One or many? Multi-species livestock grazing influences soil microbiome community structure and antibiotic resistance potential
 Gwynne Á. Mhuireach
Gwynne Á. Mhuireach Leslie Dietz
Leslie Dietz Thomas Gillett2
Thomas Gillett2- 1Biology and the Built Environment Center, University of Oregon, Eugene, OR, United States
- 2Black Tansy Farm, Springfield, OR, United States
Soil health has been highlighted as a key dimension of regenerative agriculture, given its critical importance for food production, carbon sequestration, water filtration, and nutrient cycling. Microorganisms are critical components of soil health, as they are responsible for mediating 90% of soil functions. Multi-species rotational grazing (MSRG) is a promising strategy for maintaining and improving soil health, yet the potential effects of MSRG on soil microbiomes are poorly understood. To address this knowledge gap, we collected soil microbial samples at three timepoints during the 2020 grazing season for 12 total paddocks, which were equally split into four different grazing treatments—cattle only, sheep only, swine only, or multi-species. Shallow shotgun metagenomic sequencing was used to characterize soil microbial community taxonomy and antibiotic resistome. Results demonstrated broad microbial diversity in all paddock soil microbiomes. Samples collected early in the season tended to have greater archaeal and bacterial alpha diversity than samples collected later for all grazing treatments, while no effect was observed for fungi or viruses. Beta diversity, however, was strongly influenced by both grazing treatment and month for all microbial kingdoms, suggesting a pronounced effect of different livestock on microbial composition. Cattle-only and swine-only paddocks were more dissimilar from multi-species paddocks than those grazed by sheep. We identified a large number of differentially abundant taxa driving community dissimilarities, including Methanosarcina spp., Candidatus Nitrocosmicus oleophilus, Streptomyces spp., Pyricularia spp., Fusarium spp., and Tunggulvirus Pseudomonas virus ϕ-2. In addition, a wide variety of antibiotic resistance genes (ARGs) were present in all samples, regardless of grazing treatment; the majority of these encoded efflux pumps and antibiotic modification enzymes (e.g., transferases). This novel study demonstrates that grazing different species of livestock, either separately or together, can impact soil microbial community structure and antibiotic resistance capacity, though further research is needed to fully characterize these impacts. Increasing the knowledge base about soil microbial community structure and function under real-world grazing conditions will help to construct metrics that can be incorporated into traditional soil health tests and allow producers to manage livestock operations for optimal soil microbiomes.
Introduction
The agricultural industry bears substantial responsibility for many global environmental issues, including greenhouse gas emissions; soil and water pollution from fertilizers, pesticides, and herbicides; and emerging antibiotic resistance from overuse of subclinical antibiotic use in livestock feed (Hoelzer et al., 2017; Poore and Nemecek, 2018). To mitigate these and other problems, the concept of “regenerative agriculture” has been proposed (Pearson, 2007; Rhodes, 2012; Provenza et al., 2019). Regenerative agriculture includes a broad suite of practices that aim to protect and preserve natural resources, improve animal welfare, and return to closed-loop nutrient cycles in the food system (Horrigan et al., 2002). Soil health is at the crux of the regenerative agriculture paradigm, being closely linked with agricultural productivity, economic prosperity, and environmental quality (Schreefel et al., 2020).
Soil health depends to a large extent on microorganisms, which are responsible for mediating 90% of soil functions, including nutrient cycling, organic matter decomposition, and plant growth and health maintenance (Aislabie and Deslippe, 2013; Teague, 2018; Yang et al., 2019). Thus, soil biodiversity metrics, including microbial community analyses, have been suggested as potentially useful indicators of soil health (Schloter et al., 2018; Lehmann et al., 2020; Norris et al., 2020; Fierer et al., 2021). A number of recent soil studies have found that farm management practices are a highly important factor driving microbial abundance, diversity, composition, and function. Application of manure, for instance, has a significant positive effect on soil microbial biomass and activity (Esperschütz et al., 2007; Sun et al., 2016; van der Bom et al., 2018). Plant species' identity and composition are other factors that affect soil microbial community diversity and composition near the root zone, due to exudates that can encourage beneficial microorganisms or discourage pathogens (Lei et al., 2019; Schmid et al., 2019). Fields that have undergone monocropping for long periods often develop disease-suppressive qualities, likely driven by soil microbial community structure, against specific plant pathogens, like Fusarium wilt (caused by F. oxysporum), wheat take-all (caused by Gaeumannomyces graminis), and potato scab (caused by Streptomyces scabies) (Weller et al., 2002).
Compared with cropping systems, however, little is known about how grazing management impacts soil microbial communities in pastures and rangelands (Dignam et al., 2016; Acharya et al., 2021). Multi-species rotational grazing (MSRG) is a management-intensive alternative to continuous monospecies stocking that may hold promise for improving environmental sustainability of animal agriculture (Kleppel, 2020). In MSRG systems, pasture is subdivided into smaller cells or paddocks through which two or more livestock species are rotated in series, followed by a long rest period to allow forage regrowth. This strategy attempts, in a more constrained and human-managed manner, to mimic natural grassland ecosystems where ungulates and other herbivorous/omnivorous animals evolved under competitive pressure to share the same landscape (Walker, 1997). Proponents of MSRG posit that pastures can remain healthy and productive under higher grazing intensity with two or more types of herbivores, because of their different dietary habits, than with a single species (Anderson et al., 2012; Jerrentrup et al., 2020; Martin et al., 2020).
Since different livestock species preferentially graze different plant species and have different manure chemical and microbiological compositions, soil microbial communities may demonstrate variable responses dependent on the grazing species (Sitters and Andriuzzi, 2019). One of the few studies on how rotational grazing affects the soil microbiome reports that microbial biomass and activity increase markedly (average 200 and 500%, respectively) in pastures that transition from conventional grazing management to a rotational system (Teague, 2018). Another compared long-term continuously grazed and rotational pasture management, finding that continuously grazed pastures had greater soil microbial richness (Yang et al., 2019), although more antimicrobial resistance genes (ARGs) were found under continuous grazing (Yang et al., 2020). Lastly, pastures that transitioned from conventional to organic management tended to have higher richness and diversity in soil microbial communities (Acharya et al., 2021). Notably, both of these studies only tested the impact of cattle grazing; to our knowledge, no research currently exists on the effects of MSRG on soil microbial communities.
This study sought to further our understanding of how livestock grazing management influences soil health, with special focus on microbial community structure and antibiotic resistome. We hypothesized that, in comparison to monospecies grazing, MSRG would lead to: 1) greater microbial α diversity; 2) dissimilar taxonomic composition; and 3) fewer putative antibiotic resistance genes (ARGs).
Methods
Overview
The study took place on a 22-acre diversified livestock farm located in Springfield, Oregon, USA (Figure 1; 44.029728 N -122.953615 W). Soil type for the pasture is Newberg fine sandy loam (33.8%) and Newberg loam (66.2%), which are deep well-drained soil found on flood plains (https://websoilsurvey.sc.egov.usda.gov/App/WebSoilSurvey.aspx). The prior history of the property included approximately 50 years of intensive use as a vegetable farm followed by almost 20 years of disuse. The current livestock operation was established in 2014 with six Dexter breed cattle and ten Icelandic sheep; in 2018, Red Wattle and Berkshire pigs were added to the livestock operation. The farm is transitioning to organic practices, therefore, no chemical fertilizers, herbicides, or pesticides have been applied to pastures since 2013. In addition, antibiotic use on this farm is strictly limited to treatment of acute illness and any animals requiring treatment are temporarily housed in the barn rather than on pasture. Therefore it is expected that little to no antibiotic residue would have contaminated the pasture in the 6 years preceding this study. Immediately prior to the onset of this study, the entire operation comprised 15 cattle, 40 sheep, and 25 breeding/finishing pigs, all of which were contained within a single large pasture area.
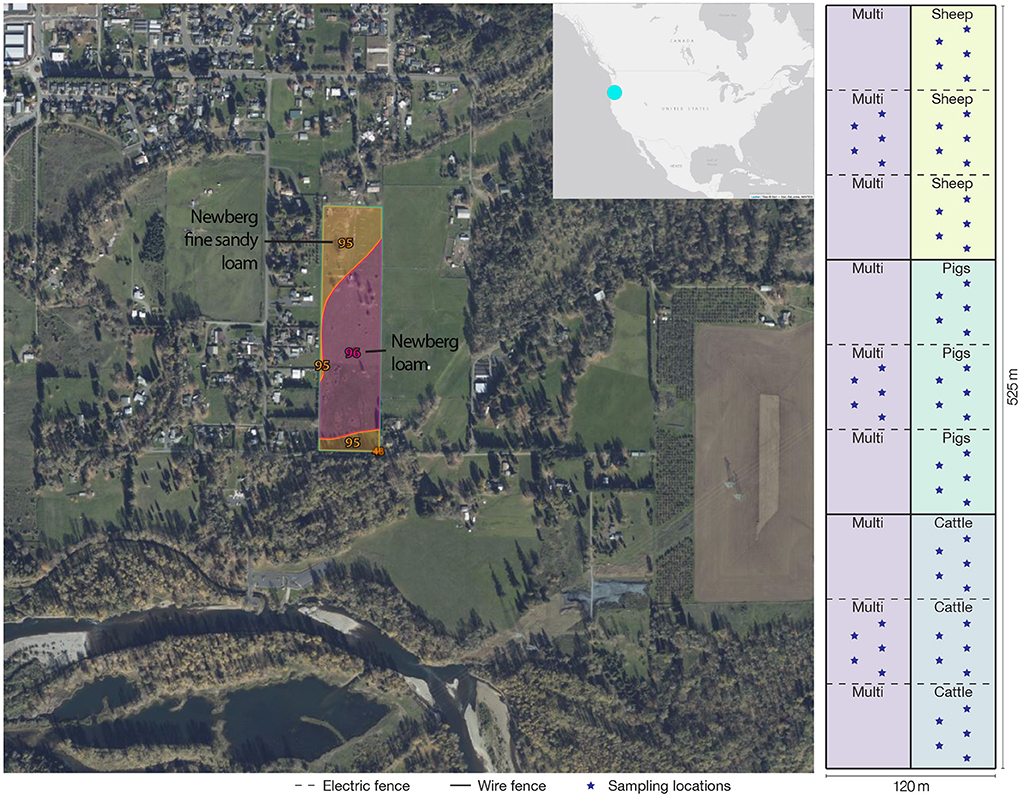
Figure 1. Study farm location and diagram of paddock layout, grazing treatments, and approximate sample collection locations. To maintain a balanced analytical design, only three of the nine multi-species paddocks were sampled.
In May 2020, fences were constructed to enable multi-species rotational grazing, which was implemented on the western half of the pasture, while the eastern half was dedicated to monospecies grazing. Each half was divided into thirds, one for each livestock species on the eastern half, and then each third was further divided into thirds again, providing 12 small paddocks for each species to rotate through (Figure 1). We use the term “paddock” in reference to the smaller subdivided areas; these are contained within the larger pasture system. In the grazing management plan, sheep would graze each multi-species paddock first, as they have the highest nutritional needs, followed by cattle, and pigs last, since they were provided with additional feed comprising mixed milled, rolled, and whole grains, field peas, organic non-GMO okara, and third-cutting alfalfa. Due to poor grass growth during late summer and fall, the cattle also received supplemental feed in the form of baled grassy alfalfa hay. The length of each rotation period varied, depending on forage height, aiming to leave 10–15 cm of forage standing after each complete rotation. Forage plants in all paddocks included annual and perennial ryegrasses (Lolium multiflorum and L. perenne, respectively), white clover (Trifolium repens), dandelion (Taraxacum officinale), narrowleaf plantain (Plantago lanceolata), chicory (Cichorium intybus), and other voluntary forbs and grasses. Temperature, relative humidity, and daily precipitation measurements from the local airport climatological station (Mahlon Sweet Field, Eugene, Oregon, USA) are shown in Figure 2. Due to a combination of water rights limitations and severe–extreme drought conditions (https://www.drought.gov/historical-information?state=oregon&countyFips=41039), only the northern two-thirds of the pasture, containing sheep, pig, and two of the multi-species paddocks, received periodic irrigation during the study.
