Geranylgeranylacetone Ameliorates Beta-Amyloid Toxicity and Extends Lifespan via the Heat Shock Response in Caenorhabditis elegans
 Isiah Mossiah
Isiah Mossiah  Sabrina M. Perez
Sabrina M. Perez  Taylor R. Stanley
Taylor R. Stanley  Michaela K. Foley Karen S. Kim Guisbert
Michaela K. Foley Karen S. Kim Guisbert  Eric Guisbert*
Eric Guisbert*- Department of Biomedical and Chemical Engineering and Sciences, Florida Institute of Technology, Melbourne, FL, United States
Activation of a cytoprotective cellular pathway known as the heat shock response (HSR) is a promising strategy for the treatment of Alzheimer’s disease and other neurodegenerative diseases. Geranylgeranylacetone (GGA) is a commonly used anti-ulcer drug in Japan that has been shown to activate the HSR. Here, we establish C. elegans as a model system to investigate the effects of GGA. First, we show that GGA-mediated activation of the HSR is conserved in worms. Then, we show that GGA can ameliorate beta-amyloid toxicity in both muscle and neuronal worm Alzheimer’s disease models. Finally, we find that exposure to GGA is sufficient to extend the lifespan of wild-type worms. Significantly, the beneficial effects of GGA on both beta-amyloid toxicity and lifespan are dependent on HSR activation. Taken together, this research supports further development of GGA as a therapeutic for Alzheimer’s disease, provides evidence that HSR activation is a relevant therapeutic mechanism, and indicates that the beneficial effects of GGA are not limited to disease.
Introduction
Translation of biomedical science discoveries into disease-modifying therapeutics is a slow and laborious process that requires enormous financial resources. The estimated cost of developing a new pharmaceutical is between 1 and 3 billion dollars (DiMasi et al., 2016; Nosengo, 2016). One analysis suggests that the returned value for investment in the pharmaceutical industry may be less than the value of the initial investments (Pushpakom et al., 2019).
Repurposing of current therapeutics is a strategy that can substantially reduce development costs (Pushpakom et al., 2019). Drug repurposing significantly reduces the attrition of candidate molecules during therapeutic development due to safety concerns, rapid metabolism, and lack of bioavailability. Therefore, drug repurposing has the potential to facilitate the development of new disease treatments. One successful example of this approach is Viagra, which was originally an antihypertensive drug that was repurposed for the treatment of erectile dysfunction.
Alzheimer’s disease (AD) is a devastating neurodegenerative disease with an urgent and desperate need for therapeutic development (Sierksma et al., 2020; Walsh and Selkoe, 2020). Two of the key pathological features of AD, senile plaques and neurofibrillary tangles, are associated with protein misfolding and aggregation. In the brains of AD patients, beta-amyloid peptides accumulate in senile plaques while Tau proteins accumulate in neurofibrillary tangles. Distinct protein aggregates also accumulate in a stunning variety of other neurodegenerative diseases (Hipp et al., 2019). While the specific connections between protein folding and pathology remain an active area of research, the consequences of protein misfolding accumulation include general loss of cellular protein folding homeostasis, synaptic loss, and neuronal death. Therefore, preventing or reversing protein misfolding is being pursued as a therapeutic strategy for AD.
The heat shock response (HSR) is a cytoprotective, cellular stress response that responds to protein misfolding induced by temperature and other stresses (Guisbert and Morimoto, 2012). The HSR is mediated by the highly conserved HSF1 transcription factor. HSF1 regulates a suite of beneficial heat shock genes, including molecular chaperones that directly facilitate the refolding and/or degradation of misfolded proteins.
Genetic activation of HSR genes has been shown to have beneficial effects in disease models for Alzheimer’s disease and other neurodegenerative diseases. For example, overexpression of the HSP70 heat shock gene suppresses toxicity in a mouse AD model (Hoshino et al., 2011). Furthermore, HSR activation has been shown to extend lifespan even in the absence of disease (Hsu et al., 2003; Morley and Morimoto, 2004). These results have prompted efforts to identify and develop small molecule HSR inducers (Calamini et al., 2011; Neef et al., 2011; West et al., 2012).
Geranylgeranylacetone (GGA), also known as Teprenone, is a drug that has had widespread use in Japan as an anti-ulcer agent since the 1980s (Zeng et al., 2018). While the mechanism of action for this drug remains incompletely characterized, it has been shown to activate the HSR in cultured cells (Hirakawa et al., 1996). HSR activation by GGA occurs via disruption of the interaction between the HSP70 moleculer chaperone and HSF1, as HSP70 is a key negative regulator of HSF1 (Otaka et al., 2007).
