 Zhen Xiao1,2†
Zhen Xiao1,2† Yaqi Li1†
Yaqi Li1† Piao Zhao1Xiangyue Wu1Guoqing Luo1Shuijiao Peng1Hongrong Liu2
Piao Zhao1Xiangyue Wu1Guoqing Luo1Shuijiao Peng1Hongrong Liu2 Cheng Tang1*
Cheng Tang1* Zhonghua Liu1*
Zhonghua Liu1*- 1The National and Local Joint Engineering Laboratory of Animal Peptide Drug Development, College of Life Sciences, Hunan Normal University, Changsha, China
- 2Key Laboratory for Matter Microstructure and Function of Hunan Province, Key Laboratory of Low-dimensional Quantum Structures and Quantum Control, School of Physics and Electronics, Hunan Normal University, Changsha, China
The bacterial sodium channel NaChBac is the prokaryotic prototype for the eukaryotic NaV and CaV channels, which could be used as a relatively simple model to study their structure–function relationships. However, few modulators of NaChBac have been reported thus far, and the pharmacology of NaChBac remains to be investigated. In the present study, we show that the spider toxin κ-LhTx-1, an antagonist of the KV4 family potassium channels, potently inhibits NaChBac with an IC50 of 491.0 ± 61.7 nM. Kinetics analysis revealed that κ-LhTx-1 inhibits NaChBac by impeding the voltage-sensor activation. Site-directed mutagenesis confirmed that phenylalanine-103 (F103) in the S3–S4 extracellular loop of NaChBac was critical for interacting with κ-LhTx-1. Molecular docking predicts the binding interface between κ-LhTx-1 and NaChBac and highlights a dominant hydrophobic interaction between W27 in κ-LhTx-1 and F103 in NaChBac that stabilizes the interface. In contrast, κ-LhTx-1 showed weak activity on the mammalian NaV channels, with 10 µM toxin slightly inhibiting the peak currents of NaV1.2–1.9 subtypes. Taken together, our study shows that κ-LhTx-1 inhibits the bacterial sodium channel, NaChBac, using a voltage-sensor trapping mechanism similar to mammalian NaV site 4 toxins. κ-LhTx-1 could be used as a ligand to study the toxin–channel interactions in the native membrane environments, given that the NaChBac structure was successfully resolved in a nanodisc.
Introduction
The mammalian voltage-gated sodium (NaV) channels are the molecular determinants of action potential generation and propagation in excitable cells (Hodgkin and Huxley, 1990; Hille, 2001). To date, nine NaV channel genes have been identified. NaV1.4 and NaV1.5 are expressed mainly in the skeletal muscle and myocardial muscle cells, respectively. NaV1.1-NaV1.3 and NaV1.6-NaV1.9 are the dominant subtypes in neurons (Goldin, 1999; Catterall et al., 2005; de Lera Ruiz and Kraus, 2015). Their dysfunction causes hyper- or hypo-excitability of the cell types they are expressed in. The altered excitability leads to myotonia (NaV1.4), arrhythmia (NaV1.5), epilepsy (NaV1.1-1.3, NaV1.6), and pain (NaV1.7-1.9) (Andavan and Lemmens-Gruber, 2011; Huang et al., 2017). Therefore, pharmacological modulators regulating the activity of NaV channels are valuable drug candidates for treating these diseases (Bagal et al., 2015; Wulff et al., 2019). Topologically, the pore-forming NaV alpha subunit contains 24 transmembrane segments, which could be divided into four homologous domains (DI-DIV). The S1–S4 segments from each domain construct the voltage sensor module (VSM), and the S5–S6 segments make the pore module (PM). The four PMs in DI-DIV form the central Na+ conducting pathway, with the four VSMs surrounding the pore (Pan et al., 2018; Pan et al., 2019; Shen et al., 2019; Jiang et al., 2020; Pan et al., 2021). Driven by membrane depolarization, the S4 segments, which carry an array of positively charged arginine/lysine residues move outwardly, the local conformation changes were then transmitted into the central pore, resulting in pore opening (de Lera Ruiz and Kraus, 2015). The tremendous progress in cryo-EM structural studies of mammalian NaV channels has enhanced our understanding of their structure–function relationships (Pan et al., 2018; Pan et al., 2019; Shen et al., 2019; Jiang et al., 2020; Pan et al., 2021; Wisedchaisri et al., 2021).
The prokaryotic NaV channels, counterparts of the mammalian ones in prokaryotes, were proposed as evolutionary ancestors of mammalian NaV and CaV channels (Charalambous and Wallace, 2011). These channels share the key features of mammalian NaV channels, such as voltage-gating and Na+ selectivity. They are involved in specialized functions in prokaryotic organisms, such as motility and chemotaxis (Ren et al., 2001; Ito et al., 2004; Koishi et al., 2004). Structurally, it is assembled by four identical subunits, each of which contains six transmembrane segments, which resembles a single domain of the mammalian NaV channel (Scheuer, 2014). Several prokaryotic NaV channels have been identified, including NaChBac from Bacillus halodurans, NaVPZ from Paracoccus zeaxanthinifaciens, NaVSP from Silicibacter pomeroyi, and NsVBa from Bacillus alcalophilus (Ren et al., 2001; Koishi et al., 2004; DeCaen et al., 2014). In light of the simplified topological structure and efficient heterologous expression of prokaryotic NaV channels, they serve as ideal models for studying the structure–function relationships of mammalian NaV and CaV channels. For example, investigating the structures of the prokaryotic NaVAb channel in both the resting and activated states uncovered the likely conserved voltage sensing and gating mechanism in mammalian and prokaryotic NaV channels (Wisedchaisri et al., 2019; Catterall et al., 2020). NaChBac is the first prokaryotic NaV channel identified from Bacillus halodurans (Ren et al., 2001). Recently, the structure of a chimeric NaChBac channel containing the domain II S3-S4 loop of NaV1.7 channel and complexed to the spider toxin HWTx-IV was resolved at high-resolution in a nanodisc. This structure shed light on understanding the toxin–channel interactions in a natural membrane environment (Gao et al., 2020).
Compared to the mammalian NaV channels, relatively few peptide antagonists have been documented for bacterial NaV channels. Most of these toxins act on both the mammalian and bacterial NaV channels. They are either pore-blockers or gating-modifiers based on their mechanisms of action. The mammalian NaV site 1 modulator, μ-Conotoxin PIIIA, was characterized as a pore-blocker of NaChBac and was suggested to directly occlude the conducting pathway with the side chain of Arg14 (Finol-Urdaneta et al., 2019). We previously purified two potent gating-modifier toxins, JZTx-27 and JZTx-14, from the venom of spider Chilobrachys jingzhao. These toxins interact with the S3–S4 loop region of NaChBac and inhibit the channel activity by impeding voltage-sensor activation (Tang et al., 2017; Zhang et al., 2018). A recent study testing the activity of several known peptide modulators of mammalian NaV channels showed that the site 4 toxins GsAF-I, GrTx1, and GsAF-II also effectively inhibit NaChBac currents, possibly by affecting the voltage-dependent gating of the channel (Zhu et al., 2020). Interestingly, the site 3-toxin BDS-I has opposite effects on the bacterial and mammalian NaV channels. BDS-I inhibits NaChBac and activates the human NaV1.7 (Zhu et al., 2020). The mechanism of action of these gating-modifier toxins, with the exception of JZTx-27 and JZTx-14, has not been defined to the best of our knowledge. Identifying novel peptide modulators of NaChBac and elucidating their mechanism of action will enhance our understanding of the pharmacology of NaChBac. Such studies will also identify and characterize useful ligands for defining toxin–channel interactions in the native membrane environment with established structural biology approaches (Gao et al., 2020).
