Andersen–Tawil Syndrome With Novel Mutation in KCNJ2: Case Report
 Jisook Yim
Jisook Yim Kyoung Bo Kim
Kyoung Bo Kim Minsun Kim1
Minsun Kim1  Myungshin Kim
Myungshin Kim- 1Department of Laboratory Medicine, College of Medicine, The Catholic University of Korea, Seoul, South Korea
- 2Catholic Genetic Laboratory Center, Seoul St. Mary's Hospital, College of Medicine, The Catholic University of Korea, Seoul, South Korea
- 3Department of Laboratory Medicine, Keimyung University School of Medicine, Daegu, South Korea
Andersen–Tawil syndrome (ATS) is a rare autosomal dominant disorder characterized by a classic symptom triad: periodic paralysis, ventricular arrhythmias associated with prolonged QT interval, and dysmorphic skeletal and facial features. Pathogenic variants of the inwardly rectifying potassium channel subfamily J member 2 (KCNJ2) gene have been linked to the ATS. Herein, we report a novel KCNJ2 causative variant in a proband and her father showing different ATS-associated symptoms. A 15-year-old girl was referred because of episodic weakness and periodic paralysis in both legs for 2–3 months. The symptoms occurred either when she was tired or after strenuous exercise. These attacks made walking or climbing stairs difficult and lasted from one to several days. She had a short stature (142 cm, <3rd percentile) and weighed 40 kg. The proband also showed orbital hypertelorism, dental crowding, mandibular hypoplasia, fifth-digit clinodactyly, and small hands. Scoliosis in the thoracolumbar region was detected by chest X-ray. Since she was 7 years old, she had been treated for arrhythmia-associated long QT interval and underwent periodic echocardiography. Brain MRI revealed cerebrovascular abnormalities indicating absence or hypoplasia of bilateral internal carotid arteries, and compensation of other collateral vessels was observed. There were no specific findings related to intellectual development. The proband's father also had a history of periodic paralysis similar to the proband. He did not show any cardiac symptoms. Interestingly, he was diagnosed with hyperthyroidism during an evaluation for paralytic symptoms. Clinical exome sequencing revealed a novel heterozygous missense variant: Chr17(GRCh37):g.68171593A>T, NM_000891.2:c.413A>T, p.(Glu138Val) in KCNJ2 in the proband and the proband's father.
Introduction
Andersen–Tawil syndrome (ATS; MIM 170390) is a rare autosomal dominant disorder characterized by a classic triad of recurrent flaccid muscle weakness (periodic paralysis), cardiac arrhythmias, and distinctive skeletal and facial features (1). Since the first ATS case presenting with muscle weakness, extrasystoles, and multiple developmental anomalies was reported (2), ATS prevalence is estimated at 0.8–1 in 1,000,000 (3, 4). However, the actual prevalence is expected to be higher than previously reported because the reported prevalence is from a specific population and subgroup of ATS patients (5) and ATS is a rare disease with a broad phenotypic heterogeneity that is difficult to diagnose. Affected individuals present with episodes of periodic paralysis or cardiac symptoms in their first or second decade (6); however, highly variable phenotypes and incomplete penetrance make diagnosis challenging. Cardiac arrhythmias include ventricular arrhythmias associated with prolonged QT interval and prominent U waves. Skeletal and facial features include low-set ears, broad forehead, ocular hypertelorism, small mandible, dental abnormalities, fifth digit clinodactyly, syndactyly, short stature, and scoliosis (7). The KCNJ2 and KCNJ5 are causative genes of ATS. KCNJ2 on chromosome 17q24.3 encodes an inward rectifier potassium channel protein (Kir2.1), which plays an important role in setting and stabilizing the resting membrane potential primarily in the skeletal muscles, heart, and brain (8). KCNJ5 (11q24.3) encodes a G-protein-activated inward rectifier potassium channel 4 protein (Kir3.4) that induces an inhibitory effect on the inward rectifier potassium current (9). Therefore, the diagnosis of ATS is established in individuals with two or more symptoms of the classic triad or individuals with one of the classic triad and at least one family member with ATS and/or identification of pathogenic variants in KCNJ2 and KCNJ5. Here, we report a family with ATS with a novel causative missense variant in KCNJ2 c.413A>T, p.(Glu138Val) and compare their phenotypes along with a literature review.