Administration of GGA was found to have dramatic effects in a mouse Alzheimer’s disease model. GGA treated APP23 mice exhibited decreased levels of beta-amyloid peptide, beta-amyloid aggregates, and synaptic loss (Sun et al., 2017). Importantly, these benefits are accompanied by improvements in cognitive function. Furthermore, these effects occurred during oral administration of GGA, indicating that GGA can cross the blood-brain barrier (Katsuno et al., 2005).
The promising results with GGA in the mouse Alzheimer’s disease model and its established clinical use in humans makes GGA an ideal candidate for drug repurposing. Continued development of GGA would benefit from a model system that is experimentally feasible and genetically tractable. Therefore, we establish Caenorhabditis elegans as a model to study the mechanism of GGA and determine if the beneficial effects of GGA against beta-amyloid toxicity requires HSR activation.
Materials and Methods
Nematode Strains and Cultures
Nematodes were maintained using standard laboratory techniques on NGM plates seeded with OP50 bacteria at 20°C unless otherwise indicated (Brenner, 1974). Worms were age-matched by bleaching with hypochlorite (NaOCl) and hatching overnight in M9 buffer or with synchronized egg-laying. The following strains were used: 1) N2 (wild-type) (Brenner, 1974); 2) CL 2006 [pCL12 (unc-54/human Aβ peptide 3-42 minigene) + rol-6(su1006)] (Link, 1995); 3) CL2355 [pCL45 (snb-1:Aβ 1-42:3′ UTR (long) + mtl-2:GFP]I (Wu et al., 2006); 4) CL2122 [(pPD30.38) unc-54 (vector) + (pCL26) mtl-2::GFP] (Wu et al., 2006); 5) PS3551 hsf-1(sy441) (Hajdu-Cronin et al., 2004); 6) CF1038 daf-16(mu86) (Hsin and Kenyon, 1999).
Chemicals and Reagents
GGA was obtained from Santa Cruz Biotechnology. Diacetyl was obtained from TCI America. RNAi constructs were obtained from the Ahringer RNAi library and used for feeding RNAi as previously described (Kamath and Ahringer, 2003; Ma et al., 2017). The following oligos were used: hsp-16.2 forward (ACTTTACCACTATTTCCGTCCAGC), hsp-16.2 reverse (CCTTGAACCGCTTCTTTCTTTG), hsp-16.11 forward (GGCTCAGATGGAACGTCAA), hsp-16.11 reverse (GCTTGAACTGCGAGACATTG), F44E5.5 forward (CAACTGCTGGTGATACCCATCTC), F44E5.5 reverse (CTTGAAAGTGTTCTCTTGGCACG), hsp-70 forward (GTACTACGTACTCATGTGTCGGTATTTATC), hsp-70 reverse (ACGGGCTTTCCTTGTTTTCC), cdc-42 forward (TTTGCTTCTCCGTGGTTGCTCC), and cdc-42 reverse (TCCGTTGACACTGGTTTCTGCTTG).
RT-qPCR
N2 worms were grown at 20°C on NGM control or 10 μM GGA plates starting at the L1 larval stage until day one of adulthood. RNA was isolated using Trizol Reagent (Thermo Fisher) as previously described with the addition of a freeze-thaw in liquid nitrogen and use of a Direct-zol RNA kit (Zymo Research) (Golden et al., 2020). RNA was reverse transcribed with an iScript cDNA Synthesis Kit (Bio-Rad) and RT-qPCR was performed using an iTaq Universal SYBR Green Supermix (Bio-Rad) and the CFX Connect cycler (Bio-Rad). HSR gene expression was normalized to expression of cdc-42. All qPCR experiments were performed with biological triplicates and p-values were calculated using the Student’s t-test.
Paralysis Assay
Transgenic CL 2006 (muscle beta-amyloid) worms were grown at 20°C on NGM control or 10 μM GGA plates seeded with empty vector RNAi (L4440) or hsf-1 RNAi containing bacteria starting at the L1 larval stage. Worms were scored for paralysis starting on day 1 of adulthood. Individuals were considered paralyzed if the body of the worm did not move when gently prodded. Worms were transferred by picking onto new plates as needed to separate adults from eggs and larva. Data was analyzed using OASIS 2 (Han et al., 2016).
Associative Memory Assay
Transgenic CL2122 (control) and CL2355 (neuronal beta-amyloid) worms were grown at 16°C on NGM control or 10 μM GGA plates starting at the L1 larval stage until day one of adulthood. Then, worms were shifted to 25°C for 24 h to induce neuronal beta-amyloid expression. Day 2 adults were conditioned by transferring to bacteria-free NGM plates with 6 μL of 0.04% diacetyl added to the plate lid (Dosanjh et al., 2010). Control worms were transferred to bacteria-free NGM plates but not exposed to diacetyl. Plates were inverted and incubated for 2 h at 25°C. The chemotaxis assay was conducted by spotting 2 µL of 0.04% diacetyl 1 inch from the center of the assay plate on one side and 95% ethanol on the other. Worms were quickly placed at the center of each plate, followed by an additional 2 µL of odorant and 2 µL of 1 M sodium azide in each spot to paralyze the worms. Plates were then incubated at 20°C for 1 h and immediately scored. The chemotaxis index was calculated using the following formula (number of worms at the diacetyl spot—number of worms at the ethanol spot)/total number of worms.