In the present study, we identified the spider toxin κ-LhTx-1, which was previously reported as an antagonist of the KV4 family voltage-gated potassium channel channels (Xiao et al., 2021), as a novel antagonist of the NaChBac channel. Compared with the other known NaChBac peptide inhibitors, κ-LhTx-1 selectively inhibited NaChBac but greatly spared the mammalian NaV channels. Kinetics analysis demonstrated that κ-LhTx-1 inhibits NaChBac by preventing voltage-sensor activation. Site-directed mutagenesis showed that phenylalanine-F103 (F103) in the S3–S4 extracellular loop of NaChBac was crucial for interacting with κ-LhTx-1. Molecular docking revealed that κ-LhTx-1 bound to NaChBac mainly by the hydrophobic interaction, with W27 in toxin being registered with F103 in the channel. This study describes κ-LhTx-1, a new peptide modulator of NaChBac. This peptide could be used as a tool for characterizing toxin–channel interactions in a native membrane environment using established structural biology methods.
Materials and methods
Toxin
Chemical synthesis and refolding of the linear κ-LhTx-1 was conducted as previously described (Xiao et al., 2021). The purity of synthesized toxins was determined to be >98% by analytic HPLC and MALDI-TOF MS analysis.
Constructs and site-directed mutations
The cDNA of mammalian NaV1.2-1.8 channels was kindly gifted from Professor Theodore R. Cummins (Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN, United States) and subcloned in the pCDNA3.1 or pCMV-blank vectors. The cDNA of NaV1.9 was obtained by gene synthesis and cloned into the pEGFP-N1 vector as previously reported (Zhou et al., 2017). The NaChBac and NaVPZ expression plasmids were kind gifts from Professor David E. Clapham (Janelia Research Campus, Howard Hughes Medical Institute, Ashburn, VA, United States). NaChBac mutants were made by site-directed mutations. Briefly, the NaChBac plasmid was amplified using a pair of oppositely directed primers harboring the designed mutation site with KOD Fx (TOYOBO Co., Ltd., Osaka, Japan), and the PCR products were treated with FastDigest DpnI (ThermoFisher, Waltham, MA, United States) to remove the methylated template. 10 µl digested product was used to transform 100 µl E. coli DH5α competent cells. Transformants were randomly picked for DNA sequencing to obtain the correct mutants.
Cell culture and transient transfection
HEK293T and ND7/23 cells were cultured in a DMEM medium, and CHO-K1 in a DMEM-F12 medium, supplemented with 10% FBS and 1% PS (all from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, United States), in standard cell culture conditions (37°C, 5% CO2, saturated humidity). The NaV expression plasmid was co-transfected with pEGFP-N1 plasmid into HEK293T (NaChBac and its mutants, NaVPZ and NaVPZ/P92F, and NaV1.2–1.7 channels) or ND7/23 (NaV1.8 channel) cells using Lipofectamine 2000 following manufacturer’s instructions (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, United States). NaV1.9-GFP plasmid was transfected into ND7/23 using the X-treme GENE HP DNA Transfection Reagent (Roche, Basel, Switzerland). After 4–6 h transfection, the cells were then seeded onto poly-lysine-coated coverslips. Note that the NaV1.9 transfected ND7/23 cells were maintained at 28°C for additional 20 h to promote the functional expression of the channel after the initial culture at 37°C for 24 h.
Electrophysiology
Whole-cell patch clamp recordings were performed using a MultiClamp 700B amplifier equipped with the Axon Digidata 1550 AD/DA digitizer (Molecular devices, San Jose, CA, United States) at room temperature. The EGFP fluorescence was used to identify the positively transfected cells. The bath solution for sodium currents recording contains (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 Glucose, and 10 HEPES; adjust pH to 7.3 with NaOH. The corresponding pipette solution contains (in mM): 140 CsF, 1 EGTA, 10 NaCl, and 10 HEPES; adjust pH to 7.2 with CsOH. All the chemicals were obtained from Sigma-Aldrich (Sigma-Aldrich, Saint Louis, MO, United States). For NaV1.8 and NaV1.9 currents recording, 1 μM TTX was added to the bath solution to eliminate the contamination of endogenous TTX-sensitive NaV currents in ND7/23 cells. The recording pipettes were made from glass capillaries using a PC-10 puller (NARISHIGE, Tokyo, Japan), and the pipette resistance was adjusted to be between 2 and 3 MΩ after filling with the pipette solution. Whole-cell configuration was established following a standard program. The fast and slow capacitance was sequentially canceled using a computer-controlled circuit of the amplifier. Moreover, series resistance was compensated by 80% to reduce the voltage error, and the cells with series resistance larger than 10 MΩ after break-in were discarded. The steady-state activation curve of NaChBac channels was obtained by calculating the conductance (G) at each depolarizing voltage (V) using the equation: G = I/(V–Vrev), and plotting G/Gmax as a function of V, where I, Gmax, and Vrev represent the current amplitude, the maximum conductance, and the reversal potential of the channel, respectively.The G–V curve was fitted using the Boltzmann equation: y = 1/{1+exp[(Va–V)/Ka]}, in which Va, V, and Ka represent the half maximum activation voltage, test voltage and slope factor, respectively. The steady-state inactivation curve of NaChBac channels was measured using a classical two-pulse protocol: cell was held at −120 mV, and a series of 1 s pre-pulses (−120 to 0 mV, in 10 mV increment) were applied to induce the channel inactivation, followed by a −20 mV/500 ms test pulse to assess the availability of non-inactivated channels (sweep interval was set to 10 s). Currents at the test pulse (I) were normalized to the maximum one (Imax) and plotted as a function of the conditional voltage (V), the curve was fitted by the Boltzmann equation: I/Imax = A + (1−A)/{1 + exp[(V−Vh)/Kh]}, where Vh is the half-maximum inactivation voltage, A represents the minimum channel availability, and Kh is the slope factor. The binding and unbinding kinetics of κ-LhTx-1 with NaChBac were measured by monitoring the time-dependent current rundown after toxin application and recovery upon bath solution perfusion. The association time constant (τon) was calculated by fitting the decay phase with the one exponential decay equation: y = ysteady+ae−x/τ; and the dissociation time constant (τoff) was calculated by fitting the recovery phase with one exponential rising equation: y = y(0)+a(1−e−x/τ). The concentration-response curves were fitted using the following Hill logistic equation: y = fmax−(fmax−fmin)/(1+([Tx]/IC50)n), where fmax and fmin represent the maximum and minimum response of the channel to toxin, [Tx] represents the toxin concentration, IC50 represents the half maximum inhibition concentration, and n is an empirical Hill coefficient, respectively. Data were acquired using Clampex 10.5 (Molecular devices, San Jose, CA, United States).
