 Mona Dawood
Mona Dawood Sami Hamdoun
Sami Hamdoun Thomas Efferth
Thomas Efferth- Department of Pharmaceutical Biology, Institute of Pharmacy and Biochemistry, Johannes Gutenberg University, Mainz, Germany
Arsenic trioxide is a traditional remedy in Chinese Medicine since ages. Nowadays, it is clinically used to treat acute promyelocytic leukemia (APL) by targeting PML/RARA. However, the drug’s activity is broader and the mechanisms of action in other tumor types remain unclear. In this study, we investigated molecular modes of action by classical and network pharmacological approaches. CEM/ADR5000 resistance leukemic cells were similar sensitive to As2O3 as their wild-type counterpart CCRF-CEM (resistance ratio: 1.88). Drug-resistant U87.MG ΔEGFR glioblastoma cells harboring mutated epidermal growth factor receptor were even more sensitive (collateral sensitive) than wild-type U87.MG cells (resistance ratio: 0.33). HCT-116 colon carcinoma p53-/- knockout cells were 7.16-fold resistant toward As2O3 compared to wild-type cells. Forty genes determining cellular responsiveness to As2O3 were identified by microarray and COMPARE analyses in 58 cell lines of the NCI panel. Hierarchical cluster analysis-based heat mapping revealed significant differences between As2O3 sensitive cell lines and resistant cell lines with p-value: 1.86 × 10-5. The genes were subjected to Galaxy Cistrome gene promoter transcription factor analysis to predict the binding of transcription factors. We have exemplarily chosen NF-kB and AP-1, and indeed As2O3 dose-dependently inhibited the promoter activity of these two transcription factors in reporter cell lines. Furthermore, the genes identified here and those published in the literature were assembled and subjected to Ingenuity Pathway Analysis for comprehensive network pharmacological approaches that included all known factors of resistance of tumor cells to As2O3. In addition to pathways related to the anticancer effects of As2O3, several neurological pathways were identified. As arsenic is well-known to exert neurotoxicity, these pathways might account for neurological side effects. In conclusion, the activity of As2O3 is not restricted to acute promyelocytic leukemia. In addition to PML/RARA, numerous other genes belonging to diverse functional classes may also contribute to its cytotoxicity. Network pharmacology is suited to unravel the multifactorial modes of action of As2O3.
Introduction
Cancer represents a major cause of mortality. It accounted for about 7 Mio deaths in 2000 (Guilbert, 2001) and 8.2 million deaths in 2012 worldwide (Ferlay et al., 2015). In 2008, 12.7 Mio new cancer cases were estimated (Jemal et al., 2010). Recently, 1,688,780 new cancer cases and about 600,920 cancer deaths were estimated to occur in the United States (Siegel et al., 2017). Although, significant progress has been made in cancer therapy during the past years, treatment outcome is still largely hampered by drug resistance and severe side effects (Wiench et al., 2012). Various mechanisms of anticancer drug resistance have been described, e.g., point mutations in drug targets, drug efflux pumps, xenobiotic detoxification mechanisms, resistance to apoptosis, repair DNA damage, alteration of cell proliferation features and others (Cree and Charlton, 2017). For this reason, novel compounds are urgently required for the improvement of treatment outcomes and survival prognosis of patients (Wiench et al., 2012).
Arsenic is a semimetal naturally existing in earth, water and air. It is found in many chemical forms such as yellow arsenic (As2S3), red arsenic (As2S2), white arsenic (As2O3) etc. (Miller et al., 2002). Since ages, As2O3 is used in traditional Chinese medicine. In the 1970s, Chinese scientists tested the potential of As2O3 to treat acute PML (Rao et al., 2013). Today, As2O3 is clinically used worldwide to treat APL and multiple myeloma (Soignet et al., 1998; Breccia and Lo-Coco, 2012; Iland et al., 2014). A major driver of tumorigenesis of APL is the PML/RARA oncogene. Interestingly, As2O3 targets this oncogene, which explains its strong activity in this tumor entity. In combination with retinoic acid, As2O3 can cure up to 90% of APL patients (Lallemand-Breitenbach et al., 2012). After binding of As2O3 to PML/RARA, sumoylation and ubiquitination takes place. Further events included in As2O3’s modes of action include reactive oxygen species generation, inducing apoptosis and cell cycle arrest, activating caspases 8 and 9, down-regulating VEGF thus suppress the angiogenesis as well as inhibition of tumor invasion and metastasis (Zhao et al., 1997; Huang et al., 1999; Perkins et al., 2000; Roboz et al., 2000; Anderson et al., 2002; Hayashi et al., 2002; Liu et al., 2003).
Independent from the activity of As2O3 in APL, other tumor types may also be affected via other mechanisms, e.g., the hedgehog signaling pathway in medulloblastoma (Beauchamp and Uren, 2012; Klinger et al., 2017), indicating that the full range of mechanisms of As2O3 has not been elucidated yet.
Recently, network pharmacology becomes an important bioinformatics tools for identifying the mechanism of action of traditional Chinese medicine (TCM). Several methodologies including proteomics, metabolomics, genomics and serum pharmacokinetics are used to identify molecular target and mechanisms of TCM formulas. Applying this methods will lead to a shift from one drug- one target model to network target-multi-components models (Liang et al., 2014). In addition, applying network analysis (protein–protein interaction) may identify drug-target-related proteins (Li and Zhang, 2013).
In an endeavor to study the cytotoxic activity of As2O3 in cell lines of other tumor types than APL and to identify possible novel modes of action, we undertook the present project. The aims of this study were firstly to investigate whether classical drug resistance mechanisms such as P-glycoprotein, the tumor suppressor p53 and the EGFR may decrease cellular responsiveness to As2O3. The anti-proliferation activity of As2O3 and predominantly its effects against colon carcinoma resistant cell lines HCT116 (p53-/-), U87M. ΔEGFR glioblastoma multiforme cancer cells is reported here for the first time. Furthermore, we performed COMPARE and hierarchical cluster analyses for 58 cell lines of the National Cancer Institute (NCI, United States) from nine different tumor types1. Then, we run bioinformatical gene promoter binding motif analyses for those genes obtained by COMPARE and hierarchical cluster analyses. Since AP-1 and NF-κB binding sites were predominant in the gene promoter sequences subjected to binding motif analyses, we used reporter cell lines for these two transcription factors and indeed observed that As2O3 inhibited promoter binding of these two transcription factors. Finally, we performed interactome network analysis using IPA software to identify cellular pathways that might be affected upon As2O3 treatment.
Materials and Methods
Cell Lines and Reagents
We tested cell lines expressing different modes of drug resistance and their corresponding parental, drug sensitive cell lines. These included the leukemia CCRF-CEM (drug sensitive), and the drug resistant CEM/ADR5000 (multidrug resistant), the human glioblastoma U87.MG (wild type) and U87.MG ΔEGFR (mutated type), and the colon carcinoma HCT-116 (p53+/+) and HCT-116 (p53-/-) cell lines. The details about the cell lines were described previously by our group (Saeed et al., 2014). Glioblastoma were provided from Dr. W. K. Cavenee (Ludwig Institute for Cancer Research, San Diego, CA, United States). While, colon cancer cell lines were provided by Dr. B. Vogelstein and H. Hermeking (Howard Hughes Medical Institute, Baltimore, MD, United States).
Tumor cell lines were routinely cultured in proper medium (Sigma–Aldrich, Taufkirchen, Germany) with 10% FBS (Sigma–Aldrich, Taufkirchen, Germany), 1% penicillin/streptomycin Sigma–Aldrich) (Bunz et al., 1998; Efferth et al., 2003). Moreover, doxorubicin (5000 ng/mL) was used to maintain the resistance of CEM/ADR5000 cells (Kimmig et al., 1990). While U87.MG ΔEGFR and HCT-116 (p53-/-) was treated with geneticin to maintain expression of the transcript. Cells were incubated at 37°C under 5% CO2 in a humidified atmosphere. As2O3 and geneticin were purchased from Sigma–Aldrich, Germany. Doxorubicin was kindly provided by the University Medical Center, Johannes Gutenberg University (Mainz, Germany).
Cytotoxicity Assays
The cytotoxicity of As2O3 was assessed using the resazurin reduction assay method as previously described (Kuete et al., 2016, 2017). Cells were treated with different concentrations of As2O3, ranging from 0.003 to 500 μM. The assay principle is that resazurin (Sigma–Aldrich) is reduced by viable cells to the highly fluorescent resorufin (O’Brien et al., 2000). Dead cells have no capability to convert resazurin to resorufin which lack the fluorescence property. Infinite M2000 proplate reader (Tecan, Germany) detected the fluorescent signal that emitted by the viable cells. Experiments were done three times, with six replicates for each concentration. The viability of the cells treated with As2O3 was calculated as percentage in compared with untreated control cells. The obtained cell viability was plotted against As2O3 concentration using Microsoft Excel 2016. Dose response curve was used to calculate the IC50 values of every cell line.
NF-κB Reporter Assay
HEK-Blue-Null1 cells were obtained from Invivogen (San Diego, CA, United States). It is derived from embryonic kidney-293 (HEK-293) cell line. HEK-Blue-Null1 cells culture conditions were previously described (Kadioglu et al., 2016b; Seo et al., 2016; Hamdoun and Efferth, 2017). Briefly, HEK-Blue-Null1 cells constantly transcribe the SEAP protein under control of the NF-κB promoter. Cells were treated with varying concentrations of As2O3. NF-κB pathway was activated using 100 ng/mL of TNF and inducted for 24 h. Then, detection using pre-warmed Quanti-Blue reagent (Invivogen) was performed according to the manufacturer’s protocol. SEAP was measured spectrophotometrically at 630 nm to detect NF-κB activity. The fold change was calculated in compare with untreated control cells. The NF-κB inhibitor triptolide (1 μM, Invivogen) was used as positive control. The experiments were repeated three times.
AP-1 Reporter Assay
HEK293 cells were trypsinized and evenly distributed into 96 well plate polystyrene U96. The cells were then transfected with AP-1 luciferase reporter construct (CCS-011L, Qiagen, Germantown, MD, United States) using Attractene transfection reagent (Promega, Germany) according to the manufacturer’s protocols. After 24 h of the transfection process, cells were incubated with different doses of As2O3. After 18 h of incubation time with As2O3, AP-1 activation was induced by of 50 ng/ml phorbol 12-myristate 13-acetate PMA (Sigma–Aldrich) for 24 h. AP-1 promoter activity was measured by Dual-Luciferase Reporter Assay System (Promega, Madison, WI, United States) following the manufacturer’s recommendations. Both firefly and Renilla luciferase luminescences were measured using Infinite M2000 Pro plate reader (Tecan). The firefly luciferase luminescence ratio to Renilla luciferase luminescence for each sample was calculated to obtain the relative luciferase. Normalization of AP-1 activity was done using the following equation: relative luciferase of sample over the relative luciferase of the untreated control cells (Kadioglu et al., 2016a). AP-1 luciferase assay experiments were repeated twice.
Bioinformatical Methods
In the present manuscript, we applied several methods of systems biology. While conventional medicine prefers a reductionist approach with one (or few) targets for an investigational drug, traditional medicine always emphasized the multi-specific nature of natural products (Efferth and Koch, 2011). The advent of genome-wide expression profiling techniques was estimated as specifically promising for natural product research, as complex cascades, pathways, and gross gene alteration patterns can be measured in a single experiment (Kadioglu and Efferth, 2014; Quan et al., 2014; Dos Santos et al., 2016; Fang et al., 2017). Molecular pharmacology with the investigation of single pathways has been enlarged by the new field of network pharmacology (Poornima et al., 2016; Schmidt and Efferth, 2016; Efferth et al., 2017). As myriads of data points are collected with genome-wide methods, the data evaluation requires the application of bioinformatics to uncover relevant biological mechanisms of drugs. The “-omics” technology in conjunction with bioinformatical methods allow the generation of hypothetical predictions that could be tested and verified in experimental and clinical settings.
COMPARE Analysis
A panel of 58 cell lines from National Cancer Institute (NCI), United States were used to perform COMPARE and hierarchical cluster analyses. Logarithmic IC50 values (log10IC50) of As2O3 have been deposited at the NCI database2. The mRNA expression values of NCI cell lines were determined via microarray analyses were deposited at the NCI website2 as well. These data were used to generate rank ordered lists of genes expressed in the NCI cell lines panel using COMPARE analyses (Paull et al., 1989).
To extract the most meaningful results from a non-relevant “background noise” of transcriptome-wide microarray-based mRNA hybridizations, we applied the COMPARE analysis, which has been developed by Paull et al. (1989) from the NCI (United States). During the past decades, NCI has investigated more than 300,000 compounds for their cytotoxic capability against a panel of 60 cell lines from different tumor origin. The still growing NCI drug repository does not contain only synthetic compounds but also natural products. Paull and his team observed that drugs with similar molecular modes of action reveal similar patterns of growth inhibition based on their log10IC50. This correlation was used to develop an automated algorithm based on the Pearson correlation rank test. The details of the COMPARE methodology have been described (Paull et al., 1989, 1995; Zaharevitz et al., 2002). The use of the COMPARE algorithm to analyze data from the NCI human tumor cell line screen emerged more and more as a standard tool in drug discovery.
COMPARE analysis can be applied to identify inhibitors for a given mechanism of interest (e.g., inhibitors of EGFR, tubulins, DNA topoisomerases and others) (Wosikowski et al., 1997; Pfister et al., 2009). Vice versa, COMPARE analyses can also be used to suggest possible modes of action and/or determinants of resistance for compounds of interest (e.g., P-glycoprotein, p53 or Ras) (Lee et al., 1994; Alvarez et al., 1995; Koo et al., 1996; O’Connor et al., 1997).
COMPARE analyses were performed with software implemented into the web site of the NCI3. COMPARE analyses yielded rank-ordered lists of compounds. To obtain COMPARE rankings, a scale index of similarity between log10IC50 values of As2O3 and the transcriptome-based mRNA-based gene for the NCI panel of cell lines has been generated. The mRNA microarray hybridization of the NCI cell lines has been reported and deposited at the NCI Web site4 (Scherf et al., 2000; Amundson et al., 2008).
COMPARE analyses were performed to produce rank-ordered lists of genes expressed in the NCI cell lines. The results are sorted by the correlation coefficient (R-values). The methodology has been described previously in detail as a tool to identify candidate genes for drug resistance and sensitivity (Evans et al., 2008; Fagan et al., 2012; Luzina and Popov, 2012). Greater mRNA expression correlated with enhanced drug resistance in the standard COMPARE approach, whereas greater mRNA expression in cell lines indicated drug sensitivity in reverse COMPARE analyses. Pearson’s correlation test was used to calculate significance values and rank correlation coefficients as a relative measure for the linear dependency of two variables.
Hierarchical Cluster Analyses
Another wide used methodology to extract relevant results from transcriptomic data sets are cluster analyses, which represent distance/proximity-based approaches to unravel structures in large data sets. In systems biology, cluster analyses are valuable to define profiles of genes (“gene signatures”), whose expression is linked to biological phenomena, i.e., histological subtypes of tumors, resistance or sensitivity of tumors toward anticancer treatments, survival chances of cancer patients etc. Among the numerous clustering methods (e.g., topological interaction models, influence maps, physical regulatory maps, self-organizing maps, principal component analysis etc.), supervised, hierarchical and aggregative techniques provide advantages for pharmacological questions in cancer biology and pharmacology, because of their flexibility, possibility to include biological knowledge with different weighting, and detection of higher-order relationships between clusters of profiles (Mocellin et al., 2005). Aggregative hierarchical clustering represents a frequently method to investigate gene expression signatures (Eisen et al., 1998; Perou et al., 2000; Ross et al., 2000).
