Genetically Modified Mouse Models of Congenital Diaphragmatic Hernia: Opportunities and Limitations for Studying Altered Lung Development
 Florian Friedmacher
Florian Friedmacher Udo Rolle
Udo Rolle Prem Puri
Prem Puri- 1Department of Pediatric Surgery, University Hospital Frankfurt, Goethe University Frankfurt, Frankfurt, Germany
- 2Beacon Hospital, University College Dublin, Dublin, Ireland
- 3Conway Institute of Biomolecular and Biomedical Research, School of Medicine, University College Dublin, Dublin, Ireland
Congenital diaphragmatic hernia (CDH) is a relatively common and life-threatening birth defect, characterized by an abnormal opening in the primordial diaphragm that interferes with normal lung development. As a result, CDH is accompanied by immature and hypoplastic lungs, being the leading cause of morbidity and mortality in patients with this condition. In recent decades, various animal models have contributed novel insights into the pathogenic mechanisms underlying CDH and associated pulmonary hypoplasia. In particular, the generation of genetically modified mouse models, which show both diaphragm and lung abnormalities, has resulted in the discovery of multiple genes and signaling pathways involved in the pathogenesis of CDH. This article aims to offer an up-to-date overview on CDH-implicated transcription factors, molecules regulating cell migration and signal transduction as well as components contributing to the formation of extracellular matrix, whilst also discussing the significance of these genetic models for studying altered lung development with regard to the human situation.
Introduction
Congenital diaphragmatic hernia (CDH) represents a relatively common and life-threatening birth defect with an estimated global prevalence of 2.3 in 10,000 live births (1, 2). It is characterized by incomplete formation and/or muscularization of the primordial diaphragm, which allows herniation of abdominal viscera into the thoracic cavity, thereby filling space usually reserved to hold the growing lung (3, 4). Hence, pulmonary development is disrupted, leading to immature and hypoplastic lungs (5–7). Today, more than 70% of CDH cases are diagnosed prenatally based on maternal-fetal ultrasound or magnetic resonance imaging in the second trimester of pregnancy, thus potentially altering future outcome (8, 9). Depending on the extent of pulmonary hypoplasia, newborns with CDH often present with severe respiratory distress at birth, requiring immediate and complex treatment (10, 11). Although significant advances have been achieved in postnatal resuscitation and ventilation strategies over the past decades (12, 13), CDH continues to be one of the major challenges in neonatal intensive care with mortality rates ranging between 30 and 50% (14–16). Surgical repair of CDH is generally performed after clinical stabilization either by primary closure or in larger defects by reconstruction using a prosthetic patch or muscle flap (17, 18). While newer therapeutic measures such as gentle ventilation techniques, high-frequency oscillation and extracorporeal membrane oxygenation have improved overall survival rates (19–21), this has led to substantial long-term morbidity in CDH patients (22, 23), including chronic lung disease, gastroesophageal reflux, scoliosis, sensorineural hearing loss and neurodevelopmental deficits (24–26).
A defect in the posterolateral diaphragm (also referred to as Bochdalek hernia) is the most common type of CDH and comprises approximately 80–90% of all cases, with the majority being left-sided (85%), less often right-sided (10%), or bilaterally (<5%) (27). The dual-hit hypothesis explains CDH-associated pulmonary hypoplasia by an initial disruption in bilateral lung organogenesis before diaphragm closure, in combination with a second ipsilateral insult resulting from the intrathoracic herniation and subsequent restriction of fetal breathing movements (28). Typical features of pulmonary hypoplasia in CDH are structural immaturity and smaller lung volume with a significantly reduced number of terminal airways, disrupted alveologenesis, diminished alveolar airspaces, thickened alveolar walls accompanied by increased interstitial tissue and decreased gas-exchange surface area (29). These findings have indicated that the pulmonary anomalies in CDH are at least partially independent of the diaphragmatic defect, suggesting a potential developmental linkage between both organs at a molecular level. Much of our present knowledge on the morphogenetic lung abnormalities in CDH has derived from experimental animal research (30–33). Because diaphragm and pulmonary evolution is remarkably similar between mice and humans, mouse models represent a crucial aspect in advancing our insight into the pathogenic mechanisms underlying CDH and associated lung hypoplasia.
This article aims to offer a comprehensive overview of genetically modified mouse models of CDH, resultant candidate genes and signaling pathways, whilst also discussing new opportunities and limitations for studying altered lung development in relation to the human situation.
Overview of Genetically Modified Mouse Models of Congenital Diaphragmatic Hernia
A large variety of genetic factors have been found to play key roles during the pathogenesis of CDH and pulmonary hypoplasia. Currently, genetic causes are detected in about 30% of CDH patients (34–36). Through recent advances in genetic engineering technologies, genetically modified mouse models of CDH are now frequently used in basic science research (37), offering several potential genes and signaling pathways involved in the etiology of diaphragmatic defects and allied lung anomalies (Table 1).
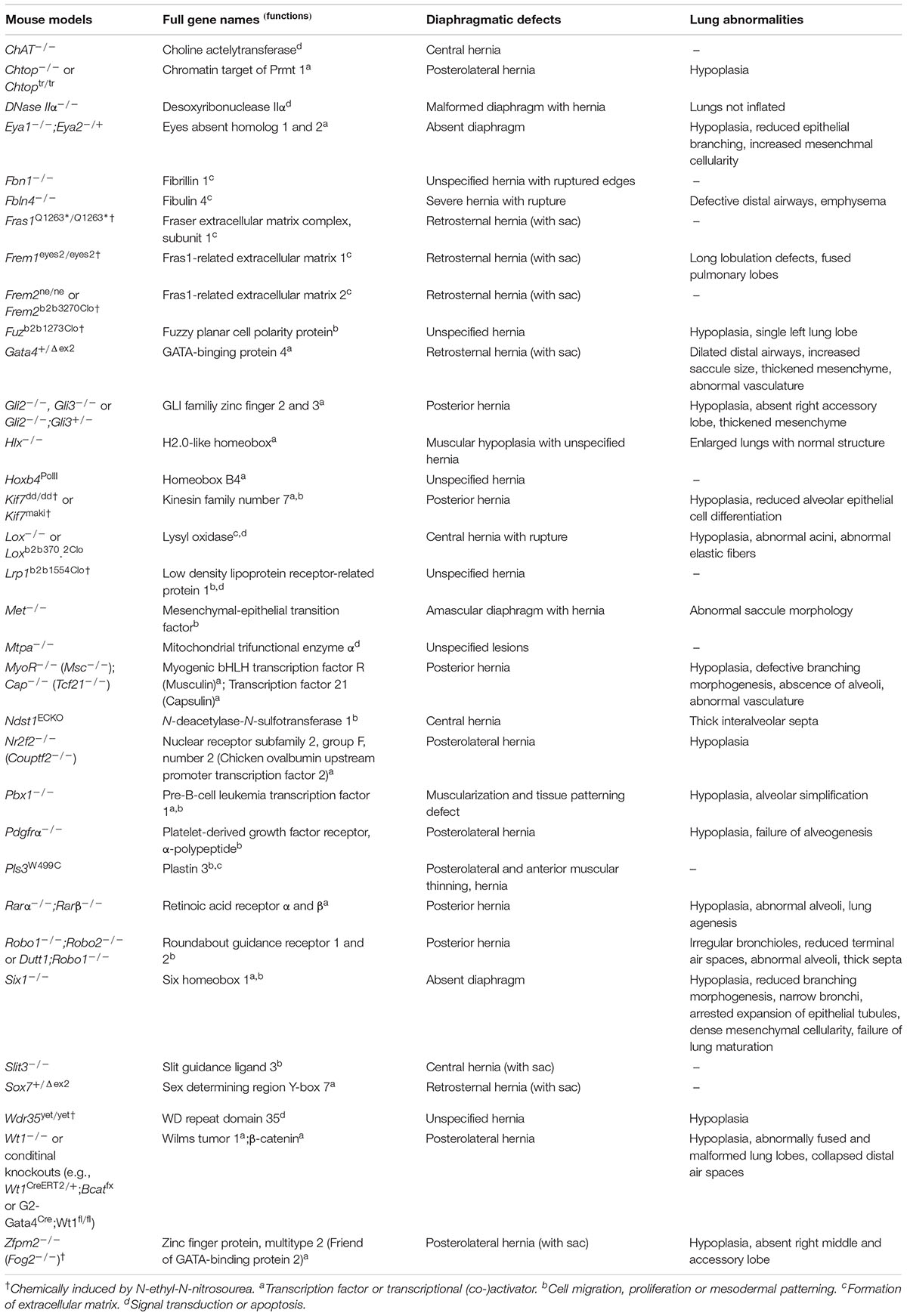
Table 1. Genetically modified mouse models of congenital diaphragmatic hernia.
