jacc.2017.01.043
32. Campuzano O, Fernández-Falgueras A, Sarquella-Brugada G, Sanchez O, Cesar S, Mademont I, et al. A genetically vulnerable myocardium may predispose to myocarditis. J Am Coll Cardiol. (2015) 66:2913–4. doi: 10.1016/j.jacc.2015.10.049
33. Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. Eur Heart J. (2007) 28:3076–93. doi: 10.1093/eurheartj/ehm456
34. Canter CE, Simpson KP. Diagnosis and treatment of myocarditis in children in the current era. Circulation. (2014) 129:115–28. doi: 10.1161/CIRCULATIONAHA.113.001372
35. Shamszad P, Hall M, Rossano JW, Denfield SW, Knudson JD, Penny DJ, et al. Characteristics and outcomes of heart failure-related intensive care unit admissions in children with cardiomyopathy. J Card Fail. (2013) 19:672–7. doi: 10.1016/j.cardfail.2013.08.006
36. Hsu DT, Lamour JM, Canter CE. Heart diseases leading to pediatric transplantation: cardiomyopathies and congenital heart diseases. Pediatr Heart Transplant. ISHLT Monogr Series 2. Addison, TX: Int Soc Heart Lung Transp. (2008) 2.
37. Everitt MD, Sleeper LA, Lu M, Canter CE, Pahl E, Wilkinson JD, et al. Recovery of echocardiographic function in children with idiopathic dilated cardiomyopathy: results from the pediatric cardiomyopathy registry. J Am Coll Cardiol. (2014) 63:1405–13. doi: 10.1016/j.jacc.2013.11.059
38. den Boer SL, Rizopoulos D, du Marchie Sarvaas GJ, Backx APCM, ten Harkel ADJ, van Iperen GG, et al. Usefulness of serial N-terminal pro-B-type natriuretic peptide measurements to predict cardiac death in acute and chronic dilated cardiomyopathy in children. Am J Cardiol. (2016) 118:1723–9. doi: 10.1016/j.amjcard.2016.08.053
39. Pahl E, Sleeper LA, Canter CE, Hsu DT, Lu M, Webber SA, et al. Incidence of and risk factors for sudden cardiac death in children with dilated cardiomyopathy: a report from the pediatric cardiomyopathy registry. J Am Coll Cardiol. (2012) 59:607–15. doi: 10.1016/j.jacc.2011.10.878
40. Singh RK, Canter CE, Shi L, Colan SD, Dodd DA, Everitt MD, et al. Survival without cardiac transplantation among children with dilated cardiomyopathy. J Am Coll Cardiol. (2017) 70:2663–73. doi: 10.1016/j.jacc.2017.09.1089
41. Marston NA, Han L, Olivotto I, Day SM, Ashley EA, Michels M, et al. Clinical characteristics and outcomes in childhood-onset hypertrophic cardiomyopathy. Eur Heart J. (2021) 42:1988–96. doi: 10.1093/eurheartj/ehab148
42. Wilkinson JD, Lowe AM, Salbert BA, Sleeper LA, Colan SD, Cox GF, et al. Outcomes in children with noonan syndrome and hypertrophic cardiomyopathy: a study from the pediatric cardiomyopathy registry. Am Heart J. (2012) 164:442–8. doi: 10.1016/j.ahj.2012.04.018
43. Norrish G, Cantarutti N, Pissaridou E, Ridout DA, Limongelli G, Elliott PM, et al. Risk factors for sudden cardiac death in childhood hypertrophic cardiomyopathy: a systematic review and meta-analysis. Eur J Prev Cardiol. (2017) 24:1220–30. doi: 10.1177/2047487317702519
44. Norrish G, Ding T, Field E, Ziółkowska L, Olivotto I, Limongelli G, et al. Development of a novel risk prediction model for sudden cardiac death in childhood hypertrophic cardiomyopathy (HCM risk-kids). JAMA Cardiol. (2019) 4:918–27. doi: 10.1001/jamacardio.2019.2861
45. Miron A, Lafreniere-Roula M, Steve Fan CP, Armstrong KR, Dragulescu A, Papaz T, et al. A validated model for sudden cardiac death risk prediction in pediatric hypertrophic cardiomyopathy. Circulation. (2020) 142:217–29. doi: 10.1161/CIRCULATIONAHA.120.047235