In the present study, we performed hierarchical cluster Analysis to group heterogeneous objects into clusters of homogeneous objects. All objects are assembled into a cluster tree (dendrogram). Thus, objects with tightly related features appear together, whereas the separation in the cluster tree increases with progressive dissimilarity. The merging of objects with similar features leads to formation of a cluster, the shortest the distance of the branch the closest degree of relatedness (Kadioglu and Efferth, 2015). Hierarchical clustering and heat map analysis were conducted using clustered image map (CIM) miner software. One matrix CIM4 and Ward method were performed. Importantly, COMPARE analyses and cluster models have been validated for gene expression profiling, identification of candidate genes for drug resistance and sensitivity and for understanding molecular biology of cancer (Efferth et al., 1997; Scherf et al., 2000).
Interactome Network Analysis
The biomedical literature has been flooded with genomic and transcriptomic data, which urgently need to be organized in bioregulatory networks comparable to electronic circuit programs. The rationale to generate regulatory network models is that pathophysiological processes may be associated with unexpected traits that are not amenable to standard statistical techniques. Building reliable regulatory networks from large-scale genomic and transcriptomic will considerably improve the identification of novel signaling routes and therapeutic targets, since both may differ in healthy and diseased cells (Lefebvre et al., 2012). Hence network approaches will be indispensable for patient-tailored precision medicine to predict drug response and overcome drug resistance (Ganter and Giroux, 2008).
Therefore, a number of software programs have been developed to address this problem, including programs developed at academia such as network molecular interaction maps (MIMs) (Kohn, 1999, 2001; Kohn et al., 2006a,b; Luna et al., 2011), Cytoscape, (Ideker et al., 2001), CellDesigner (Funahashi et al., 2003), TranscriptomeBrowser (Lopez et al., 2008; Lepoivre et al., 2012), PathVisio-Validator (Chandan et al., 2012), Systems Biology Graphical Notation (Ma et al., 2014) as well as commercial products, e.g., Biocarta, Ingenuity Pathway analysis, Pathway Studio, MetaCore and many others. All these programs reveal advantages and disadvantages (Thomas and Bonchev, 2010; Wang et al., 2013).
In the present investigation, genes described in the literature as factors determining cellular responsiveness to As2O3 (Table 5) plus the genes identified via compare analysis of our present and previous investigations on As2O3 (Efferth and Kaina, 2004) were chosen to perform network analyses using Ingenuity Pathway Analysis (IPA) (Qiagen Bioinformatics, Redwood City, CA, United States), as IPA is one of the most used programs to generate bioregulatory networks. We performed network analysis using two different approaches: manually curated pathways and software-based analyses. Published information from literature was used to generate the network and to cluster the genes. IPA Pathway Designer was used to construct the network and the pathways where our genes contributed to.
Gene Promoter Transcription Factor Motif Analysis
Forty genes from the COMPARE analyses results significantly associated with log10IC50 values of As2O3 for the 58 cell lines. UCSC Genome Browser Gene5 was used to analyze the 40 genes to obtain gene’s promoter sequences up to 50 kilobases upstream of the transcription start site. The results were saved on BED (Browser Extensible Data) files for further use (Seo et al., 2016).
The bed file was uploaded on Galaxy, Cistrome software (Liu et al., 2011) to detect the possible transcription factor binding motifs for the selected genes. The software is available at http://cistrome.dfci.harvard.edu/ap/root. Integrative analysis using SeqPos tool was applied to screen the motif binding site on the uploaded genes promoter sequences. Both, UCSC Genome Browser and Galaxy, Cistrome are free softwares. SeqPos tool scans all the motifs that are available on the following databases Transfac, JASPAR, UniPROBE (pbm), hPDI database. The result of the screening was ordered by -log10 (P-value).
Statistical Analysis
Pearson’s correlation test was applied to calculate significance correlation in COMPARE and hierarchical cluster analyses. The chi-squared test was performed to investigate the linear dependency between the resistance and the sensitivity of the cell lines panel. Student’s t-test (Microsoft Excel 2016) was applied to determine the statistical significance of As2O3 effect on AP-1 and NF-κB transcription factors. A level of P < 0.05 was revealed as statistically significant. All data were shown as mean values ± SD.
Results
Cytotoxicity
The cytotoxic effect of As2O3 was determined using resazurin reduction assay. As2O3 was applied on three different resistance mechanisms (P-glycoprotein overexpression, mutant EGFR, knockout p53). The IC50 values for CCRF-CEM cells and CEM/ADR5000 cells were 0.48 ± 0.22 μM and 0.90 ± 0.38 μM, respectively. For CEM/ADR5000 the degrees of resistance was 1.88. Additionally, As2O3 was tested against HCT116 p53 knockout cells (p53-/-) and their wild-type cells, HCT116 (p53+/+). The HCT116 (p53-/-) cells exhibited resistance toward As2O3 with an IC50 value of 90.83 ± 0.83 μM, which was higher than the IC50 value of wild-type HCT116 (p53+/+) cells (12.68 ± 0.78 μM). Interestingly, glioblastoma cell line (U87.MG) were even more sensitive to As2O3 than U87.MG wild-type cells with IC50 values of 15.60 ± 0.93 and 47.82 ± 1.25 μM, respectively. The degree of resistance was 0.33. The Dose response curves and IC50 values for the above-mentioned cell lines are shown in Figure 1 and Table 1.
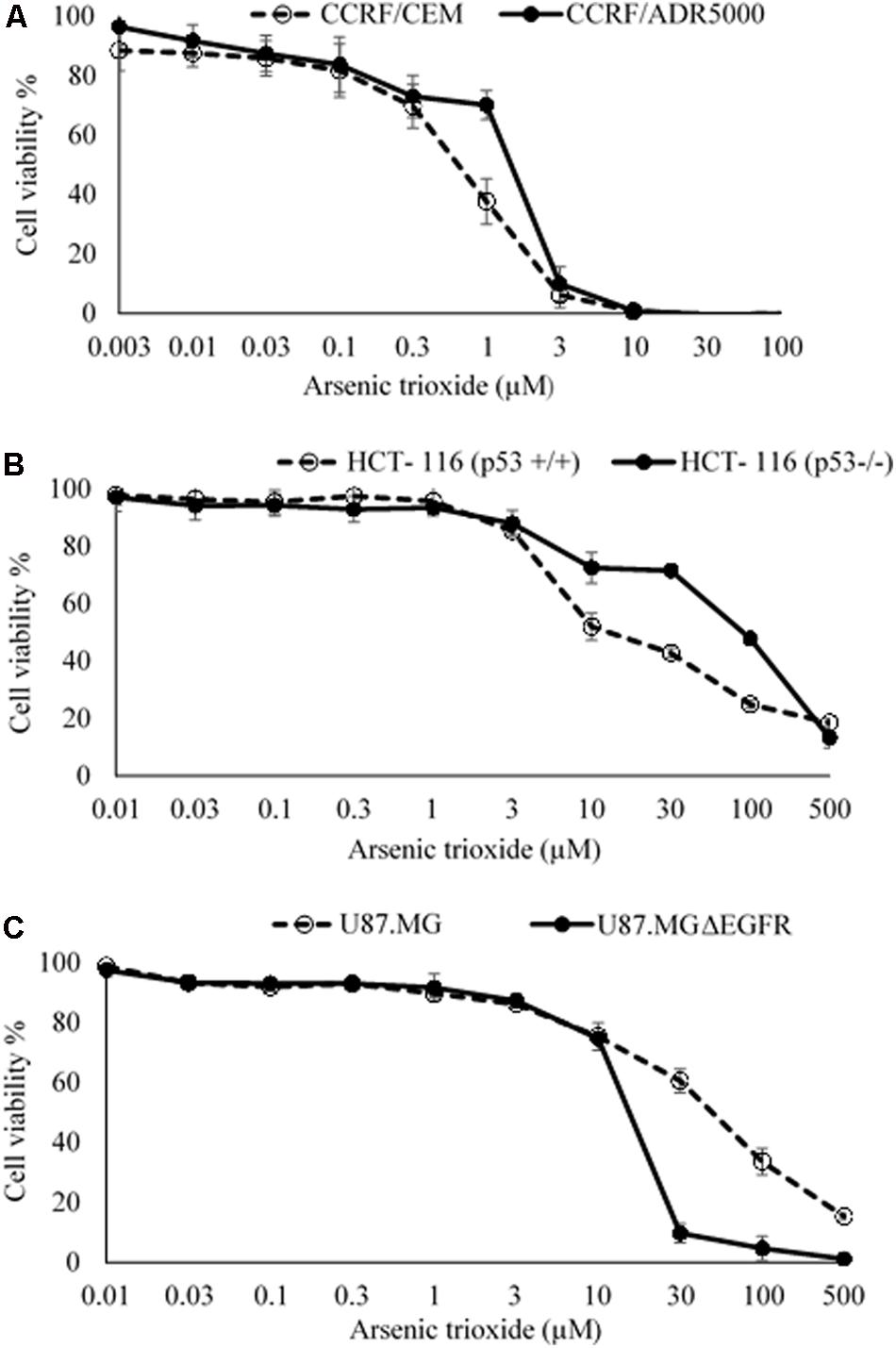
FIGURE 1. Growth inhibition of sensitive and drug-resistance cancer cell lines treated with different concentrations of As2O3. (A). CCRF-CEM and P-glycoprotein-expressing CEM/ADR5000 leukemia cell lines; (B) HCT-116 (p53+/+) wild-type and HCT-116 (p53-/-) knockout colon carcinoma cell lines; (C) U87.MG wild-type and EGFR-transfected U87.MG ΔEGFR glioblastoma cell lines. The dose response curves show mean values ± SD of three independent experiments with each six parallel measurements.
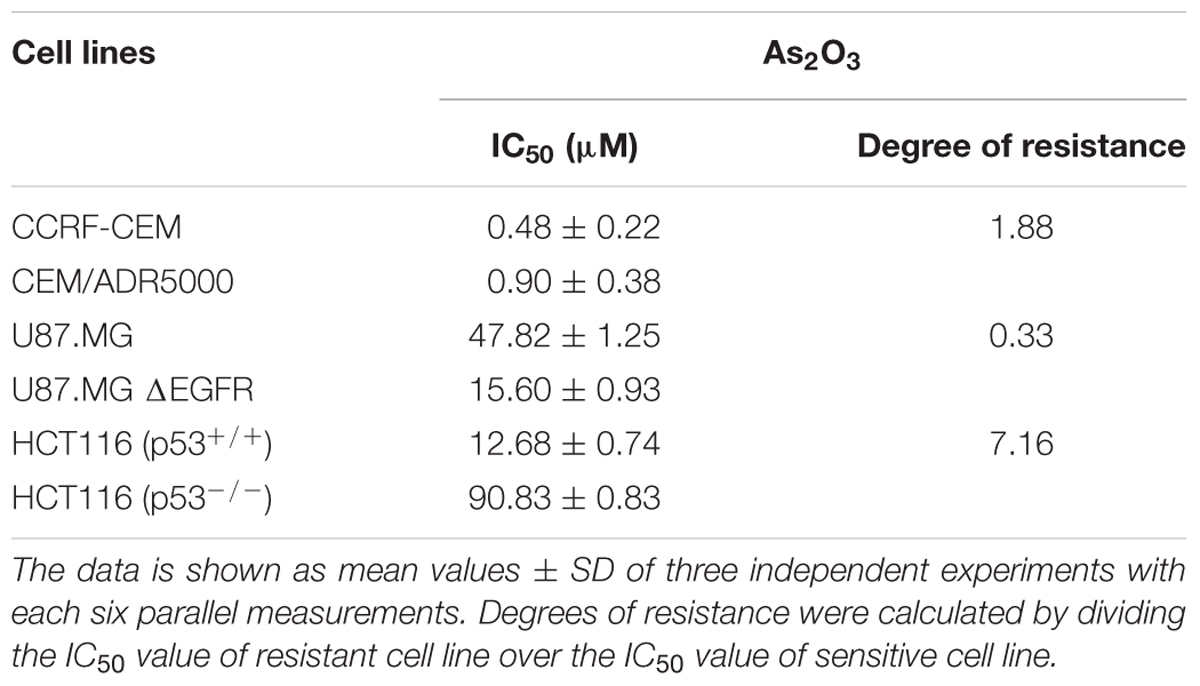
TABLE 1. IC50 values of As2O3 toward various tumor cell lines.
COMPARE and Hierarchical Cluster Analyses
The transcriptome-wide mRNA expression of the 58 NCI cell lines were subjected to COMPARE analyses based on Pearson’s rank correlation tests. In this test, the microarray expression data were correlated with the log10IC50 values of As2O3 for the 58 cell lines. This bioinformatics approach permitted the identification of molecular factors that liked to cellular response to As2O3. The top 20 genes with positive correlation coefficients and top 20 genes with negative correlation coefficients are shown in Table 2. These 40 genes were used to conduct hierarchical cluster analysis (cluster image mapping) to predict, whether these genes may play a role in sensitivity or resistance of the cells to As2O3. The resulting dendrogram with the 58 cell lines applied cluster analyses depicted on the left of the heat-map, while distribution of the 40 genes are shown on the top of the heat-map (Figure 2). The dendrogram can be divided into four major clusters for As2O3. Based on chi-square test we looked whether the clustering of cell was associated with cellular responsiveness of the cell lines to As2O3. Indeed, the branching of sensitive or resistant cell lines was statistically significant (Table 3). The genes identified via this approach belong to diverse groups such as cell cycle progression and proliferation genes including (PRKACA, ARHGAP19, CDKN2D etc.), tumor suppressor related genes (RBBP4, CHMP1A, and GPRC5A), cytoskeleton related genes (PTPRC, ABLIM1, EPB41L1 and PLS1), metabolic pathway genes (ME1, COASY, and ALDH3A2), signal transduction genes (PPAP2C and PKP3) and apoptosis-regulating gene (ID1).
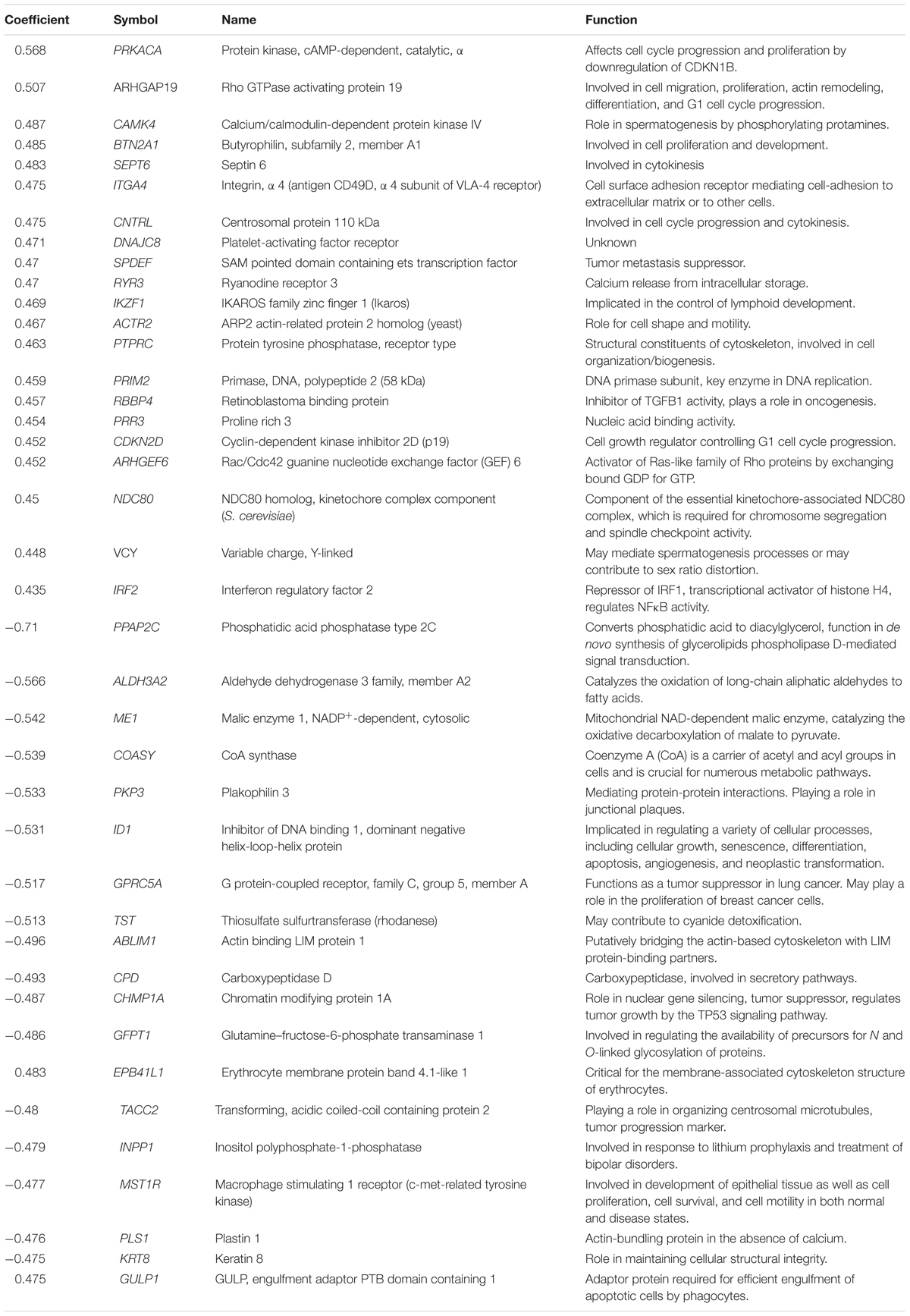
TABLE 2. Correlation coefficients of mRNA expression to log10 IC50 values identified by COMPARE analyses for 58 NCI tumor cell lines and functions of the identified genes.