Molecular docking
The structure of κ-LhTx-1 was predicted by C-I-TASSER by using a contact-guided iterative threading assembly refinement (Zheng et al., 2021). The resting-state conformation model of NaChBac channel was generated by modifying the partially activated conformation NaChBac structure (PDB ID: 6VWX) according to the resting-state NaVAb/NaV1.7-VS2A chimeric channel structure (PDB ID: 7K48) (Wisedchaisri et al., 2021). The binding mode between NaChBac and κ-LhTx-1 was simulated by ZDOCK (ZDOCK 3.0.2) (Mintseris et al., 2007; Pierce et al., 2014). The residues which buried into the membrane were blocked in protein-peptide docking. F103 was selected as the binding site residue according to the results of the patch clamp assay.
Data analysis
Data were presented as the MEAN ± SEM, where n represents the number of separate experimental cells. Data were analyzed by using the software Clampfit 10.5 (Axon Instruments, Irvine, CA, United States), Graphpad Prism 5.01 (GraphPad Software, La Jolla, CA, United States), and Excel 2010 (Microsoft Corporation, Redmond, WA, United States). Statistical difference was assessed using the unpaired t-test, and the significant difference was accepted at p < 0.05.
Results
κ-LhTx-1 is a novel NaChBac antagonist
We screened RP-HPLC fractions of the venom of spider Pandercetes sp (the lichen huntsman spider) for potential modulators of NaChBac. We identified κ-LhTx-1, an antagonist of KV4 family potassium channels, as a potent inhibitor of NaChBac (Figure 1B) (Xiao et al., 2021). The amino acid sequence of κ-LhTx-1 shows relatively low homology to the other known NaChBac peptide inhibitors, including GrTx1 (45%), GsAF-I (42%), JZTx-27 (29%), and JZTx-14 (20%). However, they all share a conserved cysteine framework, which is predicted to fold as an ICK motif (Figure 1A) (Pallaghy et al., 1994). Due to the low abundance of κ-LhTx-1 in the venom, we used the synthetic toxin, which was shown to be correctly refolded as the native one (Xiao et al., 2021), for further experiments. κ-LhTx-1 dose-dependently inhibited the peak current of NaChBac with an IC50 of 491.0 ± 61.7 nM at −20 mV (Figures 1B,D). Interestingly, κ-LhTx-1 did not modulate another bacterial sodium channel, NaVPZ, even at 10 μM (Figure 1C). Toxin-binding kinetics assays showed that κ-LhTx-1 rapidly inhibited NaChBac currents, with a time constant of 2.0 ± 0.2 s for toxin association. Dissociation of κ-LhTx-1 from NaChBac was slow with a time constant of 34.1 ± 2.5 s (Figure 1E).
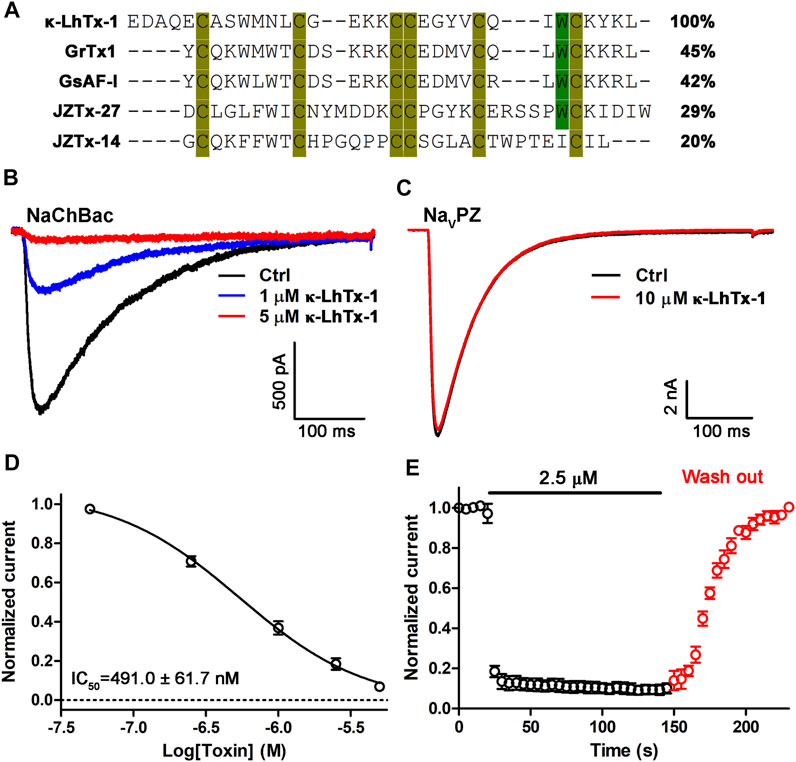
FIGURE 1. Characterization of κ-LhTx-1 as a novel NaChBac antagonist. (A) Sequence alignment of κ-LhTx-1 with other known NaChBac peptide antagonists. (B) Representative traces showing κ-LhTx -1 dose-dependently inhibited NaChBac currents elicited by depolarization to −20 mV from the holding potential of −100 mV (n = 5). (C) Representative traces showing that NaVPZ currents were not affected by 10 μM κ-LhTx-1 treatment (n = 4). (D) Dose-response curve of κ-LhTx-1 inhibiting NaChBac at −20 mV, and the IC50 value was determined as 491.0 ± 61.7 nM (n = 5). (E) Time course of NaChBac current inhibition by 2.5 μM κ-LhTx-1 and subsequent washing off with bath solution. The association time constant (τon) and the dissociation time constant (τoff) were determined as 2.0 ± 0.2 s and 34.1 ± 2.5 s, respectively (n = 5).
Effects of κ-LhTx-1 on the gating kinetics of NaChBac
The peptide toxins inhibit NaV channels possibly by physically occluding the ion conducting pathway or by impeding the voltage-dependent activation of the voltage sensor, which could be assessed by comparing the channels’ gating kinetics before and after toxin treatment. We first tested the effect of κ-LhTx-1 on the current–voltage (I-V) relationships of NaChBac. Figure 2A shows the representative current traces of the same cell before and after 1 μM κ-LhTx-1 treatment, demonstrating that the inward currents were partially inhibited, while the outward currents were unaffected. This result was further validated by the I–V curves shown in Figure 2B. Between the depolarization voltages of −50 mV and +30 mV, NaChBac currents were differently inhibited by 1 μM κ-LhTx-1, whereas the inhibition was absent at depolarizing voltages higher than +40 mV. These data suggested that those toxin-bound NaChBac channels were reopened as toxin-free channels at stronger depolarizations, resulting in the phenotype of voltage-dependent inhibition. When normalizing the currents at each depolarizing voltage in the toxin-treated group to its maximum peak current, the I–V relationship showed apparent right-forward shift when compared with the control group. Likewise, 1 μM κ-LhTx-1 caused a significant shift of the G-V curve toward more depolarized direction, and the half maximum activation voltage (Va) was determined as −35.0 ± 1.3 mV and −22.0 ± 2.8 mV for control and toxin-treated channels, respectively (Figure 2C; p < 0.001, unpaired t-test). Meanwhile, κ-LhTx-1 treatment also remarkably reduced the slope of the G–V curve (Figure 2C; Ka = 7.3 ± 0.4 mV and 12.5 ± 0.9 mV for control and toxin-treated channels, respectively; p < 0.001, unpaired t-test), indicating toxin greatly attenuated the channel voltage sensitivity during the voltage-dependent activation. These data strongly suggested that κ-LhTx-1 trapped the voltage sensor of the NaChBac channel in a deactivated state, while the energy barrier of voltage sensor outward movement caused by toxin binding could be counteracted by strengthening membrane depolarization. The steady-state inactivation of NaChBac channels, however, was not affected by κ-LhTx-1 (Figure 2D). Altogether, these results argued that κ-LhTx-1 trapped the voltage sensor of the NaChBac channel in the deactivated state, functioning as a gating modifier.
