Fellfields of the Kerguelen Islands harbour specific soil microbiomes and rhizomicrobiomes of an endemic plant facing necrosis
 Lorène Julia Marchand1,2*
Lorène Julia Marchand1,2*  Françoise Hennion1
Françoise Hennion1  Michèle Tarayre1
Michèle Tarayre1  Marie-Claire Martin1
Marie-Claire Martin1  Benoit Renaud Martins1,3
Benoit Renaud Martins1,3  Cécile Monard1
Cécile Monard1- 1University of Rennes 1, CNRS, ECOBIO (Ecosystèmes, Biodiversité, Evolution), OSUR UMR 6553, Rennes, France
- 2Research Group PLECO (Plants and Ecosystems), Department of Biology, University of Antwerp, Antwerp, Belgium
- 3Helmholtz Zentrum München, Research Unit Comparative Microbiome Analysis, Neuherberg, Germany
Polar regions are characterized by rocky terrains with sparse vegetation and oligotrophic soils, i.e. “fellfields”. In such ecosystems, microbial communities should be essential for soil-plant functioning but their diversity is poorly explored. The sub-Antarctic Kerguelen Islands fellfields are characterized by an endemic long-lived cushion plant, Lyallia kerguelensis which rhizosphere may be a shelter for microbes in this harsh environment. Cushions are affected by necrosis and we expect the rhizomicrobiome composition to be related to plant necrosis. We analysed bacterial and fungal communities in bulk- and rhizospheric soils from L. kerguelensis in five different fellfields across the Kerguelen Islands using 16S rRNA and ITS1 metabarcoding. We found that soil microbial communities were composed of both restricted and cosmopolitan taxa. While all sites were dominated by the same bacterial taxa (Chloroflexi, Actinobacteria, α-Proteobacteria and Acidobacteria), the relative abundance of the main fungal phyla (Ascomycota, Basidiomycota, Mortierellomycota and Rozellomycota) highly differed between sites. L. kerguelensis rhizomicrobiome was at least as diverse as the bulk soil, making the rhizosphere a possible reservoir of microbial diversity. It was composed of the same main bacterial phyla than detected in the bulk soil while the composition of the rhizosphere fungal communities was specific to each plant. No common microorganisms were identified regarding cushion necrosis extent across plants and sites, but several microbial putative functions were shared, suggesting a possible shift in soil functioning with cushion necrosis increase. Our study brings new information on the diversity and composition of the microbial communities of fellfield soils in a sub-Antarctic Island and the rhizomicrobiome of a characteristic endemic cushion plant.
Introduction
Fellfields are habitats characteristic of polar, sub-polar and alpine regions and they consist in a form of tundra ecosystem made of rocks with sparse vegetation and infertile soil (1). Microbial diversity in fellfield soils remains understudied despite the potentially meaningful implication of microbes in soil functioning, nutrient cycling and plant survival in such harsh habitats. For example, due to the rarity of herbivores and invertebrates (in most fellfields), the food webs are mainly driven by decomposition processes that involve microbial activity (2, 3). Within the sub-Antarctic, the Kerguelen Islands are located in the southern Indian Ocean and are isolated by 3 500 km to the nearest vegetated continent and by 2 000 km to the Antarctic. According to these geographical properties, the soil microbial communities they harbour might be shaped by dispersal limitation, leading to narrow microbial diversity as observed in soils from the Antarctic continent (2). However, strong winds that occur in this region may also transport microorganisms over long distances (4, 5), leading to complex assemblages of restricted and cosmopolitan soil microbes shaped by both dispersal limitation and aeolian transport. Moreover, within the archipelago, fellfields show contrasted edaphic properties which may induce strong environmental filtering that drive the soil microbiome assemblages, as already observed in the heterogeneous Arctic landscape (6).
In the fellfields of the Kerguelen Islands, the vegetation is composed of small forbs, short grasses and cushion plants and among them, stands Lyallia kerguelensis (Montiaceae) (7), an endemic and long-lived (more than 16 years) cushion plant. For most cushion plants, their dense prostrate shape increases the beneath soil organic matter content, generally making this rhizospheric environment a favorable habitat for soil microorganisms, compared to the surrounding nutrient-poor soil (8, 9). Although the rhizomicrobiome is known to be less diverse with a specific composition compared to the bulk soil microbiome in many ecosystems (e.g. grassland and forest) (10). Indeed, the assembly of the rhizomicrobiome, governed by plants, is directed toward beneficial microorganisms that enhance the plant’s capacity to resist to various stresses and to adapt to its environment (11). The composition of the rhizomicrobiome of L. kerguelensis might thus result from both plant-driven selection, corresponding to co-evolved species assemblage favoured by i) the longevity of this plant species, and ii) the modifications of the habitat conditions beneath it (12, 13).
The involvement of the rhizomicrobiome in plant nutrition, water uptake and stress resistance (e.g., drought, starvation, frost) was observed in many cases, enhancing plant fitness and homeostasis (e.g., its ability to resist to abiotic stresses) (11, 12, 14–16). These microorganisms might be of great importance for the plants to tolerate climate change that is rapidly occurring in polar and sub-polar regions, indeed, higher winter temperatures and lower winter precipitations are currently observed (17). These changes induce abiotic stresses to the inhabiting vegetation leading to plant dieback (i.e. the impossibility of the plant to generate new foliage) (18–20). In the Kerguelen Islands, necrotic parts in the cushions of L. kerguelensis are described since the early 1990s (7) and are now observed in many sites across the archipelago (7, 21). Thus, considering the harsh environmental conditions that prevail in the fellfields of the Kerguelen Islands and the long lifespan of this plant, we expect L. kerguelensis to recruit within its rhizosphere beneficial microorganisms and this rhizomicrobiome would be involved in the plant’s ability to resist to the abiotic stresses that this it faces, i.e. limiting necrosis extent.
Using amplicon sequencing that allows access to the total microbial diversity including uncultivable microorganisms (22), we analysed the diversity of bacterial and fungal communities, within the Kerguelen Islands, in both fellfield soils and rhizospheres of L. kerguelensis cushions randomly selected for their necrosis extent. We believed that, in fellfield soils, i) an assemblage of both restricted and cosmopolitan soil microorganisms occurred, shaped in particular by dispersal limitation and aeolian transport, and ii) that the soil properties could lead to environmental filtering, inducing site-specificity of the soil microbiome. Then, we thought that the buffered climatic conditions under the plant cushion offer a favorable habitat for soil microorganisms in these particular harsh environmental conditions, leading to a larger diversity within the rhizosphere than in the surrounding bulk soil. Finally, we hypothesised that the rhizomicrobiome might be involved in plant’s vigour facing necrosis and studied linear relations between microbial composition and plant necrosis extent.
Materials and methods
Study sites and model plant
The Kerguelen Islands (48°30’ - 50°00’ S, 68°27’ - 70°35’ E) are located in the southern Indian Ocean. The landscape is hilly and there are 22 species of native phanerogams (23), with no shrubs or trees and numerous fellfields are observed from the shore to the highest elevations exposed to wind (24). The climate is chronically cold with a mean annual temperature of 4.6°C and a constant wind (around 10 m.s-1), inducing sea-salt spray in a large part of the islands (25, 26). Our study took place during two summer field campaigns (2017-2018 and 2018-2019). In total, five fellfield sites were studied in “Ile Mac Murdo” (MAC1), “Ile Longue” (LON30), “Presqu’île Jeanne d’Arc” (PJDA2 and PJDA6) and “Ile Australia” (AUS25) (Figure 1A and Table 1. Each site (50 m²) comprised at least twenty (up to a hundred) cushions of L. kerguelensis scattered on a scale of meters.
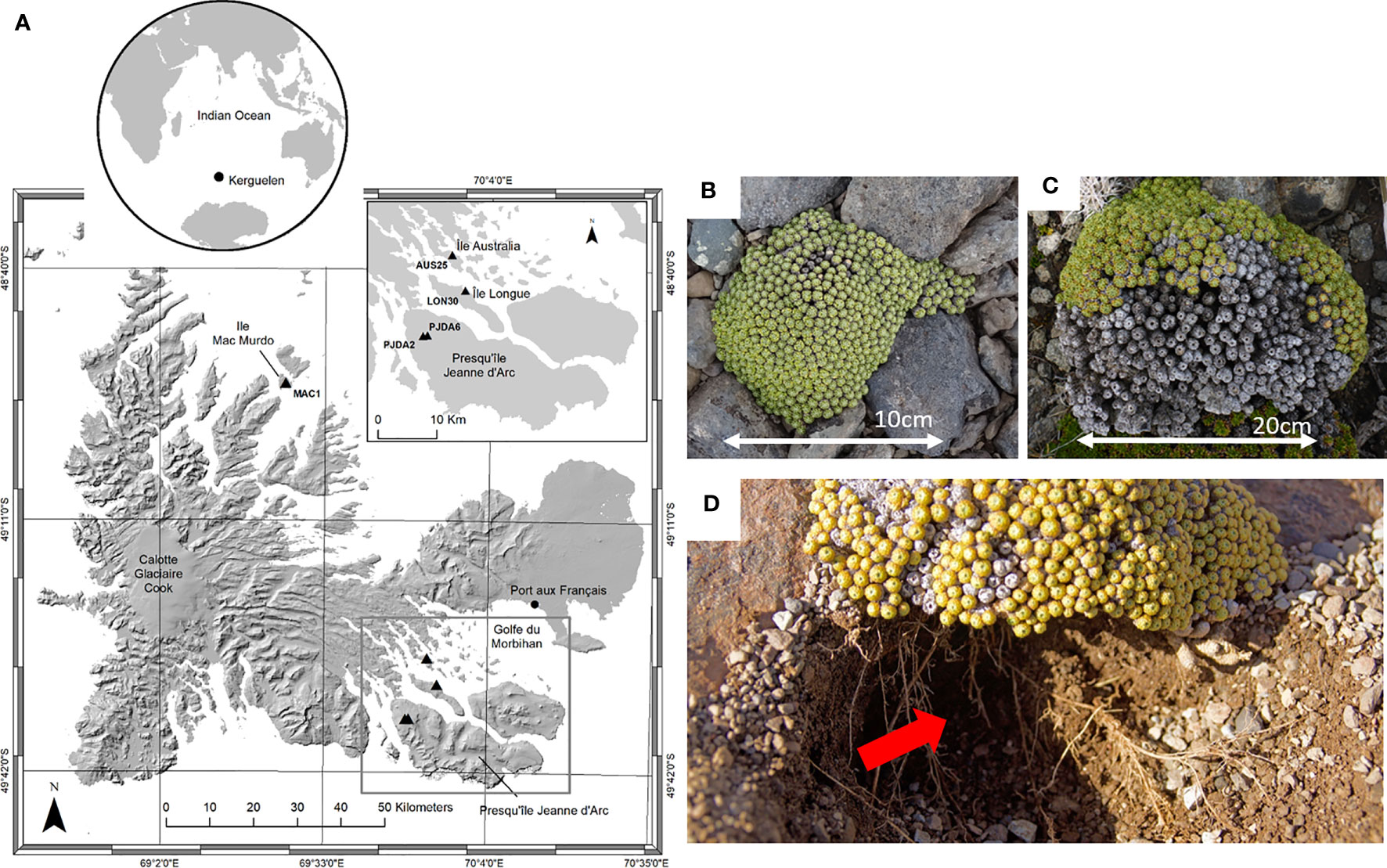
Figure 1 (A) Locations of the five sampling sites (black triangles) in Kerguelen Islands on an altitudinal background (NASA–SRTM 30 M, 2005). The black dots indicate the research stations, labels indicate the site names and important features in the island, the grey rectangle shows the zoom into presented on the top right of the figure. Top right map: zoom into the Southeast part with four out of the five studied sites. The map background comes from IGN 1/200 000 georeferenced with spatial adjustment (D. Fourcy, unpublished). The small globe sets the Kerguelen Islands in the South Indian Ocean. ArcGIS 10.8.1. (B) Picture (from above) of Lyallia kerguelensis with only a few necrotic apices and (C) with more than half of the cushion necrotic. (D) Lyallia kerguelensis and its root system, location of the soil sampling (red arrow), the cushion shows necrosis.