Transcription Factors and Transcriptional (Co-)Activators
Numerous transcription factors and transcriptional (co-)activators have been suggested in the development of the primordial diaphragm and lungs. Many of them are associated with retinoid or sonic hedgehog signaling pathways.
Retinoid Signaling Pathway
Vitamin A (i.e., retinol) and its derivates (i.e., retinoids) are indispensable for various aspects of early embryogenesis. Over the years, several knockout models have indicated a role of the retinoid signaling pathway and its downstream targets in the pathogenesis of CDH (38). For instance, mice lacking both subtypes of retinoic acid receptors α and β (Rarα and Rarβ) have been demonstrated to generate offspring with posterolateral diaphragmatic defects identical to those observed in human patients (39–44), and similar to the vitamin A-deficient CDH mouse model as previously reported by Anderson (45, 46). Surprisingly, single Rar null mutant mice did not display any of the predicted malformations that were seen in rats with vitamin A deficiency (31). Nevertheless, when the activity of several receptors was inhibited, various deformities were noted including right-sided CDH in Rarα/β2 and left-sided CDH in Rarα/β2+/– animals. Moreover, these mice exhibited severe pulmonary hypoplasia (31). Despite the convincing data, these genetically modified mouse models manifest only a comparatively low incidence of diaphragmatic defects and a high rate of additional comorbidities (e.g., cranial, cardiac, vertebral and limb), which do not accurately depict the human situation (39, 42). Still, mutations in the stimulated by retinoic acid gene 6 (STRA6), a membrane receptor that controls the cellular uptake of vitamin A and cellular retinoic acid binding protein 1 (CRABP1), which is located on chromosome 15, have been found to lead to a spectrum of developmental anomalies including CDH and hyperplastic lungs (47, 48).
Nr2f2 (Couptf2)
Another important gene that is linked with the retinoid signaling pathway is chick ovalbumin upstream promoter transcription factor II (COUP-TFII), a transcription factor that is affiliated with the nuclear steroid/thyroid hormone receptor superfamily, whose DNA-binding site has been shown to reduce the induction of retinoic acid receptors (49–51). COUP-TFII was recently renamed as nuclear receptor subfamily 2 group F (NR2F2), which is expressed in the diaphragm and lungs during early gestation (34). Mapped to chromosome 15q26 in humans, the NR2F2 gene is situated on a recognized CDH hotspot region, thus making it a likely contributor to the etiology of diaphragmatic defects. On the basis of this observation, You et al. (52) have created a tissue-specific Nr2f2–/– mouse model that features left-sided Bochdalek-type CDH and pulmonary hypoplasia similar to the human situation. Through targeted ablation of Nr2f2 in the foregut mesenchyme and pleuroperitoneal folds (PPFs), posterolateral diaphragmatic defects presumably arise because of the failure of the posthepatic mesenchymal plate to merge with the lateral body wall, thus enabling stomach and liver to protrude into the chest (52).
Wt1
The creation of genetic mouse models for various other applications has revealed several genes, which one would not necessarily immediately associate with CDH. Initially introduced as a model for the investigation of early urogenital organogenesis (53), Wilm’s tumor 1 (Wt1) null mutant mice die during mid-gestation, displaying posterolateral diaphragmatic defects and lung hypoplasia alongside urogenital abnormalities (54–56). Heterozygous mutations of the WT1 gene, which encodes a transcription factor that contains four zinc finger motifs, is known to produce distinct syndromes with clinical overlap that include CDH (e.g., Denys-Drash syndrome or Meacham syndrome) (57, 58). Wt1–/–, vitamin A-deficient and nitrofen mouse models of CDH each implied a mutual pathomechanism for the formation of diaphragmatic defects with several analogies to the condition in humans (59). More recently, Carmona et al. (60) and Cleal et al. (61) have reported that conditional deletion of Wt1 in the mesenchyme of the septum transversum can cause CDH in mice. Today, it is proven that Wt1 and Couptf2 both interact with the retinoid signaling pathway during embryonic development (3). Surprisingly, Wt1 and Couptf2 are not found in the muscle precursors but in the non-muscular mesenchymal compartment of the PPFs (3). Paris et al. (62) have developed a novel genetically modified mouse model of CDH, demonstrating that Wt1-induced β-catenin loss-of-function produces posterior diaphragmatic defects, bilateral pulmonary hypoplasia and liver herniation, comparable to the phenotypes associated with CDH in human patients. Additionally, a decreased mesothelial proliferation and increased rate of cell death was identified in the posterior diaphragm mesenchyme, and all mouse pups died postnatally with malformed lung lobes and collapsed distal air spaces (62). Loss of Wt1 has also been associated with lung branching defects before diaphragm closure in another genetic model of CDH (63).
Sonic Hedgehog Signaling Pathway
GLI-Kruppel family member 2 (Gli2) and Gli3 and are both members of a highly conserved morphogenetic family, belonging to the sonic hedgehog (Shh) signaling pathway (64). This pathway is thought to be crucial during normal diaphragmatic development (36). A murine model of the VACTERL-like syndrome (i.e., vertebral, anorectal, cardiac, tracheoesophageal, renal and limb anomalies) created by Kim et al. (65) involved Gli2–/–;Gli3–/– and Gli2–/–;Gli3+/– mice that developed left-sided posterior CDH and pulmonary hypoplasia besides the observed VACTERL components. This was the first experimental model that reproduced the human VACTERL association, indicating that disruptions in Shh signaling might contribute to the pathogenesis of VACTERL syndrome. Likewise, as Gli2, Gli3 and Wt1 all encode important zinc finger proteins, further transcription factors of this type have been hypothesized through the generation of newer genetic animal models of CDH. For example, kinesin family member 7 (Kif7) and pre-B-cell leukemia transcription factor 1 (Pbx1) were recently recognized as indispensable components of the Shh signaling pathway, functioning as regulators during early embryogenesis (66, 67). Kif7 encodes a motor protein that functions downstream of the transmembrane receptor smoothened, and interacts with both Gli2 and Gli3 (68). Furthermore, Kif7 was found to coordinate cell proliferation, central tendon patterning and differentiation of the primordial diaphragm in a genetically modified mouse model of CDH (69). Homozygous Kif7dd/dd mutant mice and Pbx1–/– knockout mice both display left-sided posterior diaphragmatic defects and hypoplastic lungs (36, 69–71). In turn, haploinsufficieny of PBX1 has been associated with various congenital anomalies including CDH (72). Moreover, two predicted variants in the KIF7 gene were recently detected in patients with CDH (73). Additionally, mice lacking chromatin target of protein arginine methyltransferase 1 (Chtop) have numerous developmental abnormalities including posterolateral defects in the diaphragm, pulmonary hypoplasia and liver herniation (74–76). High-resolution 3D imaging further characterized these diaphragmatic defects in Chtop–/– mice embryos (77).
Zfpm2 (Fog2), Gata4 and Sox7
Zinc finger protein 2 (ZFPM2), formerly known as friend of GATA-binding protein 2 (FOG2), encodes another zinc finger-containing protein that regulates the transcriptional activity of GATA4, hereby controlling a number of developmental mechanisms in the forming diaphragm and lung (78–81). In mice, Fog2 was initially found to be expressed in the embryonic septum transversum of the diaphragm (81). In humans, ZFPM2 is located on chromosome 8p23 and has been demonstrated to interact with COUP-TFII (82, 83). However, only a single mutation in the ZFPM2 gene has been identified in isolated patients with non-syndromic CDH to date (31). In a cohort of 275 patients with CDH, Longoni et al. (84) have recently reported the incidence of ZFPM2 mutations to be nearly 5%. In addition, their genetic analysis of a multigenerational family revealed a heritable intragenic ZFPM2 deletion with an approximated penetrance for clinical relevant diaphragmatic defects of around 37.5% (84). On the other side, mice exposed to the chemical mutagen N-ethyl-N-nitrosourea (ENU) generated Fog2–/– offspring with bilateral hypoplastic lungs and a defective posterolateral diaphragm characteristic of CDH (80), while 70% of mice heterozygous for a Gata4 deletion mutation (i.e., Gata4+/Δex2) displayed retrosternal diaphragmatic defects, dilated distal airways and thickened pulmonary mesenchyme (85). Using genetically modified mice, Merrell et al. (86) have shown that Gata4 mosaic mutations in PPF-derived muscle connective tissue fibroblasts led to the development of localized amuscular regions of the diaphragm, which were biomechanically weaker and subsequently caused CDH. GATA4 and ZFPM2 genes have been both found to be absent in humans with CDH (78), emphasizing their roles as possible candidate genes for CDH. Moreover, Zfpm2 is known to interact with Nr2f2, indicating that these two transcription factors together with Gata4 may contribute to diaphragm formation (83). Recurrent microdeletions of 8p23.1, including GATA4 and the sex determining region Y-box 7 (SOX7) gene are accompanied with a significant risk of CDH and cardiovascular anomalies (87). Even though mice lacking the Gata4 gene display both diaphragmatic and cardiac defects, no human patient with cardiac anomalies and GATA4 mutations have been identified with CDH so far (87). However, Wat et al. (87) have recently demonstrated that haploinsufficiency of Sox7 or Gata4 is enough to cause retrosternal diaphragmatic defects in mice and that haploinsufficiency of SOX7 and GATA4 may in turn be involved in the pathogenesis of CDH in patients with 8p23.1 deletions.