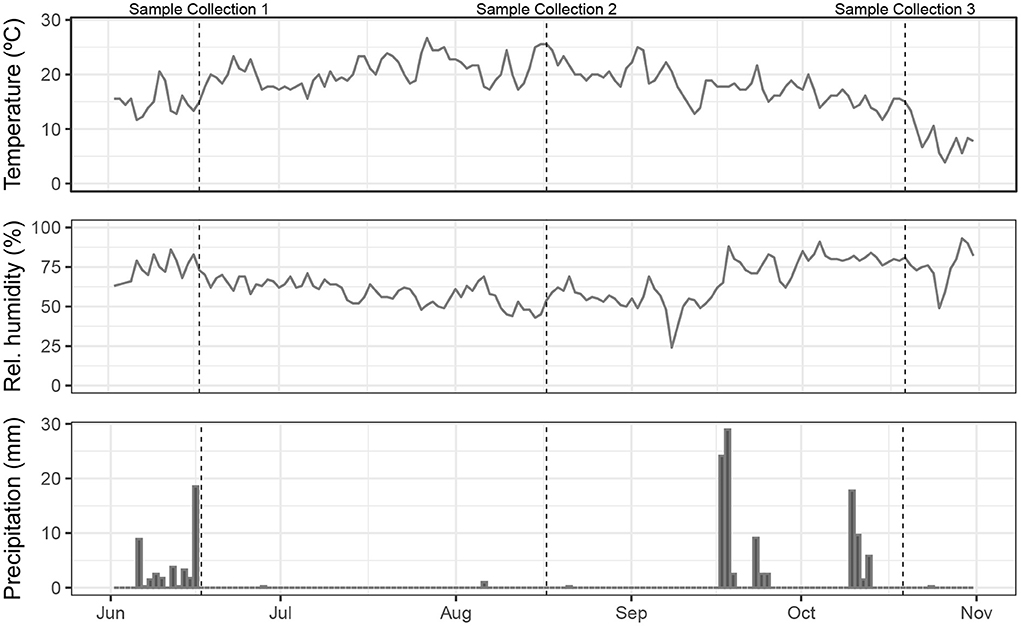
Figure 2. Weather conditions during the 2020 grazing season (June 1–October 31) as measured at Mahlon Sweet Field Airport, Eugene, Oregon, USA.
Soil sample collection and processing
Soil samples were collected from paddocks representing treatment replicates at three timepoints during the 2020 grazing season–June, August, and October, representing early, middle, and end of grazing season, respectively. Only three of the nine multi-species paddocks shown in Figure 1 were sampled, in order to maintain a balanced analytical design. Because this study was conducted on a real-world working farm, the additional (unsampled) multi-species paddocks were required for the livestock to have adequate forage and allow them to rotate frequently through the paddocks. Soil cores 2.5 cm in diameter and 10 cm deep were collected from five locations in a roughly “W” shape within each paddock (Figure 1) using sterilized stainless steel soil probes, avoiding areas immediately near livestock feces. Samples were collected in June, August, and October to assess changes in soil microbial community structure and antibiotic resistome over the course of the grazing season. For each time point, the five cores from each of the 12 paddocks were combined into a single integrated sample, which has been suggested as an effective strategy for accurately measuring microbial diversity (Li et al., 2021), resulting in a total of 36 bulk soil samples. Samples were transported on ice to the University of Oregon Biology and the Built Environment Laboratory where they were stored at −20°C until further processing.
After thawing, each soil sample was homogenized and sieved with a 2 mm sieve prior to aliquoting two 0.25 g subsamples (technical replicates) to be used for shallow shotgun metagenomic sequencing. Bulk soil from samples collected in October from the center paddock of each grazing treatment was sent to AgSource Laboratories (Lincoln, NE, USA) for soil physical and chemical analyses, including pH, organic matter, plant macro- and micronutrients, and bulk density, which was used as background information about the site.
Microbial DNA processing
Shallow shotgun metagenomics methodology was used to characterize microbial community structure and antibiotic resistance capacity, as it has been demonstrated to generate more accurate taxonomic and functional information about bacterial communities than amplicon analysis, while also providing information about viral and fungal communities (Hillmann et al., 2018; Cattonaro et al., 2020). We used QIAGEN DNeasy PowerLyzer PowerSoil Kit (Qiagen #12855-100) to purify nucleic acids from each technical replicate. DNA extractions were performed in triplicate and then pooled for each technical replicate. After extraction, genomic DNA libraries were prepared using Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA). Qubit dsDNA high sensitivity quantitative assay was used to quantify DNA and libraries were pooled to 2 nM. Libraries were sent to University of Oregon Genomics & Cell Characterization Core Facility (Eugene, OR, USA) for paired-end sequencing (2 × 150) on an Illumina HiSeq 4000 Sequencing System.
Read quality of raw sequences was inspected with FastQC (Andrews, 2010). Adapters were trimmed and contaminants, including PhiX and human sequences, were removed using BBDuk (Bushnell, 2014) with suggested parameters (ktrim=r k=23 mink=11 hdist=1 tpe tbo). Taxonomic classification of clean unassembled reads was performed using exact kmer matching, as implemented by Kraken 2 (Wood et al., 2019), against the standard database containing NCBI taxonomic information, as well as the complete bacterial, fungal, viral, and archaeal genomes in RefSeq (O'Leary et al., 2016). The resulting Kraken 2 report was converted into a BIOM table for later use in R using the kraken-biom tool (Dabdoub, 2016). We used the R package phyloseq to import the Kraken 2 BIOM table into R (McMurdie and Holmes, 2013), and the decontam package to identify and remove potential laboratory contaminants (Davis et al., 2018), resulting in elimination of 164,795 reads. Replicates from the same bulk soil samples were combined in silico. Control samples and two samples that did not meet our minimum threshold of 300,000 reads were also removed prior to downstream analysis. This minimum read threshold was chosen after inspecting rarefaction curves, which indicated that these two samples were low outliers in sampling effort.
Antibiotic resistance annotation
Clean reads from each paddock and timepoint combination were coassembled with MegaHIT (Li et al., 2015) using preset -meta-large, resulting in an average of 362,989 contigs. Open reading frames and total protein sequences were predicted from coassembled contigs using Prodigal (Hyatt et al., 2010). Antibiotic resistance features were annotated using hmmscan from HMMER3 (Eddy, 2011) against the full ResFams database, which provides a curated list of protein families associated with antibiotic resistance (Gibson et al., 2015).
Statistical analysis
We calculated the number of shared taxa across grazing treatments and sampling months using presence-absence data. Rarefaction curves were constructed using the rarecurve function in vegan to assess whether sampling depth provided adequate characterization of the true diversity. We used a two-way ANOVA followed by Tukey's honestly significant difference test to examine differences in archaeal, bacterial, fungal, and viral alpha diversity across grazing treatments and across months. Shannon index values were used as the dependent variable in the models and Box-Cox transformation was applied prior to testing, because residuals failed to meet the assumption of normality. Sample abundances were adjusted for differences in library size using the variance-stabilizing transformation in the DESeq2 package (Love et al., 2014) prior to beta diversity and differential abundance analyses. We tested whether community similarity (estimated by Morisita-Horn distance) for each microbial kingdom was related to grazing treatment and/or month using PERMANOVA, as implemented in the adonis function of vegan (Oksanen et al., 2020). Clustering effects were visualized in principal coordinates analysis (PCoA) plots, also using Morisita-Horn distances. We performed pairwise comparisons for each monospecies grazing treatment against the multi-species paddock to identify which taxa from each kingdom were enriched or depauperate by fitting negative binomial GLMs on the variance-stabilizing transformed counts using DESeq2. Results were visualized in individual volcano plots showing log2 foldchanges and p-values obtained by Wald testing for each treatment comparison. For all statistical testing, we designated significance at an alpha value of 0.05 and adjusted for multiple comparisons using the Benjamini and Hochberg method (Benjamini and Hochberg, 1995).
Results
Soil physical and chemical characteristics
Physical and chemical characteristics of soil samples collected in October are shown in Table 1. In general, soil physical and chemical characteristics fell within recommended ranges for pasture and rangelands. Only one sample from each grazing treatment was assayed, therefore, no statistical analyses were performed.
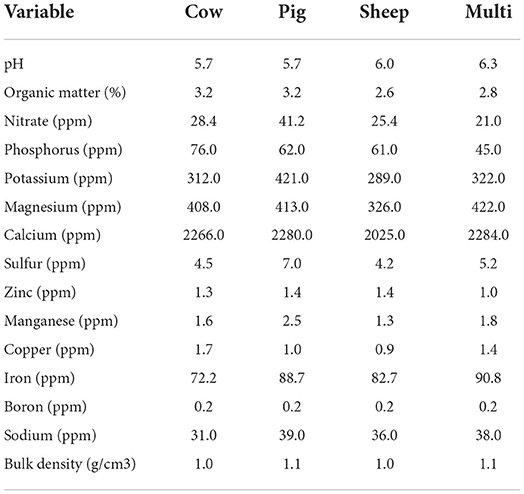
Table 1. Soil physical and chemical characteristics of integrated bulk soil samples for each grazing treatment in October.
Microbial community structure across grazing treatments and collection months
Overview
For microbial communities, a total of 267,114,103 raw paired-end reads were generated, averaging 3,709,207 reads per sample. After quality filtering, 266,949,308 clean reads remained for use in downstream analyses. Kraken 2 was able to classify 16.3% of the trimmed, quality filtered, unassembled reads, which falls into the same range reported by previous soil metagenomic surveys (Fierer et al., 2012). Possibly a large proportion of unclassified reads belonged to plants and animals (e.g., insects, worms), since the only eukaryote database used for taxonomic classification was fungal. Bacterial sequences outnumbered sequences from other kingdoms by at least two orders of magnitude and accounted for 97.7% of all reads. Bacteria also had the highest number of different taxa (7,481), closely followed by viruses (4,173), while relatively few archaeal (374) or fungal (84) taxa were identified, possibly due to limited characterization of these groups in their respective NCBI RefSeq genome databases (Loeffler et al., 2020).
The dominant phyla in archaeal communities overall were Euryarchaeota (86.0%), Thaumarchaeota (10.3%), and Crenarchaeota (2.9%). In bacterial communities, Proteobacteria (52.5%), Actinobacteria (30.7%), and Bacteroidetes (6.3%) were the most abundant phyla. Only three fungal phyla were observed in this study, with Ascomycota (95.2%) by far the most abundant, followed by Basidiomycota (4.7%) and Microsporidia (0.1%). The most abundant viral phyla were Uroviricota (62.7%), Peploviricota (16.2%), and Nucleocytoviricota (8.2%). A member of the bacterial genus Streptomyces was identified as the most abundant single taxon across the entire study.