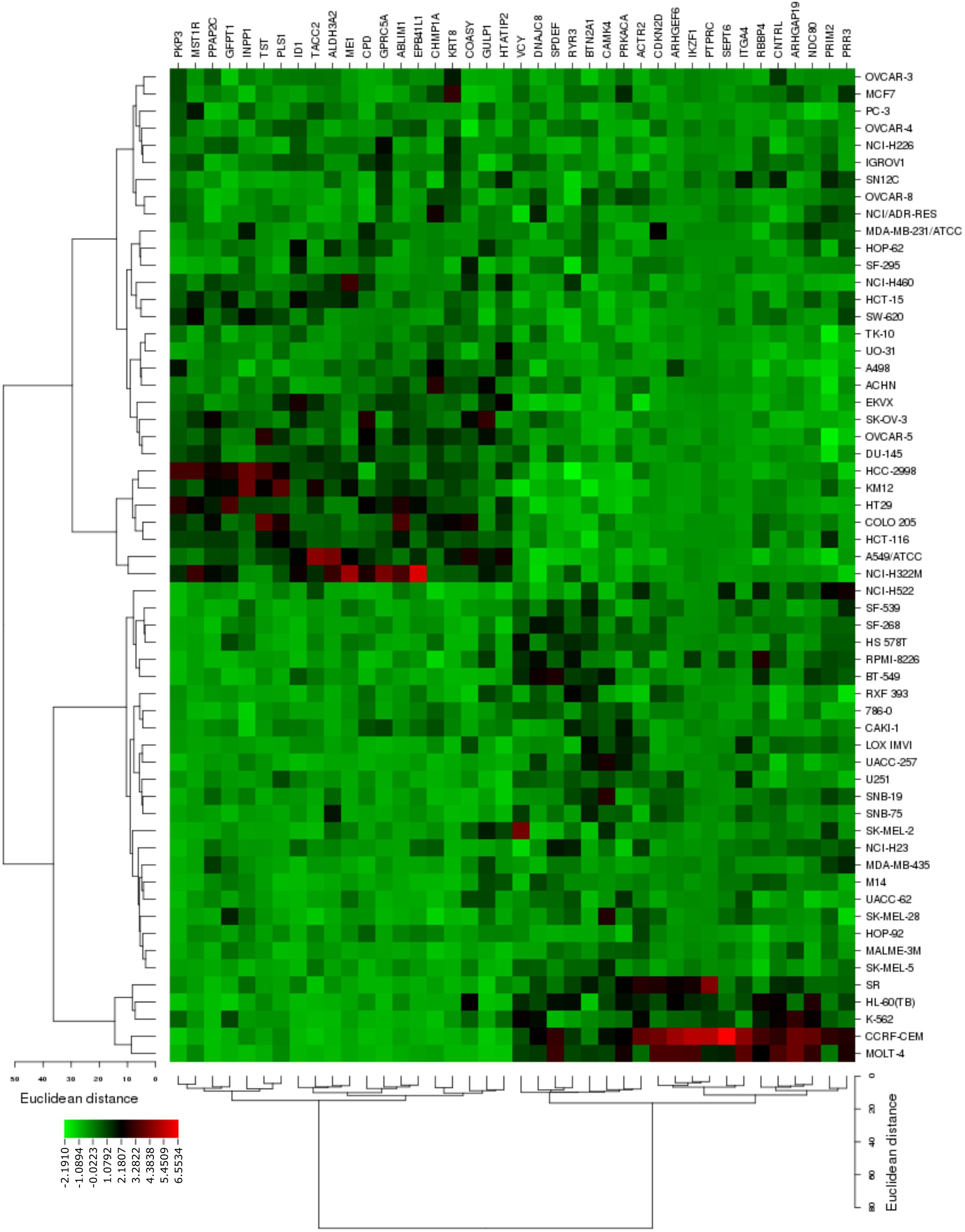
FIGURE 2. Heat map obtained by cluster analysis from microarray-based mRNA expression profiles of genes correlating with cellular responsiveness to As2O3. The analysis shows the clustering of 58 NCI tumor cell lines.

TABLE 3. Separation of clusters of NCI cell lines obtained by hierarchical cluster analyses for As2O3.
Gene Promoter Analysis for Transcription Factor Binding Motifs
Using the computational method of Galaxy Cistrome analysis, we investigated DNA promoter sequences of deregulated genes found in NCI microarray data. Forty genes were uploaded to Galaxy Cistrome for SeqPos motif scan. These genes are contribute in cell cycle progression and proliferation, cytokinesis, metabolic pathways, cell shape, apoptosis and signal transduction. The top 20 pronounced motifs are illustrated in Table 4. Interestingly, AP-1 and NF-κB were among the top transcription factors that possibly bind to the promoters with z-scores of -5.456 and -5.268, respectively. Both factors play critical role in the cancer therapy.
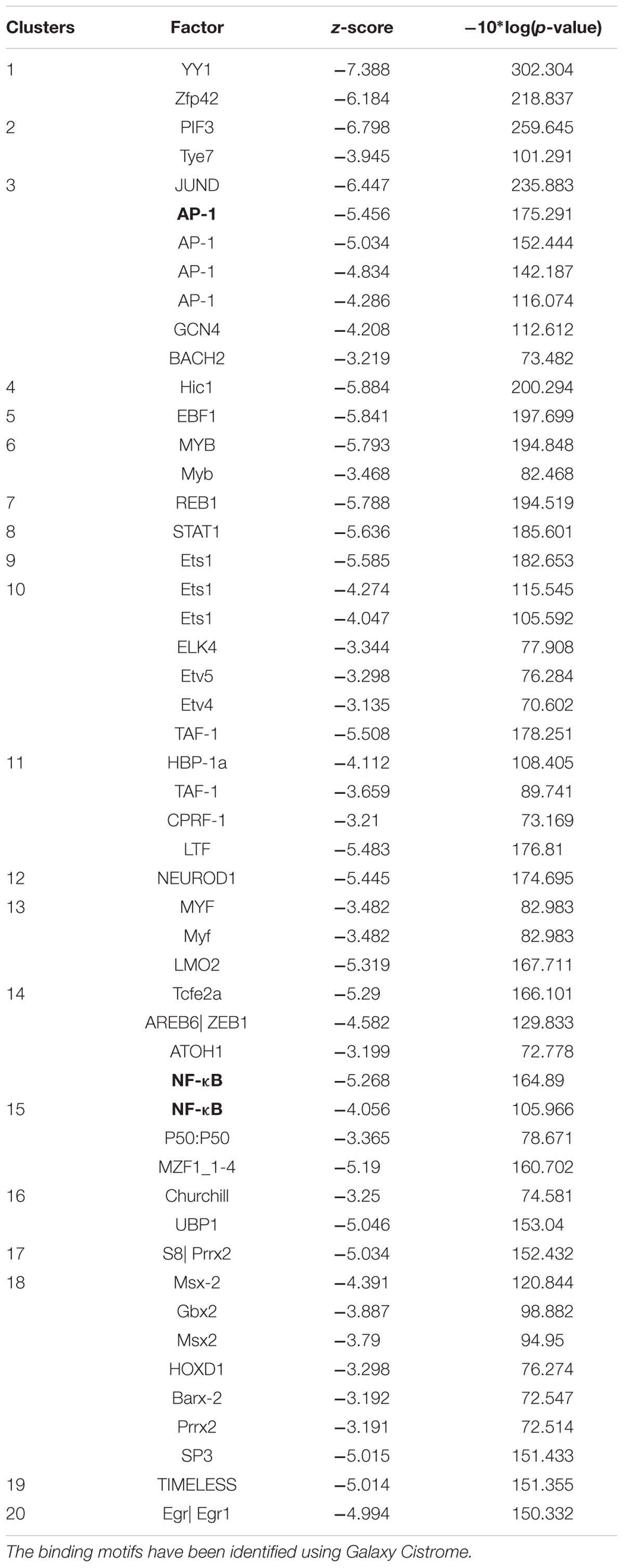
TABLE 4. Top 20 transcription factor promoter binding motifs for genes identified by COMPARE analysis (see Table 3).
AP-1 Reporter Assay
Based on this finding, we assume that As2O3 may affect AP-1. To verify this hypothesis, we generated an AP-1 reporter cell line. The transfected HEK293 was incubated with five different concentrations of As2O3 to find out, whether AP-1 promoter activity was inhibited. Treatment of transfected cells with As2O3 significantly inhibited the PMA-mediated AP-1 activity in a dose-dependent manner as shown in Figure 3.
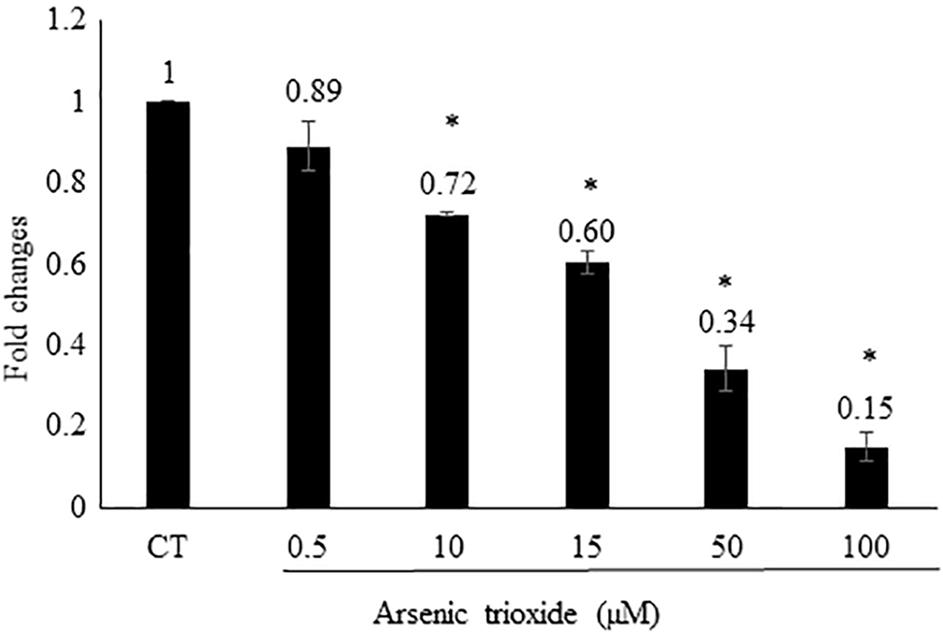
FIGURE 3. Effect of As2O3 on transfected HEK-293 cells with an AP1 luciferase reporter construct. The AP1 reporter cell line was treated with five different concentrations of As2O3. The quantification was done after 24 h incubation in comparison to untreated control cells (CT). Mean value standard deviation of triplicate measurements are shown. ∗Indicates statistical significant in compare to control cells at level of P < 0.05.
NF-κB Reporter Assay
Interestingly, the capability of As2O3 to affect NF-κB is debatable point in the literature. As2O3 can act as activator or inhibitor NF-κB (Woo et al., 2004; Han et al., 2005). From our motif analysis, we obtained NF-κB among the top 20 transcription factors. Therefore, we investigated whether As2O3 is able to inhibit NF-κB signaling. To achieve this, we performed a SEAP-driven NF-κB reporter cell line. Indeed, As2O3 inhibit NF-κB in a comparable manner as the NF-κB inhibitor, triptolide. The fold change was statistically significant for three As2O3 concentrations (10, 15, and 50 μM) as demonstrated in Figure 4.
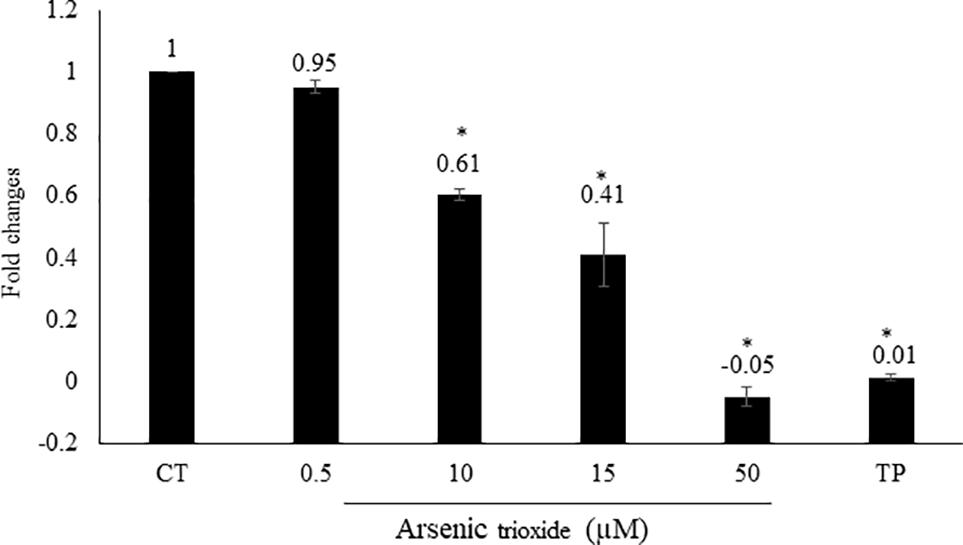
FIGURE 4. Effect of As2O3 on NF-κB activity. The HEK-BlueTM cell line was treated with four different concentrations of As2O3 or 1 μM triptolide (TP) as positive control. The quantification was done after 1 h incubation in comparison to untreated control cells (CT). Mean value standard deviation of triplicate measurements are shown. ∗Indicates statistical significant in compare to control cells at level of P < 0.05.