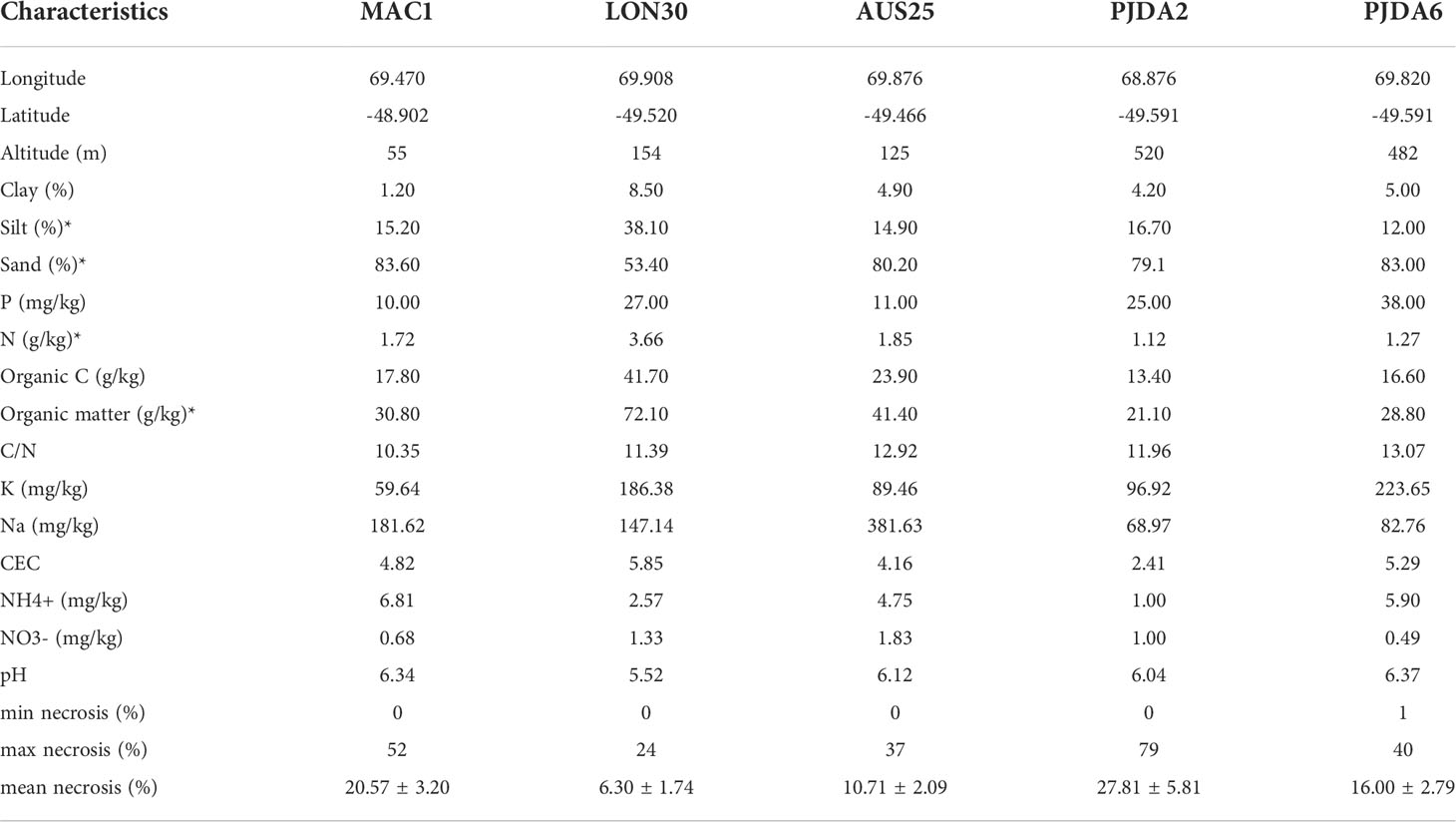
Table 1 GPS coordinates of the studied sites and their soil physico-chemical properties and plant necrosis parameters, “*” indicates the variables removed for the NMDS analysis due to correlation with other variables.
Lyallia kerguelensis Hook. f. belongs to the Montiaceae (27) and has a hemispherical shape of 20 – 40 cm in diameter (7), (Figures 1B, C). This species is endemic to the Kerguelen Islands and has a sparse distribution in fellfields (7). The root system is deep with a main taproot and numerous fine roots branching out (28), (Figure 1D). Within each site, ten plants of L. kerguelensis were randomly selected. We know that L. kerguelensis’ flowering phenology is synchronous within and among populations (28), we carefully selected plants in vegetative stage or with a small part (approximately less than 10% of the cushion surface area) showing fruit or flowers. The necrotic surface area of each plant studied (further called necrosis extent) was measured by photointerpretation (21) (Table 1). Necrosis ranged from 0% to 79% depending on the cushions studied, and the PJDA2 and MAC1 sites showed more variability in necrosis extent than LON30 and AUS25.
Soil sampling
For each plant, a single sample of soil in contact with roots, further referred to as rhizospheric soil, was in situ collected beneath the cushion, at 2 - 8 cm depth (fig. 1D). Around 6 mL of soil was collected under each plant, avoiding roots and rocks. For the bulk soil, we collected surface soil, from 0 to 8 cm depth, at different places assumed to be free of root influence, a single composite bulk soil sample of 200 mL was then made for each site. A subsample of 100 mL was used for soil physico-chemical analyses (Laboratoire d’Analyse des Sols, INRAE, Arras, France), more details on the analyses and parameters are described in Marchand et al., 2021 (23), and in Table 1. All samples were stored at -20°C until DNA extraction. A total of 5 bulk soils (one per site) and 50 rhizospheric soils (10 per site) were collected.
DNA extraction
For every rhizosphere and bulk soil sample, three technical replicates of soil DNA extraction were done and processed separately. For each replicate, DNA extractions were performed from 0.5 g of fresh soil following the protocol of Griffiths et al., 2000 (29) modified by Monard et al., 2013 (30). It consisted of a manual extraction, first with a cell lysis step in lysing matrix E tubes (MP Biomedicals, Santa Ana, California) in presence of a lysing buffer containing phenol/chloroform/isoamyl alcohol and followed by nucleic acid precipitation in polyethylene glycol (PEG) 6000 buffer. DNA quality and quantity were checked using NanoDrop device (ThermoFisher Scientific, Waltham, USA) and agarose gel electrophoresis (1%, TAE 1X). DNA extracts were stored at -20°C.
PCR amplification and Illumina sequencing
The bacterial 16S rRNA gene and the fungal ITS libraries were constructed using the protocol given by the Illumina platform (31), based on a two-step PCR approach. The following primer sets were used for bacteria: 341F (32) and 785R (33) targeting the variable V3 and V4 regions of the 16S rRNA gene, and for fungi: ITS1F (34) and ITS2 (35) targeting the ITS1 region. Amplifications were performed as described in Hellequin et al. (36). Each primer set contained the overhang adapter: forward overhang (5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’) and reverse overhang (5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3’). PCRs were carried out in a total volume of 25 µL containing: 12.5 µL of high fidelity Taq polymerase (2X TransTaq HiFi TransGen Biotech, China), 1 µL of each primer (10 µM), 0.5 µL of T4gp32 (100 ng/µl) (for fungal amplification), 2 µL (for bacterial amplification) or 4 µL (fungal amplification) of DNA diluted 100 times each and ultrapure water to reach the final volume. For bacteria, the following amplification conditions were used for the first PCR: 95°C for 4 minutes, 25 cycles of 95°C, 52°C, and 72°C during 30 sec each and 10 minutes at 72°C was applied. For Fungi, amplification in the same conditions was performed but with 30 cycles. Amplifications were checked by agarose gel electrophoresis (2%, TAE 1X). Two PCRs per sample were performed and pooled, with each time PCR negative and positive controls to ensure the success of the amplification. The amplicons were handled by the EcogenO platform (Rennes, France) for normalization, barcoding, multiplexing and Illumina paired-end (2 × 300 bp) MiSeq sequencing.