Lifespan
N2, PS3551 (hsf-1 mutant), and CF1038 (daf-16 mutant) worms were grown at 20°C on NGM control or 10 μM GGA plates starting at the L1 or L4 larval stages. Viability was scored starting on day 1 of adulthood. Individuals were considered dead if they did not have a motile response to gentle prodding and lacked pharyngeal pumping. Worms were transferred by picking onto new control or GGA plates as needed to separate adults from eggs and larva. Data was analyzed using OASIS 2 (Han et al., 2016).
Results
The ability of Geranylgeranylacetone (GGA) to ameliorate beta-amyloid toxicity in mice motivated further investigation of this drug using the genetically tractable model nematode Caenorhabditis elegans. Previously, GGA has been shown to activate the cytoprotective heat shock response (HSR) in cultured human cells and in mice (Zeng et al., 2018). Therefore, we first investigated the ability of GGA to activate the HSR in worms. Worms were exposed to GGA from the first larval stage (L1) to adulthood on standard NGM agar plates or NGM plates containing GGA. It was found that incubation of worms on plates containing 10 μM GGA was sufficient to increase the expression of the hsp-16.11, hsp-70, and F44E5.5 HSR-dependent genes between 1.5-fold and 6-fold measured by RT-qPCR (Figure 1). GGA also induced a fourth HSR gene, hsp-16.2, 1.5-fold, but this effect was not statistically significant. The hsp-70 and F44E5.5 genes both encode HSP70 family proteins while hsp-16.11 and hsp-16.2 genes encode small HSP proteins. These results demonstrate that the effects of GGA on the HSR are conserved from humans and mice to worms and establish the basis for using C. elegans as model system for investigation of the effects of GGA on the HSR.
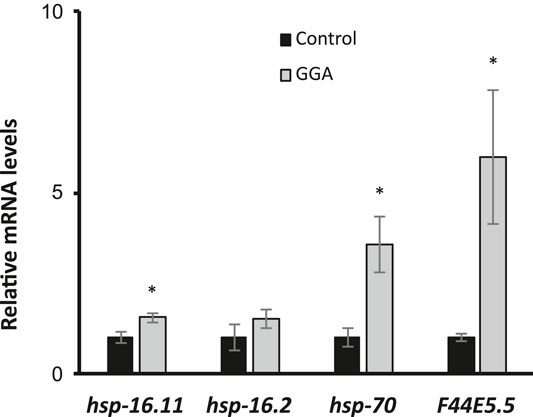
FIGURE 1. GGA activates the HSR in C. elegans. Wild-type (N2) worms were synchronized and incubated on NGM control or 10 μM GGA plates until day 1 of adulthood. RT-qPCR analysis showed a significant increase in basal expression of heat shock gene mRNA in worms exposed to GGA. Averages shown are from 3 biological replicates. Error bars represent SEM; * indicates p-value < 0.05 (Student’s t-test).
Previously, GGA was shown to have beneficial effects on a mouse model for Alzheimer’s disease (AD) containing expression of the human beta-amyloid peptide (Sun et al., 2017). Therefore, we next tested whether the effects of GGA on beta-amyloid toxicity were conserved in worms. A well-established worm AD model was used that features transgenic expression of the human A-beta peptide (Link, 1995). In this model, beta-amyloid is expressed in muscle cells and the worms develop progressive paralysis as they age with 50% of the worms becoming paralyzed on day 11 of adulthood (Figure 2). We found that incubation with GGA led to a reduction in this age-dependent paralysis with the median paralysis occurring nearly 2 days later. These results indicate that GGA can ameliorate toxicity from beta-amyloid expression in a worm disease model.
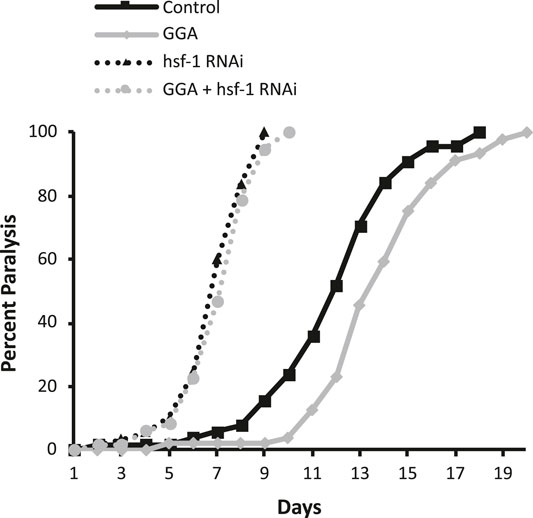
FIGURE 2. GGA alleviates paralysis in a C. elegans Alzheimer’s disease model. Transgenic CL2006 worms expressing beta-amyloid in muscle were synchronized and incubated on NGM control or 10 μM GGA plates seeded with empty vector (L4440) or hsf-1 RNAi containing bacteria. Worms were scored for paralysis starting at day 1 of adulthood. Worms exposed to GGA had significantly delayed paralysis compared to control worms (p-value < 0.05, Mantel-Cox log-rank test). Each curve represents data from 3 independent trials of n ≥ 20 individuals.
