A riboflavin transporter deficiency presenting as pure red cell aplasia: a pediatric case report
 Jingying Cheng
Jingying Cheng Jiafeng Yao
Jiafeng Yao  Lingling Fu
Lingling Fu- Department of Hematology, National Center for Children’s Health, Beijing Children’s Hospital, Capital Medical University, Beijing, China
Introduction: Riboflavin transporter deficiency (RTD) is a rare genetic disorder that affects riboflavin transport, leading to impaired red blood cell production and resulting in pure red cell aplasia. Recognizing and understanding its clinical manifestations, diagnosis, and management is important.
Case presentation: A 2-year-old patient presented with pure red cell aplasia as the primary symptom of RTD. After confirming the diagnosis, rapid reversal of anemia was achieved after high-dose riboflavin treatment.
Conclusion: RTD often has an insidious onset, and neurological symptoms appear gradually as the disease progresses, making it prone to misdiagnosis. Genetic testing and bone marrow biopsy can confirm the diagnosis.
Introduction
Riboflavin transporter deficiency (RTD), also known as Brown-Vialetto-Van Laere syndrome, is a neurological disorder caused by pathogenic variants in the riboflavin transporter protein genes SLC52A2 (encoding RFVT2) and SLC52A3 (encoding RFVT3). The most common manifestations are sensorineural hearing loss (SNHL), peripheral neuropathy, respiratory insufficiency, and medullary paralysis (1, 2). Reports of hematological manifestations as primary symptoms are extremely rare. Here, we report a case of RTD with pure red cell aplasia as the initial symptom. This study aimed to enhance our understanding of hematological manifestations and non-targeted metabolomic analysis in the treatment of RTD to achieve early detection, diagnosis, and treatment.
Case report
When the 2-year-old patient was 2 months old, he was brought to the Hematology Department of Beijing Children's Hospital outpatient clinic due to “pale complexion for over 2 months”. Initial blood tests revealed the following results: red blood cells (RBC), 1.02 × 1012/L (Normal 3.5–5.6); hemoglobin (Hb), 29 g/L (Normal 99–196); hematocrit, 9.2% (Normal 29–57); mean corpuscular volume, 90.2 fl (Normal 73–105); MCH, 28.4 pg (Normal 24–37); mean corpuscular Hb concentration, 315 g/L (Normal 305–361); Reticulocyte %, 0.48% (Normal 0.5–2.5); Reticulocyte absolute value, 4.9 × 109/L (Normal 22–139). White blood cell and platelet counts were within normal ranges. The patient received blood transfusions to correct the anemia. The patient had normal development until 6 months old, followed by delayed motor development and sensory ataxia. At 1 year old, the patient had difficulty grasping, sitting, and standing without support and did not crawl. Due to the young age of the patient, some symptoms cannot be self-reported, and cannot cooperate with auditory and visual examinations. The mother had a history of preexcitation syndrome and was prone to syncope. Mild anemia developed during pregnancy with unknown etiology. A physical examination of the patient revealed severe anemia, with no enlarged superficial lymph nodes, liver, or spleen. Bilirubin and anemia-related indicators were normal, and the patient was negative for autoimmune antibodies. No pathogenic variants were found in the genetic screening of a blood system panel. Based on the onset of symptoms within the first year of life and test results, a diagnosis of pure red cell aplasia was considered. Due to the young age of the patient and the potential impact of hormones on growth and development, symptomatic blood transfusion therapy was temporarily initiated, requiring monthly transfusions when Hb levels dropped below 70 g/L. At 6 months old, the patient experienced breath-holding spells accompanied by cyanosis of the face and lips after crying. These lasted for a few seconds and resolved spontaneously. Occasionally, episodes of limb stiffness and termor manifested, which improved after sedative therapy. No fever or infection was observed during the interictal period and no abnormalities were observed in the electroencephalogram at that time.