Interactome Network Analysis
For network analysis, we subjected the transcriptomic data of our present and previous investigations on As2O3 (Efferth and Kaina, 2004) as well as of factors determining cellular responsiveness to As2O3, which are known from the literature. In addition to the well-known target of As2O3 in APL, the PML-RARAα fusion protein (Goto et al., 2011; Tomita et al., 2013; Chendamarai et al., 2015; Lou et al., 2015; Liu J. et al., 2016), numerous other determinants of resistance have been described in the literature (Table 5). Firstly, we performed a manually curated analysis of the genes by visual inspection of the genes and assigning then to their specific functions. The main functional groups were oxidative stress response, DNA repair, regulation of cell cycle/proliferation, signal transduction, drug transporter, cytoskeletal elements, tumor suppressors/oncogenes, metabolic pathways, and apoptosis. Based on this analysis, a presumable integrative interaction network was constructed (Figure 5).

TABLE 5. Compilation of determinants of cellular responsiveness of cancer cells toward As2O3.
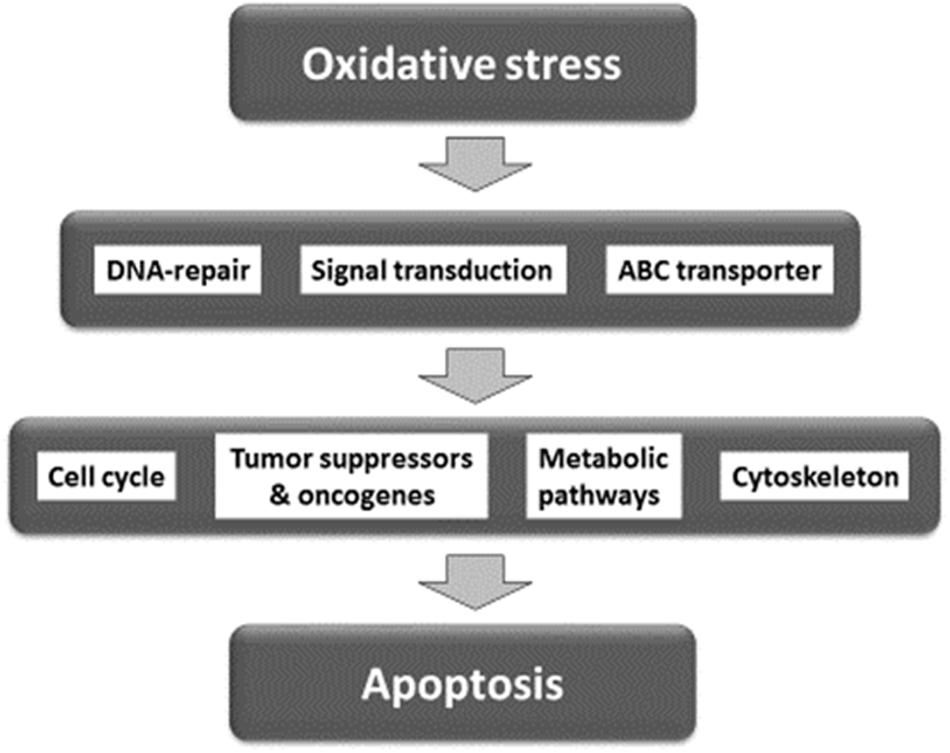
FIGURE 5. Synopsis of mechanisms involved in the activity of As2O3 against cancer cells.
As a second step, we subjected the assembled genes to Ingenuity Pathway Analysis. The interactome map obtained was rather complex (Figure 6). Therefore, we refined the analysis by analyzing the canonical pathways. The identified pathways belonged to signal transduction (phospholipase C signaling, protein kinase A signaling, signaling by Rho family GTPases, actin cytoskeleton signaling), molecular mechanisms of cancer, neurological pathways (axonal guidance signaling, neuroregulin signaling, agrin interactions with neuromuscular junctions), PXR/RXR activation, IL-7 signaling pathway and androgen signaling (Figure 7). The significance of this pathway are presented as –log (p-value) in Figure 8.
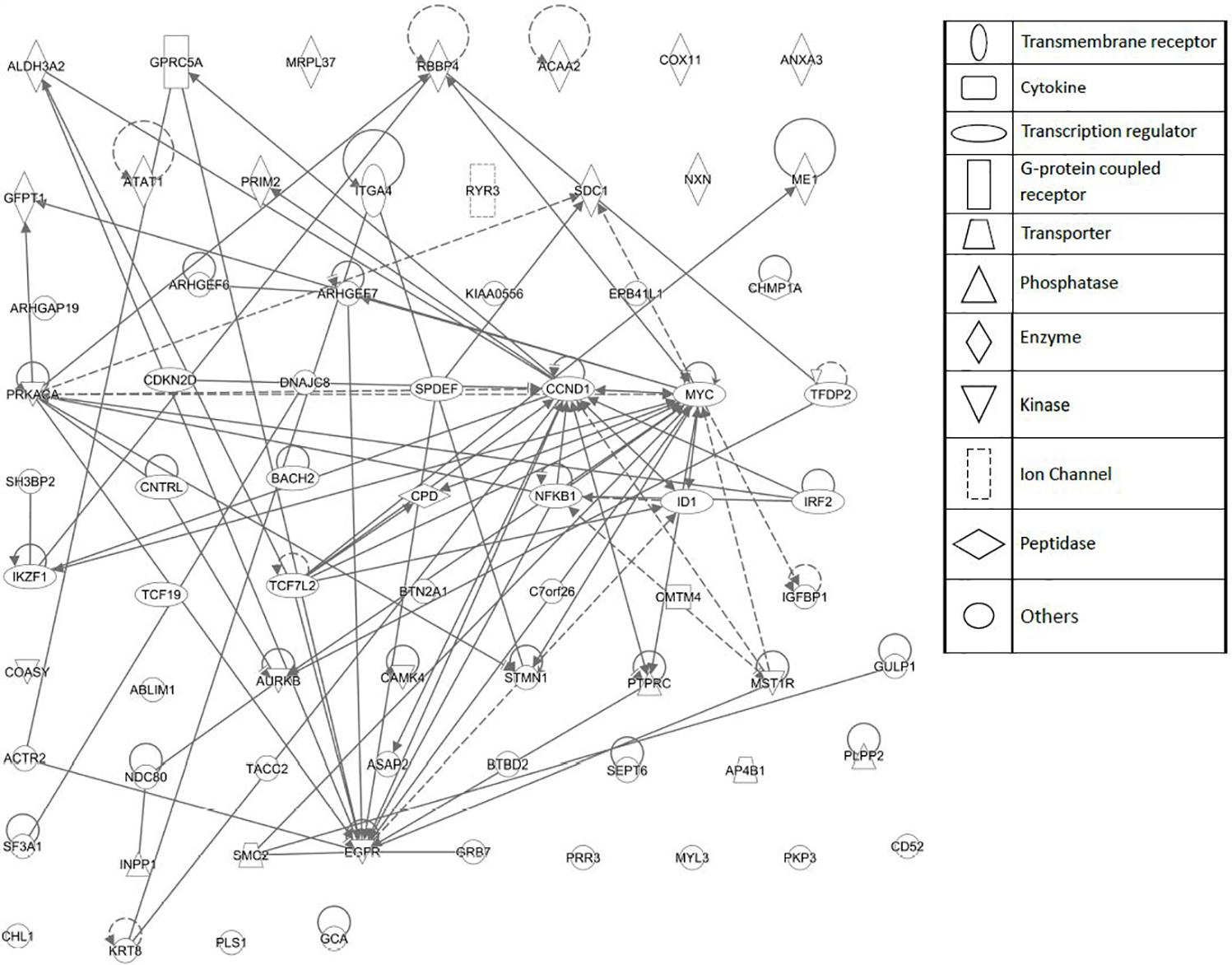
FIGURE 6. Network pharmacology for genes determining cellular responsiveness to As2O3 using Ingenuity Pathway Analysis. Dashed and solid lines indicates indirect and direct interactions, respectively.
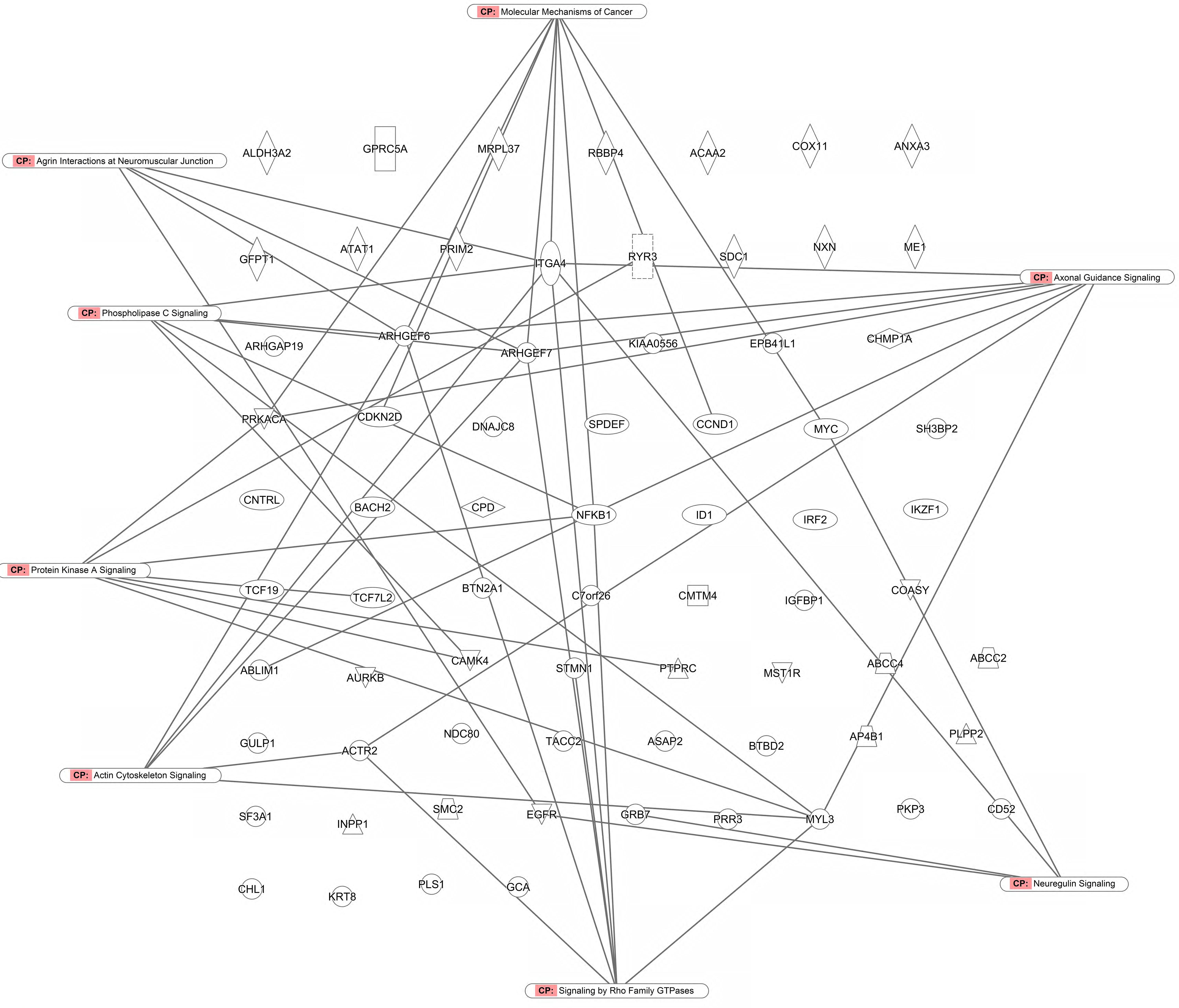
FIGURE 7. Canonical pathway analysis for genes determining cellular responsiveness to As2O3 using Ingenuity Pathway Analysis.
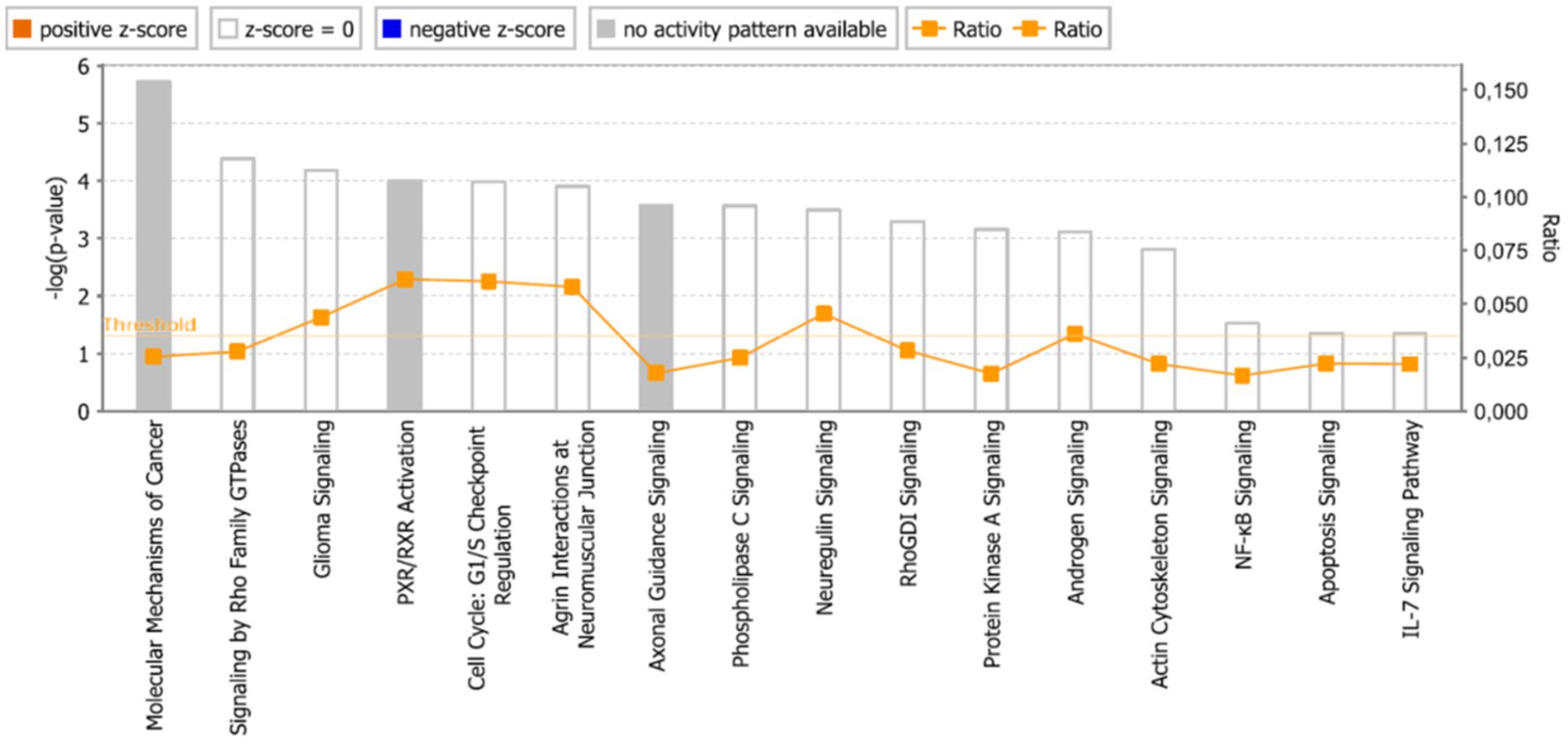
FIGURE 8. Pathways identified by IPA. Significance P-values were calculated based on the Fisher’s right tailed exact test. The –log (p-value) are shown on the y-axis of the bar chart. The color of the bars indicates the activity (orange bars) or the inhibition (blue bars) of the predicted pathways. In this analysis only significant results was shown. By default IPA applies a –log (p-value) cutoff of 1.3 (threshold). Pathways with a p-value equal to or greater than (less significant than) 0.05 are not shown. The orange and blue colored bars indicate predicted pathway activation, or predicted inhibition, respectively (z-score). White bars are those with a z-score at or very close to 0. Gray bars indicate pathways, where no prediction can currently be made.