Having established that the effects of GGA on HSR activation and beta-amyloid-induced paralysis were conserved in worms, we next tested whether the effects on beta-amyloid were dependent on the HSR. The HSR was inhibited by RNAi knockdown of the HSF1 transcription factor that is required for the response. Consistent with previous work, we found that inhibition of HSF1 alone enhanced the toxicity in this disease model, with median paralysis occurring around day 6 of adulthood. In contrast to its effects in wild-type worms, GGA did not have a significant effect on beta-amyloid toxicity when HSF-1 was inhibited. These results indicate that activation of the HSR is required for the beneficial effects of GGA.
Having shown that GGA can reverse the toxicity from beta-amyloid expression in muscle tissue, we next tested the effects of GGA in neurons, a more relevant tissue type for AD. In a second worm AD model, neuronal expression of human beta-amyloid has been shown to cause neuronal toxicity and results in a defect in associative learning (Dosanjh et al., 2010). In the associative learning assay, worms are simultaneously exposed to a chemoattractant and a lack of food. Upon subsequent exposures, worms display associative learning by reducing their chemotaxis towards the odorant. Naïve wild type worms exhibited chemotaxis towards diacetyl, measured by a chemotaxis index of 0.73 (Figure 3). When they were conditioned by exposure to diacetyl in the absence of food, they reduced their preference for this odorant to a chemotaxis index of 0.44. In the AD model, neuronal expression of beta-amyloid disrupted associative learning but did not affect chemotaxis of naïve worms (0.73 naïve verses 0.66 conditioned).
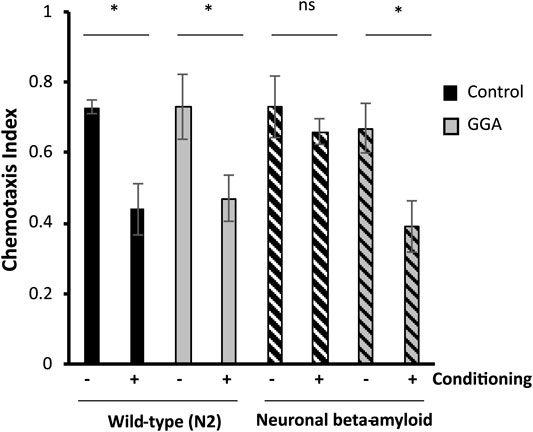
FIGURE 3. GGA restores associative learning in a neuronal Alzheimer’s disease model. Transgenic worms with or without neuronal beta-amyloid expression were synchronized and incubated on NGM control or 10 μM GGA plates at 16°C. On day one of adulthood, worms were shifted to 25°C for 24 h to induce beta-amyloid expression. Then, worms were starved in the presence or absence of the odorant diacetyl for 2 h and scored for chemotaxis. Worms expressing neuronal beta-amyloid exhibited a defect in conditioned learning that was restored upon exposure to GGA. Averages shown are from 3 biological replicates of n ≥ 30 adults each. Error bars represent SEM; * indicates p-value < 0.05; ns = non-significant (Student’s t-test).
We tested the ability of GGA to reverse the associative learning defect in worms expressing neuronal beta-amyloid. GGA did not affect chemotaxis nor associative learning in wild-type worms (0.73 naïve verses 0.47 conditioned). Significantly, exposure to GGA restored associative learning in worms with neuronal beta-amyloid (0.68 naïve verses 0.39 conditioned). Taken together, these experiments demonstrate that the beneficial effects of incubation with GGA in worms extend to neurons.