The vast majority of archaeal, bacterial, and fungal taxa were shared across all grazing treatments and sampling months (Table 2), while viral communities were less homogeneous (Supplementary Figures S1–S3).
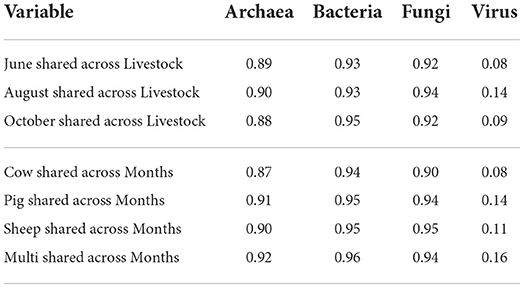
Table 2. Proportion of shared taxa for each microbial kingdom, calculated across grazing treatments within months and across months within grazing treatments.
Alpha diversity
Alpha diversity describes the number of different species (sometimes weighted by their distribution, or evenness) observed within an ecological assemblage, defined in this study as an individual soil sample, while beta diversity describes compositional similarity or differentiation across those assemblages (Jost et al., 2010; Willis, 2017). Rarefaction curves showing the number of species expected at a given library size for each soil sample are shown in Figure 3. Curves for archaeal, bacterial, and fungal communities reached an asymptote, indicating that diversity was adequately represented in most samples, whereas curves for viral communities did not approach an asymptote, suggesting that they were undersampled.
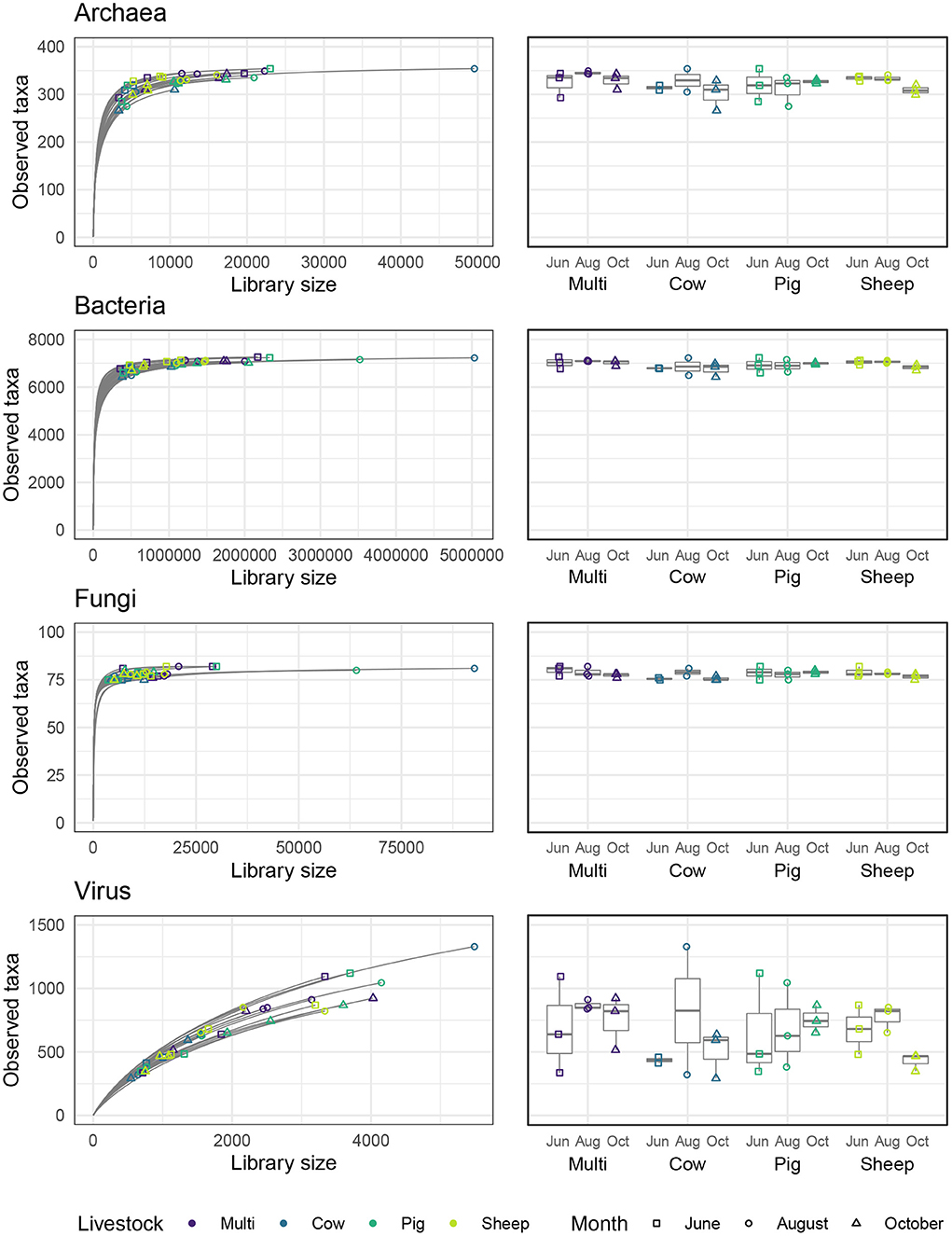
Figure 3. Rarefaction curves for each microbial kingdom and numbers of observed taxa across livestock grazing treatments and months.
Alpha diversity was quantified in three ways—observed number of species (i.e., richness), Shannon entropy, and Gini-Simpson index—corresponding to Hill numbers 0, 1, and 2, respectively, which place increasing weight on the evenness of species distribution (Chao et al., 2014). Archaeal and bacterial diversity was significantly higher in June than in August or October (Figure 4; Tukeys HSD: Archaea June_August: P = 0.01; Archaea June_October: P = 0.01; Bacteria June_August: P < 0.005; Bacteria June_October: P = 0.01). As richness was roughly similar across grazing treatments and months, these effects are likely due to a decrease in evenness during August and October for these communities. For bacterial communities only, sheep paddocks were more diverse in comparison to pig paddocks (Tukeys HSD; Bacteria Sheep_Pig: P = 0.05). There were no other significant effects of grazing treatment, nor did we find any significant patterns in fungal or viral alpha diversity.
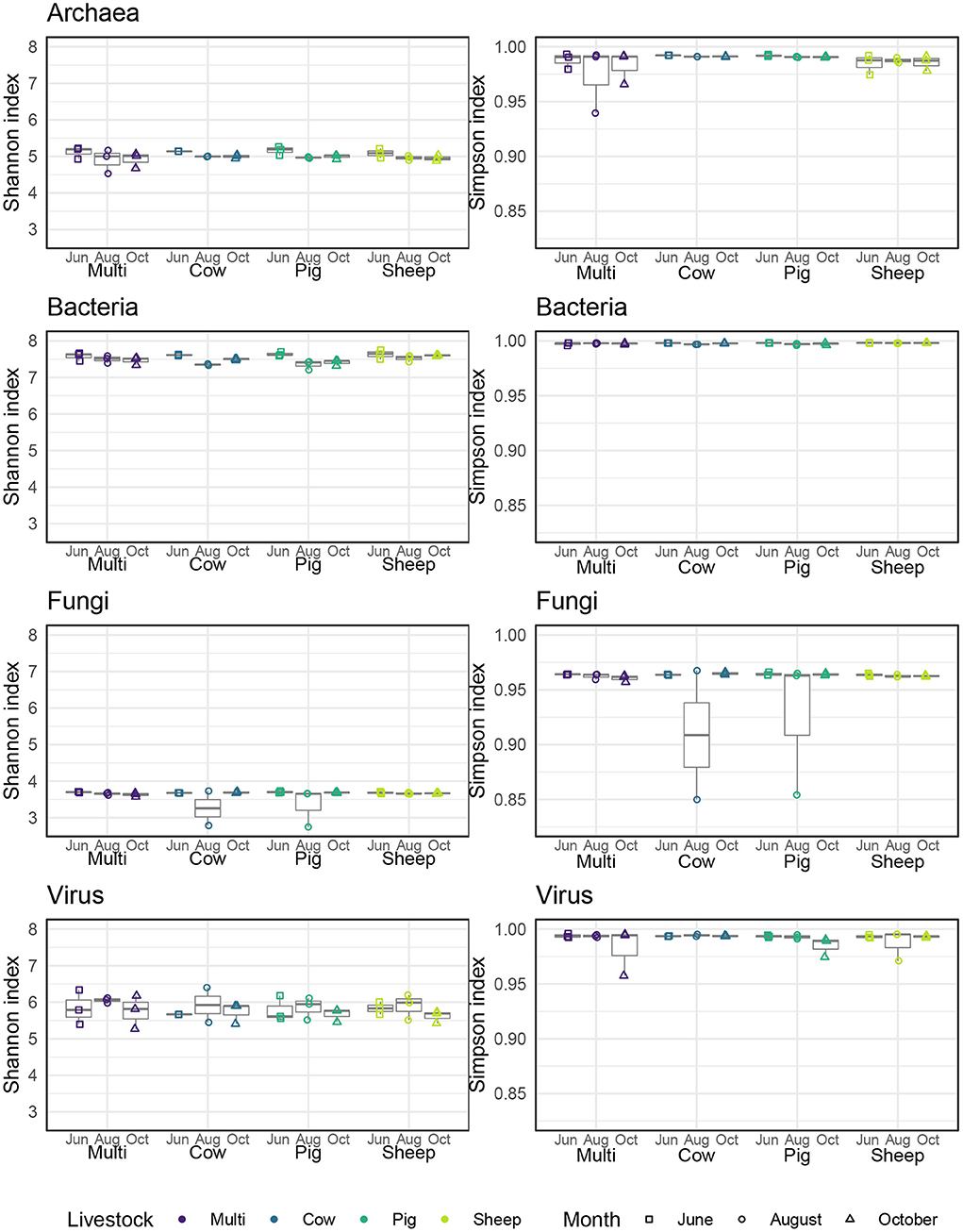
Figure 4. Alpha diversity, as measured by Shannon and Simpson indices, for each microbial kingdom across livestock grazing treatments and months.
Taxonomic composition
Across all months and grazing treatments, archaeal generic composition was characterized by high relative abundance of Halorubrum, Halosimplex, and Haloterrigena (Figure 5). Samples collected from multi-species and sheep paddocks also tended to have greater relative abundance of Candidatus Nitrosocosmicus. One multi-species paddock in June was dominated by Methanosarcina. For bacteria, Streptomyces, Bradyrhizobium, and Pseudomonas had the greatest relative abundance across all samples. Since some Pseudomonas spp. have plant growth-promoting and disease-suppressive qualities, we further investigated this group at the species level. Of the pseudomonads that could be identified at species level, the most abundant were P. stutzeri (8.6% of all Pseudomonas spp.), P. putida (7.4%), and P. fluorescens (6.2%). There was also a notable increase in Nocardioides relative abundance in one pig paddock in August. Fusarium and Pyricularia had the greatest relative abundance of all fungal genera identified in this study. Pyricularia was evenly distributed across samples, for the most part, whereas Fusarium had highest relative abundance in two August samples, which were collected from a cow and a pig paddock. Viral composition was more variable than other kingdoms. Pandoravirus had the highest relative abundance (40.7%) and tended to dominate all samples, regardless of Month or Livestock grazing treatment. Also abundant across most samples were Timquatrovirus (13.0%), Fromanvirus (7.8%) and Mimivirus (7.4%). Perisivirus had high relative abundance in one sheep paddock in August, one multi-species paddock in October, and, to a lesser degree, the same multi-species paddock in August. Also of note, Samistivirus was highly abundant in all three pig paddocks in October.