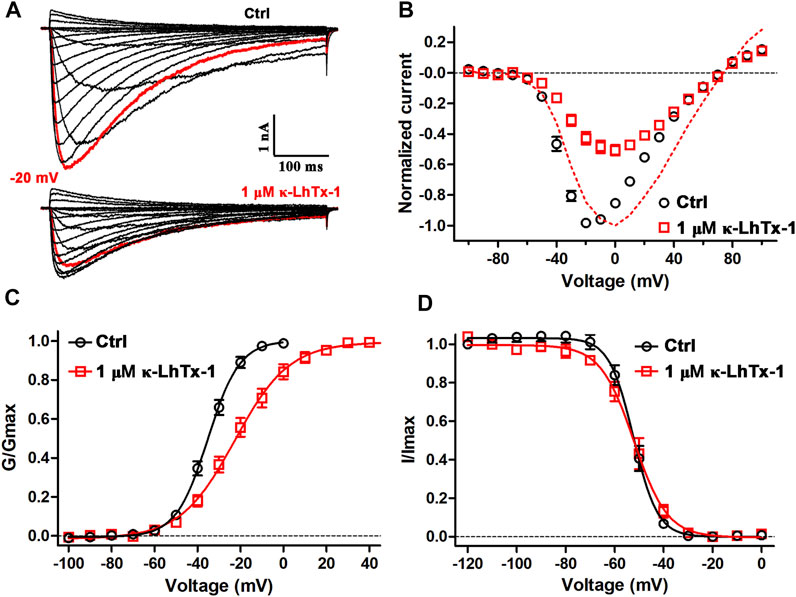
FIGURE 2. Effects of κ-LhTx-1 on the gating kinetics of NaChBac. (A) Representative NaChBac currents before (upper panel) and after (lower panel) 1 μM κ-LhTx-1 treatment. Currents were elicited by step depolarizations from −100 to +100 mV from a holding potential of −100 mV. The red traces show the currents at −20 mV (n = 6). (B) I–V relationships of NaChBac before and after 1 μM κ-LhTx-1 treatment. The red dashed line shows the I–V relationship of toxin-treated channels by normalizing the currents to their own maximum (n = 6). (C) Steady-state activation relationships of NaChBac before and after 1 μM κ-LhTx-1 treatment (Va = −35.0 ± 1.3 mV and −22.0 ± 2.8 mV, Ka = 7.3 ± 0.4 mV and 12.5 ± 0.9 mV, for control and toxin-treated channels, respectively; p < 0.001 when comparing both the Va and Ka values between the control and toxin groups; unpaired t-test; n = 6). (D) Steady-state inactivation relationships of NaChBac before and after 1 μM κ-LhTx-1 treatment (Vh = −52.3 ± 1.3 mV and −53.0 ± 1.9 mV, Kh = −4.6 ± 0.3 mV and −6.0 ± 0.6 mV, for control- and toxin treated channels, respectively; n = 5).
The molecular determinants in NaChBac for interacting with κ-LhTx-1
Since κ-LhTx-1 is a gating-modifier of NaChBac, we wondered if it might bind to the channel’s S3–S4 extracellular loop. The protein sequence-alignment through the S3–S4 region of the κ-LhTx-1-sensitive NaChBac channel and the κ-LhTx-1-resistant NaVPZ channel is shown in Figure 3A. The alignment in Figure 3A highlights F103 in NaChBac and the corresponding Proline-92 (P92) in NaVPZ. We used an alanine scan strategy to identify the key residues for interacting with κ-LhTx-1 in this region (Figure 3A). All these NaChBac mutants were functionally expressed in CHO-K1 cells (Figures 3B–F). We tested the inhibitory effect of κ-LhTx-1 on each mutant channel at voltage eliciting its maximum inward peak current. The data showed that F103A, G105A, F108A, and V109A mutations in NaChBac all attenuated the inhibitory effect of κ-LhTx-1, with 5 µM toxin not affecting the currents of NaChBac/F103A and only partially inhibiting the currents of NaChBac/G105A, NaChBac/F108A, and NaChBac/V109A by approximately 43.4 ± 8.1%, 40.7 ± 4.2%, and 64.4 ± 4.1%, respectively (Figures 3B,C,E,F). The activity of κ-LhTx-1 on NaChBac/Q107A was not changed a lot when compared with that of the wild type channel (Figure 3D). The dose–response curves in Figure 3G showed that κ-LhTx-1 inhibited the currents of NaChBac/G105A, NaChBac/Q107A, NaChBac/F108A, and NaChBac/V109A with an IC50 of 5.5 ± 1.4 μM, 1.0 ± 0.1 μM, 7.8 ± 1.0 μM, and 2.6 ± 0.4 μM, respectively, (Figure 3G). The IC50 for NaChBac/F103A could not be determined from the curve as 20 μM κ-LhTx-1 only inhibited its currents by 16.4 ± 2.6% (Figure 3G). Next, we tried to convert the κ-LhTx-1-resistant NaVPZ channel into the κ-LhTx-1-sensitive channel. Since F103 in NaChBac was deemed to be important for κ-LhTx-1 modulation, we decided to replace the corresponding residue in NaVPZ (P92) with phenylalanine. The NaVPZ/P92F mutant was potently inhibited by κ-LhTx-1 (Figure 3H), with an IC50 of 6.7 ± 2.3 μM (Figure 3I). Our successful conversion of κ-LhTx-1-resistant NaVPZ into a sensitive channel strongly supports the importance of F103 in NaChBac and the corresponding P92 in NaVPZ.