In addition, cell death and survival, cell cycle, cell morphology, cellular movement, cellular development were identified by IPA as biological functions from our selected genes (Figure 9). A network of 36 selected genes that involve in the cell death (apoptosis) as the top biological function is shown in Figure 10.
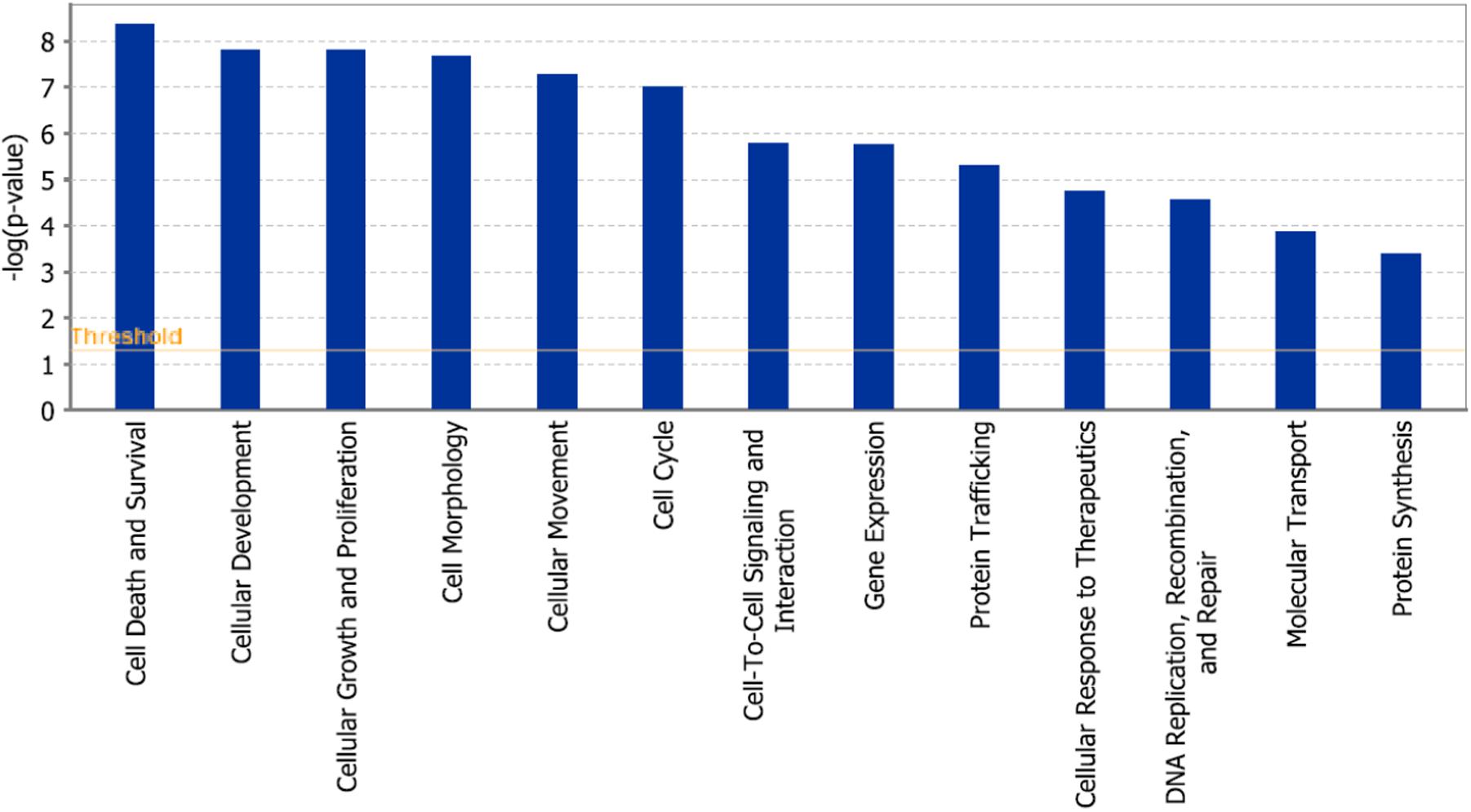
FIGURE 9. Statistically significant biological function analysis using IPA.
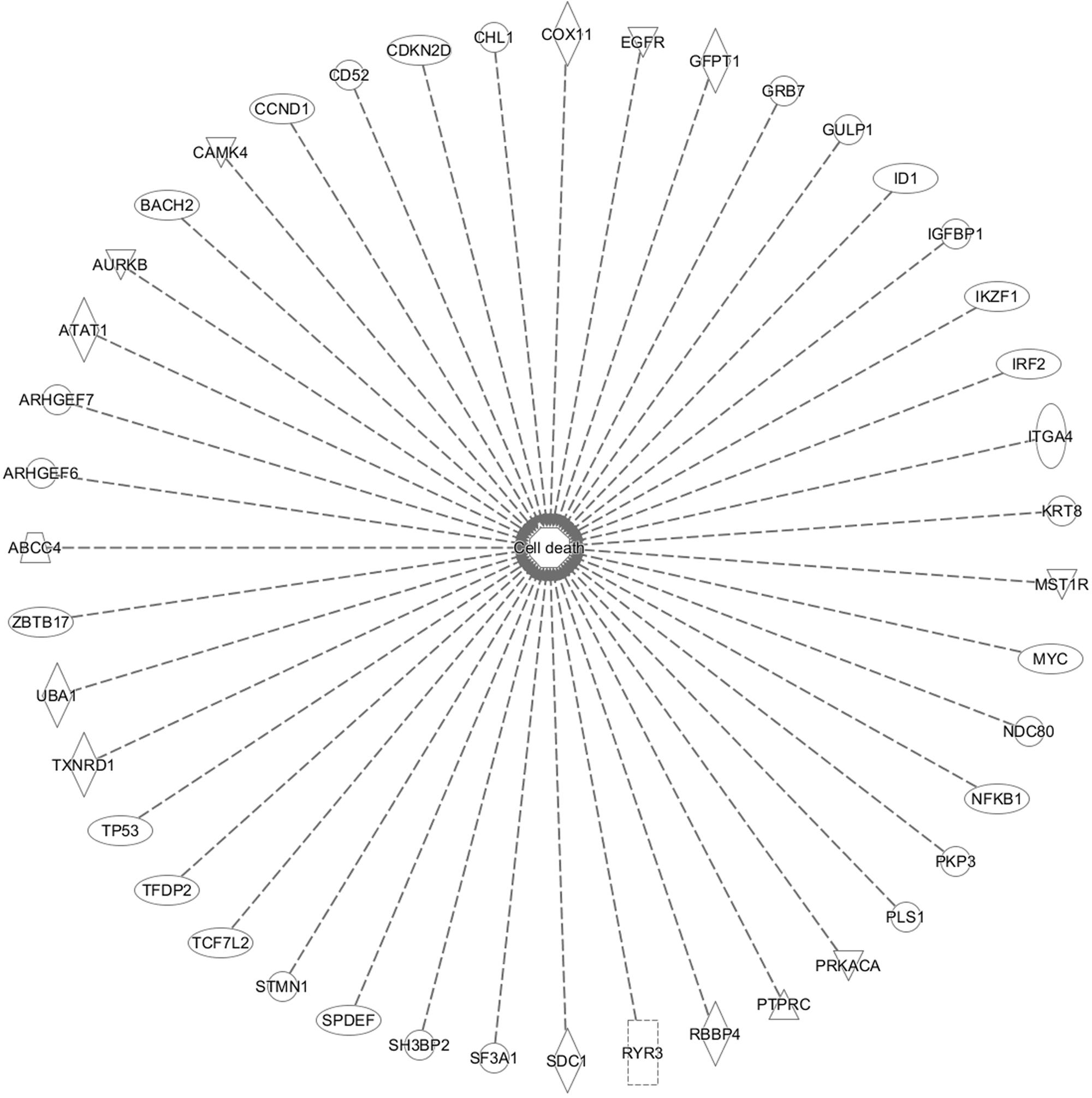
FIGURE 10. Set of 43 genes that involve in cell death function. This gene is duplicated on the inserted dataset.
While the importance of signaling processes was obvious by both visually and IPA-guided analyses, both approaches revealed additional functional groups complementing each other and highlighting the complexity of mechanisms contributing to the cytotoxicity of As2O3.
Discussion
In the current study, we investigated the potential of As2O3 to treat drug-resistant tumor cells. Tumors frequently develop resistance to a broad spectrum of anti-cancer drugs (Efferth et al., 2008). Therefore, drug resistance represents an important obstacle to defeat cancer. In this study, we showed that As2O3 revealed profound cytotoxicity toward various cancer cell lines.
Our results showed that multidrug-resistant CEM/ADR5000 cells which overexpress P-glycoprotein did not reveal cross-resistance to As2O3. Therefore, P-glycoprotein did not play an important role for resistance cellular responsiveness to As2O3. Our results are in accordance with those of (Hu et al., 2003), who also did not find cross-resistance of multidrug-resistant P-glycoprotein-expressing cell lines toward As2O3. This finding is also corroborated by data generated by Sertel et al. (2012). As2O3 counteracted multidrug resistance by interference with drug resistance genes. The expression of DNA topoisomerase 2 was increased and of glutathione S-transferase-π and Bcl-2 decreased upon treatment with As2O3 (Zhao et al., 2011). Other ATP-binding cassette (ABC) transporters than P-glycoprotein might be more important for As2O3. Treatment with As2O3 lead to the induction of ABCC1, but not P-glycoprotein (Seo et al., 2007) and As2O3-resistant cell lines overexpressed P-glycoprotein but also ABCC1, ABCC2, and ABCC4 (Chen et al., 2009, 2016; Yuan et al., 2016). ABCC1-overexpressing tumor cells accumulated less As2O3 than wild-type cells (Salerno and Garnier-Suillerot, 2003).
To the best of our knowledge, the anti-proliferative activities of As2O3 and particularly its effect against p53 knockout HCT116 (p53-/-) colon carcinoma and EGRF mutant U87.MG ΔEGFR glioblastoma multiforme cell lines are being tested for the first time.
TP53 maintains cellular integrity by regulating cell cycle arrest, DNA repair and apoptosis. Functional loss by TP53 mutations lead not only to carcinogenesis, but to drug and radio resistance too (Hientz et al., 2017). In our investigation, HCT116 (p53-/-) knockout cells exhibited resistance toward As2O3. Our data are in agreement with other investigations with isogenic cell lines (Yan et al., 2011; Zhang et al., 2013), but not with cell lines with different p53 status and different genomic background (Liu et al., 2003; Zhao et al., 2008). This indicates that the p53 mutational status in correlation studies with different cell lines may be overruled by other mechanisms of resistance to As2O3. Therefore, the targeted loss of p53 function by knockout was more suitable to undoubtedly elucidate the role of p53 as resistance factor for As2O3. A role of p53 mutations can also be deduced from cells resistant to As2O3, which acquired p53 mutations and which were re-sensitized to As2O3 by a small molecule inhibitor specifically targeting mutant p53, nutlin-3 (Zheng et al., 2014).
Furthermore, we found that U87.MG cells showed collateral sensitivity to As2O3. EGFR plays a significant role in cancer progression, not only by transducing growth signals into cancer cells, but also by triggering invasion and metastasis (Baselga, 2002; Lui and Grandis, 2002). Inhibition of EGFR leads to the induction of apoptosis. This may explain, how As2O3 suppresses EGFR-expressing tumor cells (Efferth and Kaina, 2004). The phenomenon of collateral sensitivity (hypersensitivity) offers an attractive strategy to overcome drug resistance (Pluchino et al., 2012). Moreover, collateral sensitivity may open the chance for combination therapy of known anticancer drugs with As2O3 to efficiently kill drug- resistant tumors with EGFR over expression. The reason for the collateral sensitivity is not yet known, but a direct interaction of As2O3 with EGFR could be suggested, since it has been reported that As2O3 down-regulated EGFR expression in a dose-dependent manner (Zhang et al., 2012).
We further evaluated the molecular factors of sensitivity and resistance of cancer cell lines toward As2O3. We correlated the log10IC50-values with the microarray- based mRNA expression profiles of 58 tumor cell lines by COMPARE analysis. Genes from diverse functional groups have been identified. Previously, the effect of As2O3 on mRNA expression profile of NCI cell line panel have been reported (Efferth and Kaina, 2004; Sertel et al., 2012). Here, we focused on the Novartis microarray platform. In all studies the chi-square test reveal a significant clustering of the cell lines based on their sensitivity or resistance toward As2O3. Moreover, several biological functions of the genes, e.g., cell cycle progression, and proliferation related genes, signal transduction and apoptosis-regulating gene were common in the previous and the present investigations.
Interestingly, the transcription factors binding motif analysis revealed two transcription factors (NF-κB and AP-1) which are closely connected not only to carcinogenesis but also with drug resistance (Boeckx et al., 2015). Therefore, we evaluated the potential of As2O3 to inhibit the activity of both AP-1 and NF-κB. NF-κB and AP-1 regulate numerous important cellular processes. NF-κB activity is regulated by IκB kinase, which phosphorylates the NF-κB inhibitor IκB. Fos is key constituent of AP-1 and is regulated by serum responsive factors (SRFs) as well as ternary complex factors (TCFs). AP-1 regulates cellular growth, differentiation, program cell death, transformation and invasion (McDonnell et al., 1990; Szabo et al., 1991; Brown et al., 1993). AP-1 complexes, particularly those that contain c-Jun, achieve their growth promoting function via repression of tumor suppressor genes, such as p53, p21cip1/waf1 and p16 (Shaulian and Karin, 2001). Although NF-κB and AP-1 are two distinct regulatory mechanisms, they may modulate each other (Fujioka et al., 2004).
Our results that As2O3 inhibited the DNA-binding activity in a reporter cell model are in accordance with other reports demonstrating that As2O3 inhibited NF-κB activity (Han et al., 2005; Wei et al., 2005; Mathieu and Besancon, 2006; Ghaffari et al., 2016). The ectopic expression of NF-κB p65 protected cells from As2O3 induced apoptosis (Ghaffari et al., 2016), indicating that the NF-κB pathway mediates resistance to As2O3.
Activator protein 1 (AP-1) is a transcription factor belongs to the group of trans-acting elements that consist of several proteins, including JUN, FOS, ATF (activating transcription factor) and MAF (musculoaponeurotic fibrosarcoma). AP-1 forms homodimers or heterodimers via its leucine-zipper domains, different dimerization ultimately allow that AP-1 recognizes different sequences in the promoters and enhancers of target genes (Eferl and Wagner, 2003). The AP-1 activity was causatively linked to cell transformation and it assumed to be a significant factor for tumorigenesis (Kharman-Biz et al., 2013). Therefore, AP-1 might provide opportunities for the development of novel targeted cancer treatment strategies (Angel and Karin, 1991; Milde-Langosch et al., 2004; Kharman-Biz et al., 2013). Interestingly, AP-1 drives not only carcinogenesis, but also increases the expression of multidrug resistance-conferring MDR1 gene (Daschner et al., 1999). Therefore, c-Jun is a potential drug target to combat MDR (Passegue and Wagner, 2000).
Using an AP-1-specific reporter cell line, we found that As2O3 inhibited AP-1 activity. This result fits to data of (Yedjou and Tchounwou, 2009), who observed that the As2O3-induced cell cycle arrest was correlated with repression of c-Fos (as component of the AP-1 heterodimeric complex). In another study, As2O3 treatment also downregulated c-Fos expression (Walker et al., 2016). The second partner of AP-1, c-Jun, was more phosphorylated is As2O3-resistant cells and hypophosphorylated in As2O3-sensitive cells upon As2O3 treatment (Roszak et al., 2013).