Case Report
The proband is a 15-year-old girl who presented with recurrent episodes of leg weakness and paralysis lasting several days beginning 2–3 months prior. These episodes recently became more severe and frequent, occurring at least twice a week. The severity of the weakness during the episodes varied from mild (weakness and difficulty climbing stairs) to severe (inability to walk). Triggers were intensive exercise or feeling tired. Clinical evaluation included medical and family history and previous medical record review where applicable. She had been taking flecainide for an arrhythmia associated with a long QT interval since she was 7 years old and had been receiving regular echocardiogram (ECG) follow-up. Her serum potassium levels tested during hospitalization were normal. Thyroid function test results were also within the reference interval. Electromyography (EMG) showed no specific findings. Brain magnetic resonance imaging (MRI) revealed suspicious congenital absence or hypoplasia of both internal carotid arteries (ICA) with supply to the anterior circulation via carotid-vertebrobasilar anastomoses and hypertrophy of the posterior communicating artery (PCOM). No intellectual disability was observed during development. Chest X-rays showed mild scoliosis in the thoracolumbar region. During physical examination, she was 142 cm tall (<3rd percentile) and weighed 40 kg (3rd percentile). Additional physical examination during genetic counseling revealed that the proband had dysmorphic features, including orbital hypertelorism, mandibular hypoplasia, dental crowding, clinodactyly of the fifth finger, and small hands.
Family history revealed that her 37-year-old father had similar episodes that began during adolescence. He was hospitalized for three quadriplegia attacks, and he took potassium supplement 600 mg once or twice a year when he experienced generalized weakness attacks. His potassium levels were low (lowest: 2.2 mmol/L) during his acute episodes. His medical history included transient hyperthyroidism but no cardiac symptoms. Since age 25, he had not experienced a paralysis severe enough to be hospitalized but continued to have periodic paralysis once every few years. His paralysis events were mild and improved without going to the hospital by taking a potassium supplement, applying hot compresses, and receiving massages. However, from March to May 2020, at the age of 35, he was hospitalized almost every 2 weeks with severe general paralysis during which he could not even move his fingers. His blood thyroid function test indicated hyperthyroidism. After taking thyroid medication for 2–3 months, he underwent three thyroid function tests, all of which were within the normal range. He remained well without any uncomfortable symptoms after that. However, detailed records (i.e., thyroid hormone levels) were not obtained as data by the hospital at that time. His height and weight were 163.5 cm (≈3rd percentile) and 51.25 kg, respectively. Hypertelorism, small mandible, tooth crowding, and small hands were also observed. Table 1 provides a summary of the clinical characteristics of our patients.
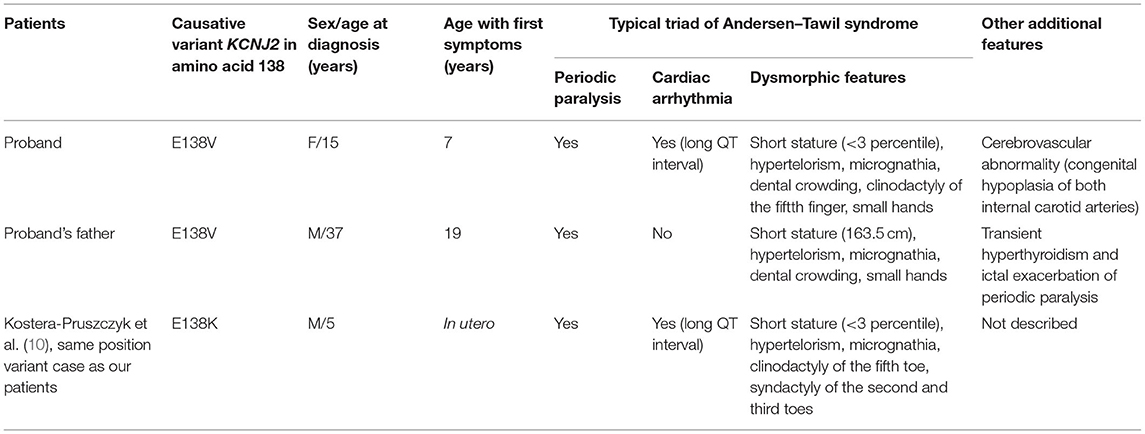
Table 1. Clinical details of ATS patients with pathogenic/likely pathogenic variants in Kir2.1 at position 138.