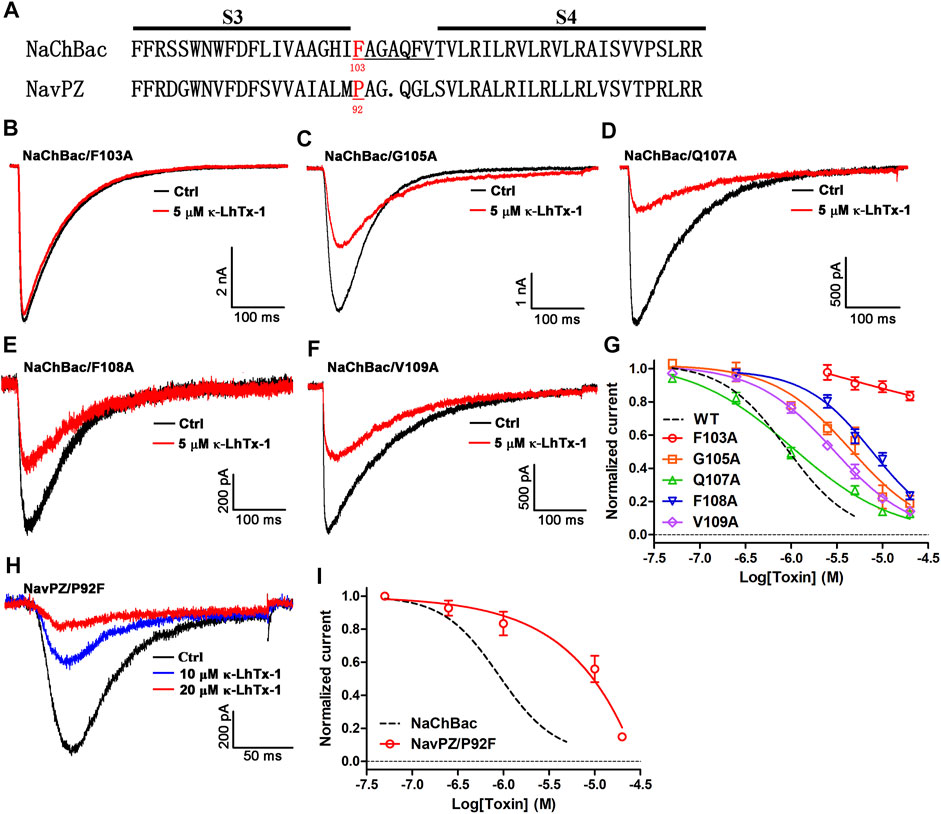
FIGURE 3. Key molecular determinants in NaChBac for interacting with κ-LhTx-1. (A) Sequence alignment of the NaChBac and NaVPZ, the number below the residue indicates its location in the sequence. (B) Representative traces showing F103A mutation in NaChBac almost fully abolished κ-LhTx-1 inhibition on the channel (n = 5). (C–F) Representative traces showing the inhibitory effects of 5 μM κ-LhTx-1 on NaChBac mutants as illustrated (n = 5–6). (G) Dose-response curves of κ-LhTx-1 inhibiting the NaChBac mutant channels, and the IC50 values were determined as 5.5 ± 1.4 μM, 1.0 ± 0.1 μM, 7.8 ± 1.0 μM, and 2.6 ± 0.4 μM for NaChBac/G105A, NaChBac/Q107A, NaChBac/F108A, and NaChBac/V109A, respectively (n = 5–6). The black dashed line shows the dose-response curve of κ-LhTx-1 against the wild-type NaChBac channel. (H) Representative traces showing the κ-LhTx-1 dose-dependently inhibited NaVPZ/P92F currents elicited by depolarization to +30 mV from the holding potential of −100 mV (n = 5). (I) Dose-response curve of κ-LhTx-1 inhibiting the currents of NaVPZ/P92F at +30 mV, the IC50 values were determined as 6.7 ± 2.3 μM (n = 4).
Effects of κ-LhTx-1 on mammalian NaV channels
The biological activity of κ-LhTx-1 on the mammalian KV channels has been systematically examined, which showed it potently inhibited the KV4 family potassium channels without affecting other KV channels, including KV1.1, KV1.3-1.5, KV2.1, and KV3.1-3.4 (Xiao et al., 2021). Herein, we tested the activity of κ-LhTx-1 on the mammalian NaV channels. As shown in Figures 4A,B, 10 μM κ-LhTx-1 showed weak inhibition on NaV1.3 (8.4 ± 2.3%), NaV1.5 (17.2 ± 7.3%), NaV1.6 (18.4 ± 15.7%), NaV1.8 (7.1 ± 1.5%), and NaV1.9 (4.8 ± 0.1%) channels and relatively stronger inhibition on NaV1.2 (41.3 ± 6.2%), NaV1.4 (38.7 ± 1.9%), and NaV1.7 (67.4 ± 3.7%) channels. Compared with the potent inhibition of κ-LhTx-1 on the KV4 family potassium channels and NaChBac channels, the toxin showed much weaker activity on the mammalian NaV channels.
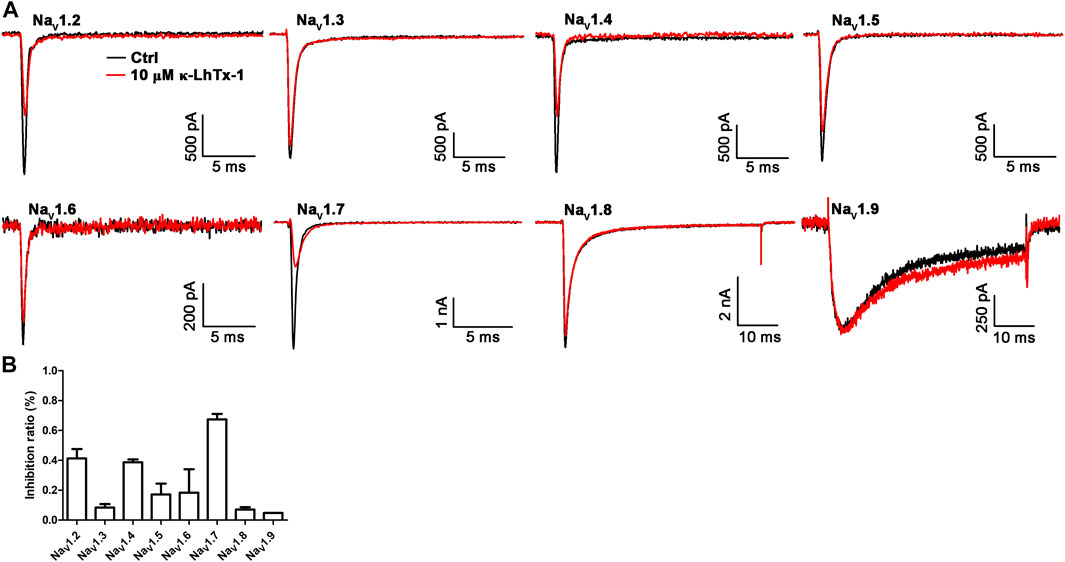
FIGURE 4. Effects of κ-LhTx-1 on the mammalian NaV channels. (A) Representative traces showing the effect of 10 μM κ-LhTx-1 on NaV1.2–1.9 channels (n = 3–5). (B) Bar graph showing the inhibition of 10 μM κ-LhTx-1 on the peak currents of Nav1.2–1.9 channels. The inhibition ratio was determined as 41.3 ± 6.2%, 8.4 ± 2.3%, 38.7 ± 1.9%, 17.2 ± 7.3%, 18.4 ± 15.7%, 67.4 ± 3.7%, 7.1 ± 1.5%, and 4.8 ± 0.1% for NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8, and NaV1.9 channels, respectively (n = 3–5).