Nuclear factor (NF-κB) is another transcription factor responsible for expression regulation of genes involve in apoptosis, drug resistance, innate and adaptive immune responses, proliferation and metastasis (Guttridge et al., 1999). It exists as inactive form in the cytoplasm by interacting with inhibitory proteins (IκB). Phosphorylation of IκB by IκB kinase (IKK) activates NF-κB. Then, NF-κB is translocated to the nucleus, where it binds to specific gene promoters to trans-activate the expression of target genes (Baud and Karin, 2009). NF-κB is constitutively active in hematologic malignancies and various types of solid tumors (Bharti et al., 2003). Besides, cancer cells become resistant to antitumor drugs and irradiations by NF-κB activation (Baud and Karin, 2009). Furthermore, decreased expression of anti-apoptotic genes is correlated with inhibition of NF-κB (Lin et al., 2010). Taken together the inhibition of NF-κB represents a promising target for cancer prevention and therapy (Nakanishi and Toi, 2005; Lin et al., 2010).
Several studies described that As2O3 exerts its anti-tumor activity by inhibiting AP-1 (Um et al., 2002; Brown et al., 2007). However, other studies showed that As2O3 stimulated AP-1 activity in cultured human fibroblasts and APL (Burnichon et al., 2003; Jang et al., 2012). This contradiction points to the fact that many arsenic compounds reveal both anti-cancer and pro-cancer activates. In this study, we used HEK293 cell lines transfected with AP-1 luciferase reporter construct to detect the activity of AP-1 after As2O3 treatment. We showed that As2O3 inhibit AP-1 in dose- dependent manner and this in line with previous study in HeLa, CaSki, and C33A cells (Wang et al., 2014).
On the other hand, many suggestions have been put forth on the mechanisms, by which As2O3 cause apoptosis. One of the proposed explanations is that As2O3 inhibits the NF-κB activity. It induces genes involve in cell survival, tumor growth, blocking apoptosis and metastasis (Berman et al., 2002). NF-κB up-regulates both intracellular adhesion molecules and vascular cell adhesion, and inhibits IL-6 secretion resulting from adhesion of multiple myeloma cells to bone marrow stroma (Miller et al., 2002).
Our results from NF-κB reporter assays are in line with other studies demonstrating that As2O3 significantly inhibits NF-κB activity in a dose- dependent manner. In HL-60 cells, As2O3 suppressed the DNA-binding activity of the NF-κB heterodimer (p65/p50) through inhibiting the degradation of IκB-α and then preventing nuclear translocation of p65 and the binding of NF-κB with their target gene consensus sequences (Han et al., 2005).
Although it is well known that natural products act rather in a multi-specific manner than mono-specifically against one single target (Efferth and Koch, 2011), it turns out more and more that synthetic drugs and even targeted small molecules affect multiple targets and pathways in diseased cells (Geromichalos et al., 2016). Therefore, network pharmacological approaches have been suggested to provide a more comprising picture of what is happening in cell upon drug treatment (Poornima et al., 2016). Hence, network pharmacology represents a valuable tool to investigate the mode of action of both synthetic and natural compounds. A conclusion that can be drawn from our network analysis of As2O3 is that rather entire gene clusters belonging to specific pathways and functional groups than single genes were involved in drug action. Given the individual differences of organisms and the biological flexibility to adapt to xenobiotic exposures, it is reasonable from an evolutionary point of view to assume cellular and molecular reactions of high flexibility and plasticity. The concept of the past (“one drug – one target”) obviously needs to be revisited and enlarged by a “one drug – multiple pathway” hypothesis.
The international sequencing projects (e.g., Cancer Genome Atlas,) unraveled a huge diversity of mutational profiles and gene expression patterns in individual tumors (Chin and Gray, 2008). Hence, it is reasonable that each patient’s tumor might react in a different manner to chemotherapy. In the case of As2O3, we found several major functional groups using manual curated network analysis to be involved in cellular responsiveness to the drug, i.e., oxidative stress response, DNA repair, regulation of cell cycle/proliferation, signal transduction, drug transporter, cytoskeletal elements, tumor suppressors/oncogenes, metabolic pathways, and apoptosis. On the other hand, we found many other important pathways using IPA network analysis, e.g., molecular mechanisms of cancer, axonal guidance signaling, signaling by Rho family GTPases, protein kinase A signaling, phospholipase signaling, actin cytoskeleton signaling etc. The manually curated and the IPA approaches revealed comparable findings for three types of pathways: signal transduction, metabolic pathways and cytoskeletal elements. Moreover, axonal guidance signaling, neuroregulin signaling and agrin interaction at neuromuscular junction pathways may demonstrate the neurotoxicity of As2O3. This is an interesting finding, since neurotoxicity is a known toxicity of arsenic (Vahidnia et al., 2007). Our IPA analysis identified three canonical pathways that might explain neurotoxic effects of As2O3. Furthermore, neurological effects of arsenic may lead to cytoskeletal framework disorganization which is a suggested mechanism of arsenic-induced neurotoxicity (Vahidnia et al., 2007). Interestingly, IPA network analysis also revealed several pathways which are directly connected with the cytoskeleton in addition to glioma signaling. It deserves further investigations, whether these pathways are also involved in the collateral sensitivity of the U87.MG. ΔEGFR glioblastoma cells to As2O3. Not surprisingly, IPA showed cell death and survival as top biological functions of the inserted genes with a p-value of 4.28E-09 and with 43 genes that contribute to this function.
Having the inter- and intra-tumor heterogeneity in mind, it is reasonable to assume that different tumors activate different genes belonging to the same main pathways and functional groups. The complexity of this process can be manage by network modeling, and corresponding bioinformatical tools are becoming more and more indispensable for pharmacology of both synthetic and natural drugs.
In conclusion, As2O3 was active against tumor cell lines. It showed collateral sensitivity toward U87.MG ΔEGFR. In addition to that, it inhibited AP-1 and NF-κB activity in a dose- dependent manner. COMPARE and cluster analyses using gene expression profiling of 58 NCI cell lines predicted the sensitivity or resistance of cancer cells to As2O3. Using a network pharmacological approach, we identified functional clusters of genes involved in the mechanisms of action in cancer cells.
Author Contributions
MD performed the cytotoxicity, COMPARE and hierarchical cluster analyses, promoter binding motif analysis. NF-KB and AP-1 reporter assays, Interactome network analysis, and drafted the paper. SH performed the cytotoxicity and drafted the paper. TE supervised the project and corrected the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful to the Ph.D. stipend provided to MD by the Ministry of Higher Education and Scientific Research as well as the Al-Neelain University, Khartoum, Sudan.
Abbreviation
APL, acute promyelocytic leukemia; AP-1, activator protein 1; As2O3, arsenic trioxide; EGFR, epidermal growth factor receptor; NF-κB, nuclear factor-kappa in B cells; PMA, phorbol 12-myristate 13-acetate; PML, promyelocytic leukemia; RARA, retinoic acid receptor alpha; SEAP, secreted embryonic alkaline phosphatase; TNF, tumor necrosis factor.
Footnotes
- ^ http://dtp.nci.nih.gov
- ^ http://dtp.cancer.gov/databases_tools/default.htm
- ^ http://dtp.nci.nih.gov
- ^ https://discover.nci.nih.gov/cimminer/oneMatrix.do
- ^ http://genome.ucsc.edu
References
Alvarez, M., Paull, K., Monks, A., Hose, C., Lee, J.-S., Weinstein, J., et al. (1995). Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the national cancer institute anticancer drug screen. J. Clin. Invest. 95, 2205–2214. doi: 10.1172/JCI117910
Amigo-Jimenez, I., Bailon, E., Aguilera-Montilla, N., Terol, M. J., Garcia-Marco, J. A., and Garcia-Pardo, A. (2015). Bone marrow stroma-induced resistance of chronic lymphocytic leukemia cells to arsenic trioxide involves Mcl-1 upregulation and is overcome by inhibiting the PI3Kdelta or PKCbeta signaling pathways. Oncotarget 6, 44832–44848. doi: 10.18632/oncotarget.6265
Amundson, S. A., Do, K. T., Vinikoor, L. C., Lee, R. A., Koch-Paiz, C. A., Ahn, J., et al. (2008). Integrating global gene expression and radiation survival parameters across the 60 cell lines of the national cancer institute anticancer drug screen. Cancer Res. 68, 415–424. doi: 10.1158/0008-5472.CAN-07-2120
Anderson, K. C., Boise, L. H., Louie, R., and Waxman, S. (2002). Arsenic trioxide in multiple myeloma: rationale and future directions. Cancer J. 8, 12–25. doi: 10.1097/00130404-200201000-00003
Angel, P., and Karin, M. (1991). The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072, 129–157. doi: 10.1016/0304-419X(91)90011-9
Baselga, J. (2002). Why the epidermal growth factor receptor? The rationale for cancer therapy. Oncologist 7(Suppl. 4), 2–8. doi: 10.1634/theoncologist.7-suppl_4-2
Baud, V., and Karin, M. (2009). Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8, 33–40. doi: 10.1038/nrd2781
Beauchamp, E. M., and Uren, A. (2012). A new era for an ancient drug: arsenic trioxide and hedgehog signaling. Vitam. Horm. 88, 333–354. doi: 10.1016/B978-0-12-394622-5.00015-8
Berman, K. S., Verma, U. N., Harburg, G., Minna, J. D., Cobb, M. H., and Gaynor, R. B. (2002). Sulindac enhances tumor necrosis factor-alpha-mediated apoptosis of lung cancer cell lines by inhibition of nuclear factor-κB. Clin. Cancer Res. 8, 354–360.
Bharti, A. C., Donato, N., Singh, S., and Aggarwal, B. B. (2003). Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-κ B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 101, 1053–1062. doi: 10.1182/blood-2002-05-1320
Boeckx, C., Blockx, L., De Beeck, K. O., Limame, R., Camp, G. V., Peeters, M., et al. (2015). Establishment and characterization of cetuximab resistant head and neck squamous cell carcinoma cell lines: focus on the contribution of the AP-1 transcription factor. Am. J. Cancer Res. 5, 1921–1938.
Breccia, M., and Lo-Coco, F. (2012). Arsenic trioxide for management of acute promyelocytic leukemia: current evidence on its role in front-line therapy and recurrent disease. Expert Opin. Pharmacother. 13, 1031–1043. doi: 10.1517/14656566.2012.677436
Brown, M., Bellon, M., and Nicot, C. (2007). Emodin and DHA potently increase arsenic trioxide interferon-alpha-induced cell death of HTLV-I-transformed cells by generation of reactive oxygen species and inhibition of Akt and AP-1. Blood 109, 1653–1659. doi: 10.1182/blood-2006-04-015537
Brown, P. H., Alani, R., Preis, L. H., Szabo, E., and Birrer, M. J. (1993). Suppression of oncogene-induced transformation by a deletion mutant of c-jun. Oncogene 8, 877–886.
Bunz, F., Dutriaux, A., Lengauer, C., Waldman, T., Zhou, S., Brown, J. P., et al. (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501. doi: 10.1126/science.282.5393.1497
Burnichon, V., Jean, S., Bellon, L., Maraninchi, M., Bideau, C., Orsiere, T., et al. (2003). Patterns of gene expressions induced by arsenic trioxide in cultured human fibroblasts. Toxicol. Lett. 143, 155–162. doi: 10.1016/S0378-4274(03)00171-1
Chandan, K., Van Iersel, M. P., Aladjem, M. I., Kohn, K. W., and Luna, A. (2012). PathVisio-Validator: a rule-based validation plugin for graphical pathway notations. Bioinformatics 28, 889–890. doi: 10.1093/bioinformatics/btr694
Chen, J., Cheng, J., Yi, J., Xie, B., Lin, L., Liu, Z., et al. (2016). Differential expression and response to arsenic stress of MRPs and ASAN1 determine sensitivity of classical multidrug-resistant leukemia cells to arsenic trioxide. Leuk. Res. 50, 116–122. doi: 10.1016/j.leukres.2016.10.003
Chen, X., Zhang, M., and Liu, L. X. (2009). The overexpression of multidrug resistance-associated proteins and gankyrin contribute to arsenic trioxide resistance in liver and gastric cancer cells. Oncol. Rep. 22, 73–80.
Chendamarai, E., Ganesan, S., Alex, A. A., Kamath, V., Nair, S. C., Nellickal, A. J., et al. (2015). Comparison of newly diagnosed and relapsed patients with acute promyelocytic leukemia treated with arsenic trioxide: insight into mechanisms of resistance. PLoS One 10:e0121912. doi: 10.1371/journal.pone.0121912
Chin, L., and Gray, J. W. (2008). Translating insights from the cancer genome into clinical practice. Nature 452, 553–563. doi: 10.1038/nature06914
Cree, I. A., and Charlton, P. (2017). Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 17:10. doi: 10.1186/s12885-016-2999-1
Daschner, P. J., Ciolino, H. P., Plouzek, C. A., and Yeh, G. C. (1999). Increased AP-1 activity in drug resistant human breast cancer MCF-7 cells. Breast Cancer Res. Treat. 53, 229–240. doi: 10.1023/A:1006138803392
Davison, K., Cote, S., Mader, S., and Miller, W. H. (2003). Glutathione depletion overcomes resistance to arsenic trioxide in arsenic-resistant cell lines. Leukemia 17, 931–940. doi: 10.1038/sj.leu.2402876
Dos Santos, B. S., da Silva, L. C., da Silva, T. D., Rodrigues, J. F., Grisotto, M. A., Correia, M. T., et al. (2016). Application of omics technologies for evaluation of antibacterial mechanisms of action of plant-derived products. Front. Microbiol. 7:1466. doi: 10.3389/fmicb.2016.01466
Eferl, R., and Wagner, E. F. (2003). AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868. doi: 10.1038/nrc1209
Efferth, T., Fabry, U., and Osieka, R. (1997). Apoptosis and resistance to daunorubicin in human leukemic cells. Leukemia 11, 1180–1186. doi: 10.1038/sj.leu.2400669
Efferth, T., and Koch, E. (2011). Complex interactions between phytochemicals. The multi-target therapeutic concept of phytotherapy. Curr. Drug Targets 12, 122–132. doi: 10.2174/138945011793591626
Efferth, T., Konkimalla, V. B., Wang, Y. F., Sauerbrey, A., Meinhardt, S., Zintl, F., et al. (2008). Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res. 14, 2405–2412. doi: 10.1158/1078-0432.CCR-07-4525
Efferth, T., Saeed, M. E. M., Mirghani, E., Alim, A., Yassin, Z., Saeed, E., et al. (2017). Integration of phytochemicals and phytotherapy into cancer precision medicine. Oncotarget 8, 50284–50304. doi: 10.18632/oncotarget.17466
Efferth, T., Sauerbrey, A., Olbrich, A., Gebhart, E., Rauch, P., Weber, H. O., et al. (2003). Molecular modes of action of artesunate in tumor cell lines. Mol. Pharmacol. 64, 382–394. doi: 10.1124/mol.64.2.382
Efferth, T, and Kaina, B. (2004). Microarray-based prediction of cytotoxicity of tumor cells to arsenic trioxide. Cancer Genomics Proteomics 1, 363–370.
Eisen, M. B., Spellman, P. T., Brown, P. O., and Botstein, D. (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U.S.A. 95, 14863–14868. doi: 10.1073/pnas.95.25.14863
Evans, A., Bates, V., Troy, H., Hewitt, S., Holbeck, S., Chung, Y.-L., et al. (2008). Glut-1 as a therapeutic target: increased chemoresistance and HIF-1-independent link with cell turnover is revealed through COMPARE analysis and metabolomic studies. Cancer Chemother. Pharmacol. 61, 377–393. doi: 10.1007/s00280-007-0480-1
Fagan, V., Bonham, S., Carty, M. P., Saenz-Méndez, P., Eriksson, L. A., and Aldabbagh, F. (2012). COMPARE analysis of the toxicity of an iminoquinone derivative of the imidazo [5, 4-f] benzimidazoles with NAD (P) H: quinone oxidoreductase 1 (Nqo1) activity and computational docking of quinones as NQO1 substrates. Bioorg. Med. Chem. 20, 3223–3232. doi: 10.1016/j.bmc.2012.03.063
Fang, J., Liu, C., Wang, Q., Lin, P., and Cheng, F. (2017). In silico polypharmacology of natural products. Brief Bioinform. doi: 10.1093/bib/bbx045 [Epub ahead of print].