Genetic Testing
Genomic DNA was extracted from peripheral blood samples using the QIAsymphony DSP DNA mini kit (Qiagen, Hilden, Germany). Sequencing was performed using a TruSight One Expanded sequencing panel (Illumina Inc., San Diego, CA, USA) consisting of 6,700 genes associated with known Mendelian genetic disorders on a NextSeq 550 (Illumina, CA, USA). The obtained sequence reads were aligned to the human reference genome sequence 19 (hg19) from University of California, Santa Cruz (UCSC) [original GRCh37 from National Center for Biotechnology Information (NCBI), February 2009] using Burrows–Wheeler Aligner (BWA version 0.7.7-isis-1.0.0). The BAM files were then processed by base quality recalibration, duplicate removal, and local realignment following the Genome Analysis Toolkit (GATK version 1.6-23-gf0210b3) for variant calling. Annotation and filtering were performed with Illumina Annotation Service (IAS version 3). Clinical exome sequencing revealed a novel causative heterozygous missense Chr17(GRCh37):g.68171593A>T, NM_000891.2:c.413A>T, p.(Glu138Val) variant in KCNJ2, which was thought to be causal of ATS syndrome. This domain (H5, intramembrane pore loop) is highly preserved, and the mutation is predicted to be damaging by the protein function prediction software SIFT and PolyPhen-2. It was not present in the general population databases such as the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/), the Single Nucleotide Polymorphism Database (dbSNP; https://www.ncbi.nlm.nih.gov/snp/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php), a public archive of the relationship between human variants and phenotypes, and the Korean Reference Genome Database (KRGDB; http://coda.nih.go.kr/coda/KRGDB/index.jsp) as an ethnic-specific Korean population database. We could not find the variant in our case in any previous reports. The identified variants were assessed according to the standards and guidelines for the interpretation of sequence variants in Mendelian disorders of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) (11), with PM2 (absent from population database), PM5 (novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before), PP2 (missense variants are a common mechanism of disease), PP3 (pathogenic computational evidence), and PP4 (phenotype specific for gene) relevant evidence. Therefore, we classified the variant as likely pathogenic. It was validated by Sanger sequencing (Figure 1A) and then confirmed diagnosed as ATS. Because a causative variant was identified in the proband, Sanger sequencing of the causative variant was also performed on her symptomatic father, and the same variant that was consistent with his history of periodic paralysis were identified (Figure 1B). Figure 1C shows the Sanger sequencing result of an unaffected control on this position. No other pathogenic variants in genes associated with other periodic paralysis disorders (CACNA1S, SCN4A, or KCNJ18) were identified in the proband. The father's parents and two brothers were asymptomatic until now. Although the proband's 11-year-old brother and 4-year-old sister had no symptoms, genetic testing was recommended to all at-risk family members during genetic counseling.