Microbial sequence analysis
Bacterial and fungal sequences were processed using the FROGS pipeline in the Galaxy project Version 3.1 (37). The reads were merged with a minimum overlap of 20 bp (FLASH). The minimal sequence length was 350 bp for bacteria and 120 bp for fungi and the maximum sequence length was 450 bp for both kinds of amplicons. The primer sequences and sequences where the two primers were not present were removed and the sequences were dereplicated. The chimeras were removed using the VSEARCH tool with the UCHIME de novo method combined with a cross-sample validation (38, 39). Clustering was performed using the Swarm method with a distance difference of 3 and OTUs with a relative abundance below 5.10-6 were removed (40). The taxonomic affiliations were done using BLAST (NCBI, USA) with the Silva_132_16S database for bacteria and the Unite_Fungi_8.0_18112018 (including a RDP assignation) for fungi. Sequences not associated with bacteria or fungal domain were discarded (e.g. archaea sequences). Only OTUs present in at least two out of the three technical replicated samples were kept for further analysis. At the end of the process, a total of 3 342 733 sequences corresponding to 6 316 OTUs for bacteria and 6 467 991 sequences corresponding to 2 623 OTUs for fungi were obtained. Six and twelve technical replicates from different samples were removed for bacteria and fungi, respectively, due to their low number of sequences (lower than 2 000 and 150 for bacteria and fungi, respectively) compared to the other technical replicates of the same sample. Moreover, for bacteria, one rhizosphere sample (all three replicates) from the AUS25 site was removed due to a too low number of sequences. All data are available in the NCBI sequence read archive (SRA) under accession number PRJNA731349.
Data analyses
Statistical analyses were done using R version 3.4.2. Graphs were created with the ggplot2 package (https://github.com/tidyverse/ggplot2). The α-error of 0.05 was used except for (i) correlations between soil physico-chemical properties and microbial community composition and (ii) correlations between necrosis extents and OTUs abundances for which a α-error of 0.01 was chosen to focus on highly significant relations.
To study the α-diversity by calculating species richness and Shannon diversity index [package Phyloseq (41)], the number of sequences was rarefied to 3 758 and 172 sequences per sample for bacteria and fungi respectively (Figure S1). These thresholds were motivated to keep as many technical replicates as possible and limit removing soil and rhizosphere samples. Non-parametric Dunn’s Kruskal-Wallis multiple comparisons were applied to study the variability of microbial diversities (species richness and Shannon index, calculated with estimated richness function of package Phyloseq) in bulk soils and rhizosphere soils across sites and between the rhizosphere and its associated bulk soil.
To study the variability of OTUs composition within and across sites, for both bulk and rhizosphere samples the unrarefied dataset was used. A core analysis (microbiome package, https://github.com/microbiome) was performed and complemented by Venn diagrams [VennDiagram package (42)]. To study a possible site effect on the composition of microbial communities in the bulk soil, we studied the bacterial community similarities between sites with a permutational multivariate analysis of variance [i.e. PERMANOVA) (function Adonis package vegan (43)] on weighted unifrac distances when the beta-dispersions were similar. When the beta-dispersions were not similar, we applied an analysis of similarities (i.e. ANOSIM) [function anosim, package vegan (43)] on weighted unifrac distances using 1000 permutations.
The correlations between bulk beta diversity (weighted unifrac distances) and soil physico-chemical properties were performed [envfit function of vegan package (43)], significantly auto-correlated soil properties were previously removed. Bacterial and fungal communities and the significant soil properties were represented with a non-metric multidimensional scaling (i.e. NMDS) [package Phyloseq (41)].
Finally, for each sampling site, Pearson correlations (package stats) were done between the relative abundance of microbial OTUs and the necrosis extent of each plant for OTUs that had a mean relative abundance higher than 0.1% and that were present in at least 85% and 65% of the rhizosphere samples of a site for bacteria and fungi, respectively. These thresholds were defined according to the distribution of OTUs and their occurrence per population.
Results
Microbial communities of fellfield soils of the Kerguelen Islands are composed of both restricted and cosmopolitan bacteria and fungi
Except in LON30, all soils from the five fellfield sites were loamy sand (from 79.1 to 83.6% of sand) and presented low contents in total nitrogen (N), organic carbon (OC) and organic matter (OM) (Table 1). The soil from LON30 contained a lower proportion of sand (53.4%) allowing higher amounts in N, OC and OM, however, all five soils presented low C:N ratios (Table 1).
On average per site, the richness of soil bacteria and fungi ranged from 337.6 to 590.3 OTUs and from 6.5 to 57.3 OTUs, respectively, and the Shannon index ranged from 4.4 to 5.8 and from 1.4 to 3.3, respectively; both presenting significant differences between sites (Figures S2A, C, E, G; Richness: Z = 3.22 and 1.96, p = 0.001 and 0.049; Shannon index: Z = 3.22 and 2.11, p = 0.001 and 0.035, for bacteria and fungi, respectively). Fungal richness was low in LON30 and PJDA2 probably due to the low amount of sequences obtained from the sequencing as observed in Figure S1. Microbial α-diversity was the highest in AUS25 and in PJDA6 for fungi (Figures S2A, E, C, G). While PJDA2 and PJDA6 fellfield sites were the closest geographically (Figure 1), they presented significant differences in the diversity of their soil microbiome (except for bacterial Shannon index), PJDA6 being always significantly richer than PJDA2 (Richness Z = 2.34 and 2.70, p = 0.019 and 0.007; Shannon index Z = 1.37 and 2.59, p = 0.17 and 0.010 for bacteria and fungi, respectively) (Figures S2A, E, C, G). Interestingly, microbial species richness and Shannon index in the soil of LON30, whose texture differed from the four other soils (Table 1), were similar to those observed in the other sites except for the bacterial species richness in AUS25 (Figures S1A, E, C, G).
Whatever the site, the dominant bacterial taxa in the bulk soils were Chloroflexi, Actinobacteria, α-Proteobacteria and Acidobacteria. However, their relative abundance differed between sites (Figure 2A). Chloroflexi was always the most represented phylum (from 33% to 49% of the total amount of sequences) excepted in the bulk soil of MAC1 that was dominated by the Actinobacteria (41% of the total amount of sequences, Figure 2A). The dominant fungal phyla detected in the bulk soils of the five sites were Ascomycota, Basidiomycota, Mortierellomycota and Rozellomycota, and their relative abundance highly differed between sites (Figure 2B). The relative abundance of Ascomycota was at least 32% in each bulk soil (Figure 2B). This phylum was dominant in MAC1, LON30 and PJDA6 sites with a relative abundance of 42%, 57% and 51%, respectively, while the Basidiomycota dominated in PJDA2 site (56% of the total amount of sequences, Figure 2B). Interestingly, the bulk soils of MAC1 and AUS25 contained a high proportion of unknown fungal sequences (37% and 48% of the total amount of sequences, respectively; Figure 2B).
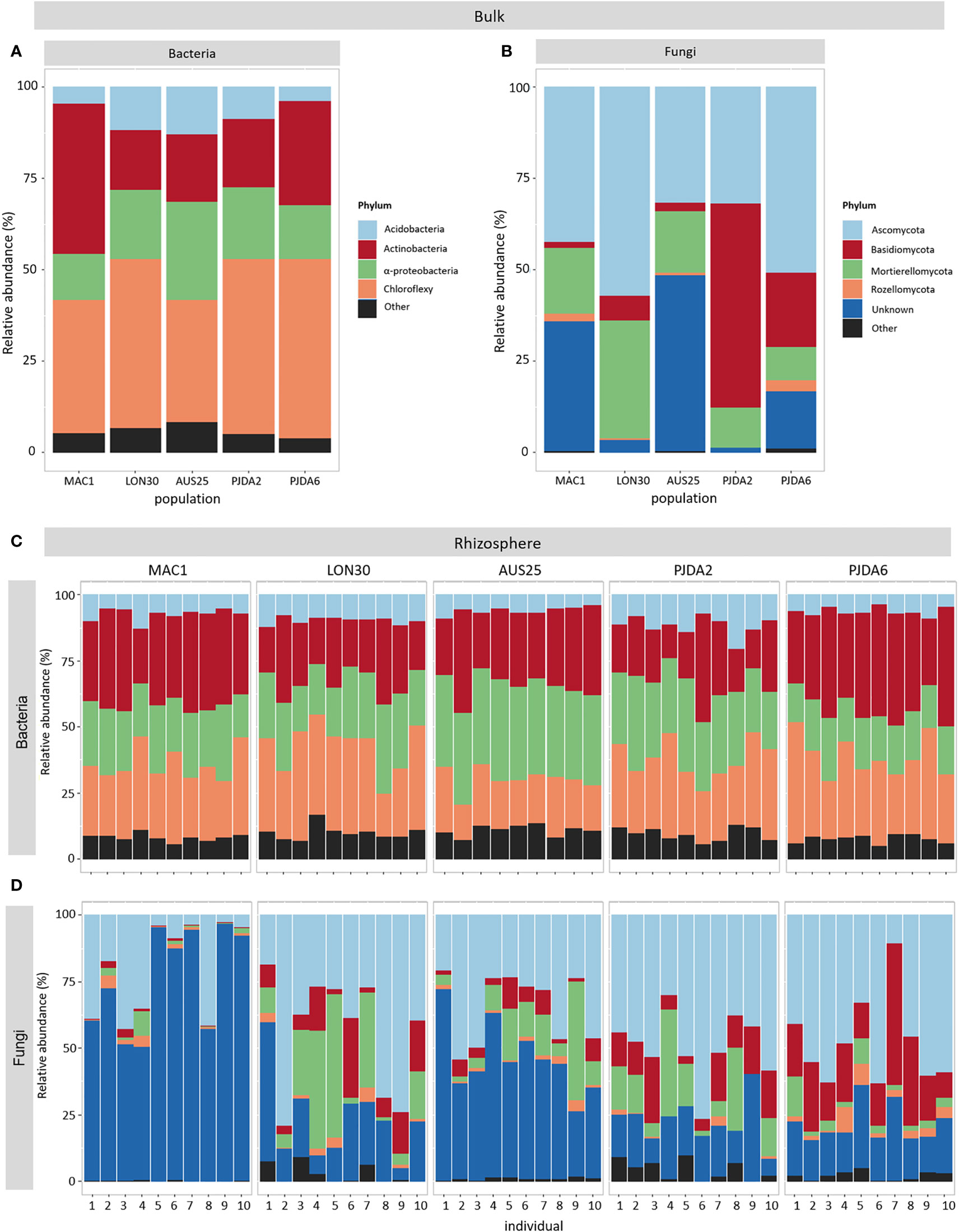
Figure 2 Relative abundance of the four dominant bacterial (A) and fungal (B) taxa present in the bulk soil of each site. (C, D) Relative abundance of the four dominant bacterial (C) and fungal (D) taxa present in the rhizosphere of each individual plant in each site.