Ferlay, J., Soerjomataram, I., Dikshit, R., Eser, S., Mathers, C., Rebelo, M., et al. (2015). Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386. doi: 10.1002/ijc.29210
Fujioka, S., Niu, J., Schmidt, C., Sclabas, G. M., Peng, B., Uwagawa, T., et al. (2004). NF-κB and AP-1 connection: mechanism of NF-κB-dependent regulation of AP-1 activity. Mol. Cell. Biol. 24, 7806–7819. doi: 10.1128/MCB.24.17.7806-7819.2004
Funahashi, A., Morohashi, M., Kitano, H., and Tanimura, N. (2003). CellDesigner: a process diagram editor for gene-regulatory and biochemical networks. Biosilico 1, 159–162. doi: 10.1016/S1478-5382(03)02370-9
Ganter, B., and Giroux, C. N. (2008). Emerging applications of network and pathway analysis in drug discovery and development. Curr. Opin. Drug Discov. Dev. 11, 86–94.
Geromichalos, G. D., Alifieris, C. E., Geromichalou, E. G., and Trafalis, D. T. (2016). Overview on the current status of virtual high-throughput screening and combinatorial chemistry approaches in multi-target anticancer drug discovery; Part I. J. BUON 21, 764–779.
Ghaffari, S. H., Yousefi, M., Dizaji, M. Z., Momeny, M., Bashash, D., Zekri, A., et al. (2016). Arsenic trioxide induces apoptosis and incapacitates proliferation and invasive properties of U87MG glioblastoma cells through a possible NF-κB-mediated mechanism. Asian Pac. J. Cancer Prev. 17, 1553–1564. doi: 10.7314/APJCP.2016.17.3.1553
Giommarelli, C., Corti, A., Supino, R., Favini, E., Paolicchi, A., Pompella, A., et al. (2009). gamma-Glutamyltransferase-dependent resistance to arsenic trioxide in melanoma cells and cellular sensitization by ascorbic acid. Free Radic. Biol. Med. 46, 1516–1526. doi: 10.1016/j.freeradbiomed.2009.03.006
Goto, E., Tomita, A., Hayakawa, F., Atsumi, A., Kiyoi, H., and Naoe, T. (2011). Missense mutations in PML-RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood 118, 1600–1609. doi: 10.1182/blood-2011-01-329433
Guilbert, J. J. (2001). The search for HR continues: WHO’s general programme of work 2002-2005 and the world health report 2001 on mental health. Educ. Health 14, 452–453.
Guttridge, D. C., Albanese, C., Reuther, J. Y., Pestell, R. G. and Baldwin, A. S. Jr. (1999). NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 19, 5785–5799. doi: 10.1128/MCB.19.8.5785
Hamdoun, S., and Efferth, T. (2017). Ginkgolic acids inhibit migration in breast cancer cells by inhibition of Nemo sumoylation and NF-κB activity. Oncotarget 8, 35103-35115 doi: 10.18632/oncotarget.16626
Han, S. S., Kim, K., Hahm, E. R., Park, C. H., Kimler, B. F., Lee, S. J., et al. (2005). Arsenic trioxide represses constitutive activation of NF-κB and COX-2 expression in human acute myeloid leukemia, HL-60. J. Cell. Biochem. 94, 695–707. doi: 10.1002/jcb.20337
Han, Y. H., Kim, S. Z., Kim, S. H., and Park, W. H. (2008). Induction of apoptosis in arsenic trioxide-treated lung cancer A549 cells by buthionine sulfoximine. Mol. Cells 26, 158–164.
Hayashi, T., Hideshima, T., Akiyama, M., Richardson, P., Schlossman, R. L., Chauhan, D., et al. (2002). Arsenic trioxide inhibits growth of human multiple myeloma cells in the bone marrow microenvironment. Mol. Cancer Ther. 1, 851–860.
Hientz, K., Mohr, A., Bhakta-Guha, D., and Efferth, T. (2017). The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 8, 8921–8946. doi: 10.18632/oncotarget.13475
Hour, T. C., Huang, C. Y., Lin, C. C., Chen, J., Guan, J. Y., Lee, J. M., et al. (2004). Characterization of molecular events in a series of bladder urothelial carcinoma cell lines with progressive resistance to arsenic trioxide. Anticancer Drugs 15, 779–785. doi: 10.1097/00001813-200409000-00007
Hu, X. M., Hirano, T., and Oka, K. (2003). Arsenic trioxide induces apoptosis equally in T lymphoblastoid leukemia MOLT-4 cells and P-gp-expressing daunorubicin-resistant MOLT-4 cells. Cancer Chemother. Pharmacol. 51, 119–126.
Huang, X. J., Wiernik, P. H., Klein, R. S., and Gallagher, R. E. (1999). Arsenic trioxide induces apoptosis of myeloid leukemia cells by activation of caspases. Med. Oncol. 16, 58–64. doi: 10.1007/BF02787360
Ideker, T., Thorsson, V., Ranish, J. A., Christmas, R., Buhler, J., Eng, J. K., et al. (2001). Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292, 929–934. doi: 10.1126/science.292.5518.929
Iland, H. J., Wei, A., and Seymour, J. F. (2014). Have all-trans retinoic acid and arsenic trioxide replaced all-trans retinoic acid and anthracyclines in APL as standard of care. Best Pract. Res. Clin. Haematol. 27, 39–52. doi: 10.1016/j.beha.2014.04.003
Jang, M., Kim, Y., Won, H., Lim, S., K R, J., Dashdorj, A., et al. (2012). Carbonyl reductase 1 offers a novel therapeutic target to enhance leukemia treatment by arsenic trioxide. Cancer Res. 72, 4214–4224. doi: 10.1158/0008-5472.CAN-12-1110
Jemal, A., Center, M. M., Desantis, C., and Ward, E. M. (2010). Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Biomarkers Prev. 19, 1893–1907. doi: 10.1158/1055-9965.EPI-10-0437
Kadioglu, O., and Efferth, T. (2014). Contributions from emerging transcriptomics technologies and computational strategies for drug discovery. Invest. New Drugs 32, 1316–1319. doi: 10.1007/s10637-014-0081-x
Kadioglu, O., and Efferth, T. (2015). Pharmacogenomic characterization of cytotoxic compounds from Salvia officinalis in cancer cells. J. Nat. Prod. 78, 762–775. doi: 10.1021/np501007n
Kadioglu, O., Fu, Y., Wiench, B., Zu, Y., and Efferth, T. (2016a). Synthetic cajanin stilbene acid derivatives inhibit c-MYC in breast cancer cells. Arch. Toxicol. 90, 575–588. doi: 10.1007/s00204-015-1480-2
Kadioglu, O., Jacob, S., Bohnert, S., Nass, J., Saeed, M. E., Khalid, H., et al. (2016b). Evaluating ancient Egyptian prescriptions today: ANti-inflammatory activity of Ziziphus spina-christi. Phytomedicine 23, 293–306. doi: 10.1016/j.phymed.2016.01.004
Kharman-Biz, A., Gao, H., Ghiasvand, R., Zhao, C., Zendehdel, K., and Dahlman-Wright, K. (2013). Expression of activator protein-1 (AP-1) family members in breast cancer. BMC Cancer 13:441. doi: 10.1186/1471-2407-13-441
Kimmig, A., Gekeler, V., Neumann, M., Frese, G., Handgretinger, R., Kardos, G., et al. (1990). Susceptibility of multidrug-resistant human leukemia cell lines to human interleukin 2-activated killer cells. Cancer Res. 50, 6793–6799.
Klinger, P. H., Andrade, A. F., Delsin, L. E., Queiroz, R. G., Scrideli, C. A., Tone, L. G., et al. (2017). Inhibition of Shh pathway mechanisms by arsenic trioxide in pediatric medulloblastomas: a comprehensive literature review. Genet. Mol. Res. 16, 1–15. doi: 10.4238/gmr16019412
Kohn, K. W. (1999). Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Mol. Biol. Cell 10, 2703–2734. doi: 10.1091/mbc.10.8.2703
Kohn, K. W. (2001). Molecular interaction maps as information organizers and simulation guides. Chaos 11, 84–97. doi: 10.1063/1.1338126
Kohn, K. W., Aladjem, M. I., Kim, S., Weinstein, J. N., and Pommier, Y. (2006a). Depicting combinatorial complexity with the molecular interaction map notation. Mol. Syst. Biol. 2:51.
Kohn, K. W., Aladjem, M. I., Weinstein, J. N., and Pommier, Y. (2006b). Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol. Biol. Cell 17, 1–13.
Koo, H.-M., Monks, A., Mikheev, A., Rubinstein, L. V., Gray-Goodrich, M., Mcwilliams, M. J., et al. (1996). Enhanced sensitivity to 1-β-D-arabinofuranosylcytosine and topoisomerase II inhibitors in tumor cell lines harboring activated ras oncogenes. Cancer Res. 56, 5211–5216.
Kuete, V., Mbaveng, A. T., Nono, E. C., Simo, C. C., Zeino, M., Nkengfack, A. E., et al. (2016). Cytotoxicity of seven naturally occurring phenolic compounds towards multi-factorial drug-resistant cancer cells. Phytomedicine 23, 856–863. doi: 10.1016/j.phymed.2016.04.007
Kuete, V., Mbaveng, A. T., Sandjo, L. P., Zeino, M., and Efferth, T. (2017). Cytotoxicity and mode of action of a naturally occurring naphthoquinone, 2-acetyl-7-methoxynaphtho[2,3-b]furan-4,9-quinone towards multi-factorial drug-resistant cancer cells. Phytomedicine 33, 62–68. doi: 10.1016/j.phymed.2017.07.010
Lallemand-Breitenbach, V., Zhu, J., Chen, Z., and De The, H. (2012). Curing APL through PML/RARA degradation by As2O3. Trends Mol. Med. 18, 36–42. doi: 10.1016/j.molmed.2011.10.001
Lee, C. C., Lin, Y. Y., Huang, M. J., Lin, C. P., Liu, C. R., Chow, J., et al. (2006). Increased cellular glutathione and protection by bone marrow stromal cells account for the resistance of non-acute promylocytic leukemia acute myeloid leukemia cells to arsenic trioxide in vivo. Leuk. Lymphoma 47, 521–529. doi: 10.1080/10428190500305851
Lee, J.-S., Paull, K., Alvarez, M., Hose, C., Monks, A., Grever, M., et al. (1994). Rhodamine efflux patterns predict P-glycoprotein substrates in the national cancer institute drug screen. Mol. Pharmacol. 46, 627–638.
Lefebvre, C., Rieckhof, G., and Califano, A. (2012). Reverse-engineering human regulatory networks. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 311–325. doi: 10.1002/wsbm.1159
Lepoivre, C., Bergon, A., Lopez, F., Perumal, N. B., Nguyen, C., Imbert, J., et al. (2012). TranscriptomeBrowser 3.0: introducing a new compendium of molecular interactions and a new visualization tool for the study of gene regulatory networks. BMC Bioinformatics 13:19. doi: 10.1186/1471-2105-13-19
Li, S., and Zhang, B. (2013). Traditional Chinese medicine network pharmacology: theory, methodology and application. Chin. J. Nat. Med. 11, 110–120. doi: 10.3724/SP.J.1009.2013.00110
Liang, X., Li, H., and Li, S. (2014). A novel network pharmacology approach to analyse traditional herbal formulae: the Liu-Wei-Di-Huang pill as a case study. Mol. Biosyst. 10, 1014–1022. doi: 10.1039/c3mb70507b
Lin, Y., Bai, L., Chen, W., and Xu, S. (2010). The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 14, 45–55. doi: 10.1517/14728220903431069
Liu, H., Zhang, Z., Chi, X., Zhao, Z., Huang, D., Jin, J., et al. (2016). Arsenite-loaded nanoparticles inhibit PARP-1 to overcome multidrug resistance in hepatocellular carcinoma cells. Sci. Rep. 6:31009. doi: 10.1038/srep31009
Liu, J., Zhu, H. H., Jiang, H., Jiang, Q., and Huang, X. J. (2016). Varying responses of PML-RARA with different genetic mutations to arsenic trioxide. Blood 127, 243–250. doi: 10.1182/blood-2015-04-637678
Liu, Q., Hilsenbeck, S., and Gazitt, Y. (2003). Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood 101, 4078–4087. doi: 10.1182/blood-2002-10-3231
Liu, T., Ortiz, J. A., Taing, L., Meyer, C. A., Lee, B., Zhang, Y., et al. (2011). Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12:R83. doi: 10.1186/gb-2011-12-8-r83
Lopez, F., Textoris, J., Bergon, A., Didier, G., Remy, E., Granjeaud, S., et al. (2008). TranscriptomeBrowser: a powerful and flexible toolbox to explore productively the transcriptional landscape of the Gene Expression Omnibus database. PLoS One 3:e4001. doi: 10.1371/journal.pone.0004001
Lou, Y., Ma, Y., Sun, J., Ye, X., Pan, H., Wang, Y., et al. (2015). Evaluating frequency of PML-RARA mutations and conferring resistance to arsenic trioxide-based therapy in relapsed acute promyelocytic leukemia patients. Ann. Hematol. 94, 1829–1837. doi: 10.1007/s00277-015-2477-x
Lui, V. W., and Grandis, J. R. (2002). EGFR-mediated cell cycle regulation. Anticancer Res. 22, 1–11.
Luna, A., Karac, E. I., Sunshine, M., Chang, L., Nussinov, R., Aladjem, M. I., et al. (2011). A formal MIM specification and tools for the common exchange of MIM diagrams: an XML-Based format, an API, and a validation method. BMC Bioinformatics 12:167. doi: 10.1186/1471-2105-12-167
Luzina, E. L., and Popov, A. V. (2012). Synthesis, evaluation of anticancer activity and COMPARE analysis of N-bis (trifluoromethyl) alkyl-N’-substituted ureas with pharmacophoric moieties. Eur. J. Med. Chem. 53, 364–373. doi: 10.1016/j.ejmech.2012.03.026
Ma, H., Groth, R. D., Cohen, S. M., Emery, J. F., Li, B., Hoedt, E., et al. (2014). γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159, 281–294. doi: 10.1016/j.cell.2014.09.019
Mathieu, J., and Besancon, F. O. (2006). Clinically tolerable concentrations of arsenic trioxide induce p53-independent cell death and repress NF-κB activation in Ewing sarcoma cells. Int. J. Cancer 119, 1723–1727. doi: 10.1002/ijc.21970
Matulis, S. M., Morales, A. A., Yehiayan, L., Croutch, C., Gutman, D., Cai, Y., et al. (2009). Darinaparsin induces a unique cellular response and is active in an arsenic trioxide-resistant myeloma cell line. Mol. Cancer Ther. 8, 1197–1206. doi: 10.1158/1535-7163.MCT-08-1072
Matulis, S. M., Morales, A. A., Yehiayan, L., Lee, K. P., Cai, Y., and Boise, L. H. (2012). Alterations in glutathione levels and apoptotic regulators are associated with acquisition of arsenic trioxide resistance in multiple myeloma. PLoS One 7:e52662. doi: 10.1371/journal.pone.0052662
McDonnell, S. E., Kerr, L. D., and Matrisian, L. M. (1990). Epidermal growth factor stimulation of stromelysin mRNA in rat fibroblasts requires induction of proto-oncogenes c-fos and c-jun and activation of protein kinase C. Mol. Cell. Biol. 10, 4284–4293. doi: 10.1128/MCB.10.8.4284
Milde-Langosch, K., Roder, H., Andritzky, B., Aslan, B., Hemminger, G., Brinkmann, A., et al. (2004). The role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res. Treat. 86, 139–152. doi: 10.1023/B:BREA.0000032982.49024.71
Miller, W. H. Jr., Schipper, H. M., Lee, J. S., Singer, J., and Waxman, S. (2002). Mechanisms of action of arsenic trioxide. Cancer Res. 62, 3893–3903.
