In-depth structural proteomics integrating mass spectrometry and polyacrylamide gel electrophoresis
 Nobuaki Takemori
Nobuaki Takemori Ayako Takemori
Ayako Takemori- Advanced Research Support Center, Institute for Promotion of Science and Technology, Ehime University, Toon, Ehime, Japan
The establishment of a highly sensitive method for obtaining structural information on proteins and protein complexes in vivo has long been a technological challenge in structural biology. In recent years, protein structure analysis approaches using top-down mass spectrometry, native mass spectrometry, and cross-linking mass spectrometry, among others, have been developed, and these techniques have emerged as the most promising methods for obtaining comprehensive structural information on the cellular proteome. However, information obtained by MS alone is derived mainly from protein components that are abundant in vivo, with insufficient data on low abundance components. For the detection of those low abundance components, sample fractionation prior to mass spectrometry is highly effective because it can reduce the complexity of the sample. Polyacrylamide gel electrophoresis (PAGE), which is widely used in biochemical experiments, is an excellent technique for protein separation in a simple straightforward procedure and is also a promising fractionation tool for structural proteomics. The difficulty of recovering proteins in gels has been an obstacle, thus far limiting its application to structural mass spectrometry. With the breakthrough of PEPPI-MS, an exceptionally efficient passive extraction method for proteins in gels that appeared in 2020, various PAGE-based proteome fractionation workflows have been developed, resulting in the rapid integration of structural mass spectrometry and PAGE. In this paper, we describe a simple and inexpensive PAGE-based sample preparation strategy that accelerates the broad use of structural mass spectrometry in life science research, and discuss future prospects for achieving in-depth structural proteomics using PAGE.
Introduction
One of the most successful application of mass spectrometry (MS) in protein science is its use in proteomics, the analysis of protein components in biological systems. Large-scale detection of protein components in vivo is made possible by the use of a liquid chromatography-MS (LC-MS) system that integrates LC and an electrospray ionization (ESI) MS (McCormack et al., 1997). Protein components extracted from biological samples are first denatured by chemical treatment and then enzymatically digested to a peptide size suitable for highly sensitive detection before LC-MS analysis. This analytical approach, called bottom-up MS, has strongly driven proteome analysis in various model organisms to date (Müller et al., 2020).
In MS-based proteomics, in-depth analysis is heavily dependent on the sample separation process prior to MS: even if the protein detection capability of LC-MS is extremely high (Hebert et al., 2014), LC alone cannot sufficiently separate complex proteome samples, rendering the detection of trace components often difficult. The now available ion mobility, an orthogonal separation process, allows for the detection of approximately 10,000 protein components in a human cultured cell sample in a single analysis (Kawashima et al., 2022), resulting in a coverage comparable to transcriptome analysis.
Information on protein components obtained by bottom-up proteomics is derived from digested peptides detected by MS. Digestion, though necessary, allows only the existence of particular proteins to be detected, not their structure. Therefore, the majority of bottom-up proteomics studies have unfortunately provided very limited information on the actual chemical structure of proteins in vivo, omitting conformational information.
Recent advances in MS technology have enabled highly sensitive and throughput protein structure analysis, leading to a significant decrease in the gap between MS-based proteomics and structural biology. To comprehensively obtain structural information on proteins in vivo, the following initiatives toward structural proteomics have emerged.
1) Top-down MS: Unlike bottom-up MS, this MS method ionizes proteins in their intact form and fragments them inside the MS instrument to obtain comprehensive chemical structural information.
2) Cross-linking MS (XL-MS): An MS method that chemically cross-links protein molecules in solution and reveals the cross-linking sites by bottom-up MS, enabling analysis of protein-protein interactions.
3) Native MS: An MS method for non-denatured proteins. Detection of protein ions that maintain higher-order structure is achieved by suppressing structural destruction of protein complexes in solution as they are transferred to the gas phase during ionization by ESI.
Common to bottom-up MS as well as any of the structural MS methods is the necessity of suitable sample preparation so that the number of detectable components can be increased as much as possible. In many cases, LC or capillary electrophoresis (CE) with on-line connectivity to MS can sufficiently fulfill this role, but for enhanced depth of analysis, additional fractionation of the sample prior to LC/CE is desirable. Polyacrylamide gel electrophoresis (PAGE), which allows protein separations despite being simple and inexpensive, has long been used in bottom-up proteomics as a powerful proteome fractionation tool. In the last several years, its use in sample preparation for structural MS has been gaining attention. In this article, we present examples of protein structure analysis combining proteome fractionation by various PAGE and structural MS techniques, and propose novel gel-based structural proteomics strategies.