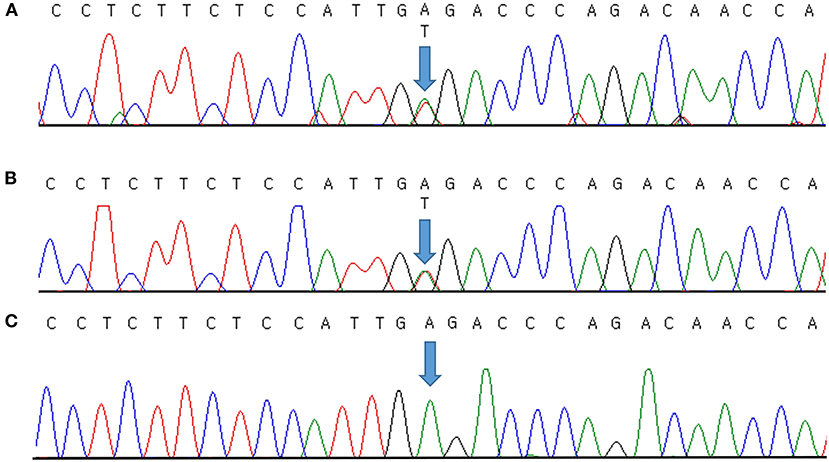
Figure 1. Molecular characterization of a family with Andersen–Tawil syndrome. Sequence chromatogram showing the position of the c.413A>T transversion (NM_000891.2) that led to the p.(Glu138Val) missense variant. The variant was heterozygous in the proband (A) and her father (B). Sanger sequencing result of an unaffected control (C). Variants are annotated according to The Human Genome Variation Society nomenclature.
Discussion
In this case report, we identified two ATS patients in a family with the novel KCNJ2 c.413A>T variant. Our proband had all triad symptoms, while her father had only two symptoms and no cardiac symptoms. It is very challenging to diagnose ATS early due to the rarity and high degree of phenotypic variability. Fifty-eight to seventy-eight percent of ATS patients present with all three symptoms (12). Moreover, patients can present with only one of the triad symptoms (5). Dysmorphic features are usually subtle in ATS patients (13), as shown in our patients (we recognized retrospectively), and are easy to overlook unless suspected of ATS. Therefore, ATS should always be considered even in patients with only periodic paralysis or cardiac arrhythmias.
Approximately 60% of ATS cases have causal variants in KCNJ2 (6, 14). Most KCNJ2 pathogenic variants in ATS are missense variants (1, 6) that result in loss of function of the Kir2.1 channel and is expressed in skeletal muscle, heart, and bone. This channel plays a role in prolonged depolarization of the action potential, and thereby cause periodic paralysis and cardiac arrhythmia (14). Additionally, KCNJ2 variants can affect skeletal dysmorphic features because KCNJ2 is expressed during the early stages of craniofacial development in Xenopus and mice (15). However, as reports by variable penetrance of ATS cases suggest, a subset of individuals with pathogenic variants do not exhibit any associated symptoms (16–18). A subset of KCNJ2 pathogenic variants that cause ATS is identified at the phosphatidylinositol-4,5-biphosphate (PIP2) binding site, a known regulator of the Kir2.1 channel. Another subset of KCNJ2 pathogenic variants is found in the pore-forming loop of Kir2.1. In addition, pathogenic variants in regions such as slide helix have also been reported (5, 19–21), suggesting various mechanisms. The p.(Glu138Val) variant in our patients was located in the pore region of the Kir2.1 channel. Another study showed the same position but other missense change (c.412G>A, p.E138K) as our case variant (c.413A>T, p.E138V) (10). Although not able to perform physiological studies to identify the nature of the alteration in channel function, other studies showed that this subunit pathogenic variant was unable to form functional homomultimeric channels, abolishing channel function (10, 18). And Yang et al. also described that position 138, a negatively charged residue in the pore loop of Kir2.1, had a strong effect on the stability of the pore loop (22, 23). Additional in silico analyses predicted deleterious (SIFT score: 0.05), probably damaging (polyphen-2 score: 1.000), pathogenic (REVEL score: 0.972), and disease-causing (MutationTaster) effects; thus, this variant may cause complete loss of channel function.
We found additional features in our patients. The proband had congenital hypoplasia of the ICA, identified by brain MRI. Although we did not find any reports that also reported vascular abnormalities in ATS, central nervous system involvement including mild learning disability was frequently observed in ATS patients. Because potassium channels contribute to angiogenesis in cancer (24), it is worth investigating a possible association between cerebrovascular development and KCNJ2 variant. Meanwhile, the proband's father had a history of transient hyperthyroidism with exacerbated periodic paralysis that was consistent with previous studies' findings. They demonstrated that the paralysis symptoms were aggravated in patients with ATS secondary to thyrotoxicosis or co-existence of both disorders (25, 26). The heterogeneous phenotypes between the proband and her father demonstrated the weak genotype–phenotype correlation in ATS. This is further evidenced by a previously reported case with the missense variant of same position 138 who had earlier onset of more severe periodic paralysis (1 year old) and arrhythmia (in utero) than our proband (7 years old).