Having established the effects of GGA on disease models, we next tested whether its beneficial effects could extend to longevity. A wide range of protein aggregates accumulate during the normal process of aging and disruption of protein folding homeostasis, or proteostasis, is considered a hallmark of aging (David et al., 2010; López-Otín et al., 2013). Overexpression of HSF1 has been shown to extend lifespan in worms while inhibition of HSF1 causes premature aging (Hsu et al., 2003; Morley and Morimoto, 2004). Therefore, we tested whether incubation with GGA was sufficient to extend lifespan in wild-type worms. Excitingly, incubation of wild-type worms with GGA extended the mean lifespan (18.7 ± 0.5 days for control versus 20.2 ± 0.7 days, p-value 0.03) (Figure 4A; Table 1). This lifespan extension did not require early developmental exposure as a lifespan extension was still observed upon exposure of worms to GGA starting at the L4 stage (16.5 ± 0.7 days for control versus 19.9 ± 1.0 days, p-value 0.002) (Figure 4B). We tested whether this phenotype was dependent on the HSR using a worm strain containing a mutation in HSF1 that blocks HSR activation. We found that GGA was no longer was able to extend lifespan in the HSF1 mutant background (12.8 ± 0.3 days versus 13.1 ± 0.3 days, p-value 0.54) (Figure 5; Table 1). This dependency was specific for the HSR as the effects of GGA were not reduced in worms containing a mutation in DAF-16, a distinct longevity pathway known as insulin-like signaling (14.8 ± 0.4 versus 16.3 ± 0.6, p-value 0.01). Together, these data indicate that GGA can extend lifespan in worms via activation of the HSR and that its beneficial effects are not limited to neurodegenerative disease models.
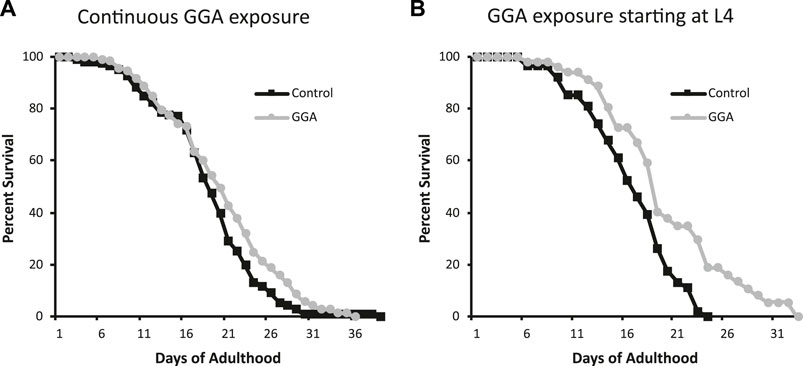
FIGURE 4. GGA extends lifespan in C. elegans. Wild-type (N2) worms were synchronized and incubated on NGM control plates or plates containing 10 μM GGA starting at the L1 (A) or L4 (B) larval stages. Worms were scored for viability starting at day 1 of adulthood. Worms exposed to GGA in either case lived significantly longer than control worms (p-value < 0.05, Mantel-Cox log-rank test). Each curve represents ≥3 pooled trials of n ≥ 20 individuals each.
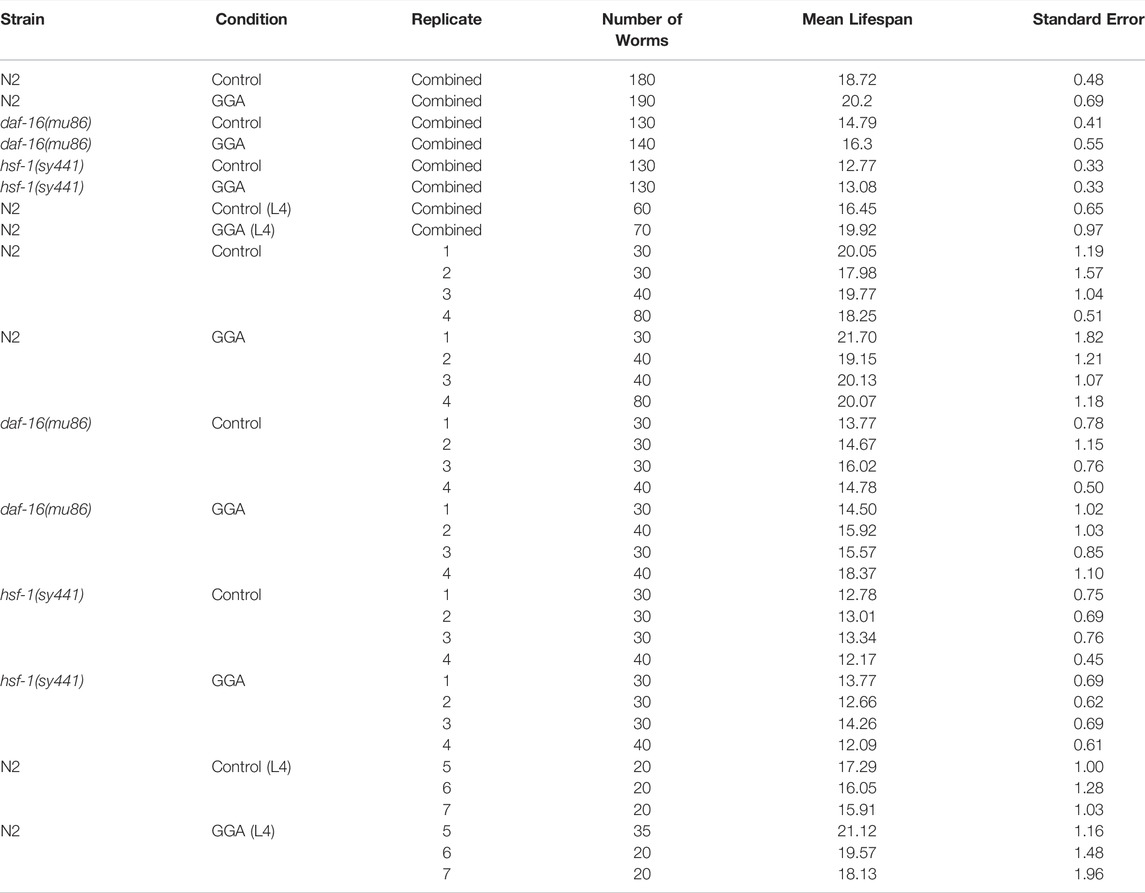
TABLE 1. Effects of GGA on lifespan.