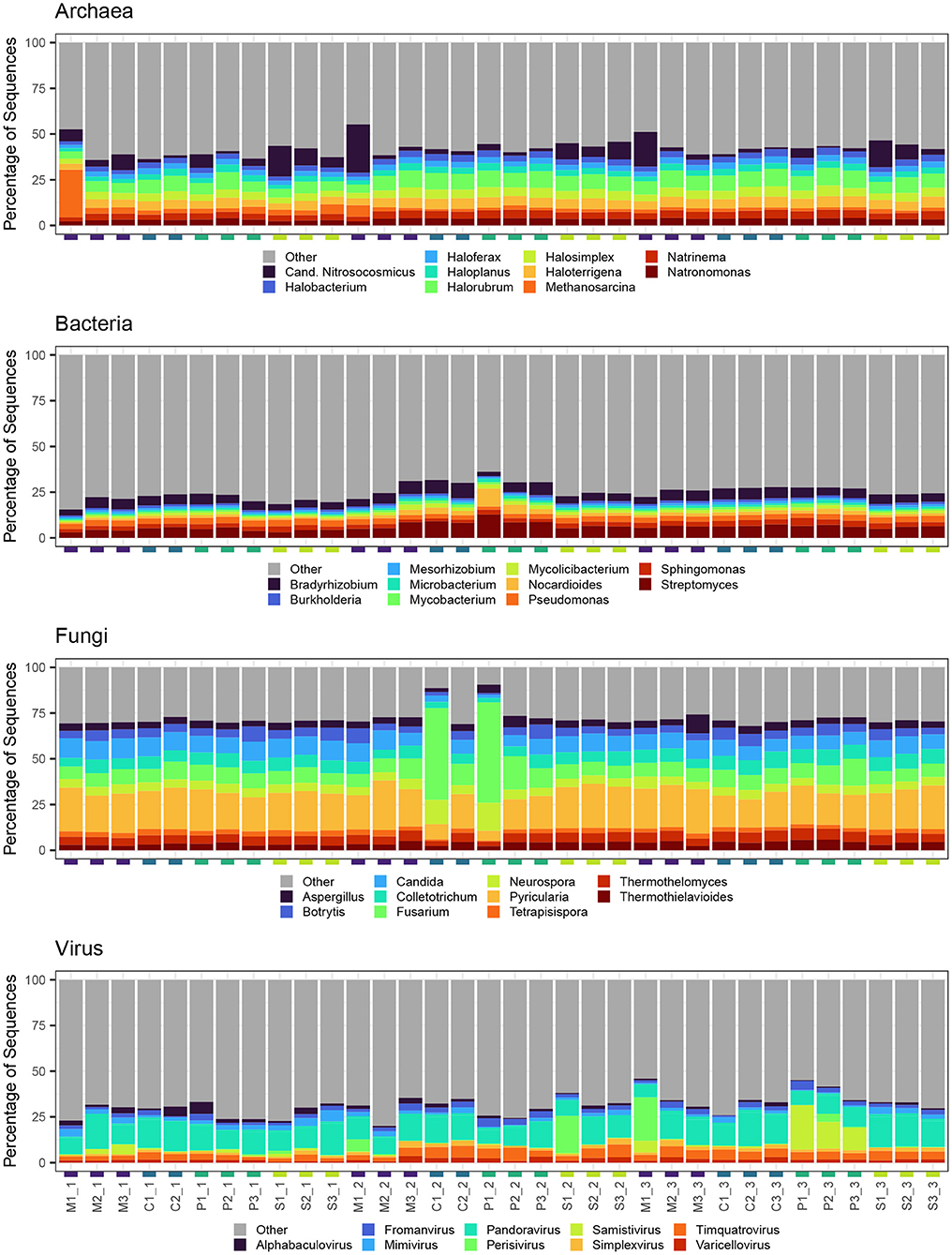
Figure 5. Top 10 most abundant genera detected in each soil sample for each microbial kingdom. All other genera were aggregated into category “Other.”
Beta diversity
We used the Morisita-Horn dissimilarity index to examine the degree of differentiation among individual samples (i.e., beta diversity). Community differentiation for all microbial kingdoms was significantly associated with both grazing treatment (Figure 6; PERMANOVA: Archaea: R2 = 0.18, P < 0.005; Bacteria: R2 = 0.23, P < 0.005; Fungi: R2 = 0.25, P = 0.01; Virus: R2 = 0.12, P < 0.005) and month (Archaea: R2 = 0.33, P < 0.005; Bacteria: R2 = 0.47, P < 0.005; Fungi: R2 = 0.24, P < 0.005; Virus: R2 = 0.09, P < 0.005). For archaea and bacteria, month had a greater influence than grazing treatment, whereas the opposite was true for fungi and viruses.
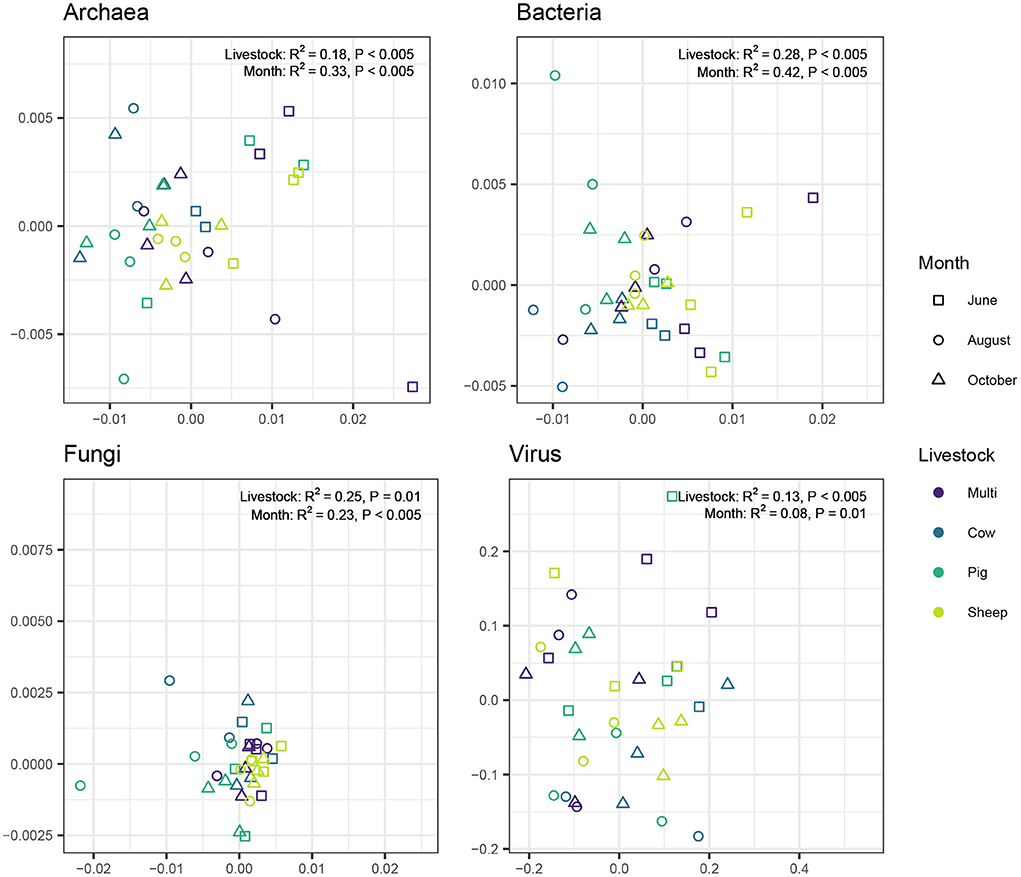
Figure 6. Principal coordinate analysis visualizing compositional similarity of archaeal, bacterial, fungal, and viral communities, based on Morisita-Horn distances. Point color indicates livestock grazing treatment and shape indicates sampling month. PERMANOVA test results are shown near the top left corner of each plot.
We used negative binomial GLMs on variance-stabilizing transformed raw counts to identify individual taxa responsible for driving observed beta diversity patterns. Fewer differentially abundant taxa were identified in the comparison between sheep and multi-species paddocks than in comparisons between other monospecies and multi-species paddocks (Figure 7). In fact, a member of bacterial genus Bradyrhizobium was the sole microbial taxa differentiating sheep paddocks from multi-species paddocks. Patterns for other pair-wise comparisons were more apparent. Notably, methanogenic archaeal taxa, such as Methanosarcina spp., were enriched in multi-species paddocks in comparison to both cow and pig paddocks. The only non-methanogenic archaeal taxa identified as differentially abundant was Candidatus Nitrocosmicus oleophilus, which was enriched in multi-species paddocks when compared with cow paddocks.
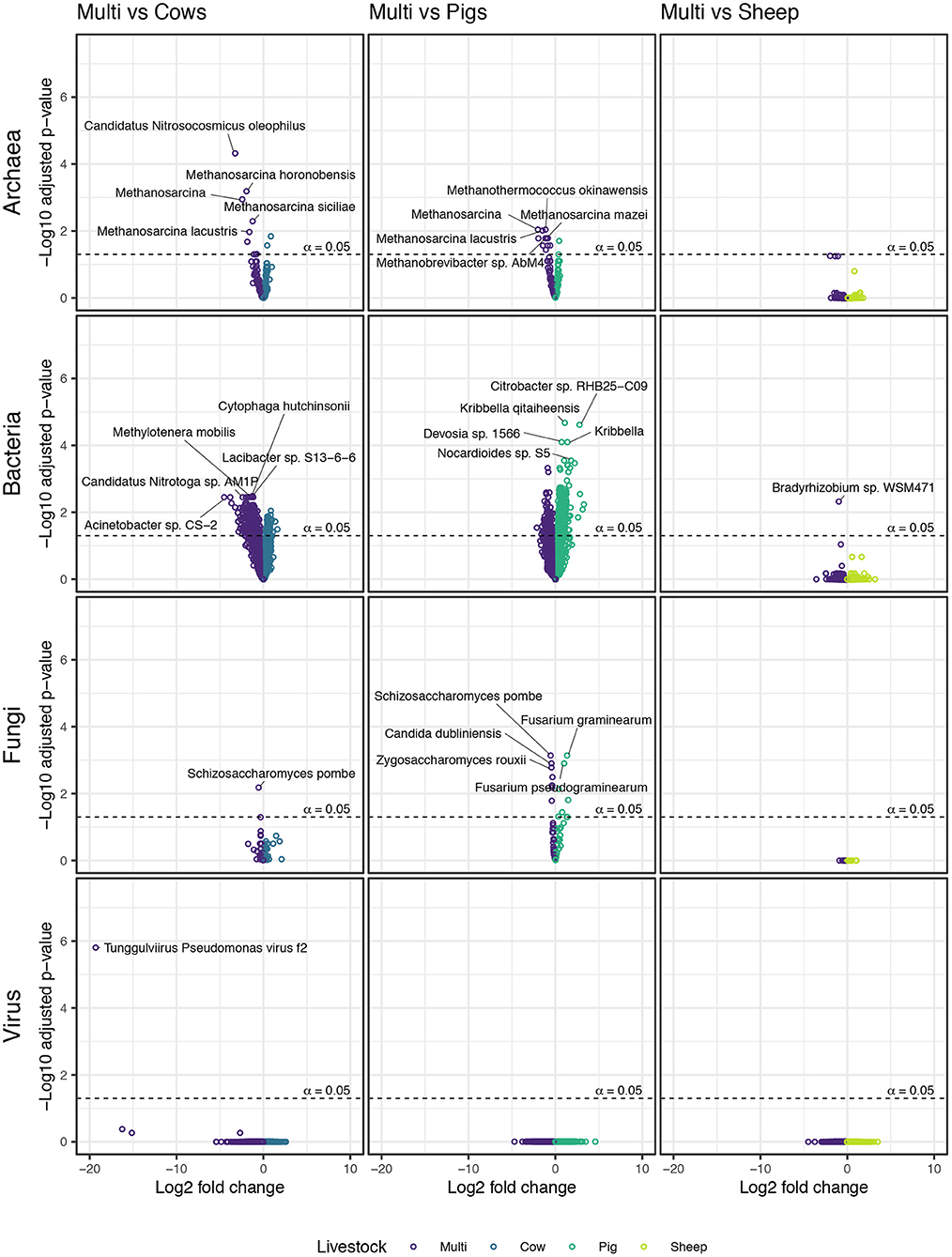
Figure 7. Taxa from each microbial kingdom that were enriched in either monospecies or multi-species paddocks.