The toxin–channel interface as revealed by molecular docking
The κ-LhTx-1 structure was simulated by C-I-TASSER (Figure 5A). The six cysteines in toxin formed three disulfide bonds as C6-C19, C13-C24, and C18-C28 (number indicates the position of cysteines in toxin sequence). Analyzing the solvent-accessible surface of κ-LhTx-1 showed it is an amphilic molecule, in which most hydrophobic residues are mainly distributed on one side, forming a hydrophobic patch, whereas charged residues are distributed on the other side, forming a hydrophilic surface (Figure 5B). The toxin–channel docking complex is shown in Figure 5C. The modeled resting-state of NaChBac and κ-LhTx-1 is depicted in yellow and green, respectively. The best scoring docking complex showed that the toxin inserted as a wedge into the cleft between S1–S2 and S3–S4 loops (Figure 5C), resembling that of α-scorpion toxin interacting with the voltage sensor of the mammalian NaV channel (Wang et al., 2011; Clairfeuille et al., 2019). In this docking model, F103 in NaChBac and W27 in κ-LhTx-1 were captured in the channel–toxin interface, in which W27 was registered with F103 by the π–π stacking interaction (Figure 5D). We used site-specific mutagenesis to experimentally verify the functional importance of the phenyl ring of F103 for κ-LhTx-1 binding. The F103A mutation abolished the modulation of NaChBac by κ-LhTx-1 (Figures 3B, 5E). Replacement of F103 with aromatic residues (F103W, F103Y) retained the inhibition by κ-LhTx-1, albeit with lower potency than the wild-type channel. These mutants were inhibited by κ-LhTx-1 with IC50s of 9.5 ± 2.9 μM for NaChBac/F103W (∼19-fold lower) and 3.7 ± 1.5 μM for NaChBac/F103Y (∼8-fold lower) (Figures 5E,F). Taken together, these results argued that the phenyl ring of F103 in NaChBac played a critical role in binding with κ-LhTx-I, and the association between κ-LhTx-1 and NaChBac likely relies on a hydrophobic interaction.
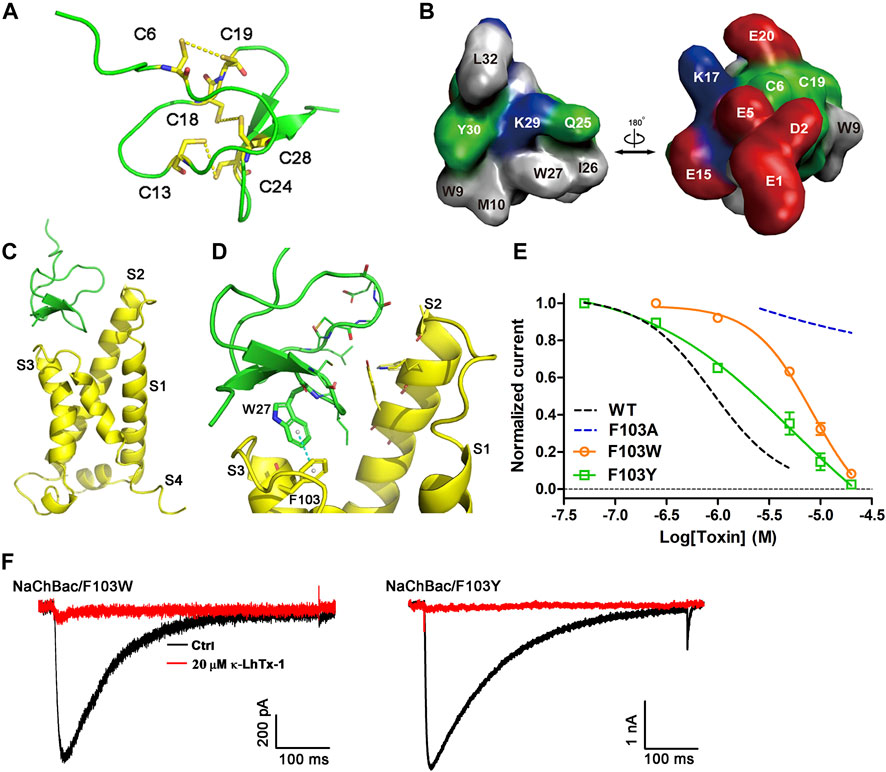
FIGURE 5. Molecular docking of κ-LhTx-1 with NaChBac. (A) Predicted structure of the κ-LhTx-1 by C-I-TASSER, the peptide toxin GsMTx2 (PDB ID: 1LUP) was used as the template. Cysteines were highlighted in yellow, and the predicted disulfide bonds were shown (C6 - C19, C13 - C24, and C18 - C28). (B) Surface presentation of κ-LhTx-1 structure. Positively charged residues are shown in blue, negatively charged residues in red, hydrophobic residues in gray, and polar uncharged residues in green. (C) Docked κ-LhTx-1-NaChBac complex with the highest score. The resting-state NaChBac channel and the κ-LhTx-1 toxin were shown in yellow and green, respectively. For simplicity, only the S1–S4 region of a single channel subunit was shown. (D) Enlarged view of the interaction interface between NaChBac and κ-LhTx-1, in which W27 in toxin captured the experimentally determined key residue in NaChBac, F103, in the toxin–channel interface. (E) Dose-response curve of κ-LhTx-1 inhibiting the NaChBac mutants as shown, and the IC50 values were determined as 9.5 ± 2.9 μM and 3.7 ± 1.5 μM for NaChBac/F103W and NaChBac/F103Y, respectively. The black and red dashed lines show the dose-response curves of κ-LhTx-1 against the wild-type NaChBac and NaChBac/F103A mutant channel, respectively (n = 5). (F) Representative traces showing the inhibition effect of 20 μM κ-LhTx-1 on the currents of NaChBac/F103W and NaChBac/F103Y mutant channels (n = 5).
Discussion
κ-LhTx-1 was demonstrated in our previous study to selectively inhibit the mammalian KV4 family potassium channels (Xiao et al., 2021). The present study confirmed that κ-LhTx-1 is also a novel NaChBac antagonist. The G–V curve of NaChBac was remarkably shifted to depolarized direction by κ-LhTx-1, indicating κ-LhTx-1 acts on NaChBac as a gating-modifier trapping the deactivated voltage sensor. Moreover, the site-directed mutagenesis and molecular docking showed κ-LhTx-1 bound to the S3–S4 loop of NaChBac, with the phenylalanine-103 (F103) being the most critical residue. Molecular docking also confirmed the W27 residue in κ-LhTx-1 was registered with F103 in NaChBac, likely by the π–π stacking interaction. The action mode of κ-LhTx-1 on NaChBac and its binding site on the channel resembles that of JZTx-27 (Tang et al., 2017). However, unlike JZTx-27, κ-LhTx-1 slightly inhibited the activation but not the inactivation of mammalian NaV channels. The structure of chimeric NaChBac channel harboring the DII S3-S4 linker of NaV1.7 channel in complex with the spider toxin HwTx-IV in a nanodisc has been successfully resolved, which has showed greatly improved resolution of the toxin–channel interacting interface and enabled visualization of their binding details (Gao et al., 2020). κ-LhTx-1 in the present study thus provided another useful ligand for studying the toxin and wild-type NaChBac interaction in the native membrane environments.