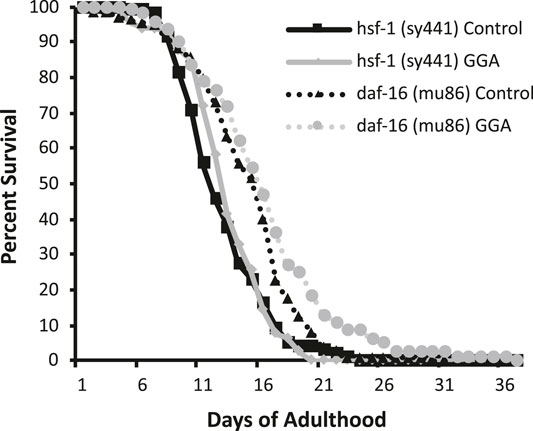
FIGURE 5. The HSR is required for GGA-mediated lifespan extension. Worms containing mutations in daf-16 (mu86) and hsf-1 (sy441) were synchronized and incubated on control NGM plates or plates containing 10 μM GGA. Worms were scored for viability starting at day 1 of adulthood. Incubation with GGA caused significant lifespan extension in daf-16 mutant worms (p-value < 0.05 Mantel-Cox log-rank test) but not worms containing a mutation in hsf-1. Each curve represents ≥4 pooled trials of n ≥ 30 individuals each.
Discussion
This manuscript establishes C. elegans as a model system for investigation of the anti-ulcer drug geranylgeranylacetone (GGA). First, conservation of the effects of GGA in worms was shown with respect to both HSR activation and amelioration of beta-amyloid toxicity. Then, genetic manipulation of the system was used to demonstrate that activation of the HSR is required for GGA’s beneficial effects. Finally, it was discovered that GGA can also extend lifespan in wild-type worms. Taken together, this research not only supports further development of GGA as a novel Alzheimer’s disease (AD) therapeutic, but also provides a foundation that can be used for new insights into HSR regulation and lifespan.
Activation of the HSR by GGA in worms establishes a new tool in the C. elegans HSR field. This activation is not surprising given the high degree of conservation in the HSF1 transcription factor that mediates this response. However, worms are resistant to the effects of many small molecules due to the barrier function of their cuticle. The ability to activate the HSR independent of temperature increases and associated protein misfolding stress will enable new experimental approaches to explore HSR regulatory mechanisms. Furthermore, as HSR-inducible promoters have been one of the mainstays for ectopic expression in C. elegans, this tool may also be useful for other fields of biology that use worms as a model organism including developmental biology and neuroscience.
Amelioration of the detrimental effects of beta-amyloid expression in worm muscle and neuronal AD models by GGA is consistent with previous results showing that GGA has beneficial effects in a mouse AD model. However, the establishment of its effects in worms facilitated an explicit test of its dependence on the HSR. The powerful genetic tools and other advantages of this model system should set the stage for further exploration of the effects of GGA on beta-amyloid toxicity. Moreover, the use of worms can facilitate investigation of GGA in other neurodegenerative disease models such as Parkinson’s disease, ALS, and Huntington’s disease.
Excitingly, we discovered that GGA can extend the lifespan of wild-type worms. This lifespan extension is also dependent on HSF1 but not dependent on insulin-like signaling, one of the major cellular pathways that influences longevity. These results are consistent with disruption of proteostasis as a hallmark of aging, further supporting that interventions targeting this pathway can have beneficial effects. To our knowledge, our results for the first time provide experimental evidence that a small molecule HSR activator can positively affect longevity in worms. Importantly, GGA is already approved for human use to treat ulcers, indicating that it is both bioavailable and does not have substantial off-target toxicity. Therefore, these findings provide strong support for future work on GGA and other HSR inducers.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Author Contributions
IM, SP, TS, MF, and EG performed the experiments. EG and KSKG supervised the project and wrote the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work was supported by National Cancer Institute Grant #R15 CA227573, a Community Foundation for Brevard Medical Research Grant from the Kenneth R. Finken and Dorothy Hallam Finken Endowment Fund, and a donation from Frank Leslie. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). The authors would like to acknowledge the students in the Developmental Biology Lab Class at the Florida Institute of Technology and their Graduate Student Assistant, Emily Wiseman, for preliminary experiments.