In summary, we identified a novel causative variant in KCNJ2 in ATS patients and provided detailed clinical findings to expand the genotype–phenotype correlation. The heterogeneous phenotype of ATS should alert physicians to perform genetic analysis for ATS even in patients with only one or two among the typical triad symptoms.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Seoul St. Mary's Hospital Institutional Review Board. Written informed consent for participation was not provided by the participants' legal guardians/next of kin: Because the patient did not return to our hospital, written consent could not be obtained. However, it has been approved by the Ethics Committee of our hospital, and the patient's identification information is not included in this case.
Author Contributions
JY and MyK wrote the manuscript. JY, KK, MiK, and GL conducted genetic analyses. All authors contributed to the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Veerapandiyan A, Statland JM, Tawil R. Andersen-Tawil syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. GeneReviews. Seattle (WA) (1993). Available at: http://www.ncbi.nlm.nih.gov/books/nbk1264/ (Last updated 2018).
2. Andersen ED, Krasilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome? Acta Paediatr Scand. (1971) 60:559–64. doi: 10.1111/j.1651-2227.1971.tb06990.x
3. Stunnenberg BC, Raaphorst J, Deenen JCW, Links TP, Wilde AA, Verbove DJ, et al. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul Disord. (2018) 28:402–7. doi: 10.1016/j.nmd.2018.03.006
4. Horga A, Raja Rayan DL, Matthews E, Sud R, Fialho D, Durran SC, et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. (2013) 80:1472–5. doi: 10.1212/WNL.0b013e31828cf8d0
5. Nguyen HL, Pieper GH, Wilders R. Andersen-Tawil syndrome: clinical and molecular aspects. Int J Cardiol. (2013) 170:1–16. doi: 10.1016/j.ijcard.2013.10.010
6. Perez-Riera AR, Barbosa-Barros R, Samesina N, Pastore CA, Scanavacca M, Daminello-Raimundo R, et al. Andersen-Tawil syndrome: a comprehensive review. Cardiol Rev. (2021) 29:165–77. doi: 10.1097/CRD.0000000000000326
7. Yoon G, Oberoi S, Tristani-Firouzi M, Etheridge SP, Quitania L, Kramer JH, et al. Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am J Med Genet A. (2006) 140:312–21. doi: 10.1002/ajmg.a.31092
8. Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, et al. Mutations in Kir21 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. (2001) 105:511–9. doi: 10.1016/S0092-8674(01)00342-7
9. Kokunai Y, Nakata T, Furuta M, Sakata S, Kimura H, Aiba T, et al. A Kir34 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir21. Neurology. (2014) 82:1058–64. doi: 10.1212/WNL.0000000000000239
10. Kostera-Pruszczyk A, Potulska-Chromik A, Pruszczyk P, Bieganowska K, Miszczak-Knecht M, Bienias P, et al. Andersen-Tawil syndrome: report of 3 novel mutations and high risk of symptomatic cardiac involvement. Muscle Nerve. (2015) 51:192–6. doi: 10.1002/mus.24293
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Statland JM, Fontaine B, Hanna MG, Johnson NE, Kissel JT, Sansone VA, et al. Review of the diagnosis and treatment of periodic paralysis. Muscle Nerve. (2018) 57:522–30. doi: 10.1002/mus.26009
13. Almuqbil M, Srour M. Child neurology: Andersen-Tawil syndrome. Neurology. (2015) 84:e78–80. doi: 10.1212/WNL.0000000000001377