Ninety-four bacterial OTUs, but no fungal ones, were detected in all five sites and constituting the soil core microbiome of Kerguelen Islands fellfields. They represented 52% of the total amount of bacterial sequences from the bulk soils (Figure 3A, Table S2). On top of the four dominant bacterial phyla described above, members of the Armatimonadetes, Planctomycetes and WSP-2 phyla were also identified as part of this soil core microbiome (Figure 3A, Table S2). Each site also harboured a specific soil microbiome, composed of 18 to 319 site-specific bacterial OTUs and up to 145 site-specific fungal OTUs (Figures 3A and C). These specific soil microbiomes induced spatial variability in the composition of microbial communities as observed with the NMDS ordination (Figures 4A and B) and, for both bacterial and fungal communities, a site effect was significantly observed (R²=0.83 and 0.96 and p-value <0.001 and 0.001 respectively). Some soil physico-chemical properties were identified as significantly correlated to microbial community structure. In particular, phosphorus and ammonium contents were significantly correlated to both bacterial and fungal community compositions (Figures 4A and B). Bacterial communities were also correlated to soil pH and contents in nitrate and sodium, while fungal communities were correlated to the CEC (Figures 4A and B).
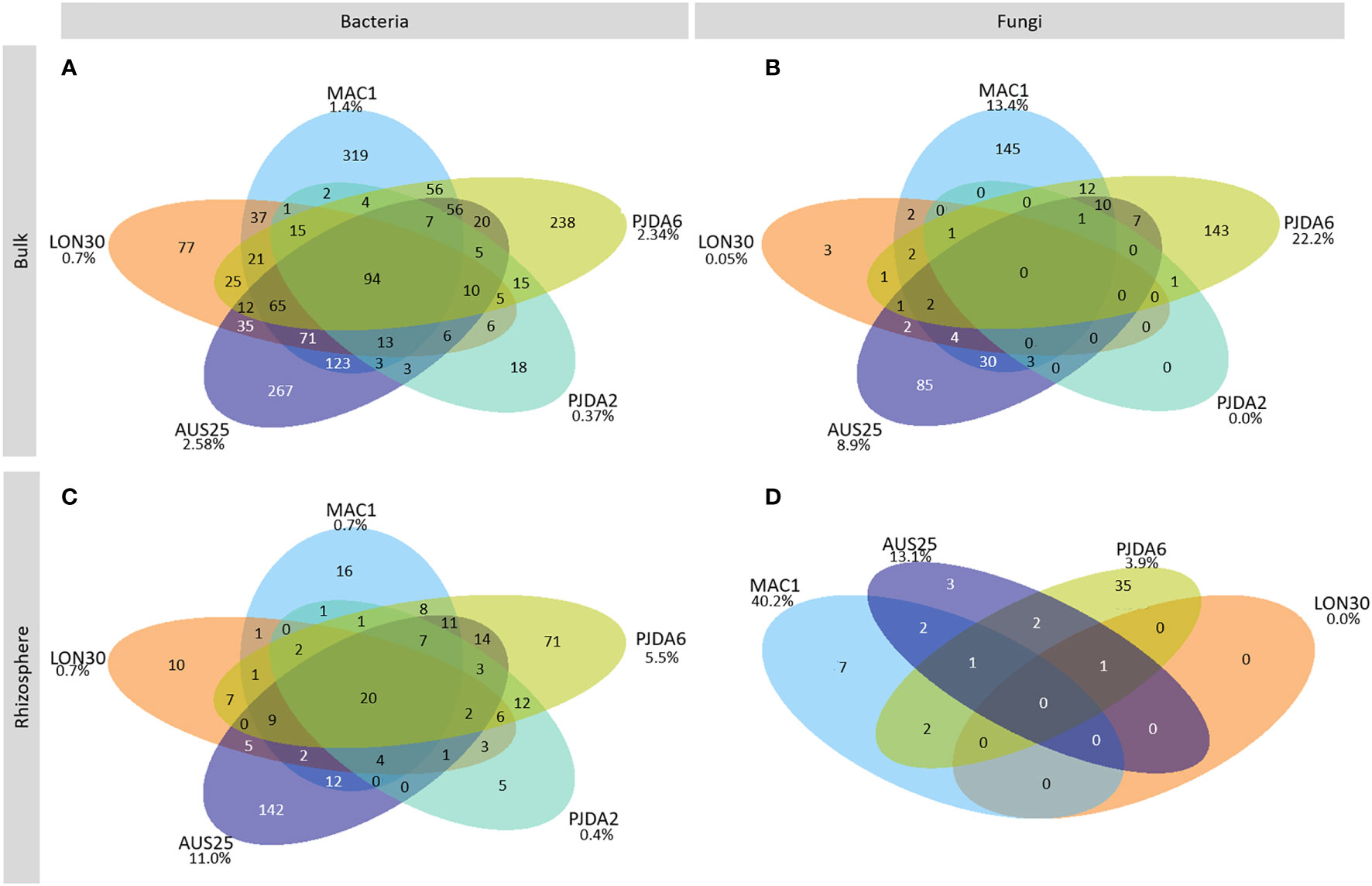
Figure 3 Venn diagrams of the soil core microbiome for (A) bacteria and (B) fungi and the core rhizomicrobiome for (C) bacteria and (D) fungi. The numbers indicate the number of OTUs in each conditions. The percentages represent the amount of OTUs specific to a site compared to the overall core soil OTUs of the same site. The soil from PJDA2 did not have any fungal core microbiome.
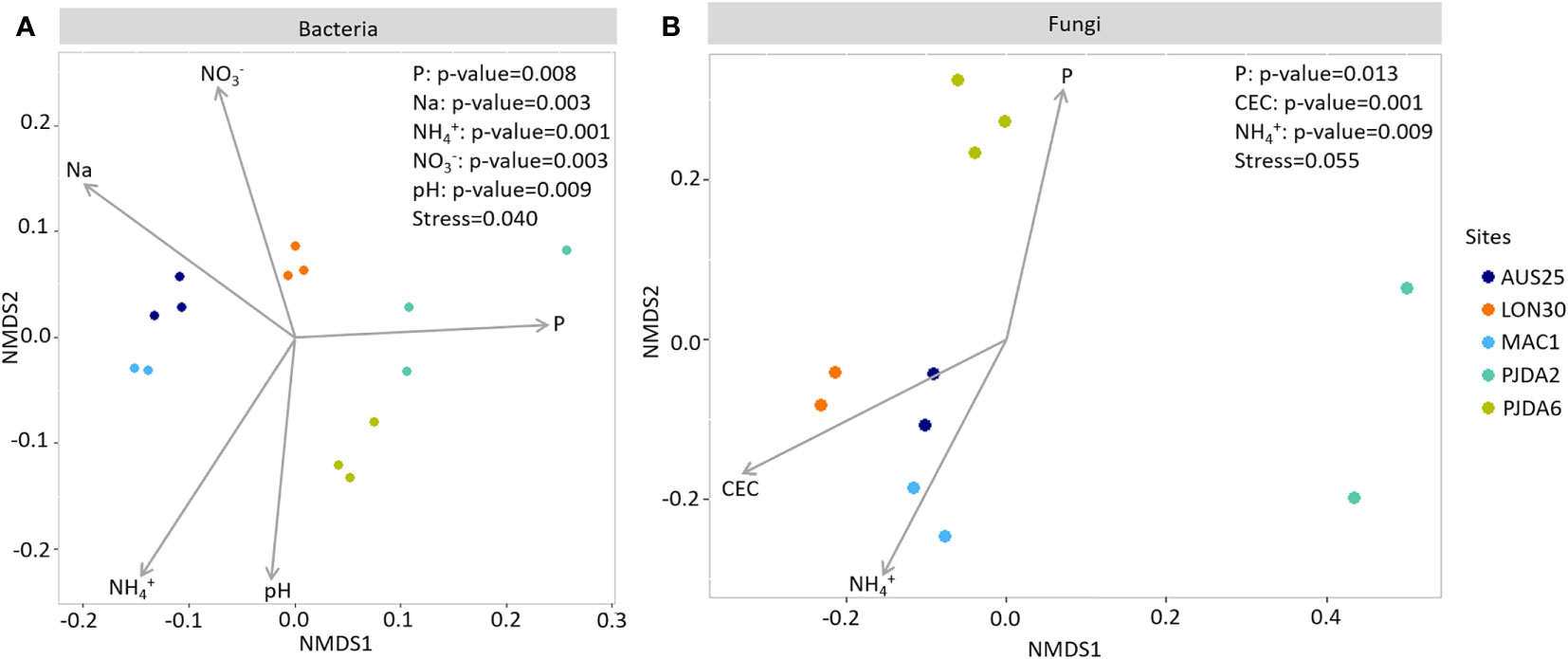
Figure 4 Non-metric multidimensional scaling (NMDS) ordination showing significant (p < 0.01) relationships between soil physico-chemical properties and the composition of (A) bacterial and (B) fungal communities in the bulk soils of each site.