References
Brenner, S. (1974). The Genetics of Caenorhabditis elegans. Genetics 77 (1), 71–94. doi:10.1093/genetics/77.1.71
Calamini, B., Silva, M. C., Madoux, F., Hutt, D. M., Khanna, S., Chalfant, M. A., et al. (2011). Small-molecule Proteostasis Regulators for Protein Conformational Diseases. Nat. Chem. Biol. 8 (2), 185–196. doi:10.1038/nchembio.763
David, D. C., Ollikainen, N., Trinidad, J. C., Cary, M. P., Burlingame, A. L., and Kenyon, C. (2010). Widespread Protein Aggregation as an Inherent Part of Aging in C. elegans. Plos Biol. 8 (8), e1000450. doi:10.1371/journal.pbio.1000450
DiMasi, J. A., Grabowski, H. G., and Hansen, R. W. (2016). Innovation in the Pharmaceutical Industry: New Estimates of R and D Costs. J. Health Econ. 47, 20–33. doi:10.1016/j.jhealeco.2016.01.012
Dosanjh, L. E., Brown, M. K., Rao, G., Link, C. D., and Luo, Y. (2010). Behavioral Phenotyping of a Transgenic Caenorhabditis Elegans Expressing Neuronal Amyloid-β. J. Alzheimer's Dis. 19(2), 681–690. doi:10.3233/JAD-2010-1267
Golden, N. L., Plagens, R. N., Kim Guisbert, K. S., and Guisbert, E. (2020). Standardized Methods for Measuring Induction of the Heat Shock Response in Caenorhabditis elegans. J. Visualized Experiments 161. doi:10.3791/61030
Guisbert, E., and Morimoto, R. I. (2012). “The Regulation and Function of the Heat Shock Response,” in Protein Quality Control in Neurodegenerative Diseases (Springer Berlin Heidelberg), 1–18. doi:10.1007/978-3-642-27928-7_1
Hajdu-Cronin, Y. M., Chen, W. J., and Sternberg, P. W. (2004). The L-type Cyclin CYL-1 and the Heat-Shock-Factor HSF-1 Are Required for Heat-Shock-Induced Protein Expression in Caenorhabditis elegans. Genetics 168 (4), 1937–1949. doi:10.1534/genetics.104.028423
Han, S. K., Lee, D., Lee, H., Kim, D., Son, H. G., Yang, J.-S., et al. (2016). OASIS 2: Online Application for Survival Analysis 2 with Features for the Analysis of Maximal Lifespan and Healthspan in Aging Research. Oncotarget 7 (35), 56147–56152. doi:10.18632/oncotarget.11269
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019). The Proteostasis Network and its Decline in Ageing. Nat. Rev. Mol. Cel Biol 20 (7), 421–435. doi:10.1038/s41580-019-0101-y
Hirakawa, T., Rokutan, K., Nikawa, T., and Kishi, K. (1996). Geranylgeranylacetone Induces Heat Shock Proteins in Cultured guinea Pig Gastric Mucosal Cells and Rat Gastric Mucosa. Gastroenterology 111 (2), 345–357. doi:10.1053/gast.1996.v111.pm8690199
Hoshino, T., Murao, N., Namba, T., Takehara, M., Adachi, H., Katsuno, M., et al. (2011). Suppression of Alzheimer's Disease-Related Phenotypes by Expression of Heat Shock Protein 70 in Mice. J. Neurosci. 31 (14), 5225–5234. doi:10.1523/JNEUROSCI.5478-10.2011
Hsin, H., and Kenyon, C. (1999). Signals from the Reproductive System Regulate the Lifespan of C. elegans. Nature 399 (6734), 362–366. doi:10.1038/20694
Hsu, A.-L., Murphy, C. T., and Kenyon, C. (2003). Regulation of Aging and Age-Related Disease by DAF-16 and Heat-Shock Factor. Science 300 (5622), 1142–1145. doi:10.1126/science.1083701
Kamath, R., and Ahringer, J. (2003). Genome-wide RNAi Screening in Caenorhabditis elegans. Methods 30 (4), 313–321. doi:10.1016/s1046-2023(03)00050-1
Katsuno, M., Sang, C., Adachi, H., Minamiyama, M., Waza, M., Tanaka, F., et al. (2005). Pharmacological Induction of Heat-Shock Proteins Alleviates Polyglutamine-Mediated Motor Neuron Disease. Proc. Natl. Acad. Sci. U.S.A. 102 (46), 16801–16806. doi:10.1073/pnas.0506249102