Cap (Tcf21) and MyoR (Msc)
Several basic helix-loop-helix transcription factors have been shown to support the development of the primordial diaphragm in mice. Capsulin (Cap) is one of those, which is strongly expressed in the fetal diaphragm and in mesenchymal cells of the lung (88). As one might expect, Cap+/– mice have severe defects in lung morphogenesis and lack alveoli (89). On the other hand, mice homozygous deficient for both cap–/– and the related myogenic bHLH transcription factor R (MyoR–/–) lack facial musculature and exhibit posterior diaphragmatic defects. This double mutant mouse model, which was generated at first to study the formation of facial muscles, displayed not only CDH and defective lung branching morphogenesis but also severe facial muscle abnormalities (90). Although these genetically modified mice died soon after birth because of pulmonary and cardiac malformations, the type of diaphragmatic defect seen in this model indicates that both Cap and MyoR are necessary for the integrity of the developing diaphragm. Previously, these genes were referred to as transcription factor 21 (Tcf21) and musculin (Msc), respectively (90, 91).
Eya1 and Six1
Eyes absent (Eya) genes and the transcription factor sine oculis homebox 1 (Six1) form an important signaling network, which plays a central role during embryonic development (92, 93). Eya1 and Six1 together constitute an evolutionary conserved transcriptional complex that coordinates multiple integrated processes needed for normal growth of the primordial diaphragm and lung (94). Further research work has confirmed that the Eya1-Six1 pathway has a key role in lung maturation by regulating its branching morphogenesis (95). Mice deficient in Eya1–/– and Eya2–/+ have no diaphragm (94), whereas single mutant Eya1 mice die shortly after birth due to respiratory failure, having severely hypoplastic lungs with reduced epithelial branching and increased mesenchymal cellularity (95). In Six1–/– mice, the diaphragm is also absent, and the Six1 deletion causes pulmonary hypoplasia with greatly reduced epithelial branching, narrow bronchi, dense mesenchyme and obvious failure of normal lung maturation (96, 97). These findings indicate that disruption of the Eya1-Six1 signaling pathway may lead to neonatal lethality as a consequence of an absent diaphragm and hypoplastic lungs.
Hlx and Hoxb4
H2.0-like homeobox (Hlx) is a protein coding gene that is relatively conserved across various species (98). This homeobox transcription factor has been found to be highly expressed during early organogenesis in the septum transversum of the diaphragm and lung mesenchyme (99, 100). Hlx–/– mice suffered early demise and showed diaphragmatic defects (101). Additionally, Farrell et al. (102) reported two human fetuses with multiple congenital anomalies including CDH that were homozygous for a missense variant in the HLX gene. The Hox gene family encodes for multiple transcription factors that have crucial regulatory functions during embryonic development (103). Targeted mutation of the homeobox B4 (Hoxb4) gene in mice resulted in offspring with poorly formed diaphragms and diaphragmatic defects, strikingly similar to the phenotype seen in humans with anterior CDH (104).
Molecules Implicated in Cell Migration, Proliferation and Mesodermal Patterning
Various genes and enzymes involved in cell migration, proliferation and mesodermal patterning have been found to be associated with embryonic diaphragm and lung development.
Slit3, Robo1/2, Ndst1 and Pdgfra
The Slit guidance ligand (Slit) family of proteins comprise a group of molecules with crucial functions in cell migration and adhesion through interaction with roundabout (Robo) receptors. Slit genes are expressed in the mesothelium of the diaphragm during embryogenesis (105). Homozygous Slit3–/– mice experience faulty detachment of the central tendon region of the diaphragm from the underlying liver due to connective tissue defects, thus causing central-type (i.e., septum transversum) CDH (105, 106). Therefore, this genetically modified model is facing the disadvantage of having the diaphragmatic defect on or near the ventral midline portion of the central tendon as opposed to the posterolateral diaphragm, thus representing less than 5% of CDH cases seen in human patients. Further malformations in this mouse model include ureteric and renal agenesis in combination with intrathoracic herniation of liver and gallbladder (106), which again occurs infrequently in humans with CDH. Until now, no SLIT3 mutations have been identified in CDH patients. Robo genes encode large transmembrane receptors that are involved together with their ligands in numerus developmental mechanisms (107–109). For example, the Slit-Robo signaling pathway has been reported to have various fundamental functions including axon guidance, neural crest cell migration, epithelial cell adhesion, embryonic heart formation as well as diaphragm and kidney development (105–107, 110–114). Inactivation of Robo1 and Robo2 genes in mice has been shown to cause diaphragmatic defects and subsequent herniation of the stomach into the thorax, which leads to poor lung inflation and perinatal death, similar to human CDH cases (107). Homozygous mice with targeted deletion in the Dutt1/Robo1 gene often die at birth due to respiratory failure, demonstrating delayed lung maturation and diaphragmatic defects in some instances (115). More recent studies identified the heparan sulfate proteoglycan as an essential part of the Slit-Robo signaling complex, which stabilizes the Slit-Robo interaction (116). Furthermore, Zhang et al. (117) have noted that absence of the heparan sulfate biosynthetic enzyme N-deacetylase-N-sulfotransferase-1 (Ndst1) in the mouse endothelium interferes with vascular development in the primordial diaphragm, resulting in hypoxia as well as diaphragmatic hypoplasia and central-type CDH. The observed phenotypes in these animals mirror the congenital anomalies seen in Slit3 knockout mice. In addition, implementation of a heterozygous mutation in the Robo4 gene, which encodes the receptor of Slit3, exacerbated the defect in vascular and diaphragmatic formation (117). Thus, these findings suggest that loss of Ndst1 may lead to abnormal vasculogenesis in the diaphragm and CDH and that heparan sulfate in turn promotes the angiogenic Slit3-Robo4 signaling cascade during normal vascular patterning. Apart from this, mice homozygous for null mutations in the platelet-derived growth factor receptor α (Pdgfra) gene exhibit not only posterolateral diaphragmatic defects, they also develop a spectrum of other comorbidities including cardiovascular anomalies, renal and urogenital malformations, facial clefts, lung hypoplasia and failure of alveogenesis (118, 119).
Fuz, Met and Pls3
Inbred C57BL/6J mice chemically mutagenized with ENU displayed a previously unknown mutation in the fuzzy planar cell polarity protein (Fuz) that was associated with CDH, liver protrusion into the chest cavity and pulmonary hypoplasia with a single left lung lobe (120). The mesenchymal-epithelial transition factor (Met) gene encodes for a receptor tyrosine kinase that is necessary for the migration of muscle precursor cells into the forming diaphragm (121), whereas fibroblast growth factor 10 (Fgf10) is crucial for early organogenesis of the lung (122). Oral administration of the herbicide nitrofen in Met–/– mice with amuscular diaphragms and Fgf10–/– mice with hypoplastic lungs resulted in CDH in both murine models, indicating that diaphragmatic defects may develop independently of myogenesis and pulmonary development (123). A novel missense variant affecting the actin-binding domains of plastin 3 (PLS3) was recently identified in eight unrelated families, causing X-linked CDH and body wall defects. A genetically modified mouse model of this Pls3W499C variant resulted in perinatal death and reproduced the main features of the human phenotype, including diaphragmatic and body wall abnormalities (124). An abnormal plastin-actin interaction is the most likely explanation for the observed congenital malformation in both humans and mice.
Components Involved in the Formation of Extracellular Matrix
Normal development of the primordial diaphragm and lung is also dependent on the proper formation of its underlying extracellular matrix (ECM). Today, several components of the ECM are known to be aberrant in CDH and associated lung defects.