GeLC-MS workflow for top-down proteomics
For proteome separations, sodium dodecyl sulfate (SDS)-PAGE is often used for size-based separations of proteins denatured with SDS, a strong anionic surfactant. Its effectiveness as sample pre-fractionation for in-depth analysis has been repeatedly demonstrated in bottom-up proteomics. In particular, GeLC-MS, a combined SDS-PAGE separation and LC-MS workflow, is widely used to realize in-depth proteome analysis (Pflieger et al., 2002; Schirle et al., 2003). However, GeLC-MS for bottom-up proteomics employs an approach in which enzymatic digestion is performed in the gel (Shevchenko et al., 2006) so the proteins are recovered as short peptide fragments. Generally, proteins after PAGE must be recovered from the gel for separation by LC, and it has long been a challenge to efficiently recover intact proteins that are tightly trapped in the insoluble N,N′-Methylenebisacrylamide (Bis) cross-linked polyacrylamide gel that are predominantly used for SDS-PAGE. For this recovery process, electroelution, in which an electric field is applied to extract proteins from the inside of the gel (Seelert and Krause, 2008; Razunguzwa et al., 2009; Hao et al., 2016; Chen et al., 2017), and passive extraction, in which proteins are recovered by diffusion from the gel through shaking, have been used (Cohen and Chait, 1997; Ehring et al., 1997; Galvani et al., 2000), but both methods often suffer from low recovery rates and long manipulation times. Since SDS-PAGE targets denatured proteins, it is more appropriate to combine SDS-PAGE with chemical structure analysis by top-down MS rather than 3D structure analysis. In particular, SDS-PAGE can demonstrate its maximum potential in the large-scale analysis of intact proteoforms by top-down MS, known as top-down proteomics (Smith and Kelleher., 2013; Smith et al., 2021). The long-standing obstacle to applying SDS-PAGE to top-down proteomics was the absence of a highly efficient protein recovery method which was finally overcome in 2020.
Passively eluting proteins from polyacrylamide gels as intact species for MS (PEPPI-MS), an innovative passive extraction technique developed by our group in 2020 (Takemori et al., 2020), successfully solves the fixation problem by using Coomassie Brilliant Blue (CBB), a reversible protein staining dye (Fazekas de St Groth et al., 1963; Diezel et al., 1972), as an extraction enhancer. Proteins are recovered from a wide range of molecular weights (MWs) with high reproducibility after 10 min of shaking (Figure 1). Most remarkably, the protein recovery rate is high, with a mean value of 68% in the MW range below 100 kDa (Takemori et al., 2020). The recovery rate for proteins larger than 100 kDa, however, is slightly lower (57%). Since the PEPPI workflow includes organic solvent precipitation for purification of recovered proteins, the difficulty of re-dissolving high MW proteins compared to low MW proteins may have affected the recovery rate. Although proteins above 50 kDa are difficult to analyze with most current top-down MS, it is necessary to introduce a purification approach that does not include precipitation treatment to improve the recovery rate of high MW regions. Rapid and highly efficient in-gel protein recovery by PEPPI-MS has enabled the introduction of GeLC-MS workflow in top-down proteomics. The use of specialized preparative electrophoresis devices such as gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) (Tran and Doucette, 2008; Tran and Doucette, 2009) is currently the mainstream for gel-electrophoresis-based sample pre-fractionation in top-down proteomics (Tran et al., 2011), but PEPPI-MS fractionation does not require any special equipment and can be easily done in most general biochemical laboratories. The first GeLC-MS for top-down proteomics was conducted using an experimental workflow combining PEPPI-MS fractionation with a powerful LC-MS system consisting of reversed-phase ultra-high performance LC and 21 T Fourier transform ion cyclotron resonance MS, and demonstrated performance that was comparable to proteoform identification results using GELFrEE fractionation (Takemori et al., 2020). A three-dimensional separation approach integrating gel, liquid, and gas phase separations dramatically increased the depth of analysis, as was reported last year in a top-down proteomics workflow combining the LC-FAIMS-Orbitrap MS system with PEPPI-MS (Takemori et al., 2022). Although PEPPI fractionation has a cost advantage over LC fractionation, it does not support the automation found in many LC fractionations. Therefore, LC currently has the advantage in processing multiple samples, and the need for the development of automated PEPPI systems will arise in the future.
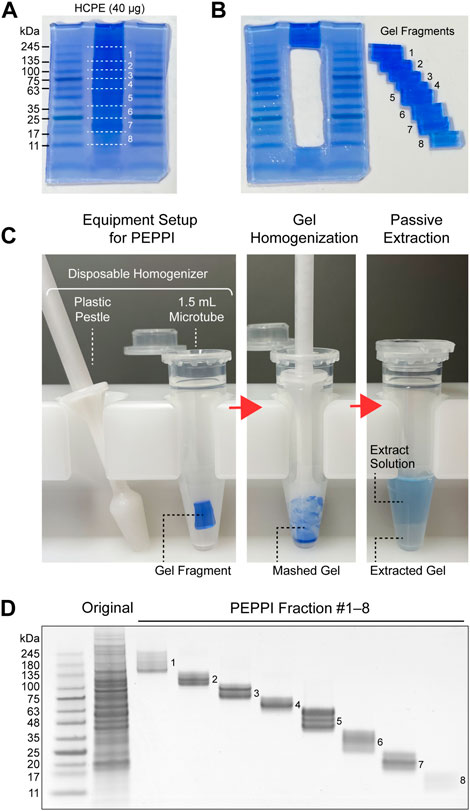
FIGURE 1. Proteome fractionation by PEPPI-MS. (A) SDS-PAGE separation of human cell protein extract (HCPE); Promega’s standard HCPE sample (40 μg) was separated by Invitrogen’s NuPAGE 4%–12% gel (Invitrogen) and stained with aqueous CBB (ATTO EzStain AQua). (B) Fractionation of the sample lanes. The molecular weight region from 245 kDa to 11 kDa was divided into eight parts by referring to the molecular weight markers on both sides (Wako WIDE-VIEW Pre-stained Protein Size Marker III). (C) In-gel protein recovery process by PEPPI-MS. Step 1: Preparation of equipment for PEPPI-MS. Protein recovery from the cut gel pieces is completed in a disposable plastic homogenizer (Nippi’s Bio Masher II). Step 2: Homogenization of gels. Thoroughly grind the gel with a pestle to facilitate extraction. Step 3: Passive extraction. By shaking the gel in 0.05% SDS/100 mM ammonium bicarbonate solution for 10 min, the proteins in the gel are released with CBB into the extraction solution. (D) SDS-PAGE image of the PEPPI fraction. The fractions were again separated by SDS-PAGE and stained with CBB.