A large number of bacteria were enriched in cow and pig paddocks compared with multi-species paddocks, and vice versa (see Supplementary material). Over twice as many bacterial taxa were enriched in multi-species paddocks (364) as in cow paddocks (165). In contrast, over four times as many taxa were enriched in pig paddocks (676) as in multi-species paddocks (160). Many (24) members of the genus Streptomyces were enriched in both cow and pig paddocks, while members of Bacteroides (6), Polaribacter (4), and Flavobacterium (4) were prevalent among taxa enriched in multi-species paddocks.
Schizosaccharomyces pombe was the only differentially abundant fungal taxon identified in the comparison between cow and multi-species paddocks; it was enriched in the multi-species paddocks. Comparing pig paddocks with multi-species paddocks revealed several species of Fusarium that were enriched in the pig paddocks, as well as Pochonia chlamydosporia, Thermothielavioides terrestris, and an unidentified member of order Hypocreales, the same order to which Fusarium belongs. In the same comparison, multi-species paddocks were enriched in Pyricularia pennisetigena, P. grisea, fission yeast S. pombe, and several genera belonging to the budding yeast family Saccharomycetaceae.
Lytic phage Tunggulvirus Pseudomonas virus ϕ2 was the only differentially abundant virus identified in any pair-wise comparison. It was enriched in multi-species paddocks, in comparison to cow paddocks. It is a dsDNA phage whose host is Pseudomonas fluorescens SBW25, a plant growth-promoting rhizobacterium.
Antibiotic resistance potential
Metagenome coassembly by grazing treatment and month recovered 4,355,870 contigs and 140,533 predicted genes associated with antibiotic resistance, using the ResFams curated database (Gibson et al., 2015). Raw counts of ARGs ranged from 1,712–20,039, and numbers of unique ARGs ranged from 39–61. The minimum and maximum abundances of both total and unique ARGs occurred in cow paddocks; multi-species paddocks tended to have a consistent, and relatively high, abundance of total and unique ARGs (Figures 8A,B). Dominant resistance mechanisms included ABC transporters, MFS transporters, β-lactamases, acetyltransferases, and aminotransferases (Figure 8C). Notably, pig and sheep paddocks in August had a similar pattern of lower relative abundance of β-lactamase and mechanisms classified as “other efflux” and higher relative abundance of MFS transporters and gene modulating resistance genes. Despite these intriguing examples, due to the low sample sizes, no statistically significant distribution patterns were found for specific ARGs, either across grazing treatments or months.
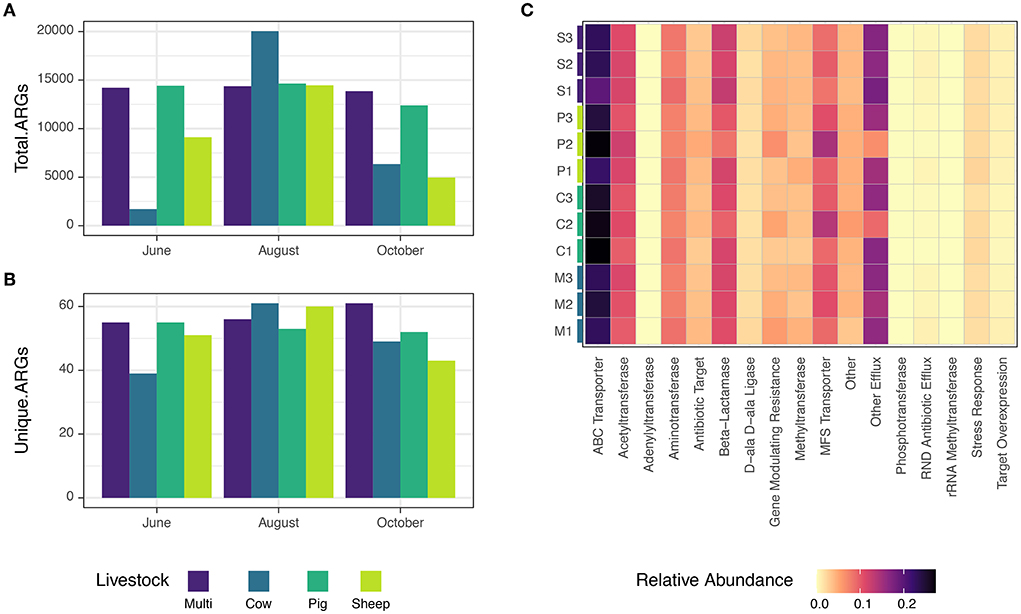
Figure 8. (A) Abundance of total and (B) unique ARGs observed for each grazing treatment and timepoint. (C) Distribution of antibiotic resistance mechanisms in this study, colored by relative abundance.
Discussion
Overview
This study used shallow shotgun metagenomics to characterize soil microbial communities in paddocks rotationally grazed by either a single livestock species (cow, pig, sheep) or by all three species in series over a 5-month period. Numbers of bacterial reads and taxa identified far outnumbered those of other kingdoms, even combined, mirroring findings from prior studies (Fierer et al., 2012; Mendes et al., 2015; Khan et al., 2020). In this study, Proteobacteria was the most abundant bacterial phylum, followed by Actinobacteria, Bacteroidetes, and Firmicutes. Other surveys of soil metagenomes have found similar dominance of these bacterial phyla (Duan et al., 2019; Khan et al., 2020). However, contrary to previous soil surveys, we found that members of phylum Euryarchaeota, rather than Thaumarchaeota, dominated the archaeal community (Bates et al., 2011; Fierer et al., 2012). Also in contrast to other soil virome studies (e.g., Khan et al., 2020; Santos-Medellin et al., 2021), we did not find high relative abundances of the 'tailed' viral families Myoviridae (0.2% in this study) or Podoviridae (0.004%), although Siphoviridae (another tailed viral family) comprised a dominant fraction (39.7%) of all identified viral sequences.
Grazing treatment had differing effects on α and β diversities
In general, we observed little to no effect of livestock grazing treatments on alpha diversity in this study for any microbial kingdom, although we did find an association with sampling month for archaea and bacteria only. Like other studies of soil bacterial communities conducted at a similar latitude in the US [Montana (Ishaq et al., 2020), Michigan (Lauber et al., 2013)], soil samples collected in early summer (June) had greater α-diversity than samples collected in late summer (August). This seasonal difference in α-diversity may be explained by soil temperature, soil moisture, and/or senescence of pasture forage plants, all of which might be expected to affect microbial communities (Waldrop and Firestone, 2006). The linkage between microbial alpha diversity and soil health remains an active area of research. More diverse communities, when combined with high biomass, may be better able to exploit soil resources and provide greater contributions to ecosystem services (Ferris and Tuomisto, 2015), although it may not always be desirable in all contexts (Fierer et al., 2021).
In contrast, beta diversity was strongly influenced by both grazing treatment and month for all microbial kingdoms, which also agrees with other studies showing that spatial and/or management factors tend to have more impact on community composition (Lauber et al., 2013; Ishaq et al., 2020). This finding supports our initial hypothesis that MSRG would result in dissimilar community structure, compared with monospecies-grazed paddocks.
Soil microbial community composition across grazing treatments
Archaea
The most abundant archaeal genera observed in this study belong to family Halobacteriaceae (Supplementary Figure S4), which are generally characterized by high salt requirements (Oren, 2014). Based on physicochemical assays performed on the October bulk soil samples, soils in this study had relatively low sodium (31–42 ppm) and soluble salt concentrations (0–0 mmhos/cm). However, some genera, such as Halorubrum have also been found on pig farms (Bønløkke et al., 2019) and in pig and calf intestines (Zhou et al., 2014; Deng et al., 2021).
While Halobacteriaceae were relatively abundant in this study, their distribution across paddocks did not vary significantly by grazing treatment or sampling month. Taxa that were more rare, on the other hand, did show an effect of grazing treatment. In particular, several members of archaeal genus Methanosarcina were enriched in multi-species paddocks in comparison to both cow and pig paddocks. Methanosarcina spp. are methane-producing anaerobic archaea that belong to the Euryarchaeota (Buan, 2018). Methanosarcina spp. have been isolated from pasture soils (Radl et al., 2007), cattle manure (Gattinger et al., 2007), and ruminant guts (Jeyanathan et al., 2011; Snelling et al., 2014). One explanation for enrichment of these methanogens in multi-species paddocks compared with cow and pig, but not sheep, paddocks may be that sheep in this study harbored greater abundance of Methanosarcina in their guts than the other livestock species. Another possibility is that soil samples from sheep and multi-species paddocks may have been inadvertently collected near sheep feces, as the fecal matter is much smaller and more broadly distributed than either cow or pig feces. The only non-methanogenic archaeal taxa identified as differentially abundant was Candidatus Nitrocosmicus oleophilus, which was enriched in multi-species paddocks when compared with cow paddocks. This taxon is a member of archaeal phylum Thaumarchaeota and is an ammonia-oxidizing archaea that exhibits plant-growth promoting potential (Jung et al., 2016; Song et al., 2019).
Bacteria
Streptomyces was the most abundant group of bacteria observed in this study, and members of this genus were overrepresented among taxa identified as enriched in cow and pig paddocks compared with multi-species paddocks (Supplementary material). Streptomyces belong to phylum Actinobacteria, which are ubiquitous in soil, accounting for 20–30% of soil microbial communities in spring and summer (Hiltner and Störmer, 1903; Barka et al., 2016) at densities of 106–109 cells per gram of soil (Goodfellow and Williams, 1983). Streptomyces are well-known for their production of vast numbers and varieties of antibiotic compounds (Hopwood, 2007). One previous study found that amending soil with lignin or cellulose led to growth in Streptomyces populations, while amending with glucose or a combination did not (Schlatter et al., 2009). We speculate that Streptomyces was enriched in cow and pig paddocks due to the necessity of supplemental feeding for these livestock groups. Cows received baled grassy alfalfa hay during late summer and fall only, while pigs were continuously fed a mixture of milled, rolled, and whole grains and seeds, third-cutting alfalfa, and non-GMO okara during the study. We also found that a very large number of bacterial taxa were enriched in pig paddocks compared with multi-species paddocks, which may also be due to the diversity of supplemental feed provided.