Fraser extracellular matrix complex subunit 1 (Fras1), Fras1-related extracellular matrix 1 (Frem1) and Frem2 form a mutually stabilizing ternary complex in the ECM, which plays a critical role in cell adhesion and intercellular signaling (125, 126). After identification of a novel FREM1 deletion in a female infant with isolated left-sided CDH and a membranous sac, Beck et al. (127) developed a Frem1-deficient mouse model that displays a comparable phenotype with retrosternal diaphragmatic defect and reduced levels of cell proliferation in the anterior portion of the growing diaphragm, hereby showing that a deficit of FREM1 can lead to CDH in both humans and mice. Because of the observed phenotypic similarities between Frem1-deficient mice and mice lacking the retinoic acid-responsive transcription factor Gata4, the same author group conducted further studies, revealing that Frem1 interacts not only with Gata4 but also with Slit3 in this mouse model of CDH and concomitant lung lobulation defects (128). More recently, Jordan et al. (129) reported that Frem2ne/ne and Fras1Q1263*/Q1263* mice developed an almost identical type of anterior midline CDH with herniated viscera covered by a thin membranous sac as seen in Frem1-defcient mice, thus concluding that loss of the Frem1/Frem2/Fras1 complex or its function results in retrosternal CDH in these animals. The cross-linking of collagens and elastin, which is essential for the structural stability of the ECM, is catalyzed by lysyl oxidase (Lox), an extracellular cuproenzyme (130). In turn, Lox–/– mice die at birth, having a ruptured diaphragm as a result of fragmentation in the central tendon (131, 132). However, no human LOX mutations have been reported so far. Another ECM protein associated with the pathogenesis of CDH is fibrillin 1 (Fbn1), an integral part of microfibrils in elastic and non-elastic connective tissues (133). A gene-targeting mutation of the mouse Fbn1 gene has been associated with diaphragmatic defects and histological examination revealed a focal inflammatory infiltrate at the ruptured edges (134). These homozygous Fbn1–/– mutant mice died postnatally due to pulmonary insufficiency, exhibiting CDH and herniation of abdominal viscera into the thoracic cavity (134). Fibulin 4 (Fbln4) also belongs to a family of ECM proteins, which controls fiber assembly and is known to bind Lox (135). Fbln4 null mutant mice die just after birth with severe CDH and rupture of the diaphragm, in addition to defective distal airways and lung emphysema (136).
Additional Genes and Enzymes Participating in Signal Transduction and Apoptosis
Several other genes and enzymes responsible for signal transduction, intracellular signaling and apoptosis have been discovered in association with diaphragmatic defects in genetically modified mouse models.
The acetylcholine-synthesizing enzyme choline acetyltransferase (ChAT) has been reported to be implicated in various morphogenetic processes during embryonic development (137, 138). In fact, cross-sections of the diaphragm from ChAT–/– mice showed liver herniation through the tendinous center of the diaphragm (139), presumably as a consequence of impaired muscle formation. Deoxyribonuclease IIα (DNase IIα) belongs to a large group of endonucleases involved in DNA digestion during apoptosis. DNase IIα–/– mice displayed a malformed diaphragm with hernia and non-inflated lungs, suggesting that these animals suffer perinatal lethality because of a dysfunctional diaphragm and associated respiratory insufficiency (140). Low-density lipoprotein receptor-related protein 1 (Lrp1) is crucial for proper embryonic development through regulation of intracellular signaling cascades (141). ENU-induced mutation in the Lrp1 gene of mice resulted in body wall closure defects with CDH and liver protruding outside of the abdominal cavity (142). Mitochondrial trifunctional protein (Mtp) is a multi-enzyme complex of four α and four β subunits that catalyzes oxidation of long-chain fatty acids, which is essential for normal embryogenesis (143). Mtpα–/– knockout mice suffer neonatal death with cardiac and diaphragmatic defects, indicating that deficiency of Mtpa may cause dysfunction of the diaphragm and subsequent respiratory insufficiency (143). WD repeat domain 35 (Wdr35) is a protein coding gene, which participates in intracellular trafficking, cargo recognition and binding during embryonic development (144). Following a recessive ENU mutagenesis screen for genes affecting embryogenesis, Mill et al. (144) noticed that mutant Wdr35yet/yet mice embryos died before birth, displaying diaphragmatic defects and hypoplastic lungs.
Conclusion and Future Directions
Over the years, experimental animal models of CDH have not only permitted us to investigate the pathogenesis of this relatively common but complex birth defect in more detail, they have also led to a better understanding of the molecular genetic basis of the underlying tissue defects. Therefore, animals with CDH in which this congenital anomaly develops naturally represent the ideal research models to study disease pathomechanisms and related lung abnormalities, as there is minimal interference to the animal before the examination. Furthermore, genetically modified animal models of CDH not only resemble the natural development of this malformation, they also provide new insights into the participating genes and signaling pathways, and how their modification can potentially change the course of this life-threatening condition. With the recent advent of novel molecular techniques including biomedical engineering and ENU mutagenesis screens, we hopefully may identify additional CDH-related mutations that are linked with abnormal diaphragm and lung development in other genetic mouse models (145–147).
Author Contributions
FF, UR, and PP critically revised the initial manuscript draft for important intellectual content and performed the literature search for the work. FF outlined and wrote the initial manuscript draft. All authors approved the final version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was supported by the German Research Foundation and the Institutional Open Access Fund of the Goethe University Frankfurt within the program of Open Access Publishing. The funders had no role in the literature search, preparation of the manuscript or decision to publish.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Politis MD, Bermejo-Sánchez E, Canfield MA, Contiero P, Cragan JD, Dastgiri S, et al. Prevalence and mortality in children with congenital diaphragmatic hernia: a multicountry study. Ann Epidemiol. (2021) 56: 61–69.e3. doi: 10.1016/j.annepidem.2020.11.007
2. Paoletti M, Raffler G, Gaffi MS, Antounians L, Lauriti G, Zani A. Prevalence and risk factors for congenital diaphragmatic hernia: a global view. J Pediatr Surg. (2020) 55:2297–307.
3. Greer JJ. Current concepts on the pathogenesis and etiology of congenital diaphragmatic hernia. Respir Physiol Neurobiol. (2013) 189:232–40. doi: 10.1016/j.resp.2013.04.015
4. Clugston RD, Greer JJ. Diaphragm development and congenital diaphragmatic hernia. Semin Pediatr Surg. (2007) 16:94–100. doi: 10.1053/j.sempedsurg.2007.01.004
5. Ameis D, Khoshgoo N, Keijzer R. Abnormal lung development in congenital diaphragmatic hernia. Semin Pediatr Surg. (2017) 26:123–8. doi: 10.1053/j.sempedsurg.2017.04.011
6. Keijzer R, Puri P. Congenital diaphragmatic hernia. Semin Pediatr Surg. (2010) 19:180–5. doi: 10.1053/j.sempedsurg.2010.03.001
7. Rottier R, Tibboel D. Fetal lung and diaphragm development in congenital diaphragmatic hernia. Semin Perinatol. (2005) 29:86–93.
8. Russo FM, Cordier AG, De Catte L, Saada J, Benachi A, Deprest J, et al. Proposal for standardized prenatal ultrasound assessment of the fetus with congenital diaphragmatic hernia by the European reference network on rare inherited and congenital anomalies (ERNICA). Prenat Diagn. (2018) 38:629–37. doi: 10.1002/pd.5297
9. Style CC, Mehollin-Ray AR, Verla MA, Lau PE, Cruz SM, Espinoza J, et al. Timing of prenatal magnetic resonance imaging in the assessment of congenital diaphragmatic hernia. Fetal Diagn Ther. (2020) 47:205–13. doi: 10.1159/000501556
10. Horn-Oudshoorn EJJ, Knol R, Te Pas AB, Hooper SB, Cochius-den Otter SCM, Wijnen RMH, et al. Perinatal stabilisation of infants born with congenital diaphragmatic hernia: a review of current concepts. Arch Dis Child Fetal Neonatal Ed. (2020) 105:449–54. doi: 10.1136/archdischild-2019-318606
11. Snoek KG, Reiss IK, Greenough A, Capolupo I, Urlesberger B, Wessel L, et al. Standardized postnatal management of infants with congenital diaphragmatic hernia in Europe: the CDH EURO consortium consensus – 2015 update. Neonatology. (2016) 110:66–74. doi: 10.1159/000444210
12. O’Rourke-Potocki A, Ali K, Murthy V, Milner A, Greenough A. Resuscitation of infants with congenital diaphragmatic hernia. Arch Dis Child Fetal Neonatal Ed. (2017) 102:F320–3. doi: 10.1136/archdischild-2016-311432
13. Williams E, Greenough A. Respiratory support of infants with congenital diaphragmatic hernia. Front Pediatr. (2021) 9:808317. doi: 10.3389/fped.2021.808317
14. Gupta VS, Harting MT, Lally PA, Miller CC, Hirschl RB, Davis CF, et al. Mortality in congenital diaphragmatic hernia: a multicenter registry study of over 5000 patients over 25 years. Ann Surg. (2021). doi: 10.1097/SLA.0000000000005113 [Epub ahead of print].