While the use of PEPPI-MS for sample pre-fractionation in top-down proteomics is expected to spread rapidly in the near future, it is forecast to also expand the scope of targets for analysis when combined with various SDS-PAGE applications. For example, the Bis-Tris gel currently used in PEPPI cannot separate proteoforms below 10 kDa, but the combination of a Tris-Tricine gel that can separate proteoforms down to 1 kDa may be instrumental in the attempt to improve detection of proteoforms in the low MW range (Schägger and von Jagow, 1987; Schägger, 2006). In addition to size-based separation, SDS-PAGE also allows separation based on specific post-translational modifications by adding functional molecules to acrylamide. In particular, Phos-Tag PAGE (Kinoshita et al., 2006), a technique allowing electrophoretic separation of phosphorylated and non-phosphorylated proteins, is of particular interest in examining phosphorylated proteins, which are the primary target of top-down proteomics analysis. The approach of two-dimensional electrophoretic separation by a combination of SDS-PAGE and Phos-Tag PAGE (Kinoshita et al., 2009) will be of significant value when considering the construction of GeLC-MS top-down workflow for proteome-wide phosphorylation analysis.
Protein-protein interaction analysis combining XL-MS and PEPPI-AnExSP
SDS-PAGE is frequently used for XL-MS analysis of protein complexes because SDS-PAGE allows the separation of samples that have been cross-linked in solution (Iacobucci et al., 2018). Data quality is improved through selectively recovering the MW of interest. An alternative is the use of Blue-native (BN)-PAGE, which separates protein complexes in a non-denaturing state (Schägger et al., 1994). In 2020, A. Heck’s group reported sample preparation for XL-MS whereby protein complexes are encapsulated in a gel by BN-PAGE, which can then be replaced by a suitable solvent for cross-linking reactions, allowing the protein complexes to be directly cross-linked in the gel (Hevler et al., 2021). After separation with SDS-PAGE or BN-PAGE, cross-linked protein complexes are generally analyzed by a bottom-up approach, whereby the cross-linking sites are identified after enzymatic digestion in the gel. However, the large size and high hydrophobicity of cross-linked peptides greatly reduce the recovery rate from the gel. With large amounts of cross-linked proteins, reduced recovery rate is not a crucial problem, but with smaller amounts, it leads to an increase in undetectable cross-linked sites. Further drawbacks include the complexity of the process that comprises decolorization and washing as well as time-consuming overnight digestion reactions.
In-gel protein recovery by PEPPI-MS, which we developed in 2020, can be an effective solution to the above problems in bottom-up MS-based XL-MS analysis because it can shift the field of digestion from in-gel to in-solution with greater degrees of freedom. However, samples recovered by PEPPI contain large amounts of CBB and SDS and cannot be used directly for downstream processes. Purification by organic solvent precipitation is effective in removing these contaminants (Wessel and Flugge, 1984; Nickerson and Doucette, 2020), but re-dissolution of precipitated samples often becomes difficult, and the risk of loss is high when applied to small sample volumes. In 2022, we developed an enhanced sample preparation method, AnExSP (anion-exchange disc-assisted sequential sample preparation) (Takemori et al., 2022), which can perform enzymatic digestion while removing CBB and SDS by using an anion-exchange stop-and-go-extraction tip (StageTip) (Rappsilber et al., 2007), a microliter-size spin column with an anion-exchange solid-phase extraction (SPE) disc fitted inside a disposable pipette tip. When a PEPPI-MS recovery solution is loaded onto the StageTip, proteins, CBB, and SDS in the solution are captured on the SPE disc. Proteins are enzymatically digested directly on the disc, and only the digested peptides are recovered from the disc through elution with .5% formic acid/30% acetonitrile solution, while the CBB and SDS are retained on the disc. Enzymatic digestion with AnExSP is characterized by a very fast reaction time compared to that normally performed overnight in solution, with trypsin/Lys-C digestion of a 10 µg sample taking only 1–4 h.