During the evaluation of the 1-year-old patient, a follow-up bone marrow morphological examination revealed active bone marrow hyperplasia with relatively normal erythroid proliferation, mainly consisting of intermediate and young RBCs. The granulocyte-erythrocyte ratio was slightly higher, and the morphology was generally normal. Early erythroid cells accounted for 1% (Normal 0–5.8), intermediate erythroid cells for 8% (Normal 5–34), and late erythroid cells for 2.5% (Normal 1.6–21.5). A bone marrow biopsy showed reduced erythroid proliferation. Therefore, pure red cell aplasia was not ruled out. Chromosomal karyotyping revealed 46, XY, del(5)(q33)[2]/46, XY[18]. Comprehensive testing for myelodysplastic syndrome and myeloid leukemia-related genes did not reveal any pathogenic variants. Based on the patient's medical history, symptoms, signs, and auxiliary examinations, pure red cell aplasia was diagnosed. Therefore, starting on October 1, 2022, the patient was treated with prednisone 10 mg BID for 2 months, followed by methylprednisolone 16 mg QD for 1 month. On December 21, 2022, oral cyclosporine was added, and methylprednisolone was discontinued after 1 month. The cyclosporine treatment lasted 6 months, but the treatment was ineffective, and the patient still required transfusions. At the age of 22 months, the patient developed progressive upper limb weakness, especially in the hands. The patient was subsequently examined at the neurology outpatient departments of Beijing Children's Hospital and Beijing Xuanwu Hospital. Cranial magnetic resonance imaging showed bilateral symmetric punctate high signals in the midbrain and dorsal pons in T2W imaging, along with high signals in DWI and low signals in ADC. Additionally, there was bilateral ventricular dilatation and slightly elevated T2 FLAIR signal in the periventricular white matter (Figures 1–3). Magnetic resonance imaging of the cervical spine showed patchy, long T1 and T2 signals in the spinal cord. Genetic metabolic screening revealed no significant abnormalities. Whole-exome sequencing revealed compound heterozygous variants in SLC52A2: c.588C > G (variant inherited from the father, heterozygous variant) and c.593G > A (variant inherited from the mother, heterozygous variant). Previous genetic tests did not include analysis of SLC52A2 genes. According to the ACMG guidelines, the variant c.588C > G is of uncertain clinical significance (PM1, PM2, PP3_Supporting), whereas variant c.593G > A is suspected to be pathogenic (PVS1_VeryStrong, PM2). The REVEL protein function damage prediction for c.588C > G is 0.718, and for c.593G > A is 0. The patient was started on oral riboflavin (vitamin B2) at a dose of 10 mg/d (0.8 mg/kg/d) on July 17, 2023, gradually increasing to 75 mg/d (6.25 mg/kg/d). Consequently, the vitamin B2 blood concentration increased from 8.18 ug/L to 11 ug/L. After one week of treatment, the Hb level increased to 116 g/L and fluctuated between 126 and 137 g/L over the next two weeks. Upper limb muscle strength improved from grade 2 to grade 3+, with the ability to perform simple grasping. Unfortunately, nystagmus developed, indicating deterioration of the patient's condition. Due to difficulty in sedating the patient, electromyography could not be completed. The patient is currently under follow-up in the hematology and neurology outpatient departments.
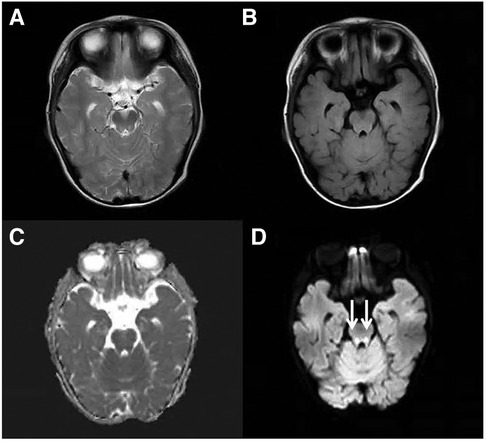
Figure 1. Cranial MRI at the level of the midbrain. An axial T2-weighted (A), an axial FLAIR (B), a dADC (C) and a DWI (D) scans are shown and abnormally high signals in DWI in the midbrain can be seen (white arrow).