15. Guner YS, Delaplain PT, Zhang L, Di Nardo M, Brogan TV, Chen Y, et al. Trends in mortality and risk characteristics of congenital diaphragmatic hernia treated with extracorporeal membrane oxygenation. ASAIO J. (2019) 65:509–15. doi: 10.1097/MAT.0000000000000834
16. Bent DP, Nelson J, Kent DM, Jen HC. Population-based validation of a clinical prediction model for congenital diaphragmatic hernias. J Pediatr. (2018) 201: 160–165.e1. doi: 10.1016/j.jpeds.2018.05.027
17. Heiwegen K, de Blaauw I, Botden SMBI. A systematic review and meta-analysis of surgical morbidity of primary versus patch repaired congenital diaphragmatic hernia patients. Sci Rep. (2021) 11:12661. doi: 10.1038/s41598-021-91908-7
18. Aydın E, Nolan H, Peiró JL, Burns P, Rymeski B, Lim FY. When primary repair is not enough: a comparison of synthetic patch and muscle flap closure in congenital diaphragmatic hernia? Pediatr Surg Int. (2020) 36:485–91. doi: 10.1007/s00383-020-04634-y
19. Kays DW, Langham MR Jr, Ledbetter DJ, Talbert JL. Detrimental effects of standard medical therapy in congenital diaphragmatic hernia. Ann Surg. (1999) 230:340–51. doi: 10.1097/00000658-199909000-00007
20. Snoek KG, Capolupo I, van Rosmalen J, Hout Lde J, Vijfhuize S, Greenough A, et al. Conventional mechanical ventilation versus high-frequency oscillatory ventilation for congenital diaphragmatic hernia: a randomized clinical trial (the VICI-trial). Ann Surg. (2016) 263:867–74. doi: 10.1097/SLA.0000000000001533
21. Rafat N, Schaible T. Extracorporeal membrane oxygenation in congenital diaphragmatic hernia. Front Pediatr. (2019) 7:336. doi: 10.3389/fped.2019.00336
22. Puligandla PS, Grabowski J, Austin M, Hedrick H, Renaud E, Arnold M, et al. Management of congenital diaphragmatic hernia: a systematic review from the APSA outcomes and evidence based practice committee. J Pediatr Surg. (2015) 50:1958–70. doi: 10.1016/j.jpedsurg.2015.09.010
23. Losty PD. Congenital diaphragmatic hernia: where and what is the evidence? Semin Pediatr Surg. (2014) 23:278–82. doi: 10.1053/j.sempedsurg.2014.09.008
24. Gerall CD, Stewart LA, Price J, Kabagambe S, Sferra SR, Schmaedick MJ, et al. Long-term outcomes of congenital diaphragmatic hernia: a single institution experience. J Pediatr Surg. (2021) 57:563–9. doi: 10.1016/j.jpedsurg.2021.06.007
25. Hollinger LE, Buchmiller TL. Long term follow-up in congenital diaphragmatic hernia. Semin Perinatol. (2020) 44:151171. doi: 10.1053/j.semperi.2019.07.010
26. Morini F, Valfrè L, Bagolan P. Long-term morbidity of congenital diaphragmatic hernia: a plea for standardization. Semin Pediatr Surg. (2017) 26:301–10. doi: 10.1053/j.sempedsurg.2017.09.002
27. Chandrasekharan PK, Rawat M, Madappa R, Rothstein DH, Lakshminrusimha S. Congenital diaphragmatic hernia – a review. Matern Health Neonatol Perinatol. (2017) 3:6. doi: 10.1186/s40748-017-0045-1
28. Keijzer R, Liu J, Deimling J, Tibboel D, Post M. Dual-hit hypothesis explains pulmonary hypoplasia in the nitrofen model of congenital diaphragmatic hernia. Am J Pathol. (2000) 156:1299–306. doi: 10.1016/S0002-9440(10)65000-6
29. Brandsma AE, ten Have-Opbroek AA, Vulto IM, Molenaar JC, Tibboel D. Alveolar epithelial composition and architecture of the late fetal pulmonary acinus: an immunocytochemical and morphometric study in a rat model of pulmonary hypoplasia and congenital diaphragmatic hernia. Exp Lung Res. (1994) 20:491–515. doi: 10.3109/01902149409031734
30. Chiu PP. New insights into congenital diaphragmatic hernia – a surgeon’s introduction to CDH animal models. Front Pediatr. (2014) 2:36. doi: 10.3389/fped.2014.00036
31. van Loenhout RB, Tibboel D, Post M, Keijzer R. Congenital diaphragmatic hernia: comparison of animal models and relevance to the human situation. Neonatology. (2009) 96:137–49. doi: 10.1159/000209850
32. Beurskens N, Klaassens M, Rottier R, de Klein A, Tibboel D. Linking animal models to human congenital diaphragmatic hernia. Birth Defects Res A Clin Mol Teratol. (2007) 79:565–72. doi: 10.1002/bdra.20370
33. Mortell A, Montedonico S, Puri P. Animal models in pediatric surgery. Pediatr Surg Int. (2006) 22:111–28. doi: 10.1007/s00383-005-1593-4
34. Kardon G, Ackerman KG, McCulley DJ, Shen Y, Wynn J, Shang L, et al. Congenital diaphragmatic hernias: from genes to mechanisms to therapies. Dis Model Mech. (2017) 10:955–70. doi: 10.1242/dmm.028365
35. Yu L, Sawle AD, Wynn J, Aspelund G, Stolar CJ, Arkovitz MS, et al. Increased burden of de novo predicted deleterious variants in complex congenital diaphragmatic hernia. Hum Mol Genet. (2015) 24:4764–73. doi: 10.1093/hmg/ddv196
36. Russell MK, Longoni M, Wells J, Maalouf FI, Tracy AA, Loscertales M, et al. Congenital diaphragmatic hernia candidate genes derived from embryonic transcriptomes. Proc Natl Acad Sci USA. (2012) 109:2978–83. doi: 10.1073/pnas.1121621109
37. Nakamura H, Doi T, Puri P, Friedmacher F. Transgenic animal models of congenital diaphragmatic hernia: a comprehensive overview of candidate genes and signaling pathways. Pediatr Surg Int. (2020) 36:991–7. doi: 10.1007/s00383-020-04705-0
38. Montedonico S, Nakazawa N, Puri P. Congenital diaphragmatic hernia and retinoids: searching for an etiology. Pediatr Surg Int. (2008) 24:755–61. doi: 10.1007/s00383-008-2140-x
39. Lohnes D, Mark M, Mendelsohn C, Dolle P, Decimo D, LeMeur M, et al. Developmental roles of the retinoic acid receptors. J Steroid Biochem Mol Biol. (1995) 53:475–86. doi: 10.1016/0960-0760(95)00094-g
40. Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, et al. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. (1994) 120:2749–71.
41. Lohnes D, Mark M, Mendelsohn C, Dollé P, Dierich A, Gorry P, et al. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development. (1994) 120:2723–48.
42. Mendelsohn C, Mark M, Dolle P, Dierich A, Gaub MP, Krust A, et al. Retinoic acid receptor beta 2 (RAR beta 2) null mutant mice appear normal. Dev Biol. (1994) 166:246–58. doi: 10.1006/dbio.1994.1311
43. Lufkin T, Lohnes D, Mark M, Dierich A, Gorry P, Gaub MP, et al. High postnatal lethality and testis degeneration in retinoic acid receptor alpha mutant mice. Proc Natl Acad Sci USA. (1993) 90:7225–9. doi: 10.1073/pnas.90.15.7225
44. Li E, Sucov HM, Lee KF, Evans RM, Jaenisch R. Normal development and growth of mice carrying a targeted disruption of the alpha 1 retinoic acid receptor gene. Proc Natl Acad Sci USA. (1993) 90:1590–4. doi: 10.1073/pnas.90.4.1590
45. Andersen DH. Incidence of congenital diaphragmatic hernia in the young of rats bred on a diet deficient in vitamin A. Am J Dis Child. (1941) 62:888–9.