14. Maggi L, Bonanno S, Altamura C, Desaphy JF. Ion channel gene mutations causing skeletal muscle disorders: pathomechanisms and opportunities for therapy. Cells. (2021) 10:1521. doi: 10.3390/cells10061521
15. Adams DS, Uzel SG, Akagi J, Wlodkowic D, Andreeva V, Yelick PC, et al. Bioelectric signalling via potassium channels: a mechanism for craniofacial dysmorphogenesis in KCNJ2-associated Andersen-Tawil Syndrome. J Physiol. (2016) 594:3245–70. doi: 10.1113/JP271930
16. Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. (2002) 71:663–8. doi: 10.1086/342360
17. Deeb R, Veerapandiyan A, Tawil R, Treidler S. Variable penetrance of Andersen-Tawil syndrome in a family with a rare missense KCNJ2 mutation. Neurol Genet. (2018) 4:e284. doi: 10.1212/NXG.0000000000000284
18. Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest. (2002) 110:381–8. doi: 10.1172/JCI15183
19. Donaldson MR, Jensen JL, Tristani-Firouzi M, Tawil R, Bendahhou S, Suarez WA, et al. PIP2 binding residues of Kir21 are common targets of mutations causing Andersen syndrome. Neurology. (2003) 60:1811–6. doi: 10.1212/01.WNL.0000072261.14060.47
20. Bendahhou S, Donaldson MR, Plaster NM, Tristani-Firouzi M, Fu YH. Ptacek LJ. Defective potassium channel Kir21 trafficking underlies Andersen-Tawil syndrome. J Biol Chem. (2003) 278:51779–85. doi: 10.1074/jbc.M310278200
21. Barron-Diaz DR, Totomoch-Serra A, Escobar-Cedillo RE, Garcia-Gutierrez A, Reyes-Quintero AE, Villegas Daviran SE, et al. Andersen-Tawil syndrome with high risk of sudden cardiac death in four mexican patients. Cardiac and extra-cardiac phenotypes. Rev Invest Clin. (2021) 73::145–53. doi: 10.24875/RIC.20000310
22. Zangerl-Plessl EM, Qile M, Bloothooft M, Stary-Weinzinger A, van der Heyden MAG. Disease associated mutations in KIR proteins linked to aberrant inward rectifier channel trafficking. Biomolecules. (2019) 9:650. doi: 10.3390/biom9110650
23. Yang J, Yu M, Jan YN, Jan LY. Stabilization of ion selectivity filter by pore loop ion pairs in an inwardly rectifying potassium channel. Proc Natl Acad Sci USA. (1997) 94:1568–72. doi: 10.1073/pnas.94.4.1568
24. Umaru B, Pyriochou A, Kotsikoris V, Papapetropoulos A, Topouzis S. ATP-sensitive potassium channel activation induces angiogenesis in vitro and in vivo. J Pharmacol Exp Ther. (2015) 354:79–87. doi: 10.1124/jpet.114.222000
25. Diaz-Manera J, Querol L, Alejaldre A, Rojas-Garcia R, Ramos-Fransi A, Gallardo E, et al. Severe exacerbation of Andersen-Tawil syndrome secondary to thyrotoxicosis. J Hum Genet. (2014) 59:465–6. doi: 10.1038/jhg.2014.43
Keywords: KCNJ2 gene, KIR2.1, periodic paralysis, long QT syndrome, potassium channel, genetic disorder, dysmorphic feature, Andersen–Tawil syndrome
Citation: Yim J, Kim KB, Kim M, Lee GD and Kim M (2022) Andersen–Tawil Syndrome With Novel Mutation in KCNJ2: Case Report. Front. Pediatr. 9:790075. doi: 10.3389/fped.2021.790075
Received: 06 October 2021; Accepted: 22 November 2021;
Published: 31 January 2022.
Edited by:
Natália Duarte Linhares, Genuity Science, IrelandReviewed by:
Aleksandra Jezela-Stanek, National Institute of Tuberculosis and Lung Diseases, PolandRonald Wilders, University of Amsterdam, Netherlands
Copyright © 2022 Yim, Kim, Kim, Lee and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Myungshin Kim, microkim@catholic.ac.kr