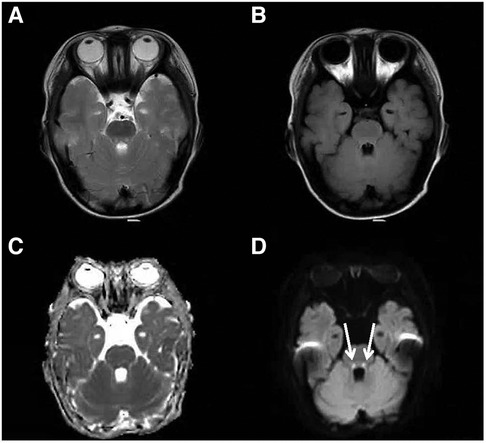
Figure 2. Cranial MRI at the level of the dorsal pons. An axial T2-weighted (A), an axial FLAIR (B), a dADC (C) and a DWI (D) scans are shown and abnormally high signals in DWI in the dorsal pons can be seen (white arrow).
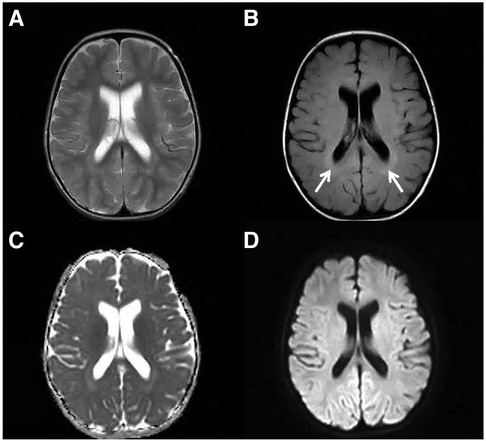
Figure 3. Elevated T2 FLAIR signal in the periventricular white matter. An axial T2-weighted (A), an axial FLAIR (B), a dADC (C) and a DWI (D) scans are shown and slightly elevated T2 FLAIR signal in the periventricular white matter can be seen (white arrow).
Discussion
RTD is an autosomal recessive progressive neurological disorder characterized by early-onset SNHL, bulbar palsy, optic atrophy, severe diffuse muscle weakness, and atrophy of the upper and lower limbs and axial muscles, leading to respiratory insufficiency (1, 2). Before diagnosis, patients may experience optic atrophy, sleep apnea, breath-holding spells, or swallowing difficulties (3). RTD is mainly caused by genetic variants in SLC52A2 or SLC52A3, which results in riboflavin transporter function deficiencies (4, 5). These disorders are referred to as RTD2 and RTD3, respectively. RTD3 typically presents with more generalized weakness. In approximately 92% of RFVT2-deficient patients, the upper limb muscles are more affected than the lower limb muscles, and most patients experience weakness in the shoulder girdle and distal hand muscles (6). These findings are consistent with those of the male patient described here, who presented with breath-holding spells at six months of age, followed by delayed motor development and tremors in both hands at 22 months old.
SLC52A2 encodes a transmembrane protein (RFVT2) that mediates the cellular uptake of riboflavin. The most common pathogenic variants are missense mutations (94.4%), followed by nonsense (2.4%), splice-site (0.8%), deletion (1.6%), and insertion mutations (0.8%) (7, 8). In this case, the patient had compound heterozygous pathogenic variants in SLC52A2. The c.588C > G variant is a novel pathogenic variant that has not been previously reported. The c.593G > A variant was reported in a 2019 study discussing the etiology of pediatric SNHL (9).
RTDs have a low incidence rate and significant clinical heterogeneity. Patients often present with neurological symptoms or deafness, and approximately 70% of individuals have an abnormal acylcarnitine profile before riboflavin supplementation (10). Due to impaired iron absorption resulting from riboflavin deficiency, some patients may experience mild-to-moderate decreases in RBC counts and/or Hb levels. Riboflavin-responsive macrocytic anemia was first reported in a patient with RTD in 2020 (3). However, rapid correction of anemia was achieved with high-dose riboflavin treatment. Subsequently, Pillai et al. reported the case of a 2-year-old boy diagnosed with RTD who had a history of seizures and breath-holding spells at 21 months of age. Bone marrow biopsy revealed active bone marrow proliferation with significantly delayed RBC maturation and maturation arrest. Subsequently, the patient developed ataxia and developmental regression. After two weeks of treatment with riboflavin, the patient's hematological abnormalities completely resolved, and the neurological symptoms eventually improved (11). In 2021, a patient presenting with pure red cell aplastic anemia was reported in China and was found to have novel SLC52A2 pathogenic variants. The patient's symptoms significantly improved with supplementation of low-dose riboflavin (20 mg/d) (12).