46. Andersen DH. Effect of diet during pregnancy upon the incidence of congenital hereditary diaphragmatic hernia in the rat: failure to produce cystic fibrosis of the pancreas by maternal vitamin A deficiency. Am J Pathol. (1949) 25:163–85.
47. Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. (2007) 315:820–5. doi: 10.1126/science.1136244
48. Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nürnberg G, et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet. (2007) 80:550–60. doi: 10.1086/512203
49. Cooney AJ, Tsai SY, O’Malley BW, Tsai MJ. Chicken ovalbumin upstream promoter transcription factor (COUP-TF) dimers bind to different GGTCA response elements, allowing COUP-TF to repress hormonal induction of the vitamin D3, thyroid hormone, and retinoic acid receptors. Mol Cell Biol. (1992) 12:4153–63. doi: 10.1128/mcb.12.9.4153-4163.1992
50. Kliewer SA, Umesono K, Heyman RA, Mangelsdorf DJ, Dyck JA, Evans RM. Retinoid X receptor-COUP-TF interactions modulate retinoic acid signaling. Proc Natl Acad Sci USA. (1992) 89:1448–52. doi: 10.1073/pnas.89.4.1448
51. Tran P, Zhang XK, Salbert G, Hermann T, Lehmann JM, Pfahl M. COUP orphan receptors are negative regulators of retinoic acid response pathways. Mol Cell Biol. (1992) 12:4666–76. doi: 10.1128/mcb.12.10.4666-4676.1992
52. You LR, Takamoto N, Yu CT, Tanaka T, Kodama T, Demayo FJ, et al. Mouse lacking COUP-TFII as an animal model of bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci USA. (2005) 102:16351–6. doi: 10.1073/pnas.0507832102
53. Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, et al. WT-1 is required for early kidney development. Cell. (1993) 74:679–91. doi: 10.1016/0092-8674(93)90515-r
54. Moore AW, Schedl A, McInnes L, Doyle M, Hecksher-Sorensen J, Hastie ND. YAC transgenic analysis reveals Wilms’ tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech Dev. (1998) 79:169–84. doi: 10.1016/s0925-4773(98)00188-9
55. Ijpenberg A, Pérez-Pomares JM, Guadix JA, Carmona R, Portillo-Sánchez V, Macías D, et al. Wt1 and retinoic acid signaling are essential for stellate cell development and liver morphogenesis. Dev Biol. (2007) 312:157–70. doi: 10.1016/j.ydbio.2007.09.014
56. Patek CE, Brownstein DG, Fleming S, Wroe C, Rose L, Webb A, et al. Effects on kidney disease, fertility and development in mice inheriting a protein-truncating denys-drash syndrome allele (Wt1tmT396). Transgenic Res. (2008) 17:459–75. doi: 10.1007/s11248-007-9157-0
57. Antonius T, van Bon B, Eggink A, van der Burgt I, Noordam K, van Heijst A. Denys-drash syndrome and congenital diaphragmatic hernia: another case with the 1097G > A(Arg366His) mutation. Am J Med Genet A. (2008) 146A:496–9. doi: 10.1002/ajmg.a.32168
58. Suri M, Kelehan P, O’Neill D, Vadeyar S, Grant J, Ahmed SF, et al. WT1 mutations in meacham syndrome suggest a coelomic mesothelial origin of the cardiac and diaphragmatic malformations. Am J Med Genet A. (2007) 143A:2312–20. doi: 10.1002/ajmg.a.31924
59. Clugston RD, Klattig J, Englert C, Clagett-Dame M, Martinovic J, Benachi A, et al. Teratogen-induced, dietary and genetic models of congenital diaphragmatic hernia share a common mechanism of pathogenesis. Am J Pathol. (2006) 169:1541–9. doi: 10.2353/ajpath.2006.060445
60. Carmona R, Cañete A, Cano E, Ariza L, Rojas A, Muñoz-Chápuli R. Conditional deletion of WT1 in the septum transversum mesenchyme causes congenital diaphragmatic hernia in mice. Elife. (2016) 5:e16009. doi: 10.7554/eLife.16009
61. Cleal L, McHaffie SL, Lee M, Hastie N, Martínez-Estrada OM, Chau YY. Resolving the heterogeneity of diaphragmatic mesenchyme: a novel mouse model of congenital diaphragmatic hernia. Dis Model Mech. (2021) 14:dmm046797. doi: 10.1242/dmm.046797
62. Paris ND, Coles GL, Ackerman KG. Wt1 and β-catenin cooperatively regulate diaphragm development in the mouse. Dev Biol. (2015) 407:40–56. doi: 10.1016/j.ydbio.2015.08.009
63. Gilbert RM, Schappell LE, Gleghorn JP. Defective mesothelium and limited physical space are drivers of dysregulated lung development in a genetic model of congenital diaphragmatic hernia. Development. (2021) 148:dev199460. doi: 10.1242/dev.199460
64. Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet. (2000) 67:1047–54. doi: 10.1016/S0002-9297(07)62934-6
65. Kim PC, Mo R, Hui C. Murine models of VACTERL syndrome: role of sonic hedgehog signaling pathway. J Pediatr Surg. (2001) 36:381–4. doi: 10.1053/jpsu.2001.20722
66. Cheung HO, Zhang X, Ribeiro A, Mo R, Makino S, Puviindran V, et al. The kinesin protein Kif7 is a critical regulator of gli transcription factors in mammalian hedgehog signaling. Sci Signal. (2009) 2:ra29. doi: 10.1126/scisignal.2000405
67. Schnabel CA, Selleri L, Jacobs Y, Warnke R, Cleary ML. Expression of Pbx1b during mammalian organogenesis. Mech Dev. (2001) 100:131–5. doi: 10.1016/s0925-4773(00)00516-5
68. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. (2001) 15:3059–87. doi: 10.1101/gad.938601
69. Coles GL, Ackerman KG. Kif7 is required for the patterning and differentiation of the diaphragm in a model of syndromic congenital diaphragmatic hernia. Proc Natl Acad Sci USA. (2013) 110:E1898–905. doi: 10.1073/pnas.1222797110
70. Coles GL, Baglia LA, Ackerman KG. KIF7 controls the proliferation of cells of the respiratory airway through distinct microtubule dependent mechanisms. PLoS Genet. (2015) 11:e1005525. doi: 10.1371/journal.pgen.1005525
71. McCulley DJ, Wienhold MD, Hines EA, Hacker TA, Rogers A, Pewowaruk RJ, et al. PBX transcription factors drive pulmonary vascular adaptation to birth. J Clin Invest. (2018) 128:655–67. doi: 10.1172/JCI93395
72. Slavotinek A, Risolino M, Losa M, Cho MT, Monaghan KG, Schneidman-Duhovny D, et al. De novo, deleterious sequence variants that alter the transcriptional activity of the homeoprotein PBX1 are associated with intellectual disability and pleiotropic developmental defects. Hum Mol Genet. (2017) 26:4849–60. doi: 10.1093/hmg/ddx363
73. Longoni M, High FA, Russell MK, Kashani A, Tracy AA, Coletti CM, et al. Molecular pathogenesis of congenital diaphragmatic hernia revealed by exome sequencing, developmental data, and bioinformatics. Proc Natl Acad Sci USA. (2014) 111:12450–5. doi: 10.1073/pnas.1412509111
74. Gillemans N, Szumska D, Veenma D, Esteghamat F, Hou J, van Ijcken W, et al. Complex congenital diaphragmatic hernia in mice lacking chromatin target of PRMT1. In: DCM Veenma editor. Genetic and Epigenetic Interplay in Congenital Diaphragmatic Hernia. (Rotterdam: Erasmus University Rotterdam Repository) (2012).