Historically, NaChBac was shown to pharmacologically resemble the mammalian NaV and CaV channels, as revealed by its inhibition by lots of CaV and NaV channel modulators, such as lidocaine, nifedipine, and various peptide toxins (Ren et al., 2001; Lee et al., 2012; Tang et al., 2017; Zhang et al., 2018; Zhu et al., 2020). κ-LhTx-1 was previously shown to inhibit the KV4 family potassium channels in a voltage-dependent manner (Xiao et al., 2021). Consequently, the present study provided the first example that NaChBac also shares similar pharmacology properties with the KV channels. Gating currents analysis confirmed that κ-LhTx-1 trapped the voltage sensor of KV4 channels in a deactivated state (Xiao et al., 2021). Therefore, we concluded that κ-LhTx-1 inhibited the KV4 and NaChBac channels using the same voltage-sensor trapping mechanism.
Mechanistically, the association between gating-modifier toxins and ion channels relies on electrostatic force and/or hydrophobic interaction between their interface (Bosmans et al., 2008). As that of the HpTx2 binding with the KV4.3 channel solely by hydrophobic force (Zhang et al., 2007; DeSimone et al., 2009), κ-LhTx-1 might also interact with NaChBac mainly by a hydrophobic interaction, as revealed by mutation and molecular docking analyses. Indeed, hydrophobic residues on the S3–S4 loop of NaChBac have been commonly characterized as key molecular determinants in interacting with its gating-modifier toxins, such as F103 for κ-LhTx-1 and JZTx-27 and F108 for JZTx-14 (Tang et al., 2017; Zhang et al., 2018). Interestingly, the hydrophobic residues LF/LV in the S3b region of KV4 channels were also identified as the key binding determinants for κ-LhTx-1 (Xiao et al., 2021), and it can be rationally presumed that hydrophobic interactions also underlie the interaction between κ-LhTx-1 and KV4 channels. Additionally, sequence alignment revealed that κ-LhTx-1 and other NaChBac peptide antagonists from spider venom share a conserved cysteine framework, and the critical W27 residue in κ-LhTx-1 is also conserved in most of them (Figure 1A, except for JZTx-14); therefore it is reasonable to speculate that these toxins used this conserved W residue to interact with NaChBac via hydrophobic interaction. Indeed, molecular docking analysis in our previous study also showed that this W residue in JZTx-27 interacts with F98 in another bacteria sodium channel, NsVBa (Tang et al., 2017).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author contributions
CT, ZX, and ZL designed the study and wrote the manuscript. ZX, YL, PZ, XW, GL, and HL performed the experiments and the data analysis. SP performed the experiments and the data analysis.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31600669, 32171271, 32071262, and 31872718), the Science and Technology Innovation Program of Hunan Province (2020RC4023), the Natural Science Foundation of Hunan Province (Grant No. 2018JJ3339), the Research Foundation of the Education Department of Hunan Province (Grant No.18B015), and the Hunan Provincial Innovation Foundation for Postgraduate (CX20190395).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andavan, G. S., and Lemmens-Gruber, R. (2011). Voltage-gated sodium channels: Mutations, channelopathies and targets. Curr. Med. Chem. 18, 377–397. doi:10.2174/092986711794839133
Bagal, S. K., Marron, B. E., Owen, R. M., Storer, R. I., and Swain, N. A. (2015). Voltage gated sodium channels as drug discovery targets. Channels (Austin) 9, 360–366. doi:10.1080/19336950.2015.1079674
Bosmans, F., Martin-Eauclaire, M. F., and Swartz, K. J. (2008). Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456, 202–208. doi:10.1038/nature07473
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409. doi:10.1124/pr.57.4.4
Catterall, W. A., Wisedchaisri, G., and Zheng, N. (2020). The conformational cycle of a prototypical voltage-gated sodium channel. Nat. Chem. Biol. 16, 1314–1320. doi:10.1038/s41589-020-0644-4
Charalambous, K., and Wallace, B. A. (2011). NaChBac: The long lost sodium channel ancestor. Biochemistry 50, 6742–6752. doi:10.1021/bi200942y
Clairfeuille, T., Cloake, A., Infield, D. T., Llongueras, J. P., Arthur, C. P., Li, Z. R., et al. (2019). Structural basis of α-scorpion toxin action on Na(v) channels. Science 363, eaav8573. doi:10.1126/science.aav8573
de Lera Ruiz, M., and Kraus, R. L. (2015). Voltage-gated sodium channels: Structure, function, pharmacology, and clinical indications. J. Med. Chem. 58, 7093–7118. doi:10.1021/jm501981g
DeCaen, P. G., Takahashi, Y., Krulwich, T. A., Ito, M., and Clapham, D. E. (2014). Ionic selectivity and thermal adaptations within the voltage-gated sodium channel family of alkaliphilic Bacillus. eLife 3, e04387. doi:10.7554/eLife.04387
DeSimone, C. V., Lu, Y., Bondarenko, V. E., and Morales, M. J. (2009). S3b amino acid substitutions and ancillary subunits alter the affinity of Heteropoda venatoria toxin 2 for Kv4.3. Mol. Pharmacol. 76, 125–133. doi:10.1124/mol.109.055657
Finol-Urdaneta, R. K., McArthur, J. R., Korkosh, V. S., Huang, S., McMaster, D., Glavica, R., et al. (2019). Extremely potent block of bacterial voltage-gated sodium channels by µ-conotoxin PIIIA. Mar. Drugs 17, 510. doi:10.3390/md17090510
Gao, S., Valinsky, W. C., On, N. C., Houlihan, P. R., Qu, Q., Liu, L., et al. (2020). Employing NaChBac for cryo-EM analysis of toxin action on voltage-gated Na(+) channels in nanodisc. Proc. Natl. Acad. Sci. U. S. A. 117, 14187–14193. doi:10.1073/pnas.1922903117
Goldin, A. L. (1999). Diversity of mammalian voltage-gated sodium channels. Ann. N. Y. Acad. Sci. 868, 38–50. doi:10.1111/j.1749-6632.1999.tb11272.x
Hille, B. (2001). Ion channels of excitable membranes (Ion channels of excitable membranes). Sunderland, MA, United States: Sinauer Associates, Inc.