The combined PEPPI-AnExSP workflow, in which in-gel proteins after SDS-PAGE separation are recovered to solution by PEPPI-MS and rapidly digested by AnExSP, successfully solves the problems of conventional GeLC-MS that mainly stem from in-gel digestion. Comparison of the performance of PEPPI-AnExSP and conventional GeLC-MS in bottom-up proteomics of human cell extracts showed that AnExSP identified a greater number of peptides, especially in the identification of long chain peptides of 14 amino acids or more (Takemori et al., 2022). The time required for sample pretreatment was significantly lower with PEPPI-AnExSP (1–2 h), allowing MS analysis to begin within 4.5 h of sample pretreatment after completion of electrophoresis. Preliminary studies with hemoglobin complexes have shown that SDS-PAGE-separated cross-linked hemoglobin can be successfully recovered into solution by PEPPI-MS with high efficiency, and MS analysis of the AnExSP digestion products after recovery into solution showed a significant improvement in the number of detectable cross-linked peptides compared to conventional in-gel digestion. The superior performance of PEPPI-AnExSP will make it a powerful tool in sample preparation for XL-MS.
Although the current mainstream of XL-MS analysis is based on purified samples, the application of XL-MS to large-scale analysis of protein complexes in vivo, known as complexome analysis, is one of the many challenging issues in the field of structural biology (Sinz, 2018; Yu and Huang, 2018). With the advent of the latest innovative XL-MS workflow using cleavable cross-linkers, the capacity to identify cross-linking sites by MS has dramatically improved (Steigenberger et al., 2020), resulting in rapid progress in complexome research using proteome-wide XL-MS analysis (Liu et al., 2015; Arlt et al., 2016; Liu et al., 2017; Klykov et al., 2018). Despite advances in XL-MS technology, increasing the depth of analysis remains a major challenge, requiring sample pre-fractionation techniques. In addition to the current mainstream of size-exclusion chromatography, a cost-effective PAGE-based approach will be a promising alternative. An in-depth sample fractionation strategy using PAGE has not yet been reported for complexome analysis, but MW-based fractionation of a cross-linked proteome by SDS-PAGE is feasible with PEPPI-AnExSP, resulting in a dramatically increased number of detectable complexes by XL-MS.
Feasibility of gel-based native proteomics
The sample prefractionation approaches for top-down MS and XL-MS described so far involve complete denaturation of the steric structure of the protein sample. Native MS, in contrast, requires that the three-dimensional structure of the protein is maintained throughout the sample fractionation process. For example, new workflows for conformational analysis combining native MS with protein fractionation methods in non-denaturing conditions, such as native GELFrEE (Skinner et al., 2015), size-exclusion chromatography–capillary zone electrophoresis (Shen et al., 2018), or native GELFrEE-ion exchange chromatography (Skinner et al., 2018), are slowly being adopted for the characterization of protein complex structures in vivo.
Native PAGE, which has long been used for gel electrophoresis under non-denaturing conditions, is an interesting technology that has the potential to be developed into a gel-based conformational analysis workflow in combination with native MS. One type, BN-PAGE, in which proteins are mildly negatively charged by the addition of CBB and then subjected to non-denaturing PAGE separations, has been widely used in biochemical experiments because of its high resolution (Wittig et al., 2006). Although Heck’s group successfully combined BN-PAGE with XL-MS (Hevler et al., 2021), it is unfortunately not practical to combine BN-PAGE with native MS because of the difficulty in removing CBB from proteins after BN-PAGE separation without affecting its conformation. Thus, instead of CBB, the use of the other type, clear native PAGE with a mild surfactant that reversibly adds to the protein (Wittig and Schägger, 2005; Wittig et al., 2007; Wittig and Schägger, 2008), is preferable.
Nonetheless, native PAGE alone is not sufficient for fractionation of crude cell extracted samples in native proteomics, a large-scale analysis of protein complexes in vivo. Two-dimensional native PAGE, which combines isoelectric focusing (IEF) with size-based PAGE separation under non-denaturing conditions, does allow detailed complex separations (Manabe et al., 1979), but the process of transferring samples from IEF gels to native PAGE gels is technically demanding and has the disadvantage of poor throughput. PEPPI-MS may be the link between native PAGE and native MS: by providing highly efficient recovery of in-gel protein complexes separated by native PAGE, it can then be combined with gel-based fractionation approaches compatible with native MS analysis. The use of the original PEPPI-MS protocol, however, which uses CBB for recovery after separation, is not suitable for native MS due to the need for CBB removal just as in BN-PAGE. As an alternative, the mild surfactant n-Octyl-β-D-glucoside (OG) is a promising candidate as an extraction enhancer to replace CBB, as it does not affect protein ionization by ESI at low concentrations (Loo et al., 1994). Our PEPPI-MS workflow using .1% (w/v) OG instead of CBB successfully recovered hemoglobin tetramers after separation by native PAGE in a measurable state by native MS (Takemori et al., 2020). To date, we have successfully recovered complexes up to 480 kDa using PEPPI-MS by OG, and although further optimization is needed, it appears to be applicable to protein complex analyses for a wide range of MWs.