Genera Bradyrhizobium, a common rhizosphere-associated bacterium capable of fixing nitrogen, and Pseudomonas were also highly abundant across all samples. Within this genus, P. stutzeri, P. putida, and P. fluorescens were the most abundant species. Some pseudomonads, such as P. fluorescens, can help promote plant growth by competing with other microorganisms, producing antimicrobial compounds that inhibit pathogens, and inducing resistance mechanisms in the plant (Jackson et al., 2005; Couillerot et al., 2009).
Fungi
Fungal communities overall contained relatively high abundances of Pyricularia species, primarily P. pennisetigena and P. grisea. In the comparison between pig and multi-species paddocks, both P. pennisetigena and P. grisea, as well as fission yeast Schizosaccharomyces pombe and several members of budding yeast family Sacharomycetaceae were enriched in the multi-species paddocks, or depauperate in pig paddocks. Pyricularia are known to cause blast disease in various grasses. Pearl millet (Pennisetum glaucum) is a natural host of P. pennisetigena, though it can also infect barley and wheat (Reges et al., 2016); all of these grains were present in the pig feed and may have begun to establish in the pasture. Other possible explanations for the differential abundance of these taxa could be competition with other organisms associated with pigs or a disadvantage in areas where soil is bare and/or overturned due to pig rooting behavior. In support of the former rationale, Streptomyces spp., which were enriched in pig paddocks, can inhibit blast disease caused by Pyricularia spp. (Law et al., 2017).
Members of Fusarium, including F. graminearum and F. pseudograminearum were also dominant members of fungal communities in this study, particularly in pig paddocks. Fusarium spp. have been found in high relative abundances in cow and sheep feces (Yang, 2019), though we found no evidence that it is found in pig feces. Fusarium spp. can, however, infect cereal grains and produce a mycotoxin to which pigs are susceptible (D'Mello et al., 1999).
Virus
It has been suggested that most viral particles recovered in shotgun metagenomic surveys of soil likely derive from bacteriophages, since most soil bacteria host one or more phages (Williamson et al., 2017). This may have implications for nutrient cycling and carbon sequestration, as dead microbial cells represent a large proportion of soil carbon stores (Kuzyakov and Mason-Jones, 2018), as well as bacterial community structure (Braga et al., 2020). Phages may also play a significant role in antibiotic resistance spread through horizontal gene transfer (Lekunberri et al., 2017; Debroas and Siguret, 2019), although conflicting studies suggest that exploratory prediction of ARGs from phage genomes has overestimated the importance of phages to antibiotic resistance spread (Enault et al., 2017; Billaud et al., 2021).
However, less is known about viruses associated with non-bacterial hosts. Interestingly, the most abundant viral taxa observed across all samples in this study belonged to genus Pandoravirus (Figure 5), a giant dsDNA (double-stranded DNA) virus associated with Acanthamoeba (Abergel et al., 2015). Mimivirus spp., another giant virus infecting Acanthamoeba hosts, were also prevalent members of the pasture soil virome. Acanthamoeba are ubiquitous and abundant in soils globally; as major bacterial predators, they are important in the soil trophic system and control bacterial abundance and diversity (Geisen et al., 2014). Other viral genera that were abundant across most or all samples included Timquatrovirus and Fromanvirus, both of which are mycobacteriophages of the Siphoviridae family, which have long non-contracting tails. Mycobacteriophage research has been ongoing since the 1940's and viral databases contain a large number of sequences belonging to this group (Hatfull, 2018), which may, in part, explain their relative abundance in this study. We noted that Samistivirus sp., a Streptomyces phage belonging to family Siphoviridae, was highly abundant in all pig paddocks in October, whereas Perisivirus, a Brucella phage belonging to family Podoviridae, was abundant only in one sheep paddock in August and one multi-species paddock in October. Brucella spp. are known to infect livestock and are responsible for premature abortions in ruminants (Cicha et al., 2020).
The sole viral taxa identified as differentially abundant was Tunggulvirus Pseudomonas virus ϕ-2, which was enriched in multi-species paddocks in comparison to cow paddocks. The bacterial host for this dsDNA phage is Pseudomonas fluorescens, some strains of which contribute to disease-suppressive qualities and plant growth-promotion (Couillerot et al., 2009).
Antibiotic resistance elements were common across all treatments and months
Livestock manure has been highlighted as a key point of antibiotic resistance spread, given the extensive application antibiotic compounds in industrial agricultural operations (Lima et al., 2020). In this study, ARGs were found across all grazing treatments and sampling months, despite the putative lack of antibiotic contamination. We conjecture that the presence of ARGs may instead be related to the relative abundance of Streptomyces and other actinomycetes, including Microbacterium, Mycobacterium, and Nocardioides in these soils. Most clinically relevant antibiotics currently in use have come from actinomycetes, particularly Streptomyces spp. (Barka et al., 2016), which employ various resistance mechanisms, such as antibiotic modification using transferases (e.g., aminotransferase, β-lactamase, phosphotransferase) to avoid self-injury from the antibiotic compounds they so copiously produce (Peterson and Kaur, 2018). Supporting this hypothesis, a recent study analyzing soils collected directly under manure piles from various livestock species found a correlation between levels of antibiotic resistance genes and abundance of actinomycetes, including Streptomyces (Duan et al., 2019). In that study, ARG abundance was also higher on pig farms than cattle farms; due to our constrained number of samples and lack of absolute abundance quantification, we were not able to perform a similar test here. Additionally, other research on organic and conventional farm soils has shown that Pseudomonas spp., which were also abundant in our study, are largely dependent on efflux mechanisms for antibiotic resistance, especially RND transporters (Armalytė et al., 2019). The RND efflux system is capable of exporting a wide range of antibiotic compounds, as well as other toxins (Poole, 2004).
In our study, genes encoding ABC efflux pumps, ACR transport, and MFS transporters were most common, followed by a variety of transferases. Despite the abundance of these and other resistance mechanisms, it is unclear whether antibiotic resistance is their primary role or whether they are involved in more basic housekeeping and/or communication functions (Peterson and Kaur, 2018). For instance, vancomycin-resistance genes have been identified in bacterial DNA recovered from ancient permafrost sediments (D'Costa, 2011; Van Goethem et al., 2018), indicating antibiotic production and resistance dynamics in soil have a long history that predates large-scale antibiotic use by humans. Antibiotic compounds have long been hypothesized to provide a competitive advantage against other bacteria competing for resources, but may also act as a form of communication when produced in sub-inhibitory amounts (Aminov, 2009).
Future directions and limitations of the study
Despite the importance of microbial-mediated processes to agricultural productivity and environmental quality, using microbial indicators of soil health remains fraught with technical and conceptual challenges, such as lack of connection to actionable management practices and context-dependent definitions of “good” and “bad” soil microbes (Bünemann et al., 2018; Fierer et al., 2021). Fierer et al. (2021) have recently addressed these issues and provided a path forward for future research, rooted in understanding methodological and interpretability limitations of current microbiological analyses. This study contributes valuable knowledge about soil microbial communities in grazing lands, which will help build the knowledge base needed to make future microbial assessments more actionable. Nonetheless, there were several limitations to this work.
As a pilot study, this project encompassed only a single grazing season and, thus, effects of grazing management must be interpreted with caution. Soil microbial communities can be dynamic from year to year (Waldrop and Firestone, 2006) and patterns associated with different grazing regimes could become more apparent with longer-term implementation. Additionally, we used a cost-effective shallow sequencing methodology to achieve a broad characterization of the most abundant taxa from each microbial kingdom being mindful that many rare taxa would not be recovered. Deeper sequencing in future studies could reveal further differentiation of community structure. Similarly, it is likely that we did not exhaustively recover all ARGs present in the samples with our shallow shotgun metagenomics sequencing approach, as a minimum of 80 million reads per sample have been suggested to amply characterize soil metagenomic ARG content (Gweon et al., 2019). Despite this limitation, we expect that the broad patterns observed in this study reflect genuine microbial taxonomic and ARG compositions. We also caution that absolute abundance cannot be inferred from our data; future farm soil studies of archaea using quantitative measurement methods would be valuable.
Lastly, due to factors beyond control (water rights, drought), only part of the pasture was irrigated during the study, possibly contributing to microbial community variability. In particular, the cow-only paddocks and one multi-species paddock at the southern end of the property received little to no irrigation and were extremely dry during the August sampling period. Despite the low soil moisture, these two paddocks generally did not appear to be extreme outliers in either alpha or beta diversity, with the possible exception of fungal alpha diversity for one cow-only paddock in August (Figure 4), though we note that one of the irrigated pig paddocks had a similar alpha diversity value. This highlights some of the challenges involved with conducting rigorous empirical research in the context of an active commercial livestock operation.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA830987.
Author contributions
GM, LD, and TG contributed to conception and design of the study. GM collected the samples and analyzed the data and wrote the manuscript. LD prepared genomic samples for sequencing. All authors contributed to interpretation of results and manuscript revisions, read, and approved the final submitted version.
Funding
This material is based upon work that was supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, under award number 2018-38640-28418 through the Western Sustainable Agriculture Research and Education program under sub award project number G269-20-W7900. USDA is an equal opportunity employer and service provider.
Acknowledgments
We would like to acknowledge Kevin Van Den Wymelenberg for providing physical laboratory space for this work and members of his lab, especially Patrick Horve and Georgia Crone, for assisting with DNA extraction and library prep.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the U.S. Department of Agriculture.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsufs.2022.926824/full#supplementary-material
References
Abergel, C., Legendre, M., and Claverie, J.-M. (2015). The rapidly expanding universe of giant viruses: mimivirus, pandoravirus, pithovirus and mollivirus. FEMS Microbiol. Rev. 39, 779–796. doi: 10.1093/femsre/fuv037
Acharya, M., Ashworth, A. J., Yang, Y., Burke, J. M., Lee, J. A., and Sharma Acharya, R. (2021). Soil microbial diversity in organic and non-organic pasture systems. PeerJ. 9, e11184. doi: 10.7717/peerj.11184
Aislabie, J., and Deslippe, J. R. (2013). “Soil microbes and their contribution to soil services,” in Ecosystem Services in New Zealand—Conditions and Trends (Lincoln: Manaaki Whenua Press), 19.