Link, C. D. (1995). Expression of Human Beta-Amyloid Peptide in Transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 92 (20), 9368–9372. doi:10.1073/pnas.92.20.9368
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The Hallmarks of Aging. Cell 153 (6), 1194–1217. doi:10.1016/j.cell.2013.05.039
Ma, J., Grant, C. E., Plagens, R. N., Barrett, L. N., Guisbert, K. S. K., and Guisbert, E. (2017). Cellular Proteomes Drive Tissue-specific Regulation of the Heat Shock Response. G3 Bethesda, Md. 7 (3), 1011–1018. doi:10.1534/g3.116.038232
Morley, J. F., and Morimoto, R. I. (2004). Regulation of Longevity inCaenorhabditis Elegansby Heat Shock Factor and Molecular Chaperones. Mol. Biol. Cel 15 (2), 657–664. doi:10.1091/mbc.e03-07-0532
Neef, D. W., Jaeger, A. M., and Thiele, D. J. (2011). Heat Shock Transcription Factor 1 as a Therapeutic Target in Neurodegenerative Diseases. Nat. Rev. Drug Discov. 10 (12), 930–944. doi:10.1038/nrd3453
Nosengo, N. (2016). Can You Teach Old Drugs New Tricks? Nature 534 (7607), 314–316. doi:10.1038/534314a
Otaka, M., Yamamoto, S., Ogasawara, K., Takaoka, Y., Noguchi, S., Miyazaki, T., et al. (2007). The Induction Mechanism of the Molecular Chaperone HSP70 in the Gastric Mucosa by Geranylgeranylacetone (HSP-Inducer). Biochem. Biophysical Res. Commun. 353 (2), 399–404. doi:10.1016/j.bbrc.2006.12.031
Pushpakom, S., Iorio, F., Eyers, P. A., Escott, K. J., Hopper, S., Wells, A., et al. (2019). Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 18 (1), 41–58. doi:10.1038/nrd.2018.168
Sierksma, A., Escott-Price, V., and De Strooper, B. (2020). Translating Genetic Risk of Alzheimer's Disease into Mechanistic Insight and Drug Targets. Science 370 (6512), 61–66. doi:10.1126/science.abb8575
Sun, Y., Zhang, J. R., and Chen, S. (2017). Suppression of Alzheimer's Disease-related P-henotypes by the H-eat S-hock P-rotein 70 I-nducer, Geranylgeranylacetone, in APP/PS1 Transgenic Mice via the ERK/p38 MAPK Signaling Pathway. Exp. Ther. Med. 14 (6), 5267–5274. doi:10.3892/etm.2017.5253
Walsh, D. M., and Selkoe, D. J. (2020). Amyloid β-protein and beyond: the Path Forward in Alzheimer's Disease. Curr. Opin. Neurobiol. 61, 116–124. doi:10.1016/j.conb.2020.02.003
West, J. D., Wang, Y., and Morano, K. A. (2012). Small Molecule Activators of the Heat Shock Response: Chemical Properties, Molecular Targets, and Therapeutic Promise. Chem. Res. Toxicol. 25 (10), 2036–2053. doi:10.1021/tx300264x
Wu, Y., Wu, Z., Butko, P., Christen, Y., Lambert, M. P., Klein, W. L., et al. (2006). Amyloid- -Induced Pathological Behaviors Are Suppressed by Ginkgo Biloba Extract EGb 761 and Ginkgolides in Transgenic Caenorhabditis elegans. J. Neurosci. 26 (50), 13102–13113. doi:10.1523/JNEUROSCI.3448-06.2006
Keywords: geranylgeranylacetone, HSF1, heat shock response, Alzheimer’s disease, aging, longevity, lifespan, drug repurposing
Citation: Mossiah I, Perez SM, Stanley TR, Foley MK, Kim Guisbert KS and Guisbert E (2022) Geranylgeranylacetone Ameliorates Beta-Amyloid Toxicity and Extends Lifespan via the Heat Shock Response in Caenorhabditis elegans. Front. Aging 3:846977. doi: 10.3389/fragi.2022.846977
Received: 31 December 2021; Accepted: 01 April 2022;
Published: 27 April 2022.
Edited by:
Cindy Voisine, Northeastern Illinois University, United StatesReviewed by:
Johnathan Paul Labbadia, Northwestern University, United StatesStephan N. Witt, Louisiana State University Health Shreveport, United States
Copyright © 2022 Mossiah, Perez, Stanley, Foley, Kim Guisbert and Guisbert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Guisbert, eguisbert@fit.edu