The composition of the rhizomicrobiome of Lyallia kerguelensis differed from the soil microbiome
The diversity (richness and Shannon index) of the L. kerguelensis rhizomicrobiome across the five sites showed, for most of the sites, similar patterns as previously observed for the bulk soil microbiomes (Figures S2B, F, D, H). Among all the rhizospheric soil samples, 22.4% of them presented significant changes in their microbial species richness and Shannon index compared to their corresponding bulk soil microbiome (Table S1). Interestingly, in the present study, among the 50 rhizospheres of L. kerguelensis analyzed, significant depletions were only observed for fungi in MAC1 and PJDA6 sites (Table S1). The opposite trend was even observed for some rhizomicrobiomes (13 for bacteria, 6 for fungi) that were highly significantly (p<0.001) more diverse than the bulk soil microbiome.
Within each site, whatever the individual rhizomicrobiome considered, the bacterial communities were dominated by the same phyla as observed in the corresponding bulk soils and showed minor changes in their relative abundance between sites (Figure 2C). On the contrary, in each site, the composition of the rhizosphere fungal communities was specific to each plant (Figure 2D). We observed that from 24 to 75% of the bacterial OTUs and from 28 to 81% of the fungal OTUs were shared between the bulk soil and the rhizosphere. Conversely, in each individual rhizosphere, some OTUs were specific and not detected in the corresponding bulk soil (Figures S3 and S4). For bacteria, their relative abundance varied from 26% to 69% of the total amount of sequences in a single rhizosphere. For fungi, these OTUs specific to the individual rhizospheres represented at least 43% of the total amount of sequences of a single plant in PJDA6 and could reach up to 94% and 96% in LON30 and PJDA2 sites, respectively. At each site, we identified a specific rhizomicrobiome composed of 5 to 142 site-specific bacterial OTUs and up to 35 site-specific fungal OTUs (Figures 3C and D). On top of these site-specific rhizomicrobiomes, some OTUs were detected in all the rhizomicrobiomes (50 plants among the five sites) and considered cosmopolitan of L. kerguelensis rhizosphere. This core rhizomicrobiome of L. kerguelensis was composed of 20 bacterial OTUs (relative abundance of 18% of the total amount of bacterial rhizosphere sequences) but no fungal OTU (Figures 3C and D, Table S2). Among these bacterial OTUs, 18 also belonged to the core microbiome, while two OTUs identified as Acidimicrobiia were only detected in the core rhizomicrobiome (Table S2).
Relationship between plant necrosis extent and composition of the rhizomicrobiome
When considering all five sites, the necrosis extent value of the studied plants (i.e. 50 plants in total) varied between 0% and 79% (Table 1) and correlative Spearman analyses were performed to identify relationships between bacterial and fungal OTUs abundance in the rhizomicrobiome of L. kerguelensis and necrosis extent. We identified 14 and 15 bacterial OTUs and 10 and 4 fungal OTUs positively and negatively correlated to the necrosis extent, respectively (Table 2). All of them were site-specific, avoiding to observe any general pattern across sites (Table 2). However, regardless of the direction of the correlation, several OTUs related to putative microbial functions such as N2 fixation, organic matter degradation and phosphate solubilisation were observed (Table 2).
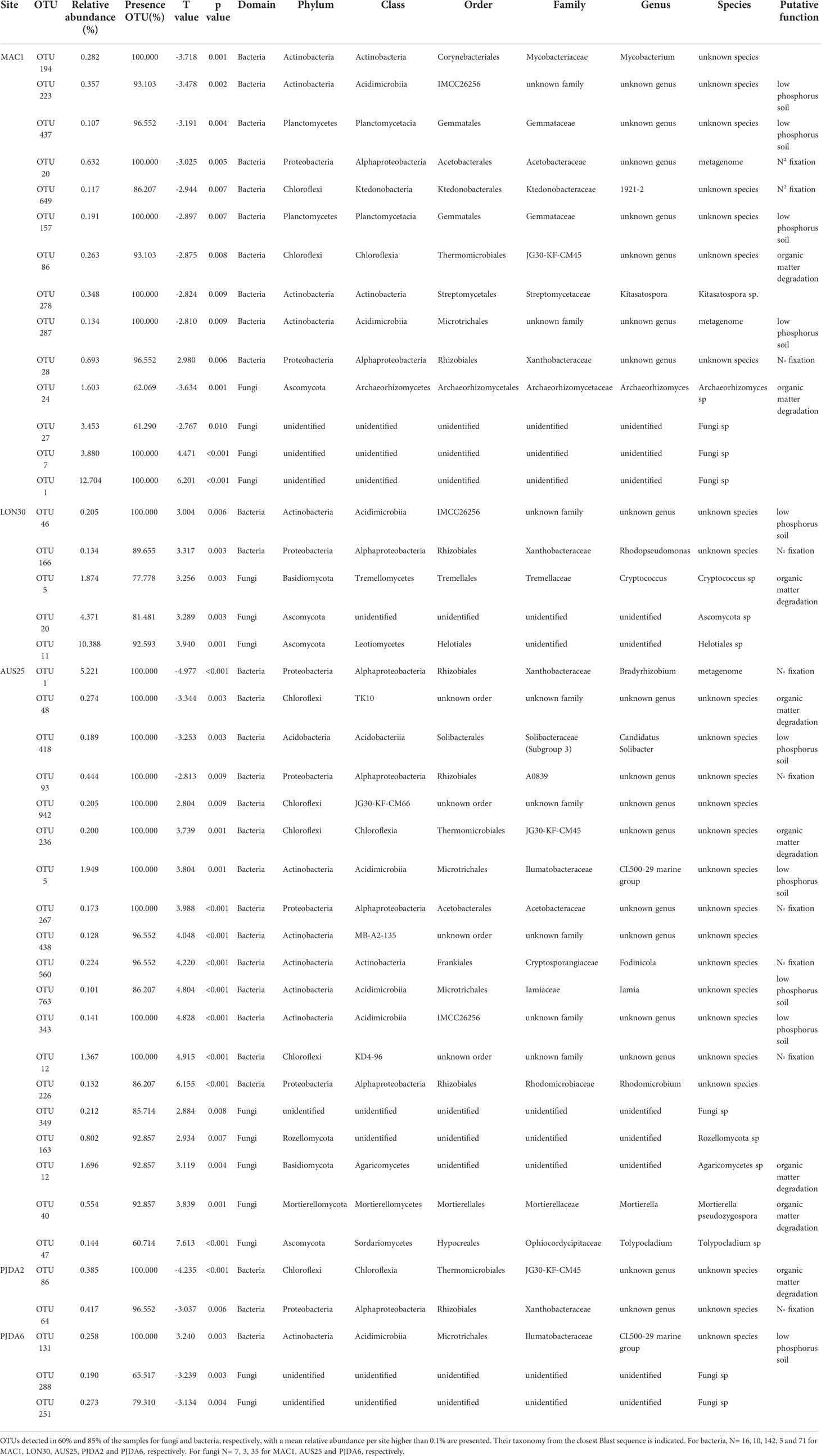
Table 2 Rhizospheric bacterial and fungal OTUs presenting significant (α = 0.01) Pearson correlation between their relative abundance and the necrotic extent of the corresponding plant. OTUs detected in 60 % and 85 % of the samples for fungi and bacteria, respectively, with a mean relative abundance per site higher than 0.1 % are presented. Their taxonomy from the closest Blast sequence is indicated. For bacteria, N= 16, 10, 142, 5 and 71 for MAC1, LON30, AUS25, PJDA2 and PJDA6, respectively. For fungi N= 7, 3, 35 for MAC1, AUS25 and PJDA6, respectively.
Discussion
Microbial diversity and dominant phyla in the fellfield soils of the Kerguelen Islands
In our study, the soil bacterial diversity (Shannon index) was similar to the one observed in other temperate ecosystems (cropland, grassland and forest) (10). It was higher than previously detected in some studies located in the Arctic and Alpine sites (44, 45) but lower than studies performed in Antarctic, Arctic and Himalayan sites (46–48). Furthermore, the composition of soil bacterial communities in the fellfields of the Kerguelen Islands differed from those reported in other sub-polar, polar and alpine fellfields by a higher proportion of Chloroflexi and an almost total absence of Planctomycetes, β-Proteobacteria, Firmicutes and Cyanobacteria (44–47, 49–52). Interestingly, a large proportion of Chloroflexi is known to fix CO2 through photosynthesis (53) and among them are the Chloroflexaceae and Roseiflexaceae families that have been detected in our samples. In these habitats where the vegetation is scarce and thus contributes poorly to soil carbon inputs, the dominance of the Chloroflexi (in four out of the five fellfield soils studied) indicates that soil microorganisms may stand in for fixing C-CO2. This might lead the fellfield soils from the Kerguelen Islands to act as an active carbon sink. As previously observed in alpine and Antarctic fellfields, the soil microbiome of the Kerguelen Island fellfields was also composed of a high proportion of Actinobacteria (47, 51, 54). These bacteria are ubiquitous and are known to have beneficial roles in soil functioning and plant fitness. They are actively involved in soil organic matter decomposition, producing a broad range of metabolites that could promote plant growth and mitigate plant abiotic stress (55, 56). Even if being among the dominant bacterial phyla, the relative abundance of Acidobacteria in the fellfield soils of the Kerguelen Islands varied from only 4 to 13% (6, 49, 57). Such a reduced proportion of Acidobacteria might have been to the advantage of α-Proteobacteria, which constituted one of the most abundant soil bacterial taxa. As observed experimentally by Yergeau et al., 2012 (54), in the Antarctic, this shift in the composition of bacterial communities might result from the increase in mean temperature, which is the case in the Kerguelen Islands over the past 70 years (26).