Aminov, R. I. (2009). The role of antibiotics and antibiotic resistance in nature. Environ. Microbiol. 11, 2970–2988. doi: 10.1111/j.1462-2920.2009.01972.x
Anderson, D. M., Fredrickson, E. L., and Estell, R. E. (2012). Managing livestock using animal behavior: Mixed-species stocking and flerds. Animal 6, 1339–1349. doi: 10.1017/S175173111200016X
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Armalytė, J., Skerniškyt e, J., Bakien e, E., Krasauskas, R., Šiugždinienė, R., Kareivien e, V., et al. (2019). Microbial diversity and antimicrobial resistance profile in microbiota from soils of conventional and organic farming systems. Front. Microbiol. 10, 12. doi: 10.3389/fmicb.2019.00892
Barka, E. A., Vatsa, P., Sanchez, L., Gaveau-Vaillant, N., Jacquard, C., Klenk, H.-P., et al. (2016). Taxonomy, physiology, and natural products of actinobacteria. Microbiol. Mol. Biol. Rev. 80, 43. doi: 10.1128/MMBR.00019-15
Bates, S. T., Berg-Lyons, D., Caporaso, J. G., Walters, W. A., Knight, R., and Fierer, N. (2011). Examining the global distribution of dominant archaeal populations in soil. ISME J. 5, 908–917. doi: 10.1038/ismej.2010.171
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Billaud, M., Lamy-Besnier, Q., Lossouarn, J., Moncaut, E., Dion, M. B., Moineau, S., et al. (2021). Analysis of viromes and microbiomes from pig fecal samples reveals that phages and prophages rarely carry antibiotic resistance genes. ISME Commun. 1, 55. doi: 10.1038/s43705-021-00054-8
Bønløkke, J. H., Duchaine, C., Schlünssen, V., Sigsgaard, T., Veillette, M., and Basinas, I. (2019). Archaea and bacteria exposure in danish livestock farmers. Ann. Work Exposures Health 63, 965–974. doi: 10.1093/annweh/wxz058
Braga, L. P. P., Spor, A., Kot, W., Breuil, M.-C., Hansen, L. H., Setubal, J. C., et al. (2020). Impact of phages on soil bacterial communities and nitrogen availability under different assembly scenarios. Microbiome 8, 52. doi: 10.1186/s40168-020-00822-z
Buan, N. R. (2018). Methanogens: Pushing the boundaries of biology. Emerg. Top. Life Sci. 2, 629–646. doi: 10.1042/ETLS20180031
Bünemann, E. K., Bongiorno, G., Bai, Z., Creamer, R. E., De Deyn, G., de Goede, R., et al. (2018). Soil quality–A critical review. Soil Biol. Biochem. 120, 105–125. doi: 10.1016/j.soilbio.2018.01.030
Bushnell, B. (2014). BBMap: A Fast, Accurate, Splice-Aware Aligner. Available online at: https://www.osti.gov/servlets/purl/1241166
Cattonaro, F., Spadotto, A., Radovic, S., and Marroni, F. (2020). Do you cov me? Effect of coverage reduction on metagenome shotgun sequencing studies. F1000Research 7, 1767. doi: 10.12688/f1000research.16804.4
Chao, A., Gotelli, N. J., Hsieh, T. C., Sander, E. L., Ma, K. H., Colwell, R. K., et al. (2014). Rarefaction and extrapolation with Hill numbers: a framework for sampling and estimation in species diversity studies. Ecol. Monogr. 84, 45–67. doi: 10.1890/13-0133.1
Cicha, C., Hedges, J., Novak, I., Snyder, D., Jutila, M., and Wiedenheft, B. (2020). Complete genome sequence of brucella abortus phage EF4, determined using long-read sequencing. Microbiol. Resour. Announ. 9, e00212–e00220. doi: 10.1128/MRA.00212-20
Couillerot, O., Prigent-Combaret, C., Caballero-Mellado, J., and Moënne-Loccoz, Y. (2009). Pseudomonas fluorescens and closely-related fluorescent pseudomonads as biocontrol agents of soil-borne phytopathogens. Lett. Appl. Microbiol. 48, 505–512. doi: 10.1111/j.1472-765X.2009.02566.x
Dabdoub, S. M. (2016). Kraken-Biom: Enabling Interoperative Format Conversion for Kraken Results (Version 1.2).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A., and Callahan, B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226. doi: 10.1186/s40168-018-0605-2
Debroas, D., and Siguret, C. (2019). Viruses as key reservoirs of antibiotic resistance genes in the environment. ISME J. 13, 2856–2867. doi: 10.1038/s41396-019-0478-9
Deng, F., Li, Y., Peng, Y., Wei, X., Wang, X., Howe, S., et al. (2021). The diversity, composition, and metabolic pathways of archaea in Pigs. Animals 11, 2139. doi: 10.3390/ani11072139
Dignam, B. E., O'Callaghan, M., Condron, L. M., Raaijmakers, J. M., Kowalchuk, G. A., and Wakelin, S. A. (2016). Challenges and opportunities in harnessing soil disease suppressiveness for sustainable pasture production. Soil Biol. Biochem. 95, 100–111. doi: 10.1016/j.soilbio.2015.12.006
D'Mello, J., Placinta, C., and Macdonald, A. (1999). Fusarium mycotoxins: a review of global implications for animal health, welfare and productivity. Anim. Feed Sci. Technol. 8, 183–205. doi: 10.1016/S0377-8401(99)00059-0
Duan, M., Gu, J., Wang, X., Li, Y., Zhang, R., Hu, T., et al. (2019). Factors that affect the occurrence and distribution of antibiotic resistance genes in soils from livestock and poultry farms. Ecotoxicol. Environ. Saf. 180, 114–122. doi: 10.1016/j.ecoenv.2019.05.005
Eddy, S. R. (2011). Accelerated Profile HMM Searches. PLoS Comput. Biol. 7, e1002195. doi: 10.1371/journal.pcbi.1002195
Enault, F., Briet, A., Bouteille, L., Roux, S., Sullivan, M. B., and Petit, M.-A. (2017). Phages rarely encode antibiotic resistance genes: a cautionary tale for virome analyses. ISME J. 11, 237–247. doi: 10.1038/ismej.2016.90
Esperschütz, J., Gattinger, A., Mäder, P., Schloter, M., and Fließbach, A. (2007). Response of soil microbial biomass and community structures to conventional and organic farming systems under identical crop rotations: response of soil microbial biomass and community structures. FEMS Microbiol. Ecol. 61, 26–37. doi: 10.1111/j.1574-6941.2007.00318.x
Ferris, H., and Tuomisto, H. (2015). Unearthing the role of biological diversity in soil health. Soil Biol. Biochem. 85, 101–109. doi: 10.1016/j.soilbio.2015.02.037
Fierer, N., Leff, J. W., Adams, B. J., Nielsen, U. N., Bates, S. T., Lauber, C. L., et al. (2012). Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. U.S.A. 109, 21390–21395. doi: 10.1073/pnas.1215210110
Fierer, N., Wood, S. A., and Bueno de Mesquita, C. P. (2021). How microbes can, and cannot, be used to assess soil health. Soil Biol. Biochem. 153, 108111. doi: 10.1016/j.soilbio.2020.108111
Gattinger, A., Höfle, M. G., Schloter, M., Embacher, A., Böhme, F., Munch, J. C., et al. (2007). Traditional cattle manure application determines abundance, diversity and activity of methanogenic Archaea in arable European soil. Environ. Microbiol. 9, 612–624. doi: 10.1111/j.1462-2920.2006.01181.x
Geisen, S., Fiore-Donno, A. M., Walochnik, J., and Bonkowski, M. (2014). Acanthamoeba everywhere: High diversity of Acanthamoeba in soils. Parasitol. Res. 113, 3151–3158. doi: 10.1007/s00436-014-3976-8
Gibson, M. K., Forsberg, K. J., and Dantas, G. (2015). Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 9, 207–216. doi: 10.1038/ismej.2014.106
Goodfellow, M., and Williams, S. T. (1983). Ecology of actinomycetes. Annu. Rev. Microbiol. 37, 189–216. doi: 10.1146/annurev.mi.37.100183.001201
Gweon, H. S., Shaw, L. P., Swann, J., De Maio, N., AbuOun, M., Niehus, R., et al. (2019). The impact of sequencing depth on the inferred taxonomic composition and AMR gene content of metagenomic samples. Environ. Microbiome 14, 7. doi: 10.1186/s40793-019-0347-1
Hatfull, G. F. (2018). Mycobacteriophages. Microbiol. Spectrum 6, 6.5.08. doi: 10.1128/microbiolspec.GPP3-0026-2018
Hillmann, B., Al-Ghalith, G. A., Shields-Cutler, R. R., Zhu, Q., Gohl, D. M., Beckman, K. B., et al. (2018). Evaluating the information content of shallow shotgun metagenomics. mSystems 3, 12. doi: 10.1128/mSystems.00069-18
Hiltner, L., and Störmer, K. (1903). Studien über die Bakterienflora des Acker- bodens, mit besonderer Berücksichtigung ihres Verhaltens nach einer Behandlung mit Schwefelkohlenstoff und nach Brache. Arb Biol Reich- sanst Land-Forstwirtsch 3, 443–545.
Hoelzer, K., Wong, N., Thomas, J., Talkington, K., Jungman, E., and Coukell, A. (2017). Antimicrobial drug use in food-producing animals and associated human health risks: What, and how strong, is the evidence? BMC Vet. Res. 13, 211. doi: 10.1186/s12917-017-1131-3
Hopwood, D. A. (2007). Streptomyces in Nature and Medicine: The Antibiotic Makers. Oxford: Oxford University Press.
Horrigan, L., Lawrence, R. S., and Walker, P. (2002). How sustainable agriculture can address the environmental and human health harms of industrial agriculture. Environ. Health Perspect. 110, 12. doi: 10.1289/ehp.02110445
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119. doi: 10.1186/1471-2105-11-119
Ishaq, S. L., Seipel, T., Yeoman, C. J., and Menalled, F. D. (2020). Soil bacterial communities of wheat vary across the growing season and among dryland farming systems. Geoderma 358, 113989. doi: 10.1016/j.geoderma.2019.113989
Jackson, R. W., Preston, G. M., and Rainey, P. B. (2005). Genetic characterization of Pseudomonas fluorescens SBW25 rsp gene expression in the phytosphere and in vitro. J. Bacteriol. 187, 8477–8488. doi: 10.1128/JB.187.24.8477-8488.2005
Jerrentrup, J. S., Komainda, M., Seither, M., Cuchillo-Hilario, M., Wrage- Mönnig, N., and Isselstein, J. (2020). Diverse swards and mixed-grazing of cattle and sheep for improved productivity. Front. Sustain. Food Syst. 3, 125. doi: 10.3389/fsufs.2019.00125
Jeyanathan, J., Kirs, M., Ronimus, R. S., Hoskin, S. O., and Janssen, P. H. (2011). Methanogen community structure in the rumens of farmed sheep, cattle and red deer fed different diets: Rumen methanogen community. FEMS Microbiol. Ecol. 76, 311–326. doi: 10.1111/j.1574-6941.2011.01056.x
Jost, L., Chao, A., and Chazdon, R. L. (2010). “Compositional similarity and β (beta) diversity,” in Biological Diversity: Frontiers in Measurement and Assessment, eds A. Magurran and B. McGill (Oxford: Oxford University Press), 66–84.
Jung, M.-Y., Hong, H., Madsen, E. L., Islam, M. A., and Rhee, S.-K. (2016). A “Physiologically, genetically, and morphologically novel isolated ammonia-oxidizing archaeon, nitrosocosmicus oleophilus, from terrestrial sediment,” in 2016 International Meeting of the Microbiological Society of Korea (Gwangju).