Mocellin, S., Provenzano, M., Rossi, C. R., Pilati, P., Nitti, D., and Lise, M. (2005). DNA array-based gene profiling: from surgical specimen to the molecular portrait of cancer. Ann. Surg. 241, 16–26.
Nakanishi, C., and Toi, M. (2005). Nuclear factor-κB inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 5, 297–309. doi: 10.1038/nrc1588
O’Brien, J., Wilson, I., Orton, T., and Pognan, F. (2000). Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 267, 5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x
O’Connor, P. M., Jackman, J., Bae, I., Myers, T. G., Fan, S., Mutoh, M., et al. (1997). Characterization of the p53 tumor suppressor pathway in cell lines of the national cancer institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 57, 4285–4300.
Passegue, E., and Wagner, E. F. (2000). JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 19, 2969–2979. doi: 10.1093/emboj/19.12.2969
Paull, K., Hamel, E., and Malspeis, L. (1995). Prediction of Biochemical Mechanism of Action from the in Vitro Antitumor Screen of the National Cancer Institute. Washington, DC: American Chemical Society.
Paull, K. D., Shoemaker, R. H., Hodes, L., Monks, A., Scudiero, D. A., Rubinstein, L., et al. (1989). Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 81, 1088–1092. doi: 10.1093/jnci/81.14.1088
Perkins, C., Kim, C. N., Fang, G., and Bhalla, K. N. (2000). Arsenic induces apoptosis of multidrug-resistant human myeloid leukemia cells that express Bcr-Abl or overexpress MDR, MRP, Bcl-2, or Bcl-x(L). Blood 95, 1014–1022.
Perou, C. M., Sørlie, T., Eisen, M. B., Van De Rijn, M., Jeffrey, S. S., Rees, C. A., et al. (2000). Molecular portraits of human breast tumours. Nature 406, 747–752. doi: 10.1038/35021093
Pfister, T. D., Reinhold, W. C., Agama, K., Gupta, S., Khin, S. A., Kinders, R. J., et al. (2009). Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol. Cancer Ther. 8, 1878–1884. doi: 10.1158/1535-7163.MCT-09-0016
Pluchino, K. M., Hall, M. D., Goldsborough, A. S., Callaghan, R., and Gottesman, M. M. (2012). Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 15, 98–105. doi: 10.1016/j.drup.2012.03.002
Poornima, P., Kumar, J. D., Zhao, Q., Blunder, M., and Efferth, T. (2016). Network pharmacology of cancer: From understanding of complex interactomes to the design of multi-target specific therapeutics from nature. Pharmacol. Res. 111, 290–302. doi: 10.1016/j.phrs.2016.06.018
Quan, Y., Wang, Z. Y., Xiong, M., Xiao, Z. T., and Zhang, H. Y. (2014). Dissecting traditional Chinese medicines by omics and bioinformatics. Nat. Prod. Commun. 9, 1391–1396.
Rao, Y., Li, R., and Zhang, D. (2013). A drug from poison: how the therapeutic effect of arsenic trioxide on acute promyelocytic leukemia was discovered. Sci. China Life Sci. 56, 495-502. doi: 10.1007/s11427-013-4487-z
Roboz, G. J., Dias, S., Lam, G., Lane, W. J., Soignet, S. L., Warrell, R. P., et al. (2000). Arsenic trioxide induces dose- and time-dependent apoptosis of endothelium and may exert an antileukemic effect via inhibition of angiogenesis. Blood 96, 1525–1530.
Ross, D. T., Scherf, U., Eisen, M. B., Perou, C. M., Rees, C., Spellman, P., et al. (2000). Systematic variation in gene expression patterns in human cancer cell lines. Nat. Genet. 24, 227–235. doi: 10.1038/73432
Roszak, J., Smok-Pieniążek, A., Nocun, M., and Stepnik, M. (2013). Characterization of arsenic trioxide resistant clones derived from Jurkat leukemia T cell line: focus on PI3K/Akt signaling pathway. Chem. Biol. Interact. 205, 198–211. doi: 10.1016/j.cbi.2013.07.011
Saeed, M., Kuete, V., Kadioglu, O., Bortzler, J., Khalid, H., Greten, H. J., et al. (2014). Cytotoxicity of the bisphenolic honokiol from Magnolia officinalis against multiple drug-resistant tumor cells as determined by pharmacogenomics and molecular docking. Phytomedicine 21, 1525–1533. doi: 10.1016/j.phymed.2014.07.011
Salerno, M., and Garnier-Suillerot, A. (2003). Resistance to arsenic- and antimony-based drugs. Bioinorg. Chem. Appl. 1, 189–198. doi: 10.1155/S1565363303000153
Scherf, U., Ross, D. T., Waltham, M., Smith, L. H., Lee, J. K., Tanabe, L., et al. (2000). A gene expression database for the molecular pharmacology of cancer. Nat. Genet. 24, 236–244. doi: 10.1038/73439
Schmidt, F., and Efferth, T. (2016). Tumor heterogeneity, single-cell sequencing, and drug resistance. Pharmaceuticals 9:33. doi: 10.3390/ph9020033
Seo, E. J., Saeed, M., Law, B. Y., Wu, A. G., Kadioglu, O., Greten, H. J., et al. (2016). Pharmacogenomics of scopoletin in tumor cells. Molecules 21:496. doi: 10.3390/molecules21040496
Seo, T., Urasaki, Y., and Ueda, T. (2007). Establishment of an arsenic trioxide-resistant human leukemia cell line that shows multidrug resistance. Int. J. Hematol. 85, 26–31. doi: 10.1532/IJH97.05142
Sertel, S., Tome, M., Briehl, M. M., Bauer, J., Hock, K., Plinkert, P. K., et al. (2012). Factors determining sensitivity and resistance of tumor cells to arsenic trioxide. PLoS One 7:e35584. doi: 10.1371/journal.pone.0035584
Shaulian, E., and Karin, M. (2001). AP-1 in cell proliferation and survival. Oncogene 20, 2390–2400. doi: 10.1038/sj.onc.1204383
Siegel, R. L., Miller, K. D., and Jemal, A. (2017). Cancer Statistics, 2017. CA Cancer J. Clin. 67, 7–30. doi: 10.3322/caac.21387
Soignet, S. L., Maslak, P., Wang, Z. G., Jhanwar, S., Calleja, E., Dardashti, L. J., et al. (1998). Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N. Engl. J. Med. 339, 1341–1348. doi: 10.1056/NEJM199811053391901
Szabo, E., Preis, L. H., Brown, P. H., and Birrer, M. J. (1991). The role of jun and fos gene family members in 12-O-tetradecanoylphorbol-13-acetate induced hemopoietic differentiation. Cell Growth Differ. 2, 475–482.
Tabellini, G., Cappellini, A., Tazzari, P. L., Fala, F., Billi, A. M., Manzoli, L., et al. (2005). Phosphoinositide 3-kinase/Akt involvement in arsenic trioxide resistance of human leukemia cells. J. Cell. Physiol. 202, 623–634. doi: 10.1002/jcp.20153
Thomas, S., and Bonchev, D. (2010). A survey of current software for network analysis in molecular biology. Hum. Genomics 4, 353–360. doi: 10.1186/1479-7364-4-5-353
Tomita, A., Kiyoi, H., and Naoe, T. (2013). Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O 3) in acute promyelocytic leukemia. Int. J. Hematol. 97, 717–725. doi: 10.1007/s12185-013-1354-4
Tung, J. N., Cheng, Y. W., Hsu, C. H., Liu, T. Z., Hsieh, P. Y., Ting, L. L., et al. (2011). Normoxically overexpressed hypoxia inducible factor 1-Alpha is involved in arsenic trioxide resistance acquisition in hepatocellular carcinoma. Ann. Surg. Oncol. 18, 1492–1500. doi: 10.1245/s10434-010-1444-y
Um, S. J., Lee, S. Y., Kim, E. J., Myoung, J., Namkoong, S. E., and Park, J. S. (2002). Down-regulation of human papillomavirus E6/E7 oncogene by arsenic trioxide in cervical carcinoma cells. Cancer Lett. 181, 11–22. doi: 10.1016/S0304-3835(02)00039-3
Vahidnia, A., Van Der Voet, G. B., and De Wolf, F. A. (2007). Arsenic neurotoxicity - A review. Hum. Exp. Toxicol. 26, 823–832. doi: 10.1177/0960327107084539
Walker, A. M., Stevens, J. J., Ndebele, K., and Tchounwou, P. B. (2016). Evaluation of arsenic trioxide potential for lung cancer treatment: assessment of apoptotic mechanisms and oxidative damage. J. Cancer Sci. Ther. 8, 1–9. doi: 10.4172/1948-5956.1000379
Wang, H., Gao, P., and Zheng, J. (2014). Arsenic trioxide inhibits cell proliferation and human papillomavirus oncogene expression in cervical cancer cells. Biochem. Biophys. Res. Commun. 451, 556–561. doi: 10.1016/j.bbrc.2014.08.014
Wang, S., Duan, C., Zhang, F., Ma, W., and Guo, X. (2013). Regulatory gene networks and signaling pathways from primary osteoarthritis and Kashin–Beck disease, an endemic osteoarthritis, identified by three analysis software. Gene 512, 89–96. doi: 10.1016/j.gene.2012.10.006
Wei, L. H., Lai, K. P., Chen, C. A., Cheng, C. H., Huang, Y. J., Chou, C. H., et al. (2005). Arsenic trioxide prevents radiation-enhanced tumor invasiveness and inhibits matrix metalloproteinase-9 through downregulation of nuclear factor kappa B. Oncogene 24, 390–398. doi: 10.1038/sj.onc.1208192
Wiench, B., Eichhorn, T., Paulsen, M., and Efferth, T. (2012). Shikonin directly targets mitochondria and causes mitochondrial dysfunction in cancer cells. Evid. Based Complement. Alternat. Med. 2012:726025. doi: 10.1155/2012/726025
Woo, S. H., Park, I. C., Park, M. J., An, S., Lee, H. C., Jin, H. O., et al. (2004). Arsenic trioxide sensitizes CD95/Fas-induced apoptosis through ROS-mediated upregulation of CD95/Fas by NF-kappa B activation. Int. J. Cancer 112, 596–606. doi: 10.1002/ijc.20433
Wosikowski, K., Schuurhuis, D., Johnson, K., Paull, K. D., Myers, T. G., Weinstein, J. N., et al. (1997). Identification of epidermal growth factor receptor and c-erbB2 pathway inhibitors by correlation with gene expression patterns. J. Natl. Cancer Inst. 89, 1505–1515. doi: 10.1093/jnci/89.20.1505
Yan, W. S., Zhang, Y. H., Zhang, J., Liu, S., Cho, S. J., and Chen, X. B. (2011). Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic-mediated growth suppression. J. Biol. Chem. 286, 17478–17486. doi: 10.1074/jbc.M111.231639
Yang, C. H., Kuo, M. L., Chen, J. C., and Chen, Y. C. (1999). Arsenic trioxide sensitivity is associated with low level of glutathione in cancer cells. Br. J. Cancer 81, 796–799. doi: 10.1038/sj.bjc.6690766
Yedjou, C. G., and Tchounwou, P. B. (2009). Modulation of p53, c-fos, RARE, cyclin A, and cyclin D1 expression in human leukemia (Hl-60) cells exposed to arsenic trioxide. Mol. Cell. Biochem. 331, 207–214. doi: 10.1007/s11010-009-0160-z
Yih, L. H., Hsu, N. C., Wu, Y. C., Yen, W. Y., and Kuo, H. H. (2013). Inhibition of AKT enhances mitotic cell apoptosis induced by arsenic trioxide. Toxicol. Appl. Pharmacol. 267, 228–237. doi: 10.1016/j.taap.2013.01.011
Yuan, B., Yoshino, Y., Fukushima, H., Markova, S., Takagi, N., Toyoda, H., et al. (2016). Multidrug resistance-associated protein 4 is a determinant of arsenite resistance. Oncol. Rep. 35, 147–154. doi: 10.3892/or.2015.4343
Zaharevitz, D. W., Holbeck, S. L., Bowerman, C., and Svetlik, P. A. (2002). Compare: a web accessible tool for investigating mechanisms of cell growth inhibition. J. Mol. Graph. Model. 20, 297–303. doi: 10.1016/S1093-3263(01)00126-7
Zhang, L., Wang, K., Zhao, F., Hu, W., Chen, J., Lanza, G. M., et al. (2012). Near infrared imaging of EGFR of oral squamous cell carcinoma in mice administered arsenic trioxide. PLoS One 7:e46255. doi: 10.1371/journal.pone.0046255
Zhang, X., Yang, X. R., Sun, C., Hu, B., Sun, Y. F., Huang, X. W., et al. (2015). Promyelocytic leukemia protein induces arsenic trioxide resistance through regulation of aldehyde dehydrogenase 3 family member A1 in hepatocellular carcinoma. Cancer Lett. 366, 112–122. doi: 10.1016/j.canlet.2015.06.014
Zhang, X. H., Feng, R., Lv, M., Jiang, Q., Zhu, H. H., Qing, Y. Z., et al. (2013). Arsenic trioxide induces apoptosis in B-cell chronic lymphocytic leukemic cells through down-regulation of survivin via the p53-dependent signaling pathway. Leuk. Res. 37, 1719–1725. doi: 10.1016/j.leukres.2013.09.019
Zhao, C. Q., Young, M. R., Diwan, B. A., Coogan, T. P., and Waalkes, M. P. (1997). Association of arsenic-induced malignant transformation with Dna hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. U.S.A. 94, 10907–10912. doi: 10.1073/pnas.94.20.10907
Zhao, D., Jiang, Y., Dong, X., Liu, Z., Qu, B., Zhang, Y., et al. (2011). Arsenic trioxide reduces drug resistance to adriamycin in leukemic K562/A02 cells via multiple mechanisms. Biomed. Pharmacother. 65, 354–358. doi: 10.1016/j.biopha.2011.04.016
Zhao, S., Zhang, J., Zhang, X., Dong, X., and Sun, X. (2008). Arsenic trioxide induces different gene expression profiles of genes related to growth and apoptosis in glioma cells dependent on the p53 status. Mol. Biol. Rep. 35, 421–429. doi: 10.1007/s11033-007-9102-6
Zheng, T., Yin, D., Lu, Z., Wang, J., Li, Y., Chen, X., et al. (2014). Nutlin-3 overcomes arsenic trioxide resistance and tumor metastasis mediated by mutant p53 in hepatocellular carcinoma. Mol. Cancer 13:133. doi: 10.1186/1476-4598-13-133
Keywords: arsenic trioxide, drug resistance, pharmacogenomics, AP-1, NF-κB
Citation: Dawood M, Hamdoun S and Efferth T (2018) Multifactorial Modes of Action of Arsenic Trioxide in Cancer Cells as Analyzed by Classical and Network Pharmacology. Front. Pharmacol. 9:143. doi: 10.3389/fphar.2018.00143
Received: 28 September 2017; Accepted: 09 February 2018;
Published: 27 February 2018.
Edited by:
Aiping Lu, Hong Kong Baptist University, Hong KongCopyright © 2018 Dawood, Hamdoun and Efferth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Efferth, efferth@uni-mainz.de