Hodgkin, A. L., and Huxley, A. F. (1990). A quantitative description of membrane current and its application to conduction and excitation in nerve. Bull. Math. Biol. 52, 25–23. doi:10.1007/BF02459568
Huang, W., Liu, M., Yan, S. F., and Yan, N. (2017). Structure-based assessment of disease-related mutations in human voltage-gated sodium channels. Protein Cell 8, 401–438. doi:10.1007/s13238-017-0372-z
Ito, M., Xu, H., Guffanti, A. A., Wei, Y., Zvi, L., Clapham, D. E., et al. (2004). The voltage-gated Na+ channel NaVBP has a role in motility, chemotaxis, and pH homeostasis of an alkaliphilic Bacillus. Proc. Natl. Acad. Sci. U. S. A. 101, 10566–10571. doi:10.1073/pnas.0402692101
Jiang, D., Shi, H., Tonggu, L., Gamal El-Din, T. M., Lenaeus, M. J., Zhao, Y., et al. (2020). Structure of the cardiac sodium channel. Cell 180, 122–e10. e110. doi:10.1016/j.cell.2019.11.041
Koishi, R., Xu, H., Ren, D., Navarro, B., Spiller, B. W., Shi, Q., et al. (2004). A superfamily of voltage-gated sodium channels in bacteria. J. Biol. Chem. 279, 9532–9538. doi:10.1074/jbc.M313100200
Lee, S., Goodchild, S. J., and Ahern, C. A. (2012). Local anesthetic inhibition of a bacterial sodium channel. J. Gen. Physiol. 139, 507–516. doi:10.1085/jgp.201210779
Mintseris, J., Pierce, B., Wiehe, K., Anderson, R., Chen, R., and Weng, Z. (2007). Integrating statistical pair potentials into protein complex prediction. Proteins 69, 511–520. doi:10.1002/prot.21502
Pallaghy, P. K., Nielsen, K. J., Craik, D. J., and Norton, R. S. (1994). A common structural motif incorporating a cystine knot and a triple-stranded beta-sheet in toxic and inhibitory polypeptides. Protein Sci. 3, 1833–1839. doi:10.1002/pro.5560031022
Pan, X., Li, Z., Huang, X., Huang, G., Gao, S., Shen, H., et al. (2019). Molecular basis for pore blockade of human Na+ channel Na v 1.2 by the μ-conotoxin KIIIA. Science 363, 1309–1313. doi:10.1126/science.aaw2999
Pan, X., Li, Z., Jin, X., Zhao, Y., Huang, G., Huang, X., et al. (2021). Comparative structural analysis of human Na(v)1.1 and Na(v)1.5 reveals mutational hotspots for sodium channelopathies. Proc. Natl. Acad. Sci. U. S. A. 118. e2100066118. doi:10.1073/pnas.2100066118
Pan, X., Li, Z., Zhou, Q., Shen, H., Wu, K., Huang, X., et al. (2018). Structure of the human voltage-gated sodium channel Na(v)1.4 in complex with β1. Science 362, eaau2486. doi:10.1126/science.aau2486
Pierce, B. G., Wiehe, K., Hwang, H., Kim, B. H., Vreven, T., and Weng, Z. (2014). ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773. doi:10.1093/bioinformatics/btu097
Ren, D., Navarro, B., Xu, H., Yue, L., Shi, Q., and Clapham, D. E. (2001). A prokaryotic voltage-gated sodium channel. Science 294, 2372–2375. doi:10.1126/science.1065635
Scheuer, T. (2014). Bacterial sodium channels: Models for eukaryotic sodium and calcium channels. Handb. Exp. Pharmacol. 221, 269–291. doi:10.1007/978-3-642-41588-3_13
Shen, H., Liu, D., Wu, K., Lei, J., and Yan, N. (2019). Structures of human Na(v)1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308. doi:10.1126/science.aaw2493
Tang, C., Zhou, X., Nguyen, P. T., Zhang, Y., Hu, Z., Zhang, C., et al. (2017). A novel tarantula toxin stabilizes the deactivated voltage sensor of bacterial sodium channel. FASEB J. 31, 3167–3178. doi:10.1096/fj.201600882R
Wang, J., Yarov-Yarovoy, V., Kahn, R., Gordon, D., Gurevitz, M., Scheuer, T., et al. (2011). Mapping the receptor site for alpha-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. U. S. A. 108, 15426–15431. doi:10.1073/pnas.1112320108
Wisedchaisri, G., Tonggu, L., Gamal El-Din, T. M., McCord, E., Zheng, N., and Catterall, W. A. (2021). Structural basis for high-affinity trapping of the Na(V)1.7 channel in its resting state by tarantula toxin. Mol. Cell 81, 38–e4. e34. doi:10.1016/j.molcel.2020.10.039
Wisedchaisri, G., Tonggu, L., McCord, E., Gamal El-Din, T. M., Wang, L., Zheng, N., et al. (2019). Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell 178, 993–e12. e1012. doi:10.1016/j.cell.2019.06.031
Wulff, H., Christophersen, P., Colussi, P., Chandy, K. G., and Yarov-Yarovoy, V. (2019). Antibodies and venom peptides: New modalities for ion channels. Nat. Rev. Drug Discov. 18, 339–357. doi:10.1038/s41573-019-0013-8
Xiao, Z., Zhao, P., Wu, X., Kong, X., Wang, R., Liang, S., et al. (2021). Variation of two S3b residues in K(V)4.1-4.3 channels underlies their different modulations by spider toxin κ-LhTx-1. Front. Pharmacol. 12, 692076. doi:10.3389/fphar.2021.692076
Zhang, J., Tang, D., Liu, S., Hu, H., Liang, S., Tang, C., et al. (2018). Purification and characterization of JZTx-14, a potent antagonist of mammalian and prokaryotic voltage-gated sodium channels. Toxins (Basel) 10, 408. doi:10.3390/toxins10100408
Zhang, M., Liu, X. S., Diochot, S., Lazdunski, M., and Tseng, G. N. (2007). APETx1 from sea anemone Anthopleura elegantissima is a gating modifier peptide toxin of the human ether-a-go-go- related potassium channel. Mol. Pharmacol. 72, 259–268. doi:10.1124/mol.107.035840
Zheng, W., Zhang, C., Li, Y., Pearce, R., Bell, E. W., and Zhang, Y. (2021). Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 1, 100014. doi:10.1016/j.crmeth.2021.100014
Zhou, X., Xiao, Z., Xu, Y., Zhang, Y., Tang, D., Wu, X., et al. (2017). Electrophysiological and pharmacological analyses of Na(v)1.9 voltage-gated sodium channel by establishing a heterologous expression system. Front. Pharmacol. 8, 852. doi:10.3389/fphar.2017.00852
Keywords: NaChBac, spider toxin, molecular mechanism, voltage sensor trapping, molecular docking, antagonist
Citation: Xiao Z, Li Y, Zhao P, Wu X, Luo G, Peng S, Liu H, Tang C and Liu Z (2022) Molecular mechanism of the spider toxin κ-LhTx-I acting on the bacterial voltage-gated sodium channel NaChBac. Front. Pharmacol. 13:924661. doi: 10.3389/fphar.2022.924661
Received: 20 April 2022; Accepted: 27 June 2022;
Published: 04 August 2022.
Edited by:
Shujia Zhu, Institute of Neuroscience, Shanghai Institute for Biological Sciences (CAS), ChinaReviewed by:
Hongtu Zhao, St. Jude Children’s Research Hospital, United StatesGeorge Chandy, University of California, Irvine, United States
Copyright © 2022 Xiao, Li, Zhao, Wu, Luo, Peng, Liu, Tang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng Tang, chengtang@hunnu.edu.cn; Zhonghua Liu, Liuzh@hunnu.edu.cn
†These authors have contributed equally to this work