Khan, M. A. W., Bohannan, B. J. M., Meyer, K. M., Klein, A. M., Nüsslein, K., Grover, J. P., et al. (2020). Amazonian soil metagenomes indicate different physiological strategies of microbial communities in response to land use change. BioRxiv [Preprint]. doi: 10.1101/2020.09.15.299230
Kleppel, G. S. (2020). Do differences in livestock management practices influence environmental impacts? Front. Sustain. Food Syst. 4, 141. doi: 10.3389/fsufs.2020.00141
Kuzyakov, Y., and Mason-Jones, K. (2018). Viruses in soil: Nano-scale undead drivers of microbial life, biogeochemical turnover and ecosystem functions. Soil Biol. Biochem. 127, 305–317. doi: 10.1016/j.soilbio.2018.09.032
Lauber, C. L., Ramirez, K. S., Aanderud, Z., Lennon, J., and Fierer, N. (2013). Temporal variability in soil microbial communities across land-use types. ISME J. 7, 1641–1650. doi: 10.1038/ismej.2013.50
Law, J. W.-F., Ser, H.-L., Khan, T. M., Chuah, L.-H., Pusparajah, P., Chan, K.-G., et al. (2017). The potential of streptomyces as biocontrol agents against the rice blast fungus, Magnaporthe oryzae (Pyricularia oryzae). Front. Microbiol. 8, 3. doi: 10.3389/fmicb.2017.00003
Lehmann, J., Bossio, D. A., Kögel-Knabner, I., and Rillig, M. C. (2020). The concept and future prospects of soil health. Nat. Rev. Earth Environ. 1, 544–553. doi: 10.1038/s43017-020-0080-8
Lei, S., Xu, X., Cheng, Z., Xiong, J., Ma, R., Zhang, L., et al. (2019). Analysis of the community composition and bacterial diversity of the rhizosphere microbiome across different plant taxa. Microbiologyopen 8, e00762. doi: 10.1002/mbo3.762
Lekunberri, I., Subirats, J., Borrego, C. M., and Balcázar, J. L. (2017). Exploring the contribution of bacteriophages to antibiotic resistance. Environ. Pollut. 220, 981–984. doi: 10.1016/j.envpol.2016.11.059
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, S., Deng, Y., Du, X., Feng, K., Wu, Y., He, Q., et al. (2021). Sampling cores and sequencing depths affected the measurement of microbial diversity in soil quadrats. Sci. Total Environ. 767, 144966. doi: 10.1016/j.scitotenv.2021.144966
Lima, T., Domingues, S., and Da Silva, G. J. (2020). Manure as a potential hotspot for antibiotic resistance dissemination by horizontal gene transfer events. Vet. Sci. 7, 110. doi: 10.3390/vetsci7030110
Loeffler, C., Karlsberg, A., Martin, L. S., Eskin, E., Koslicki, D., and Mangul, S. (2020). Improving the usability and comprehensiveness of microbial databases. BMC Biol. 18, 37. doi: 10.1186/s12915-020-0756-z
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. doi: 10.1186/s13059-014-0550-8
Martin, G., Barth, K., Benoit, M., Brock, C., Destruel, M., Dumont, B., et al. (2020). Potential of multi-species livestock farming to improve the sustainability of livestock farms: a review. Agric. Syst. 181, 102821. doi: 10.1016/j.agsy.2020.102821
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. doi: 10.1371/journal.pone.0061217
Mendes, L. W., Tsai, S. M., Navarrete, A. A., de Hollander, M., van Veen, J. A., and Kuramae, E. E. (2015). Soil-borne microbiome: linking diversity to function. Microb. Ecol. 70, 255–265. doi: 10.1007/s00248-014-0559-2
Norris, C. E., Bean, G. M., Cappellazzi, S. B., Cope, M., Greub, K. L., Liptzin, D., et al. (2020). Introducing the North American project to evaluate soil health measurements. Agron J. 112, 3195–3215. doi: 10.1002/agj2.20234
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2020). Vegan: Community Ecology Package. R Package Version 2.5-7. Available online at: https://CRAN.R-project.org/package=vegan
O'Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Oren, A. (2014). “The family halobacteriaceae,” in The Prokaryotes, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin; Heidelberg: Springer Berlin Heidelberg), 41–121.
Pearson, C. J. (2007). Regenerative, semiclosed systems: a priority for twenty-first-century agriculture. Bioscience 57, 409–418. doi: 10.1641/B570506
Peterson, E., and Kaur, P. (2018). Antibiotic resistance mechanisms in bacteria: relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front. Microbiol. 9, 21. doi: 10.3389/fmicb.2018.02928
Poole, K. (2004). Efflux-mediated multiresistance in Gram-negative bacteria. Clin. Microbiol. Infect. 10, 12–26. doi: 10.1111/j.1469-0691.2004.00763.x
Poore, J., and Nemecek, T. (2018). Reducing food's environmental impacts through producers and consumers. Science 360, 987–992. doi: 10.1126/science.aaq0216
Provenza, F. D., Kronberg, S. L., and Gregorini, P. (2019). Is grassfed meat and dairy better for human and environmental health? Front. Nutr. 6, 26. doi: 10.3389/fnut.2019.00026
Radl, V., Gattinger, A., Chroňáková, A., Němcová, A., Čuhel, J., Šimek, M., et al. (2007). Effects of cattle husbandry on abundance and activity of methanogenic archaea in upland soils. ISME J. 1, 443–452. doi: 10.1038/ismej.2007.60
Reges, J. T. d. A., Negrisoli, M. M., Dorigan, A. F., Castroagudín, V. L., Maciel, J. L. N., et al. (2016). Pyricularia pennisetigena and P. zingibericola from invasive grasses infect signal grass, barley and wheat. Pesquisa Agropecuária Tropical 46, 206–214. doi: 10.1590/1983-40632016v4641335
Rhodes, C. J. (2012). Feeding and healing the world: through regenerative agriculture and permaculture. Sci. Prog. 95, 345–446. doi: 10.3184/003685012X13504990668392
Santos-Medellin, C., Zinke, L. A., ter Horst, A. M., Gelardi, D. L., Parikh, S. J., and Emerson, J. B. (2021). Viromes outperform total metagenomes in revealing the spatiotemporal patterns of agricultural soil viral communities. ISME J. 15, 1956–1970. doi: 10.1038/s41396-021-00897-y
Schlatter, D., Fubuh, A., Xiao, K., Hernandez, D., Hobbie, S., and Kinkel, L. (2009). Resource amendments influence density and competitive phenotypes of streptomyces in soil. Microb Ecol. 57, 413–420. doi: 10.1007/s00248-008-9433-4
Schloter, M., Nannipieri, P., Sørensen, S. J., and van Elsas, J. D. (2018). Microbial indicators for soil quality. Biol. Fertil Soils 54, 1–10. doi: 10.1007/s00374-017-1248-3
Schmid, M. W., Hahl, T., van Moorsel, S. J., Wagg, C., De Deyn, G. B., and Schmid, B. (2019). Feedbacks of plant identity and diversity on the diversity and community composition of rhizosphere microbiomes from a long-term biodiversity experiment. Mol. Ecol. 28, 863–878. doi: 10.1111/mec.14987
Schreefel, L., Schulte, R., de Boer, I., Schrijver, A. P., and van Zanten, H. (2020). Regenerative agriculture –the soil is the base. Global Food Security 26, 100404. doi: 10.1016/j.gfs.2020.100404
Sitters, J., and Andriuzzi, W. S. (2019). “Impacts of browsing and grazing ungulates on soil biota and nutrient dynamics,” in The Ecology of Browsing and Grazing II, Vol. 239, eds I. J. Gordon and H. H. T. Prins (Cham: Springer International Publishingxs), 215–236.
Snelling, T. J., Genç, B., McKain, N., Watson, M., Waters, S. M., Creevey, C. J., et al. (2014). Diversity and community composition of methanogenic archaea in the rumen of scottish upland sheep assessed by different methods. PLoS ONE 9, e106491. doi: 10.1371/journal.pone.0106491
Song, G. C., Im, H., Jung, J., Lee, S., Jung, M.-Y., Rhee, S.-K., et al. (2019). Plant growth-promoting archaea trigger induced systemic resistance in Arabidopsis thaliana against Pectobacterium carotovorum and Pseudomonas syringae. Environ. Microbiol. 21, 940–948. doi: 10.1111/1462-2920.14486
Sun, R., Dsouza, M., Gilbert, J. A., Guo, X., Wang, D., Guo, Z., et al. (2016). Fungal community composition in soils subjected to long-term chemical fertilization is most influenced by the type of organic matter: fertilizer organic matter influences soil fungal community. Environ. Microbiol. 18, 5137–5150. doi: 10.1111/1462-2920.13512
Teague, W. R. (2018). Forages and pastures symposium: cover crops in livestock production: whole-system approach: managing grazing to restore soil health and farm livelihoods. J. Anim Sci. 96, 1519–1530. doi: 10.1093/jas/skx060
van der Bom, F., Nunes, I., Raymond, N. S., Hansen, V., Bonnichsen, L., Magid, J., et al. (2018). Long-term fertilisation form, level and duration affect the diversity, structure and functioning of soil microbial communities in the field. Soil Biol. Biochem. 122, 91–103. doi: 10.1016/j.soilbio.2018.04.003
Van Goethem, M. W., Pierneef, R., Bezuidt, O. K. I., Van De Peer, Y., Cowan, D. A., and Makhalanyane, T. P. (2018). A reservoir of ‘historical' antibiotic resistance genes in remote pristine Antarctic soils. Microbiome 6, 40. doi: 10.1186/s40168-018-0424-5
Waldrop, M. P., and Firestone, M. K. (2006). Seasonal dynamics of microbial community composition and function in oak canopy and open grassland soils. Microb Ecol. 52, 470–479. doi: 10.1007/s00248-006-9100-6
Walker, J. W. (1997). “Multispecies grazing: The ecological advantage,” in Proceedings: American Society of Animal Science Western Section, Vol. 48 (New Mexico State University), 7–10.
Weller, D. M., Raaijmakers, J. M., Gardener, B. B. M., and Thomashow, L. S. (2002). Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu. Rev. Phytopathol. 40, 309–348. doi: 10.1146/annurev.phyto.40.030402.110010
Williamson, K. E., Fuhrmann, J. J., Wommack, K. E., and Radosevich, M. (2017). Viruses in soil ecosystems: an unknown quantity within an unexplored territory. Ann. Rev. Virol. 4, 201–219. doi: 10.1146/annurev-virology-101416-041639
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. doi: 10.1186/s13059-019-1891-0
Yang, J. (2019). Do different livestock dwellings on single grassland share similar faecal microbial communities? Appl. Microbiol. Biotechnol. 103, 15. doi: 10.1007/s00253-019-09849-1
Yang, Y., Ashworth, A. J., DeBruyn, J. M., Durso, L. M., Savin, M., Cook, K., et al. (2020). Antimicrobial resistant gene prevalence in soils due to animal manure deposition and long-term pasture management. PeerJ. 8, e10258. doi: 10.7717/peerj.10258
Yang, Y., Ashworth, A. J., DeBruyn, J. M., Willett, C., Durso, L. M., Cook, K., et al. (2019). Soil bacterial biodiversity is driven by long-term pasture management, poultry litter, and cattle manure inputs. PeerJ. 7, e7839. doi: 10.7717/peerj.7839
Keywords: shotgun metagenomics, soil, regenerative agriculture, antibiotic resistance, grazing management, livestock
Citation: Mhuireach GÁ, Dietz L and Gillett T (2022) One or many? Multi-species livestock grazing influences soil microbiome community structure and antibiotic resistance potential. Front. Sustain. Food Syst. 6:926824. doi: 10.3389/fsufs.2022.926824
Received: 23 April 2022; Accepted: 11 July 2022;
Published: 29 July 2022.
Edited by:
Fred Provenza, Utah State University, United StatesReviewed by:
Rick Teague, Texas A&M AgriLife Research, United StatesScott Lloyd Kronberg, Agricultural Research Service (USDA), United States
Copyright © 2022 Mhuireach, Dietz and Gillett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gwynne Á. Mhuireach, gwynhwyf@uoregon.edu