75. Veenma DC, de Klein A, Tibboel D. Developmental and genetic aspects of congenital diaphragmatic hernia. Pediatr Pulmonol. (2012) 47:534–45. doi: 10.1002/ppul.22553
76. van Dijk TB, Gillemans N, Stein C, Fanis P, Demmers J, van de Corput M, et al. Friend of Prmt1, a novel chromatin target of protein arginine methyltransferases. Mol Cell Biol. (2010) 30:260–72. doi: 10.1128/MCB.00645-09
77. Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, et al. High-throughput discovery of novel developmental phenotypes. Nature. (2016) 537:508–14. doi: 10.1038/nature19356
78. Holder AM, Klaassens M, Tibboel D, de Klein A, Lee B, Scott DA. Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet. (2007) 80:825–45. doi: 10.1086/513442
79. Ackerman KG, Wang J, Luo L, Fujiwara Y, Orkin SH, Beier DR. Gata4 is necessary for normal pulmonary lobar development. Am J Respir Cell Mol Biol. (2007) 36:391–7. doi: 10.1165/rcmb.2006-0211RC
80. Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, et al. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. (2005) 1:e10. doi: 10.1371/journal.pgen.0010010
81. Scott DA. Genetics of congenital diaphragmatic hernia. Semin Pediatr Surg. (2007) 16:88–93. doi: 10.1053/j.sempedsurg.2007.01.003
82. Doi T, Sugimoto K, Puri P. Prenatal retinoic acid up-regulates pulmonary gene expression of COUP-TFII, FOG2, and GATA4 in pulmonary hypoplasia. J Pediatr Surg. (2009) 44:1933–7. doi: 10.1016/j.jpedsurg.2009.04.027
83. Huggins GS, Bacani CJ, Boltax J, Aikawa R, Leiden JM. Friend of GATA 2 physically interacts with chicken ovalbumin upstream promoter-TF2 (COUP-TF2) and COUP-TF3 and represses COUP-TF2-dependent activation of the atrial natriuretic factor promoter. J Biol Chem. (2001) 276:28029–36. doi: 10.1074/jbc.M103577200
84. Longoni M, Russell MK, High FA, Darvishi K, Maalouf FI, Kashani A, et al. Prevalence and penetrance of ZFPM2 mutations and deletions causing congenital diaphragmatic hernia. Clin Genet. (2015) 87:362–7. doi: 10.1111/cge.12395
85. Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, et al. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol. (2007) 301:602–14. doi: 10.1016/j.ydbio.2006.09.050
86. Merrell AJ, Ellis BJ, Fox ZD, Lawson JA, Weiss JA, Kardon G. Muscle connective tissue controls development of the diaphragm and is a source of congenital diaphragmatic hernias. Nat Genet. (2015) 47:496–504. doi: 10.1038/ng.3250
87. Wat MJ, Beck TF, Hernandez-Garcia A, Yu Z, Veenma D, Garcia M, et al. Mouse model reveals the role of SOX7 in the development of congenital diaphragmatic hernia associated with recurrent deletions of 8p23.1. Hum Mol Genet. (2012) 21:4115–25. doi: 10.1093/hmg/dds241
88. Lu J, Richardson JA, Olson EN. Capsulin: a novel bHLH transcription factor expressed in epicardial progenitors and mesenchyme of visceral organs. Mech Dev. (1998) 73:23–32. doi: 10.1016/s0925-4773(98)00030-6
89. Lu J, Chang P, Richardson JA, Gan L, Weiler H, Olson EN. The basic helix-loop-helix transcription factor capsulin controls spleen organogenesis. Proc Natl Acad Sci USA. (2000) 97:9525–30. doi: 10.1073/pnas.97.17.9525
90. Lu JR, Bassel-Duby R, Hawkins A, Chang P, Valdez R, Wu H, et al. Control of facial muscle development by MyoR and capsulin. Science. (2002) 298:2378–81. doi: 10.1126/science.1078273
91. Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE. Mouse genome database group. The mouse genome database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. (2015) 43:D726–36. doi: 10.1093/nar/gku967
92. Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. (2003) 426:247–54. doi: 10.1038/nature02083
93. Kumar JP. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cell Mol Life Sci. (2009) 66:565–83. doi: 10.1007/s00018-008-8335-4
94. Grifone R, Demignon J, Giordani J, Niro C, Souil E, Bertin F, et al. Eya1 and Eya2 proteins are required for hypaxial somitic myogenesis in the mouse embryo. Dev Biol. (2007) 302:602–16. doi: 10.1016/j.ydbio.2006.08.059
95. El-Hashash AH, Al Alam D, Turcatel G, Bellusci S, Warburton D. Eyes absent 1 (Eya1) is a critical coordinator of epithelial, mesenchymal and vascular morphogenesis in the mammalian lung. Dev Biol. (2011) 350:112–26. doi: 10.1016/j.ydbio.2010.11.022
96. Laclef C, Hamard G, Demignon J, Souil E, Houbron C, Maire P. Altered myogenesis in Six1-deficient mice. Development. (2003) 130:2239–52. doi: 10.1242/dev.00440
97. El-Hashash AH, Al Alam D, Turcatel G, Rogers O, Li X, Bellusci S, et al. Six1 transcription factor is critical for coordination of epithelial, mesenchymal and vascular morphogenesis in the mammalian lung. Dev Biol. (2011) 353:242–58. doi: 10.1016/j.ydbio.2011.02.031
98. Allen JD, Lints T, Jenkins NA, Copeland NG, Strasser A, Harvey RP, et al. Novel murine homeo box gene on chromosome 1 expressed in specific hematopoietic lineages and during embryogenesis. Genes Dev. (1991) 5:509–20. doi: 10.1101/gad.5.4.509
99. Arterbery AS, Bogue CW. Endodermal and mesenchymal cross talk: a crossroad for the maturation of foregut organs. Pediatr Res. (2014) 75:120–6. doi: 10.1038/pr.2013.201
100. Lints TJ, Hartley L, Parsons LM, Harvey RP. Mesoderm-specific expression of the divergent homeobox gene Hlx during murine embryogenesis. Dev Dyn. (1996) 205:457–70. doi: 10.1002/(SICI)1097-0177(199604)205:43.0.CO;2-H
101. Hentsch B, Lyons I, Li R, Hartley L, Lints TJ, Adams JM, et al. Hlx homeo box gene is essential for an inductive tissue interaction that drives expansion of embryonic liver and gut. Genes Dev. (1996) 10:70–9. doi: 10.1101/gad.10.1.70
102. Farrell SA, Sodhi S, Marshall CR, Guerin A, Slavotinek A, Paton T, et al. HLX is a candidate gene for a pattern of anomalies associated with congenital diaphragmatic hernia, short bowel, and asplenia. Am J Med Genet A. (2017) 173:3070–4. doi: 10.1002/ajmg.a.38354
103. Montavon T, Soshnikova N. Hox gene regulation and timing in embryogenesis. Semin Cell Dev Biol. (2014) 34:76–84. doi: 10.1016/j.semcdb.2014.06.005
104. Manley NR, Barrow JR, Zhang T, Capecchi MR. Hoxb2 and hoxb4 act together to specify ventral body wall formation. Dev Biol. (2001) 237:130–44. doi: 10.1006/dbio.2001.0365
105. Yuan W, Rao Y, Babiuk RP, Greer JJ, Wu JY, Ornitz DM. A genetic model for a central (septum transversum) congenital diaphragmatic hernia in mice lacking Slit3. Proc Natl Acad Sci USA. (2003) 100:5217–22. doi: 10.1073/pnas.0730709100
106. Liu J, Zhang L, Wang D, Shen H, Jiang M, Mei P, et al. Congenital diaphragmatic hernia, kidney agenesis and cardiac defects associated with Slit3-deficiency in mice. Mech Dev. (2003) 120:1059–70. doi: 10.1016/s0925-4773(03)00161-8
107. Domyan ET, Branchfield K, Gibson DA, Naiche LA, Lewandoski M, Tessier-Lavigne M, et al. Roundabout receptors are critical for foregut separation from the body wall. Dev Cell. (2013) 24:52–63. doi: 10.1016/j.devcel.2012.11.018
108. Ypsilanti AR, Zagar Y, Chedotal A. Moving away from the midline: new developments for Slit and Robo. Development. (2010) 137:1939–52. doi: 10.1242/dev.044511
109. Long H, Sabatier C, Ma L, Plump A, Yuan W, Ornitz DM, et al. Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron. (2004) 42:213–23. doi: 10.1016/s0896-6273(04)00179-5
110. Macias H, Moran A, Samara Y, Moreno M, Compton JE, Harburg G, et al. SLIT/ROBO1 signaling suppresses mammary branching morphogenesis by limiting basal cell number. Dev Cell. (2011) 20:827–40. doi: 10.1016/j.devcel.2011.05.012
111. Ye BQ, Geng ZH, Ma L, Geng JG. Slit2 regulates attractive eosinophil and repulsive neutrophil chemotaxis through differential srGAP1 expression during lung inflammation. J Immunol. (2010) 185:6294–305. doi: 10.4049/jimmunol.1001648
112. Shiau CE, Bronner-Fraser M. N-cadherin acts in concert with Slit1-Robo2 signaling in regulating aggregation of placode-derived cranial sensory neurons. Development. (2009) 136:4155–64. doi: 10.1242/dev.034355
113. Grieshammer U, Le M, Plump AS, Wang F, Tessier-Lavigne M, Martin GR. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell. (2004) 6:709–17. doi: 10.1016/s1534-5807(04)00108-x
114. De Bellard ME, Rao Y, Bronner-Fraser M. Dual function of Slit2 in repulsion and enhanced migration of trunk, but not vagal, neural crest cells. J Cell Biol. (2003) 162:269–79. doi: 10.1083/jcb.200301041
115. Xian J, Clark KJ, Fordham R, Pannell R, Rabbitts TH, Rabbitts PH. Inadequate lung development and bronchial hyperplasia in mice with a targeted deletion in the Dutt1/Robo1 gene. Proc Natl Acad Sci USA. (2001) 98:15062–6. doi: 10.1073/pnas.251407098
116. Hussain SA, Piper M, Fukuhara N, Strochlic L, Cho G, Howitt JA, et al. A molecular mechanism for the heparan sulfate dependence of Slit-Robo signaling. J Biol Chem. (2006) 281:39693–8. doi: 10.1074/jbc.M609384200
117. Zhang B, Xiao W, Qiu H, Zhang F, Moniz HA, Jaworski A, et al. Heparan sulfate deficiency disrupts developmental angiogenesis and causes congenital diaphragmatic hernia. J Clin Invest. (2014) 124:209–21. doi: 10.1172/JCI71090
118. Bleyl SB, Moshrefi A, Shaw GM, Saijoh Y, Schoenwolf GC, Pennacchio LA, et al. Candidate genes for congenital diaphragmatic hernia from animal models: sequencing of FOG2 and PDGFRalpha reveals rare variants in diaphragmatic hernia patients. Eur J Hum Genet. (2007) 15:950–8. doi: 10.1038/sj.ejhg.5201872
119. Sun T, Jayatilake D, Afink GB, Ataliotis P, Nistér M, Richardson WD, et al. A human YAC transgene rescues craniofacial and neural tube development in PDGFRalpha knockout mice and uncovers a role for PDGFRalpha in prenatal lung growth. Development. (2000) 127:4519–29.