Through fusion with top-down MS, native MS is evolving into native top-down MS, which simultaneously obtains structural information on protein complexes and the chemical structures of their constituent subunits (Zhang et al., 2015; Li et al., 2017; Li et al., 2018; Zhou et al., 2020), and is expected to lead to the development of native proteomics for large-scale analysis of protein complexes in vivo in the future (Ruotolo, 2022). While sample pre-fractionation, which enables in-depth analysis, is indeed important in native proteomics, the combination of native electrophoresis and high-throughput PEPPI-MS fractionation has the potential to become a powerful strategy. The aforementioned OG-based PEPPI-MS, which allows protein complexes to be freely recovered from the gel matrix, will facilitate the transfer of protein complexes from IEF to PAGE, enabling a high-throughput workflow for gel-based native proteomics.
Discussion
PAGE, which has long been widely used in life science research for protein separations, is now being reevaluated as a new tool for structural analysis of proteins in vivo through integration with the latest structural MS. The effectiveness of proteome fractionation strategies combining denaturing and non-denaturing PAGE and highly efficient passive extraction of proteins in gels, as exemplified by PEPPI-MS, has been successfully demonstrated with top-down, native, and XL-MS (Figure 2), and will be further extended to other structural MS techniques.
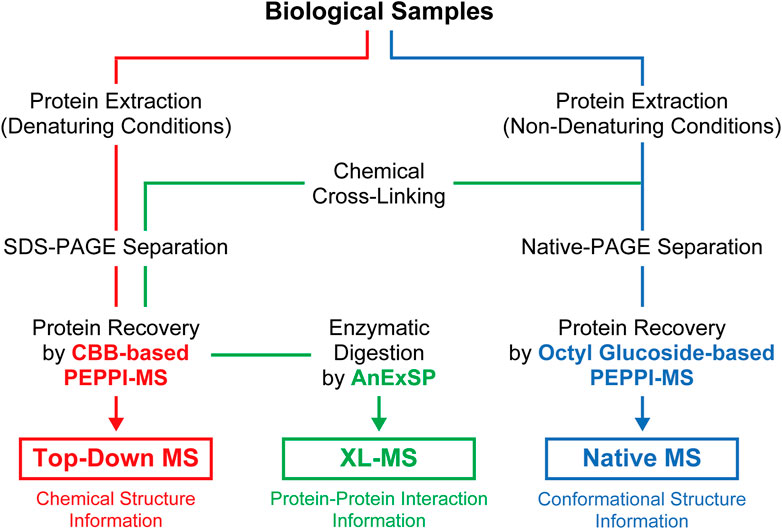
FIGURE 2. Sample preparation workflow for structural MS analyses incorporating PAGE separations under denaturing and non-denaturing conditions.
The use of gel-based sample fractionation in structural MS is still in its infancy, and more new methods are expected in the future. For the pre-fractionation processing of samples using gels, in addition to passive extraction methods described above, specialized polyacrylamide gels that can be dissolved as needed may also be a promising tool for integration with downstream structural MS. In particular, polyacrylamide gels prepared with N,N′-bis(acryloyl)cystamine (BAC) as a disulfide bonded cross-linker can be rapidly dissolved by reduction treatment and proteins in the gel can be recovered in solution without loss (Hansen, 1976; Takemori et al., 2014; Takemori et al., 2017). We have already successfully used BAC gels for sample fractionation for bottom-up proteomics (Takemori et al., 2021), and the gel dissolution process shows advantages in the recovery of high MW protein components that are difficult to recover with high efficiency by PEPPI-MS. The establishment of an efficient removal method for dissolved acrylamide is the key to practical application, and once it has been achieved, the technology is expected to be combined with top-down and XL-MS in the future.
Protein structure analysis by MS has traditionally been a difficult field for budding researchers because it requires expensive MS equipment and difficult sample preparation processes. Recently, however, the number of MS facilities available for shared use has been increasing, giving rise to the hope that some of the obstacles to entering the field are about to change for the better. In an effort to support the next-generation of researchers, the sample preparation methods introduced here are based on PAGE, which is frequently used by life science researchers, and are relatively simple and inexpensive to perform. In addition, the use of protein samples in gels that are stable at room temperature has the advantage of facilitating sample transfer between facilities, further helping to advance the widespread use of MS in protein structure studies.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by JSPS KAKENHI Grant Numbers 22KK0077, 20H04713, 19K05526.