The main fungal phyla detected in the fellfield soils of the Kerguelen Islands were Ascomycota, Basidiomycota and Mortierellomycota, which corresponded to previous observations performed in soils from alpine and polar fellfields (8, 45, 58, 59). Ascomycota are known to be more abundant in soils of moderate or low carbon contents (60) such as the fellfield soils of the present study (mean organic carbon content of 2.27%). Interestingly, the active decomposition of organic matter by Basidiomycota has been highlighted in the soils of various Arctic and Antarctic regions (61). In fellfields of the Kerguelen Islands, native herbivores and invertebrates are inexistent which implies food webs to rely mainly on microbial decomposition where this key function might partly been carried by the numerous Basidiomycota detected. The Mortierellomycota phylum was mainly composed of Mortierella sp. which are known to be a phosphorus solubilizing fungi (62, 63) and to produce high amounts of trehalose and fatty acids. These compounds are known to participate in cold stress tolerance for the soil microbiome in harsh environments (64).
Restricted and cosmopolitan soil microbiomes of the Kerguelen Islands
A large proportion of the fungal sequences (up to 48%) could not be assigned to any phylum and corresponded to unknown fungi in the database. This proportion of unknown fungi stood across a similar range as observed in soils from Antarctica (~40% detected from ITS2 region analysis) (3, 58, 59). These unknown fungal communities might be related to fungi with restricted distribution due to the remoteness of the Kerguelen Islands. These fungi might have a valuable role in soil functioning as observed in the Antarctic, where, based on RNA analyses, endemic fungi were identified as more active than cosmopolitan ones (65).
A soil core microbiome shared between the five fellfield sites was identified. It was only composed of bacterial OTUs that belonged to both widespread and more narrowly distributed taxa (66). These taxa were related to N² fixation (Rhizobiales, Frankiales, Ktedonobacterales and Acetobacterales), adapted to soil with low content in phosphorus (Gemmatales, Microtrichales and Solibacterales) and involved in soil organic matter degradation (Micrococcales and Thermomicrobiales) (67–73). Together with the unknown fungal communities, this soil core microbiome represented the specificity of the microbial communities of the fellfield soils of the Kerguelen Islands that were composed of both restricted and cosmopolitan microbes. Our results support the view of a mixed microbial assemblage in the Kerguelen Islands’ soils that would reflect both dispersal limitation due to islands’ remoteness and aeolian deposition allowing long-distance transport.
Spatial variability of microbial communities of fellfield soils at the scale of the Kerguelen Islands
In the present study, differences in the soil microbiome composition between the five studied fellfields were explained by several soil properties. Among them, nitrogen was involved in structuring both bacterial and fungal communities through soil contents in ammonium and nitrate. Nitrogen is known to be the main limiting factor in high latitude ecosystems (74, 75) and, according to the low C:N ratio observed in the fellfield soils of the Kerguelen Islands (from 10.35 to 13.07), rapid mineralisation should occur, enhancing N loss (76). Thus, soil functioning and the involved microbial communities in these ecosystems might heavily rely on the nitrogen cycle. Soil phosphorus content also appeared to structure both bacterial and fungal communities across the fellfield soils of the Kerguelen Islands. Phosphate has previously been observed to be correlated to the distribution of soil bacterial communities in Antarctica (77). Finally, even though the five soils showed slight differences in pH, it has been emphasized to shape the soil bacterial communities. This result corroborated what is widely assumed for temperate soil microbiomes, i.e. soil pH strongly structures bacterial taxa, fungal communities being less sensitive (78, 79).
The rhizosphere of Lyallia kerguelensis is a reservoir of diversity for soil microorganisms in fellfields
In each site, the relative abundances of the dominant bacterial phyla in the rhizosphere of L. kerguelensis were the same as observed in the bulk soils. Meanwhile, the composition of the fungal rhizomicrobiome at the phylum scale was plant-specific and not necessarily similar to the one observed in the corresponding bulk soil. Such plant-dependency in structuring fungal communities was already observed in several alpine fellfields (8, 49, 57) and, in the present study, translated into the absence of shared fungal OTU between all plants from the five sites. The core rhizomicrobiome of L. kerguelensis was thus only composed of bacteria, highlighting a specific bacterial assemblage within the rhizosphere of this plant species apparently little influenced by its close habitat.
It is assumed that in their rhizosphere, plants select a specific rhizomicrobiome allowing them to better deal with biotic and abiotic stresses. This selection pressure usually leads to an assemblage of less diverse microbial communities than observed in the surrounding soil (10, 12, 80). In our study, the rhizomicrobiomes were at least as diverse as the microbiome in the associated bulk soil, some of them being even more diverse (13 and 6 rhizomicrobiomes out of 50 for bacteria and fungi, respectively). The cushion shape of plants such as that of L. kerguelensis is known to buffer climatic variation to a certain extent and to increase temperature and humidity inside and beneath it. Therefore the soil underneath should be a favorable habitat for soil microorganisms in the harsh environmental conditions that prevail in fellfields, the rhizosphere of L. kerguelensis achieving to constitute a reservoir of microbial diversity (12, 81).
Relation between plant necrosis extent and the composition of its rhizomicrobiome
L. kerguelensis is affected by necrosis throughout the archipelago. Other cases of browning and dieback of long-lived plants have been documented in the Antarctic and sub-Antarctic, and several hypotheses on their origin have been raised. In the sub-Antarctic Macquarie Islands, Bergstrom et al., 2015 (18) suspected the dieback of Azorella macquariensis to result from a decrease in water availability under climate change, making the plants weaker and more susceptible to fungal pathogens. For L. kerguelensis in the Kerguelen Islands, a possible link between necrosis expansion and climate drying was previously suggested (7, 21). In the present study, by analyzing the rhizomicrobiome of L. kerguelensis, we identified some OTUs related to the necrosis extent but they did not correspond to any known plant pathogenic microorganisms. Moreover, these OTUs were not conserved across populations. More than the presence of specific microorganisms, their involvement in nutrient cycling might be important for the plant to better cope with abiotic stresses. Among the OTUs related to necrosis extent, we identified microorganisms at the order level that are known to support some key roles in soil functioning and plant mediation to stress resistance (82) such as N2 fixation (orders Acetobacterales Ktedonobacterales, Rhizobiales, Frankiales and class KD4-96), decomposition processes (orders Thermomicrobiales, Tremellale, Mortierellales and class TK10 and Agaricomycetes) and adaptation to soils with low content in phosphorus (orders IMCC26256, Gemmatales, Microtrichales, Solibacterales) (67–73, 83–86). Overall, our study tends towards the hypothesis of a combination of abiotic stresses (20, 21) and changes in the composition of the soil rhizomicrobiome, which may alter some key microbial functions.
Conclusions
Our results highlight specificities and possible critical roles of both soil microorganisms and adapted plants, such as L. kerguelensis, in the harsh environmental conditions which characterize the Kerguelen Islands’ fellfields. Soil microbial communities were composed of possibly restricted microorganisms, putatively sustaining important functions for both this endemic plant and these remote soil systems. L. kerguelensis rhizosphere might be a reservoir of microbial diversity due to its long lifespan and its capacity to buffer climatic conditions. The necrosis that is affecting this plant for decades was not related to shared microorganisms between plants but might rather be related to shift in microbial functions that sustain plant vigour. This detailed description of soil and rhizosphere microbiomes in fellfield soils from the Kerguelen Islands brings new insights on the possible role of cushion plants to sustain microbial diversity and functions in harsh isolated environments. This study constitutes a new evidence for the preservation of endemic plant species especially in ecosystems that are strongly affected by climate change.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA731349.
Author contribution
LM, FH, MT and CM conceived the ideas. LM and FH performed the fieldwork. LM, M-CM and BM performed the laboratory work. LM and CM analysed the data. LM, FH and CM led the writing with contributions from MT. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the French Polar Institute (IPEV, project 1116) and CNRS IRP grant “AntarctPlantAdapt” (F. Hennion) and AO 2018 (OSUR, Université de Rennes1). L.J. Marchand was supported by a PhD grant from the Ministry of Research and Education (France).
Acknowledgments
We are grateful to Y. Rantier (UMR ECOBIO, Université de Rennes) for creating the map.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsoil.2022.995716/full#supplementary-material
Supplementary Table 1 | Relative abundance of the OTUs from the soil core microbiome and core rhizomicrobiome (OTUs present in all the samples of the bulk or rhizosphere soil or both). Their taxonomy from the closest Blast sequence is indicated.
Supplementary Table 2 | Species richness and Shannon index of the soil microbiomes and of each individual plant rhizomicrobiome per site. The results of a Dunn’s Kruskal-Wallis multiple comparison test between the species richness or the Shannon index from each rhizosphere and the associated bulk soil are presented, the Z-value and adjusted p-value (α = 0.01) are reported.
References
1. Block W, Smith RIL, Kennedy AD. Strategies of survival and resource exploitation in the Antarctic fellfield ecosystem. Biol Rev (2009) 84:449–84. doi: 10.1111/j.1469-185X.2009.00084.x
2. Yergeau E. Fell-field soil microbiology. In: Cowan DA, editor. Antarctic Terrestrial microbiology: Physical and biological properties of Antarctic soils, vol. pp . Berlin, Heidelberg: Springer (2014). p. 115–29.
3. Cox F, Newsham KK, Bol R, Dungait JAJ, Robinson CH. Not poles apart: Antarctic soil fungal communities show similarities to those of the distant Arctic. Ecol Lett (2016) 19:528–36. doi: 10.1111/ele.12587
4. Herbold CW, Lee CK, McDonald IR, Cary SC. Evidence of global-scale aeolian dispersal and endemism in isolated geothermal microbial communities of Antarctica. Nat Commun (2014) 5:3875. doi: 10.1038/ncomms4875
5. Šantl-Temkiv T, Gosewinkel U, Starnawski P, Lever M, Finster K. Aeolian dispersal of bacteria in southwest Greenland: their sources, abundance, diversity and physiological states. FEMS Microbiol Ecol (2018) 94(4). doi: 10.1093/femsec/fiy031.