Because riboflavin must be obtained through intestinal absorption, extensive research has found that oral riboflavin supplements can effectively alleviate the clinical symptoms of patients with RTD. However, if irreversible damage has already occurred, the therapeutic effect is minimal. Therefore, early detection, definitive diagnosis, and initiation of riboflavin treatment are crucial. The literature reports a dosage range of oral riboflavin from 7 to 60 mg/kg/d, with no reports of death and few adverse reactions (13–16). After treatment, approximately 60% and 80% of patients with RFVT2 and RFVT3 defects, respectively, showed improvement in clinical symptoms, with most patients with RTD experiencing improvement in symptoms within days to months (17).
Summary
RTD often presents with an insidious onset and lacks obvious abnormalities in cranial imaging changes, making it challenging to differentiate from various neurodegenerative diseases. Although RTD is mainly characterized by early onset SNHL and optic nerve atrophy, the age of onset is young, and the patients usually cannot accurately describe visual and auditory abnormalities (18). Moreover, identifying auditory neuropathy by conventional hearing screening is generally difficult, leading to missed diagnoses (19, 20). Currently, molecular biological diagnosis and genetic testing are the main methods used for diagnosing RTD. Therefore, physicians need to enhance their understanding of the clinical manifestations of RTD and suggest a more comprehensive evaluation, along with early genetic diagnosis, for patients suspected of having RTD or patients presenting with anemia/red cell aplasia without other features of RTD. If a physician suspects RTD and patients have severe symptoms, empirical riboflavin treatment can be started immediately without waiting for molecular diagnosis results because the treatment is relatively safe (21).
Owing to the irreversibility of nerve damage, the timing of riboflavin treatment for RTD is crucial, and early initiation of treatment is key. Therefore, performing SLC52A2 genetic testing for patients suspected of having RTD should be performed as soon as possible. Applying these tests to neonatal screening should be considered to achieve rapid diagnosis and early treatment to minimize hematological and neurological damage.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committee of Beijing Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JC: Writing – original draft. JY: Writing – review & editing. SZ: Writing – review & editing. LF: Writing – review & editing. LZ: Writing – review & editing. JJ: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Woodcock IR, Menezes MP, Coleman L, Yaplito-Lee J, Peters H, White SM, et al. Genetic, radiologic, and clinical variability in Brown-Vialetto-van Laere syndrome. Semin Pediatr Neurol. (2018) 26:2–9. doi: 10.1016/j.spen.2017.03.001
2. Johnson JO, Gibbs JR, Megarbane A, Urtizberea JA, Hernandez DG, Foley AR, et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain. (2012) 135(Pt 9):2875–82. doi: 10.1093/brain/aws161
3. Amir F, Atzinger C, Massey K, Greinwald J, Hunter LL, Ulm E, et al. The clinical journey of patients with riboflavin transporter deficiency type 2. J Child Neurol. (2020) 35(4):283–90. doi: 10.1177/0883073819893159
4. Yamamoto S, Inoue K, Ohta KY, Fukatsu R, Maeda JY, Yoshida Y, et al. Identification and functional characterization of rat riboflavin transporter 2. J Biochem. (2009) 145(4):437–43. doi: 10.1093/jb/mvn181
5. Yao Y, Yonezawa A, Yoshimatsu H, Masuda S, Katsura T, Inui K. Identification and comparative functional characterization of a new human riboflavin transporter hRFT3 expressed in the brain. J Nutr. (2010) 140(7):1220–6. doi: 10.3945/jn.110.122911