Acknowledgments
The authors thank Erika Teraoka for editing and reviewing this manuscript for use of the English language and helpful discussion.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arlt, C., Götze, M., Ihling, C. H., Hage, C., Schäfer, M., and Sinz, A. (2016). Integrated workflow for structural proteomics studies based on cross-linking/mass spectrometry with an MS/MS cleavable cross-linker. Anal. Chem. 88 (16), 7930–7937. doi:10.1021/acs.analchem.5b04853
Chen, J. J., Li, G. Q., Pratush, A., Jahan, S., Kong, F. Z., Xiao, H., et al. (2017). An innovative ring-shaped electroeluter for high concentration preparative isolation of protein from polyacrylamide gel. Anal. Biochem. 523, 39–43. doi:10.1016/j.ab.2017.01.023
Cohen, S. L., and Chait, B. T. (1997). Mass spectrometry of whole proteins eluted from sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. Anal. Biochem. 247 (2), 257–267. doi:10.1006/abio.1997.2072
Diezel, W., Kopperschlager, G., and Hofmann, E. (1972). An improved procedure for protein staining in polyacrylamide gels with a new type of Coomassie Brilliant Blue. Anal. Biochem. 48 (2), 617–620. doi:10.1016/0003-2697(72)90117-0
Ehring, H., Strömberg, S., Tjernberg, A., and Norén, B. (1997). Matrix-assisted laser desorption/ionization mass spectrometry of proteins extracted directly from sodium dodecyl sulphate-polyacrylamide gels. Rapid Commun. Mass Spectrom. 11 (17), 1867–1873. doi:10.1002/(SICI)1097-0231(199711)11:17<1867:AID-RCM46>3.0.CO;2-Z
Fazekas de St Groth, S., Webster, R. G., and Datyner, A. (1963). Two new staining procedures for quantitative estimation of proteins on electrophoretic strips. Biochim. Biophys. Acta 71, 377–391. doi:10.1016/0006-3002(63)91092-8
Galvani, M., Bordini, E., Piubelli, C., and Hamdan, M. (2000). Effect of experimental conditions on the analysis of sodium dodecyl sulphate polyacrylamide gel electrophoresis separated proteins by matrix-assisted laser desorption/ionisation mass spectrometry. Rapid Commun. Mass Spectrom. 14 (1), 18–25. doi:10.1002/(SICI)1097-0231
Hansen, J. N. (1976). Electrophoresis of ribonucleic acid on a polyacrylamide gel which contains disulfide cross-linkages. Anal. Biochem. 76, 37–44. doi:10.1016/0003-2697(76)90261-x
Hao, F., Li, J., Zhai, R., Jiao, F., Zhang, Y., and Qian, X. (2016). A novel microscale preparative gel electrophoresis system. Analyst 141 (16), 4953–4960. doi:10.1039/c6an00780e
Hebert, A. S., Richards, A. L., Bailey, D. J., Ulbrich, A., Coughlin, E. E., Westphall, M. S., et al. (2014). The one hour yeast proteome. Mol. Cell. Proteomics 13 (1), 339–347. doi:10.1074/mcp.M113.034769
Hevler, J. F., Lukassen, M. V., Cabrera-Orefice, A., Arnold, S., Pronker, M. F., Franc, V., et al. (2021). Selective cross-linking of coinciding protein assemblies by in-gel cross-linking mass spectrometry. EMBO J. 40 (4), e106174. doi:10.15252/embj.2020106174
Iacobucci, C., Götze, M., Ihling, C. H., Piotrowski, C., Arlt, C., Schäfer, M., et al. (2018). A cross-linking/mass spectrometry workflow based on MS-cleavable cross-linkers and the MeroX software for studying protein structures and protein-protein interactions. Nat. Protoc. 13 (12), 2864–2889. doi:10.1038/s41596-018-0068-8
Kawashima, Y., Nagai, H., Konno, R., Ishikawa, M., Nakajima, D., Sato, H., et al. (2022). Single-shot 10K proteome approach: Over 10, 000 protein identifications by data-independent acquisition-based single-shot proteomics with ion mobility spectrometry. J. Proteome Res. 21 (6), 1418–1427. doi:10.1021/acs.jproteome.2c00023
Kinoshita, E., Kinoshita-Kikuta, E., Matsubara, M., Aoki, Y., Ohie, S., Mouri, Y., et al. (2009). Two-dimensional phosphate-affinity gel electrophoresis for the analysis of phosphoprotein isotypes. Electrophoresis 30 (3), 550–559. doi:10.1002/elps.200800386
Kinoshita, E., Kinoshita-Kikuta, E., Takiyama, K., and Koike, T. (2006). Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5 (4), 749–757. doi:10.1074/mcp.T500024-MCP200
Klykov, O., Steigenberger, B., Pektaş, S., Fasci, D., Heck, A. J. R., and Scheltema, R. A. (2018). Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat. Protoc. 13 (12), 2964–2990. doi:10.1038/s41596-018-0074-x
Li, H., Nguyen, H. H., Ogorzalek Loo, R. R., Campuzano, I. D. G., and Loo, J. A. (2018). An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat. Chem. 10 (2), 139–148. doi:10.1038/nchem.2908
Li, H., Sheng, Y., McGee, W., Cammarata, M., Holden, D., and Loo, J. A. (2017). Structural characterization of native proteins and protein complexes by electron ionization dissociation-mass spectrometry. Anal. Chem. 89 (5), 2731–2738. doi:10.1021/acs.analchem.6b02377
Liu, F., Lössl, P., Scheltema, R., Viner, R., and Heck, A. J. R. (2017). Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun. 8, 15473. doi:10.1038/ncomms15473
Liu, F., Rijkers, D. T., Post, H., and Heck, A. J. (2015). Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 12 (12), 1179–1184. doi:10.1038/nmeth.3603
Loo, R. R., Dales, N., and Andrews, P. C. (1994). Surfactant effects on protein structure examined by electrospray ionization mass spectrometry. Protein Sci. 3 (11), 1975–1983. doi:10.1002/pro.5560031109
Manabe, T., Tachi, K., Kojima, K., and Okuyama, T. (1979). Two-dimensional electrophoresis of plasma proteins without denaturing agents. J. Biochem. 85 (3), 649–659.