46. Balaji S, DiLorenzo MP, Fish FA, Etheridge SP, Aziz PF, Russell MW, et al. Risk factors for lethal arrhythmic events in children and adolescents with hypertrophic cardiomyopathy and an implantable defibrillator: an international multicenter study. Hear Rhythm. (2019) 16:1462–7. doi: 10.1016/j.hrthm.2019.04.040
47. Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, et al. Epidemiology of hypertrophic cardiomyopathy–related death. Circulation. (2000) 102:858–64. doi: 10.1161/01.CIR.102.8.858
48. Kaski JP, Syrris P, Burch M, Tomé Esteban MT, Fenton M, Christiansen M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. (2008) 94:1478–84. doi: 10.1136/hrt.2007.134684
49. Wittekind SG, Ryan TD, Gao Z, Zafar F, Czosek RJ, Chin CW, et al. Contemporary outcomes of pediatric restrictive cardiomyopathy: a single-center experience. Pediatr Cardiol. (2019) 40:694–704. doi: 10.1007/s00246-018-2043-0
50. Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. (1997) 336:267–76. doi: 10.1056/NEJM199701233360407
51. Rivenes SM, Kearney DL, Smith EOB, Towbin JA, Denfield SW. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation. (2000) 102:876–82. doi: 10.1161/01.CIR.102.8.876
52. Walsh MA, Grenier MA, Jefferies JL, Towbin JA, Lorts A, Czosek RJ. Conduction abnormalities in pediatric patients with restrictive cardiomyopathy. Circ Heart Fail. (2012) 5:267–73. doi: 10.1161/CIRCHEARTFAILURE.111.964395
53. Russo LM, Webber SA. Idiopathic restrictive cardiomyopathy in children. Heart. (2005) 91:1199–202. doi: 10.1136/hrt.2004.043869
54. Anderson HN, Cetta F, Driscoll DJ, Olson TM, Ackerman MJ, Johnson JN. Idiopathic restrictive cardiomyopathy in children and young adults. Am J Cardiol. (2018) 121:1266–70. doi: 10.1016/j.amjcard.2018.01.045
55. Weir-McCall JR, Yeap PM, Papagiorcopulo C, Fitzgerald K, Gandy SJ, Lambert M, et al. Left ventricular noncompaction: anatomical phenotype or distinct cardiomyopathy? J Am Coll Cardiol. (2016) 68:2157–65. doi: 10.1016/j.jacc.2016.08.054
56. van Waning JI, Caliskan K, Hoedemaekers YM, van Spaendonck-Zwarts KY, Baas AF, Boekholdt SM, et al. Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol. (2018) 71:711–22. doi: 10.1016/j.jacc.2017.12.019
57. Brescia ST, Rossano JW, Pignatelli R, Jefferies JL, Price JF, Decker JA, et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation. (2013) 127:2202–8. doi: 10.1161/CIRCULATIONAHA.113.002511
58. McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res. (2017) 121:722–30. doi: 10.1161/CIRCRESAHA.117.309711
59. Zimmerman MS, Smith AGC, Sable CA, Echko MM, Wilner LB, Olsen HE, et al. Global, regional, and national burden of congenital heart disease, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet Child Adolesc Heal. (2020) 4:185–200. doi: 10.1016/S2352-4642(19)30402-X
Keywords: cardiomyopathy, paediatric, epidemiology, long-term outcomes, risk factors, sudden cardiac death, heart transplantation
Citation: Rath A and Weintraub R (2021) Overview of Cardiomyopathies in Childhood. Front. Pediatr. 9:708732. doi: 10.3389/fped.2021.708732
Received: 12 May 2021; Accepted: 08 June 2021;
Published: 23 July 2021.
Edited by:
Juan Pablo Kaski, University College London, United KingdomReviewed by:
Gabrielle Norrish, Great Ormond Street Hospital Children's Charity, United KingdomAntigoni Eleni Tsatsopoulou, General Paediatrics Private Clinics, Greece
Copyright © 2021 Rath and Weintraub. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Weintraub, robert.weintraub@rch.org.au