6. Chu H, Fierer N, Lauber CL, Caporaso JG, Knight R, Grogan P. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ Microbiol (2010) 12:2998–3006. doi: 10.1111/j.1462-2920.2010.02277.x
7. Wagstaff SJ, Hennion F. Evolution and biogeography of Lyallia and Hectorella (Portulacaceae), geographically isolated sisters from the southern hemisphere. Antarct Sci (2007) 19:417–26. doi: 10.1017/S0954102007000648
8. Roy J, Bonneville J-M, Saccone P, Ibanez S, Albert CH, Boleda M, et al. Differences in the fungal communities nursed by two genetic groups of the alpine cushion plant. Silene acaulis Ecol Evol (2018) 8:11568–81. doi: 10.1002/ece3.4606
9. Rodríguez-Echeverría S, Delgado-Baquerizo M, Morillo JA, Gaxiola A, Manzano M, Marquet PA, et al. Azorella cushion plants and aridity are important drivers of soil microbial communities in Andean ecosystems. Ecosystems (2021); 24:1576–90. doi: 10.1007/s10021-021-00603-1
10. Ling N, Wang T, Kuzyakov Y. Rhizosphere bacteriome structure and functions. Nat Commun (2022) 13:836. doi: 10.1038/s41467-022-28448-9
11. Bulgarelli D, Schlaeppi K, Spaepen S, van Themaat EVL, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol (2013) 64:807–38. doi: 10.1146/annurev-arplant-050312-120106
12. Berendsen RL, Pieterse CMJ, Bakker PAHM. The rhizosphere microbiome and plant health. Trends Plant Sci (2012) 17:478–86. doi: 10.1016/j.tplants.2012.04.001
13. Nissinen R, Kumar M. 10 Plant-associated microbes in the Arctic. Microbial Life in the Cryosphere and Its Feedback on Global Change, Liebner S, Ganzert L, eds. Berlin, Boston: De Gruyter (2021), 213–26. doi: 10.1515/9783110497083-010
14. Naylor D, Coleman-Derr D. Drought stress and root-associated bacterial communities. Front Plant Sci (2018) 8:2223. doi: 10.3389/fpls.2017.02223
15. Chaudhary P, Khati P, Chaudhary A, Maithani D, Kumar G, Sharma A. Cultivable and metagenomic approach to study the combined impact of nanogypsum and pseudomonas taiwanensis on maize plant health and its rhizospheric microbiome. PloS One (2021) 16:e0250574. doi: 10.1371/journal.pone.0250574
16. Chaudhary P, Sharma A, Chaudhary A, Khati P, Gangola S, Maithani D. Illumina based high throughput analysis of microbial diversity of maize rhizosphere treated with nanocompounds and bacillus sp. Appl Soil Ecol (2021); 159. doi: 10.1016/j.apsoil.2020.103836
17. Ranasinghe R, Ruane AC, Vautard R, Arnell E, Coppola F, Cruz FA, et al. Climate change information for regional impact and for risk assessment. In: Masson-Delmotte ,V, Zhai P, Pirani A, Connors SL, Péan C, Berger S, Caud N, Chen Y, Goldfarb L, Gomis MI, Huang M, Leitzell K, Lonnoy E, Matthews JBR, Maycock TK, Waterfield T, Yelekçi O, Yu R, Zhou B, editors. Climate change 2021: The physical science basis. contribution of working group I to the sixth assessment report of the intergovernmental panel on climate change. Cambridge: Cambridge University Press.In Press (2021). p 239.
18. Bergstrom DM, Bricher PK, Raymond B, Terauds A, Doley D, McGeoch MA, et al. Rapid collapse of a sub-Antarctic alpine ecosystem: the role of climate and pathogens. J Appl Ecol (2015) 52:774–83. doi: 10.1111/1365-2664.12436
19. Bjerke JW, Treharne R, Vikhamar-Schuler D, Karlsen SR, Ravolainen V, Bokhorst S, et al. Understanding the drivers of extensive plant damage in boreal and Arctic ecosystems: Insights from field surveys in the aftermath of damage. Sci Total Environ (2017) 599–600:1965–76. doi: 10.1016/j.scitotenv.2017.05.050
20. Marchand L. Adaptive capacity to climate change in the long-lived endemic cushion plant species Lyallia kerguelensis from the sub-Antarctic Kerguelen Islands Vol. 1. Vegetal Biology. Université Rennes (2021).
21. Marchand LJ, Tarayre M, Dorey T, Rantier Y, Hennion F. Morphological variability of cushion plant Lyallia kerguelensis (Caryophyllales) in relation to environmental conditions and geography in the kerguelen islands: implications for cushion necrosis and climate change. Polar Biol (2021) 44:17–30. doi: 10.1007/s00300-020-02768-2
22. Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev MMBR (2004) 68:669–85. doi: 10.1128/MMBR.68.4.669-685.2004
23. Frenot Y, Gloaguen JC, Massé L, Lebouvier M. Human activities, ecosystem disturbance and plant invasions in subantarctic crozet, kerguelen and Amsterdam islands. Biol Conserv (2001) 101:33–50. doi: 10.1016/S0006-3207(01)00052-0
24. Hennion F, Frenot Y, Martin-Tanguy J. High flexibility in growth and polyamine composition of the crucifer Pringlea antiscorbutica in relation to environmental conditions. Physiol Plant (2006) 127:212–24. doi: 10.1111/j.1399-3054.2006.00668.x
25. de la Rüe AE. Observation sur les caractères et la répartition de la végétation des îles kerguelen. CNFRA-Biol (1964) 1:1–60.
26. Verfaillie D, Charton J, Schimmelpfennig I, Stroebele Z, Jomelli V, Bétard F, et al. Evolution of the cook ice cap (Kerguelen islands) between the last centuries and 2100 ce based on cosmogenic dating and glacio-climatic modelling. Antarct Sci (2021) 33:301–17. doi: 10.1017/S0954102021000080
27. Nyffeler R, Eggli U. Disintegrating portulacaceae: a new familial classification of the suborder portulacineae (Caryophyllales) based on molecular and morphological data. TAXON (2010) 59:220–40. doi: 10.1002/tax.591021
28. Hennion F. Etude des caracteristiques biologiques et génétiques de la flore endémique des ïles Kerguelen. Paris: Muséum National d’Histoire Naturelle (1992).
29. Griffiths RI, Whiteley AS, O’Donnell AG, Bailey MJ. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol (2000) 66:5488–91. doi: 10.1128/AEM.66.12.5488-5491.2000
30. Monard C, Gantner S, Stenlid J. Utilizing ITS1 and ITS2 to study environmental fungal diversity using pyrosequencing. FEMS Microbiol Ecol (2013) 84:165–75. doi: 10.1111/1574-6941.12046
31. Illumina. 16S metagenomic sequencing library preparation. Available at: https://www.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf.
32. Herlemann DP, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J (2011) 5:1571–9. doi: 10.1038/ismej.2011.41
33. Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res (2013) 41:e1–1. doi: 10.1093/nar/gks808
34. Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol Ecol (1993) 2:113–8. doi: 10.1111/j.1365-294X.1993.tb00005.x
35. Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson K-H. Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol Bioinforma Online (2008) 4:193–201. doi: 10.4137/ebo.s653.
36. Hellequin E, Monard C, Quaiser A, Henriot M, Klarzynski O, Binet F. Specific recruitment of soil bacteria and fungi decomposers following a biostimulant application increased crop residues mineralization. PloS One (2018) 13:e0209089. doi: 10.1371/journal.pone.0209089
37. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al. FROGS: Find, rapidly, OTUs with galaxy solution. Bioinformatics (2018) 34:1287–94. doi: 10.1093/bioinformatics/btx791
38. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (2011) 27:2194–200. doi: 10.1093/bioinformatics/btr381
39. Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ (2016) 4:e2584. doi: 10.7717/peerj.2584
40. Mahé F, Rognes T, Quince C, de Vargas C, Dunthorn M. Swarm: robust and fast clustering method for amplicon-based studies. PeerJ (2014) 2:e593. doi: 10.7717/peerj.593.
41. McMurdie PJ, Holmes S. Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PloS One (2013) 8:e61217. doi: 10.1371/journal.pone.0061217
42. Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in r. BMC Bioinf (2011) 12:35. doi: 10.1186/1471-2105-12-35
43. Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan: Community Ecology Package. R Package Version 2.2-0. (2014) Available at:http://CRAN.Rproject.org/package=vegan.
44. Mateos-Rivera A, Yde JC, Wilson B, Finster KW, Reigstad LJ, Øvreås L. The effect of temperature change on the microbial diversity and community structure along the chronosequence of the sub-arctic glacier forefield of styggedalsbreen (Norway). FEMS Microbiol Ecol (2016) 92(4). doi: 10.1093/femsec/fiw038
45. Wang C, Michalet R, Liu Z, Jiang X, Wang X, Zhang G, et al. Disentangling Large- and small-scale abiotic and biotic factors shaping soil microbial communities in an alpine cushion plant system. Front Microbiol (2020) 11:925. doi: 10.3389/fmicb.2020.00925
46. McCann CM, Wade MJ, Gray ND, Roberts JA, Hubert CRJ, Graham DW. Microbial communities in a high Arctic polar desert landscape. Front Microbiol (2016) 7. doi: 10.3389/fmicb.2016.00419
47. Chang S, Chen J, Su J, Yang Y, Sun H. Seasonal comparison of bacterial communities in rhizosphere of alpine cushion plants in the Himalayan hengduan mountains. Plant Divers (2018) 40:209–16. doi: 10.1016/j.pld.2018.09.003
48. Mapelli F, Marasco R, Fusi M, Scaglia B, Tsiamis G, Rolli E, et al. The stage of soil development modulates rhizosphere effect along a high Arctic desert chronosequence. ISME J (2018) 12:1188–98. doi: 10.1038/s41396-017-0026-4
49. Praeg N, Pauli H, Illmer P. Microbial diversity in bulk and rhizosphere soil of ranunculus glacialis along a high-alpine altitudinal gradient. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.01429
50. Jorquera MA, Maruyama F, Ogram AV, Navarrete OU, Lagos LM, Inostroza NG, et al. Rhizobacterial community structures associated with native plants grown in Chilean extreme environments. Microb Ecol (2016) 72:633–46. doi: 10.1007/s00248-016-0813-x
51. Teixeira LCRS, Yeargeau E, Balieiro FC, Piccolo MC, Peixoto RS, Greer CW, et al. Plant and bird presence strongly influences the microbial communities in soils of admiralty bay, maritime Antarctica. PloS One (2013) 8:e66109. doi: 10.1371/journal.pone.0066109
52. Ortiz M, Bosch J, Coclet C, Johnson J, Lebre P, Salawu-Rotimi A, et al. Microbial nitrogen cycling in Antarctic soils. Microorganisms (2020) 8:1442. doi: 10.3390/microorganisms8091442
53. Shih PM, Ward LM, Fischer WW. Evolution of the 3-hydroxypropionate bicycle and recent transfer of anoxygenic photosynthesis into the chloroflexi. Proc Natl Acad Sci (2017) 114:10749–54. doi: 10.1073/pnas.1710798114
54. Yergeau E, Bokhorst S, Kang S, Zhou J, Greer CW, Aerts R, et al. Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J (2012) 6:692–702. doi: 10.1038/ismej.2011.124
55. Yadav N, Nath Yadav A. Actinobacteria for sustainable agriculture. J Appl Biotechnol Bioeng (2019) 6:38–41. doi: 10.15406/jabb.2019.06.00172
56. Ranjani A, Dhanasekaran D, Gopinath PM. An introduction to actinobacteria. In: Dhanasekaran D, Jiang Y, editors. Actinobacteria - basics and biotechnological applications. London: InTech (2016).