McCormack, A. L., Schieltz, D. M., Goode, B., Yang, S., Barnes, G., Drubin, D., et al. (1997). Direct analysis and identification of proteins in mixtures by LC/MS/MS and database searching at the low-femtomole level. Anal. Chem. 69 (4), 767–776. doi:10.1021/ac960799q
Müller, J. B., Geyer, P. E., Colaço, A. R., Treit, P. V., Strauss, M. T., Oroshi, M., et al. (2020). The proteome landscape of the kingdoms of life. Nature 582 (7813), 592–596. doi:10.1038/s41586-020-2402-x
Nickerson, J. L., and Doucette, A. A. (2020). Rapid and quantitative protein precipitation for proteome analysis by mass spectrometry. J. Proteome Res. 19 (5), 2035–2042. doi:10.1021/acs.jproteome.9b00867
Pflieger, D., Le Caer, J. P., Lemaire, C., Bernard, B. A., Dujardin, G., and Rossier, J. (2002). Systematic identification of mitochondrial proteins by LC-MS/MS. Anal. Chem. 74 (10), 2400–2406. doi:10.1021/ac011295h
Rappsilber, J., Mann, M., and Ishihama, Y. (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2 (8), 1896–1906. doi:10.1038/nprot.2007.261
Razunguzwa, T. T., Biddle, A., Anderson, H., Zhan, D., and Powell, M. (2009). Development of a microfluidics-based gel protein recovery system. Electrophoresis 30 (23), 4020–4028. doi:10.1002/elps.200900485
Ruotolo, B. T. (2022). Collision cross sections for native proteomics: Challenges and opportunities. J. Proteome Res. 21 (1), 2–8. doi:10.1021/acs.jproteome.1c00686
Schägger, H., Cramer, W. A., and von Jagow, G. (1994). Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217 (2), 220–230. doi:10.1006/abio.1994.1112
Schägger, H., and von Jagow, G. (1987). Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166 (2), 368–379. doi:10.1016/0003-2697(87)90587-2
Schirle, M., Heurtier, M. A., and Kuster, B. (2003). Profiling core proteomes of human cell lines by one-dimensional PAGE and liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 2 (12), 1297–1305. doi:10.1074/mcp.M300087-MCP200
Seelert, H., and Krause, F. (2008). Preparative isolation of protein complexes and other bioparticles by elution from polyacrylamide gels. Electrophoresis 29 (12), 2617–2636. doi:10.1002/elps.200800061
Shen, X., Kou, Q., Guo, R., Yang, Z., Chen, D., Liu, X., et al. (2018). Native proteomics in discovery mode using size-exclusion chromatography-capillary zone electrophoresis-tandem mass spectrometry. Anal. Chem. 90 (17), 10095–10099. doi:10.1021/acs.analchem.8b02725
Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V., and Mann, M. (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1 (6), 2856–2860. doi:10.1038/nprot.2006.468
Sinz, A. (2018). Cross-linking/mass spectrometry for studying protein structures and protein-protein interactions: Where are we now and where should we go from here? Angew. Chem. Int. Ed. Engl. 57 (22), 6390–6396. doi:10.1002/anie.201709559
Skinner, O. S., Do Vale, L. H., Catherman, A. D., Havugimana, P. C., de Sousa, M. V., Compton, P. D., et al. (2015). Native GELFrEE: A new separation technique for biomolecular assemblies. Anal. Chem. 87 (5), 3032–3038. doi:10.1021/ac504678d
Skinner, O. S., Haverland, N. A., Fornelli, L., Melani, R. D., Do Vale, L. H. F., Seckler, H. S., et al. (2018). Top-down characterization of endogenous protein complexes with native proteomics. Nat. Chem. Biol. 14 (1), 36–41. doi:10.1038/nchembio.2515
Smith, L. M., Agar, J. N., Chamot-Rooke, J., Danis, P. O., Ge, Y., Loo, J. A., et al. (2021). The human proteoform project: Defining the human proteome. Sci. Adv. 7 (46), eabk0734. doi:10.1126/sciadv.abk0734
Smith, L. M., and Kelleher, N. L. (2013). Consortium for top down, PProteoform: A single term describing protein complexity. Nat. Methods 10 (3), 186–187. doi:10.1038/nmeth.2369
Steigenberger, B., Albanese, P., Heck, A. J. R., and Scheltema, R. A. (2020). To cleave or not to cleave in XL-MS? J. Am. Soc. Mass Spectrom. 31 (2), 196–206. doi:10.1021/jasms.9b00085
Takemori, A., Butcher, D. S., Harman, V. M., Brownridge, P., Shima, K., Higo, D., et al. (2020). PEPPI-MS: Polyacrylamide-Gel-Based prefractionation for analysis of intact proteoforms and protein complexes by mass spectrometry. J. Proteome Res. 19 (9), 3779–3791. doi:10.1021/acs.jproteome.0c00303