120. Mouse Genome Informatics. Fuzb2b1273Clo. (2011). Available online at: http://www.informatics.jax.org/allele/MGI:5311392 (accessed January 31, 2022).
121. Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. (1995) 376:768–71. doi: 10.1038/376768a0
122. Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. (1997) 124:4867–78.
123. Babiuk RP, Greer JJ. Diaphragm defects occur in a CDH hernia model independently of myogenesis and lung formation. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L1310–4. doi: 10.1152/ajplung.00257.2002
124. Petit F, Longoni M, Wells J, Maser R, Dysart MJ, Contreras HTM, et al. Missense variants affecting the actin-binding domains of PLS3 cause X-linked congenital diaphragmatic hernia and body wall defects. medRxiv. (2021) [Preprint]. doi: 10.1101/2021.07.07.21259278
125. Petrou P, Makrygiannis AK, Chalepakis G. The Fras1/Frem family of extracellular matrix proteins: structure, function, and association with fraser syndrome and the mouse bleb phenotype. Connect Tissue Res. (2008) 49:277–82. doi: 10.1080/03008200802148025
126. Wiradjaja F, Cottle DL, Jones L, Smyth I. Regulation of PDGFC signalling and extracellular matrix composition by FREM1 in mice. Dis Model Mech. (2013) 6:1426–33. doi: 10.1242/dmm.013748
127. Beck TF, Veenma D, Shchelochkov OA, Yu Z, Kim BJ, Zaveri HP, et al. Deficiency of FRAS1-related extracellular matrix 1 (FREM1) causes congenital diaphragmatic hernia in humans and mice. Hum Mol Genet. (2013) 22:1026–38. doi: 10.1093/hmg/dds507
128. Beck TF, Shchelochkov OA, Yu Z, Kim BJ, Hernández-García A, Zaveri HP, et al. Novel frem1-related mouse phenotypes and evidence of genetic interactions with gata4 and slit3. PLoS One. (2013) 8:e58830. doi: 10.1371/journal.pone.0058830
129. Jordan VK, Beck TF, Hernandez-Garcia A, Kundert PN, Kim BJ, Jhangiani SN, et al. The role of FREM2 and FRAS1 in the development of congenital diaphragmatic hernia. Hum Mol Genet. (2018) 27:2064–75. doi: 10.1093/hmg/ddy110
130. Vallet SD, Ricard-Blum S. Lysyl oxidases: from enzyme activity to extracellular matrix cross-links. Essays Biochem. (2019) 63:349–64. doi: 10.1042/EBC20180050
131. Maki JM, Sormunen R, Lippo S, Kaarteenaho-Wiik R, Soininen R, Myllyharju J. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am J Pathol. (2005) 167:927–36. doi: 10.1016/S0002-9440(10)61183-2
132. Hornstra IK, Birge S, Starcher B, Bailey AJ, Mecham RP, Shapiro SD. Lysyl oxidase is required for vascular and diaphragmatic development in mice. J Biol Chem. (2003) 278:14387–93. doi: 10.1074/jbc.M210144200
133. Bielinska M, Jay PY, Erlich JM, Mannisto S, Urban Z, Heikinheimo M, et al. Molecular genetics of congenital diaphragmatic defects. Ann Med. (2007) 39:261–74. doi: 10.1080/07853890701326883
134. Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci USA. (1999) 96:3819–23. doi: 10.1073/pnas.96.7.3819
135. Papke CL, Yanagisawa H. Fibulin-4 and fibulin-5 in elastogenesis and beyond: insights from mouse and human studies. Matrix Biol. (2014) 37:142–9. doi: 10.1016/j.matbio.2014.02.004
136. Horiguchi M, Inoue T, Ohbayashi T, Hirai M, Noda K, Marmorstein LY, et al. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci USA. (2009) 106:19029–34. doi: 10.1073/pnas.0908268106
137. Torrão AS, Carmona FM, Lindstrom J, Britto LR. Expression of cholinergic system molecules during development of the chick nervous system. Brain Res Dev Brain Res. (2000) 124:81–92. doi: 10.1016/s0165-3806(00)00113-9
138. Brandon EP, Lin W, D’Amour KA, Pizzo DP, Dominguez B, Sugiura Y, et al. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci. (2003) 23:539–49. doi: 10.1523/JNEUROSCI.23-02-00539.2003
139. Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. (2002) 36:635–48. doi: 10.1016/s0896-6273(02)01020-6
140. Krieser RJ, MacLea KS, Longnecker DS, Fields JL, Fiering S, Eastman A. Deoxyribonuclease IIalpha is required during the phagocytic phase of apoptosis and its loss causes perinatal lethality. Cell Death Differ. (2002) 9:956–62. doi: 10.1038/sj.cdd.4401056
141. Mao H, Xie L, Pi X. Low-density lipoprotein receptor-related protein-1 signaling in angiogenesis. Front Cardiovasc Med. (2017) 4:34. doi: 10.3389/fcvm.2017.00034
142. Mouse Genome Informatics. Lrp1b2b1554Clo. (2011). Available online at: http://www.informatics.jax.org/allele/MGI:5437079 (accessed January 31, 2022).
143. Ibdah JA, Paul H, Zhao Y, Binford S, Salleng K, Cline M, et al. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J Clin Invest. (2001) 107:1403–9. doi: 10.1172/JCI12590
144. Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MAM, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet. (2011) 88:508–15. doi: 10.1016/j.ajhg.2011.03.015
145. Donahoe PK, Longoni M, High FA. Polygenic causes of congenital diaphragmatic hernia produce common lung pathologies. Am J Pathol. (2016) 186:2532–43. doi: 10.1016/j.ajpath.2016.07.006
146. Yu L, Hernan RR, Wynn J, Chung WK. The influence of genetics in congenital diaphragmatic hernia. Semin Perinatol. (2020) 44:151169. doi: 10.1053/j.semperi.2019.07.008
Keywords: congenital diaphragmatic hernia, diaphragm development, lung development, pulmonary hypoplasia, pulmonary hypertension, genetic model, transgenic mice, retinoic acid
Citation: Friedmacher F, Rolle U and Puri P (2022) Genetically Modified Mouse Models of Congenital Diaphragmatic Hernia: Opportunities and Limitations for Studying Altered Lung Development. Front. Pediatr. 10:867307. doi: 10.3389/fped.2022.867307
Received: 31 January 2022; Accepted: 18 March 2022;
Published: 13 May 2022.
Edited by:
Marc Oria, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Lourenço Sbragia, University of São Paulo, BrazilJuan A. Tovar, La Paz University Hospital, Spain
Copyright © 2022 Friedmacher, Rolle and Puri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florian Friedmacher, Florian.Friedmacher@nhs.net