57. Adamczyk M, Hagedorn F, Wipf S, Donhauser J, Vittoz P, Rixen C, et al. The soil microbiome of GLORIA mountain summits in the Swiss Alps. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.01080
58. Rosa LH, da Silva TH, Ogaki MB, Pinto OHB, Stech M, Convey P, et al. DNA Metabarcoding uncovers fungal diversity in soils of protected and non-protected areas on deception island, Antarctica. Sci Rep (2020) 9(10):21986. doi: 10.1038/s41598-020-78934-7
59. da Silva TH, Câmara PEAS, Pinto OHB, Carvalho-Silva M, Oliveira FS, Convey P, et al. Diversity of fungi present in permafrost in the south Shetland islands, maritime Antarctic. Microb Ecol (2021); 83:58–67. doi: 10.1007/s00248-021-01735-6
60. Ros M, Pascual JA, Hernández MT, García C. Long-term effects of devegetation on composition and activities (including transcription) of fungal communities of a semi-arid soil. Biol Fertil Soils (2009) 45:435–41. doi: 10.1007/s00374-008-0348-5
61. Ludley KE, Robinson CH. ‘Decomposer’ basidiomycota in Arctic and Antarctic ecosystems. Soil Biol Biochem (2008) 40:11–29. doi: 10.1016/j.soilbio.2007.07.023
62. Hanson CA, Allison SD, Bradford MA, Wallenstein MD, Treseder KK. Fungal taxa target different carbon sources in forest soil. Ecosystems (2008) 11:1157–67. doi: 10.1007/s10021-008-9186-4
63. Zhang H, Wu X, Li G, Qin P. Interactions between arbuscular mycorrhizal fungi and phosphate-solubilizing fungus (Mortierella sp.) and their effects on kostelelzkya virginica growth and enzyme activities of rhizosphere and bulk soils at different salinities. Biol Fertil Soils (2011) 47:543. doi: 10.1007/s00374-011-0563-3
64. Feng J, Zeng X-M, Zhang Q, Zhou X-Q, Liu Y-R, Huang Q. Soil microbial trait-based strategies drive metabolic efficiency along an altitude gradient. ISME Commun (2021) 1:1–9. doi: 10.1038/s43705-021-00076-2
65. Cox F, Newsham KK, Robinson CH. Endemic and cosmopolitan fungal taxa exhibit differential abundances in total and active communities of Antarctic soils. Environ Microbiol (2019) 21:1586–96. doi: 10.1111/1462-2920.14533
66. Delgado-Baquerizo M, Oliverio AM, Brewer TE, Benavent-González A, Eldridge DJ, Bardgett RD, et al. A global atlas of the dominant bacteria found in soil. Science (2018) 359:320–5. doi: 10.1126/science.aap9516
67. Bian X, Xiao S, Zhao Y, Xu Y, Yang H, Zhang L. Comparative analysis of rhizosphere soil physiochemical characteristics and microbial communities between rusty and healthy ginseng root. Sci Rep (2020) 10:15756. doi: 10.1038/s41598-020-71024-8
68. Chen H, Peng W, Du H, Song T, Zeng F, Wang F. Effect of different grain for green approaches on soil bacterial community in a karst region. Front Microbiol (2020) 11. doi: 10.3389/fmicb.2020.577242
69. Cui Y, Fang L, Guo X, Wang X, Wang Y, Li P, et al. Responses of soil microbial communities to nutrient limitation in the desert-grassland ecological transition zone. Sci Total Environ (2018) 642:45–55. doi: 10.1016/j.scitotenv.2018.06.033
70. Oliverio AM, Bissett A, McGuire K, Saltonstall K, Turner BL, Fierer N. The role of phosphorus limitation in shaping soil bacterial communities and their metabolic capabilities. mBio (2020) 11:e01718–20. doi: 10.1128/mBio.01718-20
71. Reis VM, Teixeira KR dos S. Nitrogen fixing bacteria in the family Acetobacteraceae and their role in agriculture. J Basic Microbiol (2015) 55:931–49. doi: 10.1002/jobm.201400898
72. Saravanan VS, Madhaiyan M, Osborne J, Thangaraju M, Sa TM. Ecological occurrence of Gluconacetobacter diazotrophicus and nitrogen-fixing acetobacteraceae members: their possible role in plant growth promotion. Microb Ecol (2008) 55:130–40. doi: 10.1007/s00248-007-9258-6
73. Louati M, Ennis NJ, Ghodhbane-Gtari F, Hezbri K, Sevigny JL, Fahnestock MF, et al. Elucidating the ecological networks in stone-dwelling microbiomes. Environ Microbiol (2020) 22:1467–80. doi: 10.1111/1462-2920.14700
74. Smith VR. The influence of seabird manuring on the phosphorus status of Marion island (Subantarctic) soils. Oecologia (1979) 41:123–6. doi: 10.1007/BF00344842
75. Yergeau E, Kang S, He Z, Zhou J, Kowalchuk G. Functional microarray analysis of nitrogen and carbon cycling genes across an Antarctic latitudinal transect. ISME J (2007) 1:163–79. doi: 10.1038/ismej.2007.24
76. Brust GE. Management strategies for organic vegetable fertility. In: Biswas D, Micallef SA, editors. Safety and practice for organic food. Cambridge: Academic Press (2019).
77. Wang NF, Zhang T, Zhang F, Wang ET, He JF, Ding H, et al. Diversity and structure of soil bacterial communities in the fildes region (maritime Antarctica) as revealed by 454 pyrosequencing. Front Microbiol (2015) 6:1188. doi: 10.3389/fmicb.2015.01188
78. Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, et al. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J (2010) 4:1340–51. doi: 10.1038/ismej.2010.58
79. Griffiths RI, Thomson BC, James P, Bell T, Bailey M, Whiteley AS. The bacterial biogeography of British soils. Environ Microbiol (2011) 13:1642–54. doi: 10.1111/j.1462-2920.2011.02480.x
80. Vandegehuchte MW, Guyot A, Hubau M, De Groote SRE, De Baerdemaeker NJF, Hayes M, et al. Long-term versus daily stem diameter variation in co-occurring mangrove species: Environmental versus ecophysiological drivers. Agric For Meteorol (2014) 192–193:51–8. doi: 10.1016/j.agrformet.2014.03.002
81. Trivedi P, Leach JE, Tringe SG, Sa T, Singh BK. Plant–microbiome interactions: from community assembly to plant health. Nat Rev Microbiol (2020) 18:607–21. doi: 10.1038/s41579-020-0412-1
82. Liu H, Brettell LE, Qiu Z, Singh BK. Microbiome-mediated stress resistance in plants. Trends Plant Sci (2020) 25:11. doi: 10.1016/j.tplants.2020.03.014
83. Rosling A, Cox F, Cruz-Martínez K, Ihrmark K, Grelet G-A, Lindahl B, et al. Archaeorhizomycetes: unearthing an ancient class of ubiquitous soil fungi. Science (2011) 333:876–9. doi: 10.1126/science.1206958
84. Wagner L, Stielow B, Hoffmann K, Petkovits T, Papp T, Vágvölgyi C, et al. A comprehensive molecular phylogeny of the Mortierellales (Mortierellomycotina) based on nuclear ribosomal DNA. Persoonia - Mol Phylogeny Evol Fungi (2013) 30:77–93. doi: 10.3767/003158513X666268
85. Wongkiew S, Koottatep T, Polprasert C, Prombutara P, Jinsart W, Khanal SK. Bioponic system for nitrogen and phosphorus recovery from chicken manure: Evaluation of manure loading and microbial communities. Waste Manag (2021) 125:67–76. doi: 10.1016/j.wasman.2021.02.014
Keywords: sub-Antarctic, bacteria, fungi, Lyallia kerguelensis, cushion plant, dieback
Citation: Marchand LJ, Hennion F, Tarayre M, Martin M-C, Martins BR and Monard C (2022) Fellfields of the Kerguelen Islands harbour specific soil microbiomes and rhizomicrobiomes of an endemic plant facing necrosis. Front. Soil Sci. 2:995716. doi: 10.3389/fsoil.2022.995716
Received: 16 July 2022; Accepted: 07 September 2022;
Published: 26 September 2022.
Edited by:
Doongar R. Chaudhary, Central Salt & Marine Chemicals Research Institute (CSIR), IndiaReviewed by:
Salma Mukhtar, Connecticut Agricultural Experiment Station, United StatesFahad Nasir, Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences (CAS), China
Copyright © 2022 Marchand, Hennion, Tarayre, Martin, Martins and Monard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorène Julia Marchand, lorene.marchand@uantwerpen.be