Takemori, A., Ishizaki, J., Nakashima, K., Shibata, T., Kato, H., Kodera, Y., et al. (2021). BAC-DROP: Rapid digestion of proteome fractionated via dissolvable polyacrylamide gel electrophoresis and its application to bottom-up proteomics workflow. J. Proteome Res. 20 (3), 1535–1543. doi:10.1021/acs.jproteome.0c00749
Takemori, A., Kaulich, P. T., Cassidy, L., Takemori, N., and Tholey, A. (2022). Size-based proteome fractionation through polyacrylamide gel electrophoresis combined with LC-FAIMS-MS for in-depth top-down proteomics. Anal. Chem. 94 (37), 12815–12821. doi:10.1021/acs.analchem.2c02777
Takemori, A., Kawashima, Y., and Takemori, N. (2022). Bottom-up/cross-linking mass spectrometry via simplified sample processing on anion-exchange solid-phase extraction spin column. Chem. Commun. (Camb) 58 (6), 775–778. doi:10.1039/d1cc05529a
Takemori, N., Takemori, A., Ishizaki, J., and Hasegawa, H. (2014). Enzymatic protein digestion using a dissolvable polyacrylamide gel and its application to mass spectrometry-based proteomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 967, 36–40. doi:10.1016/j.jchromb.2014.07.006
Takemori, N., Takemori, A., Wongkongkathep, P., Nshanian, M., Loo, R. R. O., Lermyte, F., et al. (2017). Top-down/Bottom-up mass spectrometry workflow using dissolvable polyacrylamide gels. Anal. Chem. 89 (16), 8244–8250. doi:10.1021/acs.analchem.7b00357
Tran, J. C., and Doucette, A. A. (2008). Gel-eluted liquid fraction entrapment electrophoresis: An electrophoretic method for broad molecular weight range proteome separation. Anal. Chem. 80 (5), 1568–1573. doi:10.1021/ac702197w
Tran, J. C., and Doucette, A. A. (2009). Multiplexed size separation of intact proteins in solution phase for mass spectrometry. Anal. Chem. 81 (15), 6201–6209. doi:10.1021/ac900729r
Tran, J. C., Zamdborg, L., Ahlf, D. R., Lee, J. E., Catherman, A. D., Durbin, K. R., et al. (2011). Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 480 (7376), 254–258. doi:10.1038/nature10575
Wessel, D., and Flugge, U. I. (1984). A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138 (1), 141–143. doi:10.1016/0003-2697(84)90782-6
Wittig, I., Braun, H. P., and Schägger, H. (2006). Blue native PAGE. Nat. Protoc. 1 (1), 418–428. doi:10.1038/nprot.2006.62
Wittig, I., Karas, M., and Schägger, H. (2007). High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol. Cell. Proteomics 6 (7), 1215–1225. doi:10.1074/mcp.M700076-MCP200
Wittig, I., and Schägger, H. (2005). Advantages and limitations of clear-native PAGE. Proteomics 5 (17), 4338–4346. doi:10.1002/pmic.200500081
Wittig, I., and Schägger, H. (2008). Features and applications of blue-native and clear-native electrophoresis. Proteomics 8 (19), 3974–3990. doi:10.1002/pmic.200800017
Yu, C., and Huang, L. (2018). Cross-linking mass spectrometry: An emerging technology for interactomics and structural biology. Anal. Chem. 90 (1), 144–165. doi:10.1021/acs.analchem.7b04431
Zhang, J., Reza Malmirchegini, G., Clubb, R. T., and Loo, J. A. (2015). Native top-down mass spectrometry for the structural characterization of human hemoglobin. Eur. J. Mass Spectrom. (Chichester) 21 (3), 221–231. doi:10.1255/ejms.1340
Keywords: SDS-PAGE, native PAGE, mass spectometry, top-down proteomics, proteoform, chemical cross linking, complexome, PEPPI-MS
Citation: Takemori N and Takemori A (2023) In-depth structural proteomics integrating mass spectrometry and polyacrylamide gel electrophoresis. Front. Anal. Sci. 2:1107183. doi: 10.3389/frans.2022.1107183
Received: 24 November 2022; Accepted: 28 December 2022;
Published: 09 January 2023.
Edited by:
Mowei Zhou, Pacific Northwest National Laboratory (DOE), United StatesReviewed by:
Prashant Jethva, Washington University in St. Louis, United StatesDaoyang Chen, Merck, United States
Stephanie Thibert, Pacific Northwest National Laboratory (DOE), United States
Copyright © 2023 Takemori and Takemori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobuaki Takemori, takemori@m.ehime-u.ac.jp