6. Jaeger B, Bosch AM. Clinical presentation and outcome of riboflavin transporter deficiency: mini review after five years of experience. J Inherit Metab Dis. (2016) 39(4):559–64. doi: 10.1007/s10545-016-9924-2
7. Bosch AM, Stroek K, Abeling NG, Waterham HR, Ijlst L, Wanders RJ. The Brown-Vialetto-Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J Rare Dis. (2012) 7:83. doi: 10.1186/1750-1172-7-83
8. Zhao S, Che F, Yang L, Zheng Y, Wang D, Yang Y, et al. First report of paternal uniparental disomy of chromosome 8 with SLC52A2 mutation in brown-vialetto-van laere syndrome type 2 and an analysis of genotype-phenotype correlations. Front Genet. (2022) 13:977914. doi: 10.3389/fgene.2022.977914
9. van Beeck Calkoen EA, Engel M, van de Kamp JM, Yntema HG, Goverts ST, Mulder MF, et al. The etiological evaluation of sensorineural hearing loss in children. Eur J Pediatr. (2019) 178(8):1195–205. doi: 10.1007/s00431-019-03379-8
10. Fennessy JR, Cornett KMD, Burns J, Menezes MP. Benefit of high-dose oral riboflavin therapy in riboflavin transporter deficiency. J Peripher Nerv Syst. (2023) 28(3):308–16. doi: 10.1111/jns.12587
11. Pillai NR, Amin H, Gijavanekar C, Liu N, Issaq N, Broniowska KA, et al. Hematologic presentation and the role of untargeted metabolomics analysis in monitoring treatment for riboflavin transporter deficiency. Am J Med Genet A. (2020) 182(11):2781–7. doi: 10.1002/ajmg.a.61851
12. Liu Z, Peng Q, Li J, Rao C, Lu X. BVVLS2 Overlooked for 3 years in a pediatric patient caused by novel compound heterozygous mutations in SLC52A2 gene. Clin Chim Acta. (2021) 523:402–6. doi: 10.1016/j.cca.2021.10.031
13. Anand G, Hasan N, Jayapal S, Huma Z, Ali T, Hull J, et al. Early use of high-dose riboflavin in a case of Brown-Vialetto-Van Laere syndrome. Dev Med Child Neurol. (2012) 54(2):187–9. doi: 10.1111/j.1469-8749.2011.04142.x
14. Haack TB, Makowski C, Yao Y, Graf E, Hempel M, Wieland T, et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J Inherit Metab Dis. (2012) 35(6):943–8. doi: 10.1007/s10545-012-9513-y
15. Spagnoli C, Pitt MC, Rahman S, de Sousa C. Brown-Vialetto-van Laere syndrome: a riboflavin responsive neuronopathy of infancy with singular features. Eur J Paediatr Neurol. (2014) 18(2):231–4. doi: 10.1016/j.ejpn.2013.09.006
16. Menezes MP, Farrar MA, Webster R, Antony J, O'Brien K, Ouvrier R, et al. Pathophysiology of motor dysfunction in a childhood motor neuron disease caused by mutations in the riboflavin transporter. Clin Neurophysiol. (2016) 127(1):911–8. doi: 10.1016/j.clinph.2015.05.012
17. Foley AR, Menezes MP, Pandraud A, Gonzalez MA, Al-Odaib A, Abrams AJ, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. (2014) 137(Pt 1):44–56. doi: 10.1093/brain/awt315
18. Set KK, Weber A, Serajee FJ, Huq AM. Clinical reasoning: siblings with progressive weakness, hypotonia, nystagmus, and hearing loss. Neurology. (2018) 90(7):e625–31. doi: 10.1212/WNL.0000000000004973
19. O’Callaghan B, Bosch AM, Houlden H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J Inherit Metab Dis. (2019) 42(4):598–607. doi: 10.1002/jimd.12053
20. Sathasivam S. Brown-Vialetto-Van Laere syndrome. Orphanet J Rare Dis. (2008) 3:9. doi: 10.1186/1750-1172-3-9
Keywords: riboflavin, pure red cell aplasia, genetic testing, SLC52A2, children
Citation: Cheng J, Yao J, Zhao S, Fu L, Zhang L and Jiang J (2024) A riboflavin transporter deficiency presenting as pure red cell aplasia: a pediatric case report. Front. Pediatr. 12:1391245. doi: 10.3389/fped.2024.1391245
Received: 25 February 2024; Accepted: 4 April 2024;
Published: 17 April 2024.
Edited by:
Tomasz Szczepanski, Medical University of Silesia, PolandReviewed by:
Manoj Menezes, Kids Research Institute, The Children's Hospital at Westmead, AustraliaJulien H. Park, University of Münster, Germany
© 2024 Cheng, Yao, Zhao, Fu, Zhang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Jiang jiangjin0325@163.com